 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of a peptidic Au(I)-metalloamphiphile and its self-assembly into luminescent micelles in water†
Benedict
Kemper
ab,
Yana R.
Hristova
ab,
Sebastian
Tacke
c,
Linda
Stegemann
bd,
Laura S.
van Bezouwen
e,
Marc C. A.
Stuart
e,
Jürgen
Klingauf
c,
Cristian A.
Strassert
bd and
Pol
Besenius
*ab
aOrganic Chemistry Institute, Westfälische Wilhelms-Universität Münster, Corrensstrasse 40, 48149 Münster, Germany. E-mail: p.besenius@uni-muenster.de; Tel: +49 251 83 63928
bCeNTech, Heisenbergstrasse 11, 48149 Münster, Germany
cDepartment of Cellular Biophysics, Institute of Medical Physics and Biophysics, Westfälische Wilhelms-Universität Münster, 48149 Münster, Germany
dPhysikalisches Institut, Westfälische Wilhelms-Universität Münster, Heisenbergstrasse 11, 48149 Münster, Germany
eDepartment of Biophysical Chemistry, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands
First published on 1st July 2014
Abstract
We report a short synthetic route for the preparation of a peptidic Au(I)-metalloamphiphile which, in buffered environments of physiological ionic strength, self-assembles into luminescent micellar nanostructures of 14 nm in diameter.
Molecular self-assembly is a powerful bottom-up approach for the preparation of tailor-made nanomaterials, and offers distinct advantages due to reduced synthetic efforts and its reversibility which ensures error correction.1 Particularly in water, controlling supramolecular interactions has become an attractive feature to prepare nanosized architectures, for example rods, tubes, vesicles or micelles.2 The preparation of supramolecular (bio)organic–inorganic hybrid materials promises to combine the best of two worlds, the predictable self-assembly encoded in the (bio)organic block and the functional properties embedded in the inorganic moiety.3 We hereby report the preparation of peptidic Au(I)-metalloamphiphiles. On the one hand, we were motivated to use amphiphilic peptides because they are known to be reliable supramolecular synthons for the controlled self-assembly in water.2a,f,g,4 On the other hand, water soluble molecular Au(I)-complexes have received a renewed focus in catalysis, luminescence, and medicinal chemistry.5
The self-assembly of luminescent transition metal complexes into nanostructured materials is an effective strategy for the manipulation of their photophysical properties, such as emission wavelength, excited-state lifetime and photochemical stability.3a,6 These parameters are crucial for developments as imaging labels in biomedical applications.7 Molecular Au(I)-complexes are exceptionally appealing due to their ability to form aurophilic interactions, some of the strongest metallophilic interactions known.8 Surprisingly however, the vast majority of luminescent molecular Au(I)-complexes with short interatomic metal⋯metal distances reported is either in the solid state or in solution based on intramolecular aurophilic interactions in polynuclear complexes.8c–e,9 Rare exceptions are the report on metallophilic interactions in organogels by the Aida group10 and very recent work on hydrogels by the Rodriguez and Lima labs.11 Both have shown that in the gel state short Au⋯Au distances in the supramolecular fibres lead to luminescent properties with large Stokes shifts and long-lived emissions, which were assigned to electronic transitions from triplet-excited states.10,11
We aimed to establish a widely applicable synthetic route for equipping amphiphilic peptides with molecular Au(I)-complexes under mild reaction conditions and designed a simple strategy (Scheme 1) using the well-known oligophenylalanine supramolecular synthon as reported by Gazit, Ulijn, Xu and Adams.2f,12 First we prepared the propargyl amine functionalised Au(I)-trisulfonated-triphenylphosphane complex [H2NCH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CAuI(TPPTS)] 4 following a procedure reported by Laguna and coworkers.13 These types of Au(I)-complexes are known to be highly stable and soluble in water5 and were prepared in three steps starting from hydrogen tetrachloroaurate(III) with a 88% yield (see ESI†). Complex 4 was then reacted with the N-hydroxysuccinimide activated fluorenylmethyloxycarbonyl diphenylalanine (Fmoc-Phe-Phe-NHS) 7 to yield the peptidic Au(I)-metalloamphiphile Fmoc-Phe-Phe-HNCH2CCAuI(TPPTS) 1 with an acceptable overall yield of 26% over four steps (Scheme 1). The final reaction was performed under neutral conditions which avoided acidic degradation of the Au(I)-complex or the deprotection of the Fmoc-protecting group in a basic environment. All intermediate compounds were fully characterised by high-resolution mass spectrometry, 1H and 31P NMR spectroscopy.
CAuI(TPPTS)] 4 following a procedure reported by Laguna and coworkers.13 These types of Au(I)-complexes are known to be highly stable and soluble in water5 and were prepared in three steps starting from hydrogen tetrachloroaurate(III) with a 88% yield (see ESI†). Complex 4 was then reacted with the N-hydroxysuccinimide activated fluorenylmethyloxycarbonyl diphenylalanine (Fmoc-Phe-Phe-NHS) 7 to yield the peptidic Au(I)-metalloamphiphile Fmoc-Phe-Phe-HNCH2CCAuI(TPPTS) 1 with an acceptable overall yield of 26% over four steps (Scheme 1). The final reaction was performed under neutral conditions which avoided acidic degradation of the Au(I)-complex or the deprotection of the Fmoc-protecting group in a basic environment. All intermediate compounds were fully characterised by high-resolution mass spectrometry, 1H and 31P NMR spectroscopy.
 | ||
| Scheme 1 Synthesis of the Fmoc-protected, dipeptidic Au(I)-metalloamphiphile 1: (i) DMF, rt; 29% yield after purification with size-exclusion chromatography. | ||
The novel Fmoc-Phe-Phe-Au(I)-metalloamphiphile 1 is highly soluble in aqueous buffers. For comparison, we prepared another new 1,3,5-triaza-7-phosphaadamantane (TPA) functionalised peptidic Au(I)-complex, Ac-Phe-Phe-HNCH2CCAuI(TPA) 12, via ligand exchange of ClAuI(TPA) with Ac-Phe-Phe-HNCH2CCH under basic conditions (see ESI†).13,14 However this complex was not water soluble under neutral conditions and in millimolar concentrations, most likely due to the lower hydrophilicity of the phosphane, since the TPA ligand is only protonated in acidic water below pH 4.5.15 We therefore focussed on investigating the luminescent properties of 1 (66.7 μM) in conventional phosphate buffer (10 mM, pH 7.4), under physiological ionic strength and at room temperature (Fig. 1A, blue curve): excitation at λexc = 340 nm leads to a broad, unstructured luminescence emission band peaking at λem = 520 nm. This is indicative of the formation of nanostructures incorporating short Au⋯Au distances: such a large Stokes shift is typically observed for Au(I)-alkynyl-phosphane complexes with short interatomic metal⋯metal distances, reported for mononuclear Au(I)-complexes in the crystalline state or for polynuclear complexes in solution, which emit from long-lived triplet states.8d,9e,f,11,16
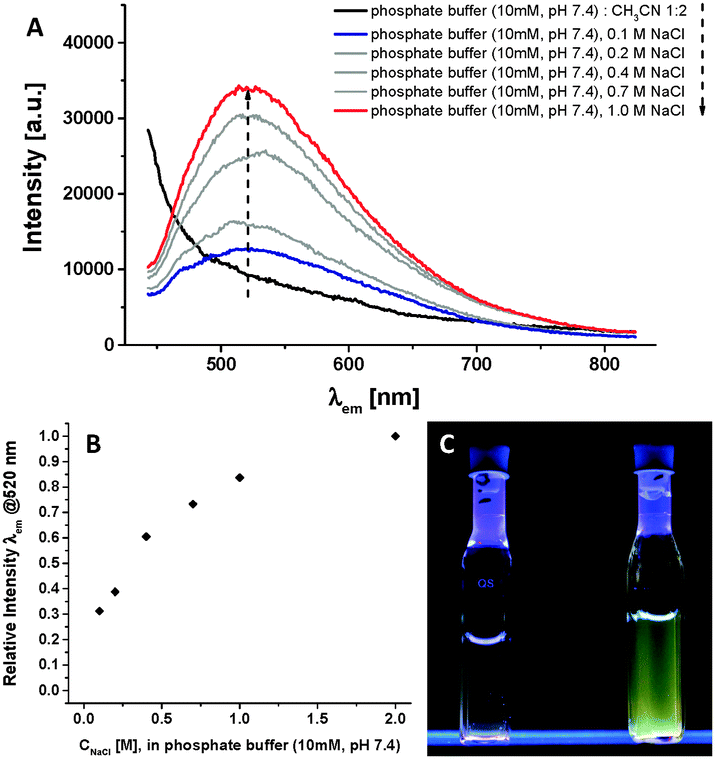 | ||
| Fig. 1 (A) Emission spectra (λexc = 340 nm) of compound 1 (66.7 μM) in 10 mM phosphate buffer (pH 7.4) at 293 K at different ionic strengths: 0.1 M NaCl (blue)–1.0 M NaCl (red) and after addition of acetonitrile to the buffer (black);‡ (B) relative emission intensity at λ = 520 nm as a function of the ionic strength; (C) images of the luminescent Au(I)-metalloamphihile 1 in 10 mM phosphate buffer (pH 7.4) (λexc = 245 nm) with 0.1 M NaCl (left) and 1.0 M NaCl (right). | ||
We obtained further evidence for the presence of nanostructures in buffered water by monitoring the luminescence after the disassembly of the aggregates. The addition of CH3CN is known to disrupt self-assembly in water, because it diminishes the hydrophobic shielding of weak intermolecular interactions.17 At the same monomer concentration of 66.7 μM, switching from the phosphate buffer to a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN:buffer mixture leads to the disappearance of the emission at λem = 520 nm, and appearance of a band at λem < 400 nm. The latter emission originates from the Fmoc unit in the molecularly dissolved amphiphile 1 (see also Fig. S4, ESI†): in Fmoc-protected oligopeptides one generally observes quenching of the organic fluorophore (λem < 400 nm) and sometimes the presence of a weak excimer band (λem = 400–450 nm) in the self-assembled state.18 Hence both features, the disappearance of the lower energy band at λem = 520 nm and the simultaneous appearance of the higher energy band (λem < 400 nm) after disassembly, support the presence of nanostructures in buffer driven by intermolecular interactions and close contacts between the Fmoc groups as well as the Au(I) moieties.
:1 CH3CN:buffer mixture leads to the disappearance of the emission at λem = 520 nm, and appearance of a band at λem < 400 nm. The latter emission originates from the Fmoc unit in the molecularly dissolved amphiphile 1 (see also Fig. S4, ESI†): in Fmoc-protected oligopeptides one generally observes quenching of the organic fluorophore (λem < 400 nm) and sometimes the presence of a weak excimer band (λem = 400–450 nm) in the self-assembled state.18 Hence both features, the disappearance of the lower energy band at λem = 520 nm and the simultaneous appearance of the higher energy band (λem < 400 nm) after disassembly, support the presence of nanostructures in buffer driven by intermolecular interactions and close contacts between the Fmoc groups as well as the Au(I) moieties.
In the self-assembled state, the emission intensity of the band at λem = 520 nm is weak as one would expect for luminescent Au(I)-complexes in water.9f,11 Furthermore, we suspected that the highly charged trisulfonated phosphane ligands on the hydrophilic head group of the metalloamphiphile 1 would hamper the self-assembly due to repulsive electrostatic interactions, and thereby weaken the luminescence. We have referred to systems like these as experiencing frustrated growth,2d,17,19 whereby attractive supramolecular interactions within the hydrophobic block of an amphiphilic peptide are balanced out by repulsive interactions in the hydrophilic periphery. By increasing the ionic strength from 0.1 M NaCl to 1 M NaCl, we observed that the intensity of the emission band at λem = 520 nm increases by a factor of three (Fig. 1A and B). The addition of NaCl simultaneously screens the repulsive Coulombic interactions and increases the hydrophobic effect originating from the apolar and aromatic moieties in the Fmoc-diphenylalanine block of amphiphile 1 and, consequently, stabilises the self-assembly of the highly charged monomer into nanostructures in water.17,19a,d The increase in the emission can then also be observed by naked eye after excitation with a 8 W standard laboratory fluorescent lamp (Fig. 1C). We also determined the luminescence quantum yield Φ = 0.01 (10 mM phosphate buffer, pH 7.4, 1 M NaCl, 293 K). Interestingly, between 0.1 M and 1 M NaCl the emission wavelength and the excited state lifetimes are not affected, thus indicating that rather the aggregation equilibrium but not the morphology of the aggregates or the nature of the excited state are influenced by the abovementioned Coulombic shielding (vide infra).
The long excited state lifetimes indicate that the emission at λem = 520 nm is originated from a triplet state (Fig. S3, ESI†). Indeed, the exponential fits of the time-resolved luminescence decay curves yield coincident values of 1.5 μs ± 0.1 μs for both the low and high ionic strength buffers. As recently highlighted, it is often difficult to unambiguously attribute the emission of Au(I)-alkynyl-phosphane complexes to excited states with a defined character: the broad and structureless emission at λem = 520 nm can be attributed to excited states that are approximately described as a 3[σ(Au–P) → π*(CC)] metal-to-ligand charge-transfer excitation, or as a metal-perturbed intra-ligand excitation 3IL [π → π*(CC)], while Au(I)⋯Au(I) interactions can contribute to the broad emission band as well.8d,9f,20 Altogether the spectroscopic investigations reveal that the aqueous self-assembly of the peptidic Au(I)-metalloamphiphile 1 into nanostructures leads to luminescent properties with large Stokes shifts and long-lived emissions, that can be assigned to triplet-excited states.
Finally, we investigated the morphology of the self-assembled nanostructures of metalloamphiphile 1 using cryogenic transmission electron microscopy (cryo-TEM). We were able to identify 10–17 nm sized spherical objects that are most probably micellar structures (Fig. S5, ESI†). Micelles are known to have a swollen hydrophobic core, which unfortunately reduces contrast in cryo-TEM images and makes their morphological characterisation difficult. We therefore switched to conventional TEM, using uranyl acetate as staining agent. The presence of spherical structures is clearly observed after depositing 2 mg ml−1 solutions of 1 on carbon film coated copper grids (Fig. 2), with an averaged size of 14 nm ± 3.5 nm (Fig. S10B, ESI†). Assuming a length of 3.5 nm for the Au(I)-metalloamphiphile 1, these structures are assigned to be micelles with a weakly packed peptide-based hydrophobic core that is swollen when dispersed in aqueous buffers, thus conforming cryo-TEM investigations. After comparing TEM images obtained from solutions of 1 in buffer and 0.1 M NaCl, with those after adding 1 M NaCl, we noticed that there is no apparent change in the diameter of the micelles (Fig. S6–S10, ESI†). These results corroborate NaCl titrations in luminescence spectroscopy: the increased ionic strength does not lead to more densely packed micelles, since a reduced Au(I)⋯Au(I) distance in supramolecular structures is known to lead to a red shift in the luminescence emission band.9a,d,f The spectroscopic and microscopic investigations therefore strongly suggest that by increasing the ionic strength from 0.1 NaCl to 1 M NaCl in phosphate buffer, the formation of self-assembled micelles in solution becomes more thermodynamically favourable, without affecting the order in and size of the prepared nanostructures (vide supra). Intriguingly, when the ionic strength is kept at a minimum (10 mM phosphate buffer, 0 M NaCl) we observed in cryo-TEM images that the metalloamphiphile 1 self-assembles in large sheet-like aggregates21 (Fig. S11, ESI†). It is known that at very low ionic strength (I < 0.02 M) clustering of counter ions can reduce the effective charge of assemblies in solution22 which in our system is the likely cause for the formation of densely packed 2D sheets. The presence of a closely packed secondary order is furthermore supported by photoluminescence spectroscopy: excitation at λexc = 340 nm leads to a shift of the emission at 0.1 M NaCl from λem = 520 nm to 550 nm at 0 M NaCl (Fig. S12, ESI†). This red shift in the luminescence emission band suggests a reduced intermolecular Au(I)⋯Au(I) distance9a,d,f from the curved micellar structure to a planar sheet-like morphology.
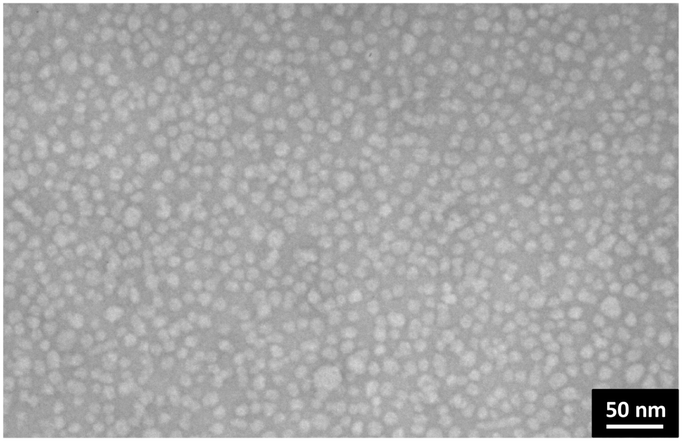 | ||
| Fig. 2 TEM image of the Au(I)-metalloamphiphile 1, deposited on carbon coated grids from a 2 mg ml−1 solution in 10 mM Tris buffer, pH 7.5 and 1 M NaCl (negative staining was performed with 2% w/v uranyl acetate). | ||
We like to point out that unlike the large variety of peptide amphiphiles reported in literature, metalloamphiphile 1 does not form rod-like materials. In intermediate to high ionic strength the triple charges in the phosphane ligand as head group, compared with the small, albeit very hydrophobic Fmoc-Phe-Phe-based peptide chain of the amphiphile, result in a high packing parameter23 which drives the materials into highly soluble micellar structures with sizes of 14 nm. Such small micelles are very rarely observed for peptidic supramolecular materials in water.2a,f,g,4,12 This is therefore a unique example where a new water soluble Au(I)-metalloamphiphile self-assembles in buffered water of physiological ionic strength to form luminescent and well-defined spherical nanoparticles.
In conclusion, we present a facile synthetic route for the preparation of a new peptidic Au(I)-metalloamphiphile, using a nucleophilic water soluble Au(I)-complex H2NCH2CCAuI(TPPTS), and a NHS activated peptide Fmoc-Phe-Phe-NHS. In buffered aqueous environments of medium to high ionic strength (0.1–1 M NaCl), Fmoc-Phe-Phe-HNCH2CCAuI(TPPTS) self-assembles into luminescent micellar nanostructures with an average diameter of 14 nm. In low ionic strength we have observed the formation of densely packed sheet-like morphologies. We assign the luminescent properties to electronic transitions from triplet-excited states due to the large Stokes and excited state life times of 1.5 μs, which are likely to be enhanced due to short Au(I)⋯Au(I) distances in the self-assembled nanostructures. The facile synthetic strategy is fully compatible with peptide protecting group chemistry and allows for the construction of more complex peptidic nanomaterials in water, using our recently reported supramolecular synthons. By adjusting the hydrophilicity and charged character of tailor-made phosphane ligands bound to the metal complex, we aim to position functional Au(I)-complexes into anisotropic nanostructures and exploit applications in bioimaging, catalysis and therapeutics.
We thank U. Malkus and A. Ricker (Institute of Medical Physics and Biophysics, Münster) for performing TEM and Dr M. Peterlechner (Institute of Material Physics, Münster) for preliminary TEM experiments. This work was supported by the DFG (SFB 858, Project B16) [B.K., Y.R.H., P.B.]; P.B. acknowledges the ‘Fonds der Chemischen Industrie’ for a Liebig Fellowship, the ‘Nordrhein-Westfälische Akademie der Wissenschaften und der Künste’ for a Fellowship via the ‘Junges Kolleg’ and COST Action CM1005 (Supramolecular Chemistry in Water). L.S. and C.A.S gratefully acknowledge the DFG for financial support, (SFB-TRR 61, project C07).
Notes and references
- (a) G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS PubMed; (b) J. M. Lehn, Science, 2002, 295, 2400 CrossRef CAS PubMed; (c) K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1304 CrossRef PubMed; (d) R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2010, 10, 14 CrossRef PubMed; (e) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- (a) M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301 CrossRef CAS PubMed; (b) L. Zhang and A. Eisenberg, Science, 1995, 268, 1728 CAS; (c) S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195 CrossRef; (d) K. J. C. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. van Esch, Angew. Chem., Int. Ed., 2004, 43, 1663 CrossRef CAS PubMed; (e) D. M. Vriezema, M. C. Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445 CrossRef CAS PubMed; (f) R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664 RSC; (g) H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1 CrossRef CAS PubMed; (h) R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe and S. Perrier, Chem. Soc. Rev., 2012, 41, 6023 RSC; (i) H. Frisch, J. P. Unsleber, D. Lüdeker, M. Peterlechner, G. Brunklaus, M. Waller and P. Besenius, Angew. Chem., Int. Ed., 2013, 52, 10097 CrossRef CAS PubMed.
- (a) C. A. Strassert, C.-H. Chien, M. D. Galvez Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946 CrossRef CAS PubMed; (b) M. J. Mayoral Munoz and G. Fernandez, Chem. Sci., 2012, 3, 1395 RSC; (c) S. Sengupta, D. Ebeling, S. Patwardhan, X. Zhang, H. von Berlepsch, C. Böttcher, V. Stepanenko, S. Uemura, C. Hentschel, H. Fuchs, F. C. Grozema, L. D. A. Siebbeles, A. R. Holzwarth, L. Chi and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6378 CrossRef CAS PubMed; (d) R. Charvet, Y. Yamamoto, T. Sasaki, J. Kim, K. Kato, M. Takata, A. Saeki, S. Seki and T. Aida, J. Am. Chem. Soc., 2012, 134, 2524 CrossRef CAS PubMed; (e) M. R. Reithofer, K.-H. Chan, A. Lakshmanan, D. H. Lam, A. Mishra, B. Gopalan, M. Joshi, S. Wang and C. A. E. Hauser, Chem. Sci., 2014, 5, 625 RSC.
- (a) J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684 CrossRef CAS PubMed; (b) L. C. Palmer, Y. S. Velichko, M. Olvera de la Cruz and S. I. Stupp, Philos. Trans. R. Soc., A, 2007, 365, 1417 CrossRef CAS PubMed; (c) I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128 CrossRef CAS PubMed; (d) A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37 CrossRef CAS; (e) X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. E. Hauser, S. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480 RSC.
- Y. R. Hristova, B. Kemper and P. Besenius, Tetrahedron, 2013, 69, 10525 CrossRef CAS PubMed.
- (a) K. Wang, M.-a. Haga, H. Monjushiro, M. Akiba and Y. Sasaki, Inorg. Chem., 2000, 39, 4022 CrossRef CAS; (b) P. C. Griffiths, I. A. Fallis, T. Chuenpratoom and R. Watanesk, Adv. Colloid Interface Sci., 2006, 122, 107 CrossRef CAS PubMed; (c) A. Guerrero-Martínez, Y. Vida, D. Domínguez-Gutiérrez, R. Q. Albuquerque and L. De Cola, Inorg. Chem., 2008, 47, 9131 CrossRef PubMed; (d) C. A. Strassert, M. Mauro and L. De Cola, in Advances in Inorganic Chemistry, ed. R. van Eldik and G. Stochel, Academic Press, 2011, vol. 63, p. 47 Search PubMed; (e) M. Mauro, G. De Paoli, M. Otter, D. Donghi, G. D'Alfonso and L. De Cola, Dalton Trans., 2011, 40, 12106 RSC; (f) Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662 CrossRef CAS PubMed.
- (a) V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186 RSC; (b) M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144 RSC.
- (a) H. Schmidbaur, Gold Bull., 2000, 33, 3 CrossRef CAS; (b) P. Pyykkö, Angew. Chem., Int. Ed., 2002, 41, 3573 CrossRef; (c) E. J. Fernandez, A. Laguna and J. M. Lopez-de-Luzuriaga, Dalton Trans., 2007, 1969 RSC; (d) V. W.-W. Yam and E. C.-C. Cheng, Chem. Soc. Rev., 2008, 37, 1806 RSC; (e) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- (a) M. J. Irwin, J. J. Vittal and R. J. Puddephatt, Organometallics, 1997, 16, 3541 CrossRef CAS; (b) J. Barberá, A. Elduque, R. Giménez, F. J. Lahoz, J. A. López, L. A. Oro and J. L. Serrano, Inorg. Chem., 1998, 37, 2960 CrossRef; (c) M. Enomoto, A. Kishimura and T. Aida, J. Am. Chem. Soc., 2001, 123, 5608 CrossRef CAS; (d) S.-K. Yip, E. C.-C. Cheng, L.-H. Yuan, N. Zhu and V. W.-W. Yam, Angew. Chem., Int. Ed., 2004, 43, 4954 CrossRef CAS PubMed; (e) R. J. Puddephatt, Chem. Soc. Rev., 2008, 37, 2012 RSC; (f) J. C. Lima and L. Rodriguez, Chem. Soc. Rev., 2011, 40, 5442 RSC.
- A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2004, 127, 179 CrossRef PubMed.
- (a) R. Gavara, J. Llorca, J. C. Lima and L. Rodriguez, Chem. Commun., 2013, 49, 72 RSC; (b) E. Aguilo, R. Gavara, J. C. Lima, J. Llorca and L. Rodriguez, J. Mater. Chem. C, 2013, 1, 5538 RSC.
- (a) M. Reches and E. Gazit, Science, 2003, 300, 625 CrossRef CAS PubMed; (b) V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611 CrossRef CAS; (c) Z. Yang, G. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038 CrossRef CAS PubMed; (d) D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856 RSC.
- S. Sanz, L. A. Jones, F. Mohr and M. Laguna, Organometallics, 2007, 26, 952 CrossRef CAS.
- E. Vergara, E. Cerrada, A. Casini, O. Zava, M. Laguna and P. J. Dyson, Organometallics, 2010, 29, 2596 CrossRef CAS.
- Z. Assefa, B. G. McBurnett, R. J. Staples, J. P. Fackler Jr., B. Assmann, K. Angermaier and H. Schmidbaur, Inorg. Chem., 1995, 34, 75 CrossRef CAS.
- N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2003, 42, 2586 CrossRef CAS PubMed.
- (a) P. Besenius, G. Portale, P. H. H. Bomans, H. M. Janssen, A. R. A. Palmans and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888 CrossRef CAS PubMed; (b) M. von Gröning, I. de Feijter, M. C. A. Stuart, I. K. Voets and P. Besenius, J. Mater. Chem. B, 2013, 1, 2008 RSC.
- (a) S. Fleming, S. Debnath, P. W. J. M. Frederix, N. T. Hunt and R. V. Ulijn, Biomacromolecules, 2014, 15, 1171–1184 CrossRef CAS PubMed; (b) Y. M. Abul-Haija, S. Roy, P. W. J. M. Frederix, N. Javid, V. Jayawarna and R. V. Ulijn, Small, 2014, 10, 973 CrossRef CAS PubMed.
- (a) H. Dong, S. E. Paramonov, L. Aulisa, E. L. Bakota and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 12468 CrossRef CAS PubMed; (b) F. Versluis, H. R. Marsden and A. Kros, Chem. Soc. Rev., 2010, 39, 3434 RSC; (c) A. Ghosh, M. Haverick, K. Stump, X. Yang, M. F. Tweedle and J. E. Goldberger, J. Am. Chem. Soc., 2012, 134, 3647 CrossRef CAS PubMed; (d) C. Schaefer, I. K. Voets, A. R. A. Palmans, E. W. Meijer, P. van der Schoot and P. Besenius, ACS Macro Lett., 2012, 1, 830 CrossRef CAS.
- (a) D. Li, X. Hong, C.-M. Che, W.-C. Lo and S.-M. Peng, J. Chem. Soc., Dalton Trans., 1993, 2929 RSC; (b) V. W.-W. Yam, S. W.-K. Choi and K.-K. Cheung, Organometallics, 1996, 15, 1734 CrossRef CAS; (c) V. W.-W. Yam, K.-L. Cheung, S.-K. Yip and K.-K. Cheung, J. Organomet. Chem., 2003, 681, 196 CrossRef CAS; (d) M. Ferrer, A. Gutiérrez, L. Rodríguez, O. Rossell, J. C. Lima, M. Font-Bardia and X. Solans, Eur. J. Inorg. Chem., 2008, 2899 CrossRef CAS; (e) X. He, E. C.-C. Cheng, N. Zhu and V. W.-W. Yam, Chem. Commun., 2009, 4016 RSC.
- J. T. van Herpt, M. C. A. Stuart, W. R. Browne and B. L. Feringa, Chem. – Eur. J., 2014, 20, 3077 CrossRef CAS PubMed.
- (a) T. Odijk, Biophys. Chem., 1991, 41, 23 CrossRef CAS; (b) P. van der Schoot, Langmuir, 1997, 13, 4926 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninha, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S12, synthetic procedures and full details about the instrumentation. See DOI: 10.1039/c4cc03868a |
| ‡ The luminescence emission spectra were recorded using a 405 nm filter to filter out the fluorescence from the Fmoc moieties that is generally observed in self-assembled Fmoc-dipeptides.18 |
| This journal is © The Royal Society of Chemistry 2015 |