
SnSe alloy as a promising anode material for Na-ion batteries†
Youngjin
Kim
 a,
Yongil
Kim
a,
Yuwon
Park
a,
Yong Nam
Jo
b,
Young-Jun
Kim
b,
Nam-Soon
Choi
a and
Kyu Tae
Lee
*a
a,
Yongil
Kim
a,
Yuwon
Park
a,
Yong Nam
Jo
b,
Young-Jun
Kim
b,
Nam-Soon
Choi
a and
Kyu Tae
Lee
*a
aSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), 100 Banyeon-ri, Eonyang-eup, Ulju-gun, Ulsan, 689-798, South Korea. E-mail: ktlee@unist.ac.kr; Fax: +82-52-217-3009; Tel: +82-52-217-2930
bAdvanced Batteries Research Center, Korea Electronics Technology Institute (KETI), 68 Yatap-dong, Bundang-gu, Seongnam-si, Gyeonggi-do, 463-816, South Korea
First published on 24th October 2014
Abstract
SnSe alloy is examined for the first time as an anode for Na-ion batteries, and shows excellent electrochemical performance including a high reversible capacity of 707 mA h g−1 and stable cycle performance over 50 cycles. Upon sodiation, SnSe is changed into amorphous NaxSn nanodomains dispersed in crystalline Na2Se, and SnSe is reversibly restored after desodiation.
In recent years, the demand for large scale energy storage devices has increased with the development of electric vehicles (EVs) and the smart grid, and Li-ion batteries are considered promising candidates because of their high energy density.1 However, there is growing concern that lithium resources are insufficient to meet the demands of large scale applications. Moreover, the recent price of lithium raw materials has shown sharp increases. Consequently, Na-ion batteries are an alternative to Li-ion batteries because sodium resources are much more abundant and inexpensive than lithium.2 However, there is a critical obstacle to their development. The energy density of Na-ion batteries is slightly lower than that of Li-ion batteries because the reversible capacity and operating voltage of currently reported electrode materials in Na-ion batteries are lower in comparison. This implies that it is not easy to replace Li-ion batteries with Na-ion batteries because the cost per energy stored ($/Wh) of Na-ion batteries does not provide much advantage.3 Therefore, new electrode materials having higher reversible capacities are necessary if one is to increase the energy density of Na-ion batteries.
Recently, there have been notable achievements in the development of high-capacity anode materials such as phosphorus,4 phosphides,5 oxides,6 sulfides,7 selenides8 and alloy-based materials such as Sn9 and Sb.10 In particular, Sn-based materials have been a focus as promising anode materials. Sn easily reacts with group VA elements (pnictogens including N, P, and Sb) and group VIA elements (chalcogens including O, S, and Se) to form binary compounds such as pnictogenides and chalcogenides. Among these binary compounds, SnSb,11 SnO2,12 SnS2,13 and Sn4P35a,b have been examined, and show promising electrochemical performance via conversion reactions forming NaαSn and NaβX (X = pnictogens or chalcogens) during sodiation. In the case of tin nitride, it was calculated that the conversion reaction with Na is not thermodynamically allowed.14 Tin selenide, however, has not been examined as an anode for Na-ion batteries. SnSe is a p-type semiconductor (band gap of 0.9–1.3 eV) with an orthorhombic crystal structure. In SnSe, Se is more electronegative than Sn whose oxidation state is +2.15
In this study, SnSe was for the first time examined as an anode material for Na-ion batteries, although a few studies have reported the electrochemical reaction mechanism of SnSe for Li-ion batteries.16 SnSe showed excellent electrochemical performance for Na-ion batteries, including a high reversible capacity of 707 mA h g−1 and very stable cycle performance with negligible capacity fading over 50 cycles.
SnSe/C composite powders were obtained via facile ball milling in an Ar atmosphere using tin selenide and Super P carbon with a weight ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. Fig. 1a shows the X-ray diffraction (XRD) pattern of as-prepared SnSe/C powders; no impurities were observed. The space group of the SnSe crystal structure is Pnma, and SnSe crystallizes in the orthorhombic lattice. A Raman spectrum of the SnSe/C composite shows that Super P carbon in the composite is a disordered carbon (Fig. 1b), where the two peaks around 1350 and 1580 cm−1 are designated as D and G bands, respectively. Fig. 1c shows a field emission scanning electron microscopy (FE-SEM) image of the SnSe/C composite powders. Primary particles with sizes tens of nm were aggregated to form secondary particles with sizes hundreds of nm. Scanning transmission electron microscopy (STEM) and energy dispersive X-ray spectroscopy (EDS) mapping images of the SnSe/C composite indicate that SnSe and carbon are homogeneously mixed at the nanoscale level (Fig. 1d).
:3. Fig. 1a shows the X-ray diffraction (XRD) pattern of as-prepared SnSe/C powders; no impurities were observed. The space group of the SnSe crystal structure is Pnma, and SnSe crystallizes in the orthorhombic lattice. A Raman spectrum of the SnSe/C composite shows that Super P carbon in the composite is a disordered carbon (Fig. 1b), where the two peaks around 1350 and 1580 cm−1 are designated as D and G bands, respectively. Fig. 1c shows a field emission scanning electron microscopy (FE-SEM) image of the SnSe/C composite powders. Primary particles with sizes tens of nm were aggregated to form secondary particles with sizes hundreds of nm. Scanning transmission electron microscopy (STEM) and energy dispersive X-ray spectroscopy (EDS) mapping images of the SnSe/C composite indicate that SnSe and carbon are homogeneously mixed at the nanoscale level (Fig. 1d).
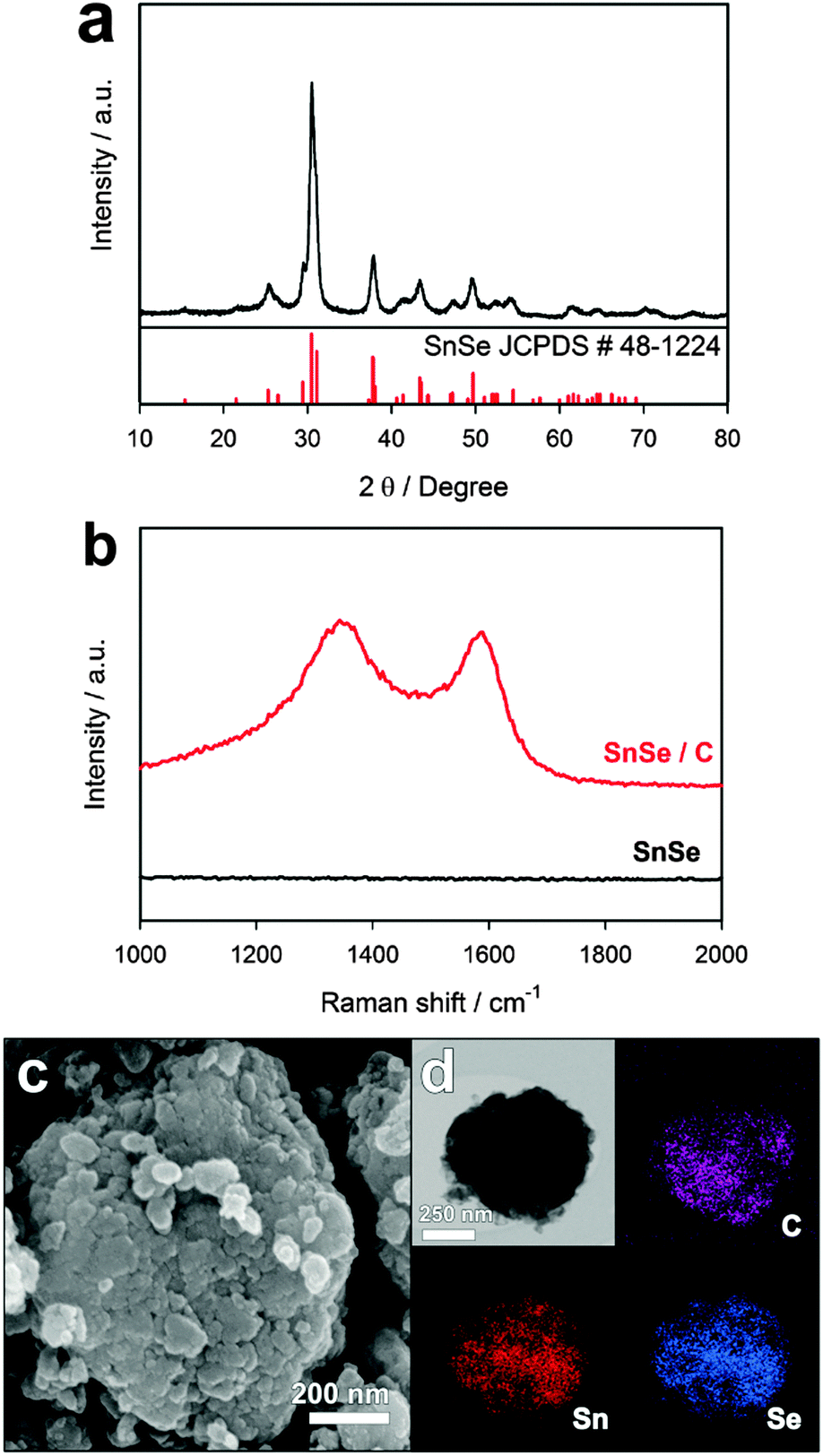 | ||
| Fig. 1 (a) X-ray diffraction pattern of SnSe/C; (b) Raman spectra of SnSe/C (red line) and bare SnSe (black line); (c) FE-SEM image and (d) STEM image and EDS mapping images of carbon, tin, and selenium. | ||
The electrochemical performance of the SnSe/C composite was evaluated using a half cell with a sodium metal electrode at a current density of 143 mA g−1 (∼0.2C). Fig. 2a shows the cycle performance of the SnSe/C composite, exhibiting negligible capacity fading over 50 cycles. Its reversible capacity is 707 mA h g−1, delivering 91% of the theoretical capacity of SnSe (780 mA h g−1). This calculation assumes that SnSe is changed into Na2Se and Na15Sn4 during sodiation. The coulombic efficiency in the first cycle was 75.3%. Fig. 2b shows galvanostatic voltage profiles of the SnSe/C electrode. The sodiation profile is characterized by two regions including a plateau located at about 0.9 V and a sloping profile between 0.6 and 0 V, which is clearly shown in the differential capacity (dQ/dV) plots (Fig. 2c). The peak area (corresponding to capacity) around 0.9 V for the first cycle decreased after subsequent cycling. This indicates that the plateau at 0.9 V for the first cycle is attributable to irreversible electrolyte decomposition such as that occurring through the formation of a solid electrolyte interphase (SEI). However, the small hump around 1 V was still observed in the 10th cycle, suggesting that the plateau at 0.9 V for the first cycle includes not only irreversible electrolyte decomposition but also reversible (de)sodiation. Furthermore, the rate performance of the composite was examined, and it delivered about 350 mA h g−1 at 1C rate (Fig. S1, ESI†).
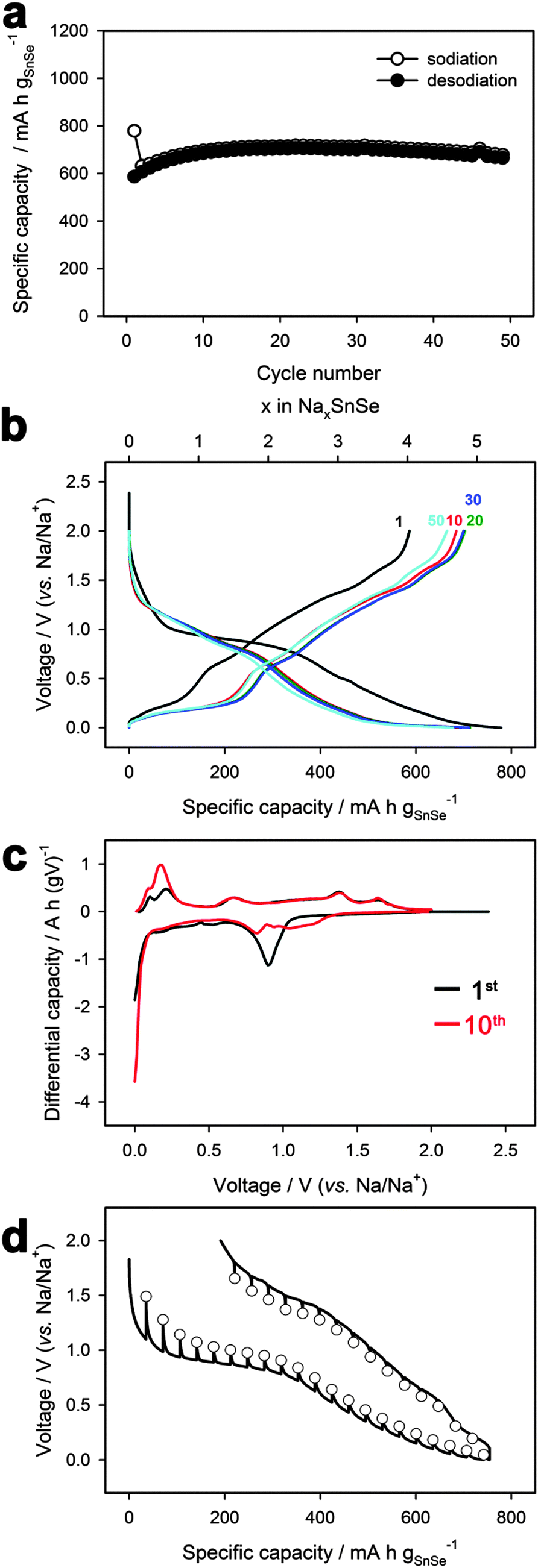 | ||
| Fig. 2 (a) Cycle performance, (b) voltage profiles, (c) differential capacity (dQ/dV) plots, and (d) GITT profiles of SnSe/C. | ||
In order to elucidate the (de)sodiation mechanism of SnSe/C during charge–discharge, ex situ XRD analysis of the SnSe/C was performed at various degrees of state of charge (SOC) in the first cycle (Fig. 3). It revealed that sodiation of the SnSe/C proceeds in two consecutive reactions. Upon approaching a redox potential of 0.6 V vs. Na/Na+, the formation of crystalline Na2Se and Sn was observed with the disappearance of SnSe, as shown in Fig. 3 (ii). This indicates that the plateau at 0.9 V is attributed to the two-phase reaction for conversion of SnSe into Na2Se and Sn. After full sodiation (at 0 V), the XRD peaks of Sn disappeared, and only those of Na2Se were observed (Fig. 3 (iii)). This indicates that the amorphous NaxSn phase was formed via the sodiation between 0.6 and 0 V. This is the one-phase reaction, because a sloping voltage profile was observed in this region, as shown in the quasi-open-circuit voltage (QOCV) profile obtained via a galvanostatic intermittent titration technique (GITT) (Fig. 2d). After full desodiation (at 2 V), a small broad XRD peak near 31° corresponding to SnSe was observed, indicating that SnSe was reversibly restored from NaxSn and Na2Se (Fig. 3 (v)). The (de)sodiation mechanism of SnSe is summarized, as follows:
 | ||
| Fig. 3 Ex situ XRD patterns of the SnSe/C composite electrode collected at various points as indicated in the corresponding voltage profile: (i) pristine, (ii) 0.6 V, (iii) 0 V, (iv) 0.5 V, (v) 1 V, and (vi) 2 V. | ||
Sodiation:
| Step 1: SnSe + 2Na+ + 2e− → c-Sn + c-Na2Se | (1) |
| Step 2: c-Sn + xNa+ + xe− + c-Na2Se → a-NaxSn + c-Na2Se | (2) |
Desodiation:
| a-NaxSn + c-Na2Se → SnSe + (2 + x)Na+ + (2 + x)e− | (3) |
(a: amorphous, c: crystalline)
Further support for the (de)sodiation mechanism of SnSe/C was given by the ex situ TEM analysis combined with EDS mapping images. Fig. 4a shows the bright-field TEM and EDS mapping images of the fully sodiated SnSe/C electrode. After full sodiation (at 0 V), NaxSn domains (dark region) with sizes between tens of and a few hundred nm dispersed in the Na2Se matrix (bright region) were observed, which is clearly supported by the EDS mapping images for the elements of Sn, Se, and Na. After full desodiation, the segregated Sn domains disappeared, and homogenously-overlapped EDS maps for Sn and Se were observed (Fig. 4b). This indicates that SnSe was reversibly formed after full desodiation. A relatively weak EDS intensity from Na was also observed even after desodiation; this is attributed to SEI layers containing Na. In addition, it is considered that the morphology change showing the confinement of NaxSn domains in the Na2Se matrix was critical in the better cycle performance of SnSe compared to previous reports for Sn.9 It is well-known that the pulverization or cracking of Sn is caused by the agglomeration of Sn particles during cycling, resulting in capacity fading of Sn electrodes. However, in the case of SnSe, Sn or NaxSn domains were embedded in the Na2Se matrix during cycling, and thus agglomeration of Sn is inhibited, leading to enhanced cycle performance. Moreover, the cycle performance of pure Sn is dependent on its operating voltage range because the electrolyte decomposition is accelerated at >0.8 V vs. Na/Na+.9a Therefore, pure Sn showed poor cycle performance in the voltage range of 0–1.5 V vs. Na/Na+. However, pure Se exhibited excellent cycle performance better than pure Sn, indicating that the electrolyte decomposition on the surface of Se is not as severe as that of Sn.17 In the case of SnSe, the surface of Sn is not exposed to electrolytes because of Sn or NaxSn domains embedded in the Na2Se matrix, and this results in the alleviated electrolyte decomposition of SnSe. Therefore, the better cycle performance of SnSe than pure Sn is partially attributed to that the surface of Se is exposed to electrolytes.
 | ||
| Fig. 4 The STEM images and EDS elemental mapping of tin, selenium, carbon, and sodium of fully (a) sodiated and (b) desodiated SnSe/C. | ||
In summary, a SnSe/C composite was obtained through facile ball milling, and we demonstrated, for the first time, its electrochemical performance as an anode material for Na-ion batteries. SnSe/C is remarkable as a promising anode candidate for Na-ion batteries because of its reversible specific capacity. In this study, SnSe/C showed excellent electrochemical performance including a high reversible capacity of 707 mA h g−1 and excellent cycle performance with negligible capacity fading over 50 cycles. The improved cycle performance of SnSe/C was attributed to the confinement effect of NaxSn domains in the Na2Se matrix during cycling. In addition, the reversible (de)sodiation mechanism of SnSe was established through ex situ XRD and TEM with EDS analyses. It is believed that understanding this reaction mechanism of SnSe will contribute to developing alloy-based materials for Na-ion batteries.
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MSIP and MOE) (No. 2010-0019408 and NRF-2013R1A1A2013446), and by Korea Electrotechnology Research Institute (KERI) Primary research program through the National Research Council of Science & Technology funded by the Ministry of Science, ICT and Future Planning (MSIP) (No. 14-12-N0101-69).
Notes and references
- (a) M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed; (b) N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994 CrossRef CAS PubMed; (c) B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691 CrossRef CAS.
- (a) B. L. Ellis and L. F. Nazar, Curr. Opin. Solid State Mater. Sci., 2012, 16, 168 CrossRef CAS PubMed; (b) S.-W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710 CrossRef CAS; (c) S. Y. Hong, Y. Kim, Y. Park, A. Choi, N.-S. Choi and K. T. Lee, Energy Environ. Sci., 2013, 6, 2067 RSC; (d) V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884 RSC; (e) M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947 CrossRef CAS; (f) H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338 RSC.
- Y. Kim, K.-H. Ha, S. M. Oh and K. T. Lee, Chem. – Eur. J., 2014, 20, 11980 CrossRef CAS PubMed.
- (a) Y. Kim, Y. Park, A. Choi, N.-S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045 CrossRef CAS PubMed; (b) J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, Angew. Chem., Int. Ed., 2013, 52, 4633 CrossRef CAS PubMed; (c) N. Yabuuchi, Y. Matsuura, T. Ishikawa, S. Kuze, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, ChemElectroChem, 2014, 1, 580 CrossRef CAS; (d) W.-J. Li, S.-L. Chou, J.-Z. Wang, H.-K. Liu and S.-X. Dou, Nano Lett., 2013, 13, 5480 CrossRef CAS PubMed.
- (a) Y. Kim, Y. Kim, A. Choi, S. Woo, D. Mok, N. S. Choi, Y. S. Jung, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2014, 26, 4139 CrossRef CAS PubMed; (b) J. Qian, Y. Xiong, Y. Cao, X. Ai and H. Yang, Nano Lett., 2014, 14, 1865 CrossRef CAS PubMed; (c) W. Li, S.-L. Chou, J.-Z. Wang, J. H. Kim, H.-K. Liu and S.-X. Dou, Adv. Mater., 2014, 26, 4037 CrossRef CAS PubMed; (d) J. Fullenwarth, A. Darwiche, A. Soares, B. Donnadieu and L. Monconduit, J. Mater. Chem. A, 2014, 2, 2050 RSC.
- (a) S. Yuan, X.-l. Huang, D.-l. Ma, H.-g. Wang, F.-z. Meng and X.-b. Zhang, Adv. Mater., 2014, 26, 2273 CrossRef CAS PubMed; (b) S. Hariharan, K. Saravanan, V. Ramar and P. Balaya, Phys. Chem. Chem. Phys., 2013, 15, 2945 RSC; (c) B. Koo, S. Chattopadhyay, T. Shibata, V. B. Prakapenka, C. S. Johnson, T. Rajh and E. V. Shevchenko, Chem. Mater., 2012, 25, 245 CrossRef; (d) M. C. Lopez, P. Lavela, G. F. Ortiz and J. L. Tirado, Electrochem. Commun., 2013, 27, 152 CrossRef CAS PubMed; (e) M. M. Rahman, A. M. Glushenkov, T. Ramireddy and Y. Chen, Chem. Commun., 2014, 50, 5057 RSC; (f) M. Valvo, F. Lindgren, U. Lafont, F. Björefors and K. Edström, J. Power Sources, 2014, 245, 967 CrossRef CAS PubMed.
- (a) G. S. Bang, K. W. Nam, J. Y. Kim, J. Shin, J. W. Choi and S.-Y. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 7084 CrossRef CAS PubMed; (b) L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759 CrossRef CAS PubMed; (c) D. Y. Yu, P. V. Prikhodchenko, C. W. Mason, S. K. Batabyal, J. Gun, S. Sladkevich, A. G. Medvedev and O. Lev, Nat. Commun., 2013, 4, 2922 Search PubMed; (d) C. Zhu, X. Mu, P. A. van Aken, Y. Yu and J. Maier, Angew. Chem., Int. Ed., 2014, 53, 2152 CrossRef CAS PubMed; (e) Y.-X. Wang, K. H. Seng, S.-L. Chou, J.-Z. Wang, Z. Guo, D. Wexler, H.-K. Liu and S.-X. Dou, Chem. Commun., 2014, 50, 10730 RSC.
- Y. N. Ko, S. H. Choi, S. B. Park and Y. C. Kang, Nanoscale, 2014, 6, 10511 RSC.
- (a) S. Komaba, Y. Matsuura, T. Ishikawa, N. Yabuuchi, W. Murata and S. Kuze, Electrochem. Commun., 2012, 21, 65 CrossRef CAS PubMed; (b) Y. Xu, Y. Zhu, Y. Liu and C. Wang, Adv. Energy Mater., 2013, 3, 128 CrossRef CAS; (c) L. D. Ellis, T. D. Hatchard and M. N. Obrovac, J. Electrochem. Soc., 2012, 159, A1801 CrossRef CAS PubMed; (d) J. W. Wang, X. H. Liu, S. X. Mao and J. Y. Huang, Nano Lett., 2012, 12, 5897 CrossRef CAS PubMed; (e) Y. Liu, Y. Xu, Y. Zhu, J. N. Culver, C. A. Lundgren, K. Xu and C. Wang, ACS Nano, 2013, 7, 3627 CrossRef CAS PubMed; (f) H. Zhu, Z. Jia, Y. Chen, N. Weadock, J. Wan, O. Vaaland, X. Han, T. Li and L. Hu, Nano Lett., 2013, 13, 3093 CrossRef CAS PubMed.
- (a) A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, J. Am. Chem. Soc., 2012, 134, 20805 CrossRef CAS PubMed; (b) J. Qian, Y. Chen, L. Wu, Y. Cao, X. Ai and H. Yang, Chem. Commun., 2012, 48, 7070 RSC; (c) L. Baggetto, P. Ganesh, C.-N. Sun, R. A. Meisner, T. A. Zawodzinski and G. M. Veith, J. Mater. Chem. A, 2013, 1, 7985 RSC; (d) W. Luo, S. Lorger, B. Wang, C. Bommier and X. Ji, Chem. Commun., 2014, 50, 5435 RSC; (e) X. Zhou, Z. Dai, J. Bao and Y.-G. Guo, J. Mater. Chem. A, 2013, 1, 13727 RSC; (f) Y. Zhu, X. Han, Y. Xu, Y. Liu, S. Zheng, K. Xu, L. Hu and C. Wang, ACS Nano, 2013, 7, 6378 CrossRef CAS PubMed; (g) M. He, K. Kravchyk, M. Walter and M. V. Kovalenko, Nano Lett., 2014, 14, 1255 CrossRef CAS PubMed.
- (a) A. Darwiche, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, Electrochem. Commun., 2013, 32, 18 CrossRef CAS PubMed; (b) L. Ji, M. Gu, Y. Shao, X. Li, M. H. Engelhard, B. W. Arey, W. Wang, Z. Nie, J. Xiao, C. Wang, J.-G. Zhang and J. Liu, Adv. Mater., 2014, 26, 2901 CrossRef CAS PubMed; (c) L. Xiao, Y. Cao, J. Xiao, W. Wang, L. Kovarik, Z. Nie and J. Liu, Chem. Commun., 2012, 48, 3321 RSC.
- (a) M. Gu, A. Kushima, Y. Shao, J.-G. Zhang, J. Liu, N. D. Browning, J. Li and C. Wang, Nano Lett., 2013, 13, 5203 CrossRef CAS PubMed; (b) D. Su, H.-J. Ahn and G. Wang, Chem. Commun., 2013, 49, 3131 RSC.
- (a) B. Qu, C. Ma, G. Ji, C. Xu, J. Xu, Y. S. Meng, T. Wang and J. Y. Lee, Adv. Mater., 2014, 26, 3854 CrossRef CAS PubMed; (b) X. Xie, D. Su, S. Chen, J. Zhang, S. Dou and G. Wang, Chem. – Asian J., 2014, 9, 1611 CrossRef CAS PubMed; (c) T. Zhou, W. K. Pang, C. Zhang, J. Yang, Z. Chen, H. K. Liu and Z. Guo, ACS Nano, 2014, 8, 8323 CrossRef CAS PubMed.
- F. Klein, B. Jache, A. Bhide and P. Adelhelm, Phys. Chem. Chem. Phys., 2013, 15, 15876 RSC.
- I. Lefebvre, M. A. Szymanski, J. Olivier-Fourcade and J. C. Jumas, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 1896 CrossRef CAS.
- M.-Z. Xue, J. Yao, S.-C. Cheng and Z.-W. Fu, J. Electrochem. Soc., 2006, 153, A270 CrossRef CAS PubMed.
- (a) C. Luo, Y. Xu, Y. Zhu, Y. Liu, S. Zheng, Y. Liu, A. Langrock and C. Wang, ACS Nano, 2013, 7, 8003 CrossRef CAS PubMed; (b) A. Abouimrane, D. Dambournet, K. W. Chapman, P. J. Chupas, W. Weng and K. Amine, J. Am. Chem. Soc., 2012, 134, 4505 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section. See DOI: 10.1039/c4cc06106c |
| This journal is © The Royal Society of Chemistry 2015 |