
“AND” logic gate regulated pH and reduction dual-responsive prodrug nanoparticles for efficient intracellular anticancer drug delivery†
Lan
Bai
,
Xiao-hui
Wang
,
Fei
Song
*,
Xiu-li
Wang
and
Yu-zhong
Wang
*
Center for Degradable and Flame-Retardant Polymeric Materials (ERCPM-MoE), College of Chemistry, State Key Laboratory of Polymer Materials Engineering, National Engineering Laboratory of Eco-Friendly Polymeric Materials (Sichuan), Sichuan University, 29 Wangjiang Road, Chengdu 610064, China. E-mail: songfei520@gmail.com; yzwang@scu.edu.cn
First published on 3rd November 2014
Abstract
A dual-responsive drug delivery system simulating an AND logic gate is developed by core-cross-linking of a disulfide-containing anticancer prodrug with Cu2+ for safe and efficient delivery of anticancer drugs. These prodrug nanoparticles are stable and exhibit nearly no premature drug release, and allow a fast drug release under simulated intracellular conditions, realizing a precise drug delivery towards cell nuclei.
Due to the superior drug loading capacity, no/slight premature burst release, and a reduced dose of inactive carriers that may induce systemic toxicity, significant progress has been made towards the design and development of anticancer prodrug-based drug delivery systems during the past decade.1 A series of prodrugs with hydrophilic components, such as poly(ethylene glycol) (PEG),2 poly(N-(2-hydroxypropyl) methacrylamide),3 dextran,4etc., have been synthesized. Although being able to self-assemble into nanoparticles in aqueous solution, the prodrugs are still facing enormous challenges and obstacles. Premature dissociation may occur under in vivo conditions for the nanoparticles upon serious dilution, and the drugs are only transported into cytoplasm but are hard to be trafficked into cell nuclei due to the poor cellular uptake resulting from the stealth effect of hydrophilic shells.5 As a predominant strategy for drug carriers, core-cross-linking showed advantages of reducing sizes and improving stability of nanoparticles in aqueous solution, and showed that environmental stimuli-responsive cleavage of the cross-links could trigger disassembly of the carriers as well as drug release.3,6 However, this strategy has never been employed in prodrug systems because the hydrophobic domain composed of anticancer drugs is difficult to be modified by cross-linkers without losing activity.
Since a porous metal–organic framework was developed for the first time as a drug carrier,7 coordination polymer based delivery systems are emerging to be new platforms for drug delivery.8 In addition, metal ions could directly form coordination bonds with some anticancer drugs, and the bonds showed pH-responsive formation and cleavage characteristics.9 As is well known, the pH value and glutathione (GSH) concentration in normal extracellular matrices are about 7.4 and 2–20 µM, respectively, whereas those in intracellular environments are 5.0 and 2–10 mM. Therefore, we wished to explore a suitable methodology based on metal–drug interaction to efficiently transport a currently used anticancer drug, doxorubicin (DOX), into cancer cells and utilize the intracellular biological signals to precisely trigger a fast and targeted release of DOX. The molecule-based “logic gate” is an interesting concept, which contains binary switches to determine the output state by controlling input conditions.10 In this work, we developed the first molecular AND logic gate modulated core-cross-linked prodrug system with joint dual stimuli-responsive properties. This AND logic gate was switched OFF under neutral pH conditions in the absence of a reducing agent and the output signal (DOX release) was suppressed; whereas the cross-links were unlocked and DOX was shed from a polymer chain once the nanoparticle was internalized into cancer cells with features of both a low pH value and a high reducing agent level, suggesting the ON state of the AND logic gate.
In a typical experiment, the conjugate of PEG and DOX with a disulfide-linkage (PEG-SS-DOX) was synthesized by a two-step reaction (Fig. S1, ESI†), and its chemical structure was confirmed by 1H NMR and high resolution electrospray ionization time-of-flight mass spectrometry (HR ESI-TOF MS) analyses (Fig. S2 and S3, ESI†), indicating the successful synthesis of the resultant prodrug. It is worth noting that a short thiol residue, still conjugated with DOX after the cleavage of the disulfide bond, would not affect the activity and spectral characteristics of DOX.4a,11
Prior to the preparation of core-cross-linked prodrug nanoparticles fit for DOX delivery, the pH-responsive formation and cleavage of coordination bonds between metal ions and PEG-SS-DOX were investigated by UV-Vis measurements. Cu(NO3)2, Zn(NO3)2, Co(NO3)2, and Fe(NO3)3 were selected as the sources of metals. Compared with PEG-SS-DOX, only the PEG-SS-DOX-Cu2+ complex showed a sharp decrease in absorbance at pH 7.4 and a reversible change in absorbance at pH 5.0 (late endosomal/lysosomal compartment conditions).12 Cu2+ was therefore chosen as the dynamic cross-linker for this prodrug system. According to previous reports, the stoichiometric ratio of the Cu2+–DOX complex is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 within the pH region from 5.6 to 8.0 and will change to 1:1 within the pH region from 4.0 to 5.6.13 The core-cross-linked PEG-SS-DOX nanoparticles (PEG-SS-DOX-Cu) were prepared accordingly under the same molar ratio as that of Cu2+/DOX. As shown in Fig. 1, the formation and cleavage of coordination bonds between Cu2+ and DOX were accompanied with changes in solution color and UV-Vis spectra. The colors of PEG-SS-DOX solution at pH 7.4 and 5.0 were found to be orange without an obvious difference, and their UV-Vis spectra were nearly the same. Upon adding Cu2+ into the neutral solution, its color became purplish-red and a decreased UV-Vis absorbance as well as a red-shift of the maximum absorbance peak was detected, which was attributed to the π–π* electron transition of the anthraquinone portion of DOX resulting from the bimolecular coordination.14 The interaction between Cu2+ and DOX was also proven by FTIR spectroscopy, because two peaks at 1643 and 1584 cm−1 (C
:2 within the pH region from 5.6 to 8.0 and will change to 1:1 within the pH region from 4.0 to 5.6.13 The core-cross-linked PEG-SS-DOX nanoparticles (PEG-SS-DOX-Cu) were prepared accordingly under the same molar ratio as that of Cu2+/DOX. As shown in Fig. 1, the formation and cleavage of coordination bonds between Cu2+ and DOX were accompanied with changes in solution color and UV-Vis spectra. The colors of PEG-SS-DOX solution at pH 7.4 and 5.0 were found to be orange without an obvious difference, and their UV-Vis spectra were nearly the same. Upon adding Cu2+ into the neutral solution, its color became purplish-red and a decreased UV-Vis absorbance as well as a red-shift of the maximum absorbance peak was detected, which was attributed to the π–π* electron transition of the anthraquinone portion of DOX resulting from the bimolecular coordination.14 The interaction between Cu2+ and DOX was also proven by FTIR spectroscopy, because two peaks at 1643 and 1584 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of quinine domain in DOX) were red-shifted to 1619 and 1572 cm−1 (Fig. S4, ESI†). Furthermore, the determination of the decreased intensity of the peaks also implied the complexation of phenolate oxygen.9a However, since the bimolecular coordination could be transferred to monomolecular coordination under acidic conditions (Fig. 1c), the solution turned weak pink and the maximum absorbance peak went back to 497 nm.
O of quinine domain in DOX) were red-shifted to 1619 and 1572 cm−1 (Fig. S4, ESI†). Furthermore, the determination of the decreased intensity of the peaks also implied the complexation of phenolate oxygen.9a However, since the bimolecular coordination could be transferred to monomolecular coordination under acidic conditions (Fig. 1c), the solution turned weak pink and the maximum absorbance peak went back to 497 nm.
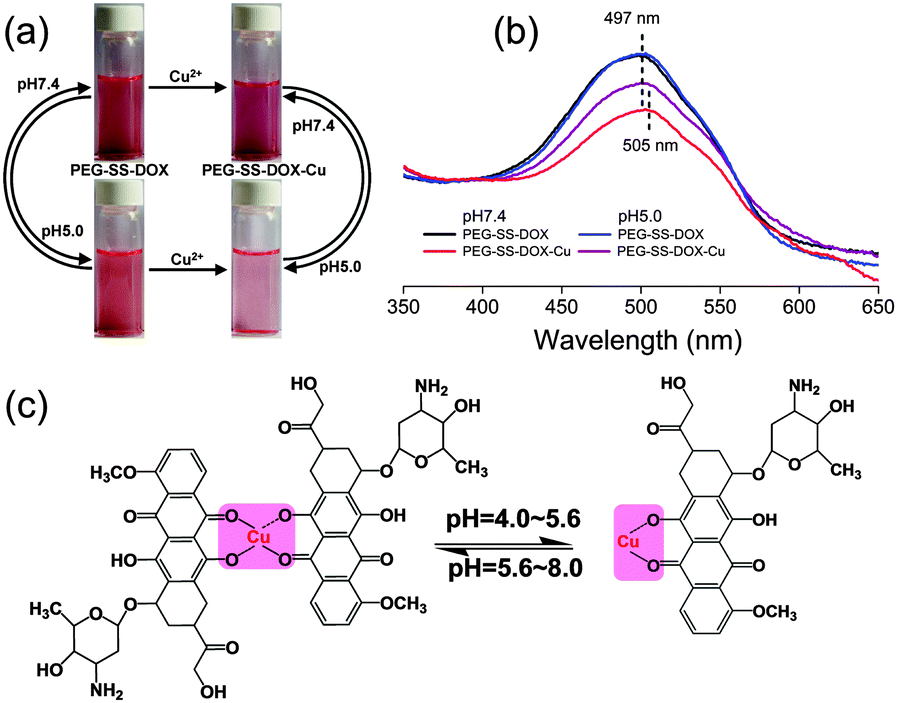 | ||
| Fig. 1 (a) Photographs and (b) UV-Vis spectra of PEG-SS-DOX and PEG-SS-DOX-Cu nanoparticles at pH 7.4 and 5.0. (c) A schematic illustration of coordination between Cu2+ and DOX at different pH values. | ||
Transmission electron microscopy (TEM) showed that the PEG-SS-DOX prodrug formed uniform spherical nanoparticles at pH 7.4 with a mean diameter of less than 100 nm, which was consistent with a hydrodynamic diameter of 114 nm determined by dynamic light scattering (DLS, Fig. 2a). Under these conditions, the critical micelle concentration (CMC) of PEG-SS-DOX was 0.23 mg mL−1, while as expected, it was obviously reduced to 0.056 mg mL−1 after core-cross-linking with Cu2+ (Fig. S5, ESI†). Moreover, the core-cross-linked nanoparticles were of irregular shapes rather than sphere-shaped and their size was decreased evidently (Fig. 2b); these changes were caused by the formation of a tighter hydrophobic core. When the pH value of solution was adjusted to 5.0, the cross-links were broken and the DOX chain became hydrophilic, resulting in disassembly of cross-linked nanoparticles. Therefore, only random aggregates with a hydrodynamic diameter of 32 nm could be observed (Fig. 2c). The characteristics such as reduced size of the drug delivery system and its disintegration in response to acid are desired for cell uptake and efficient intracellular drug release.
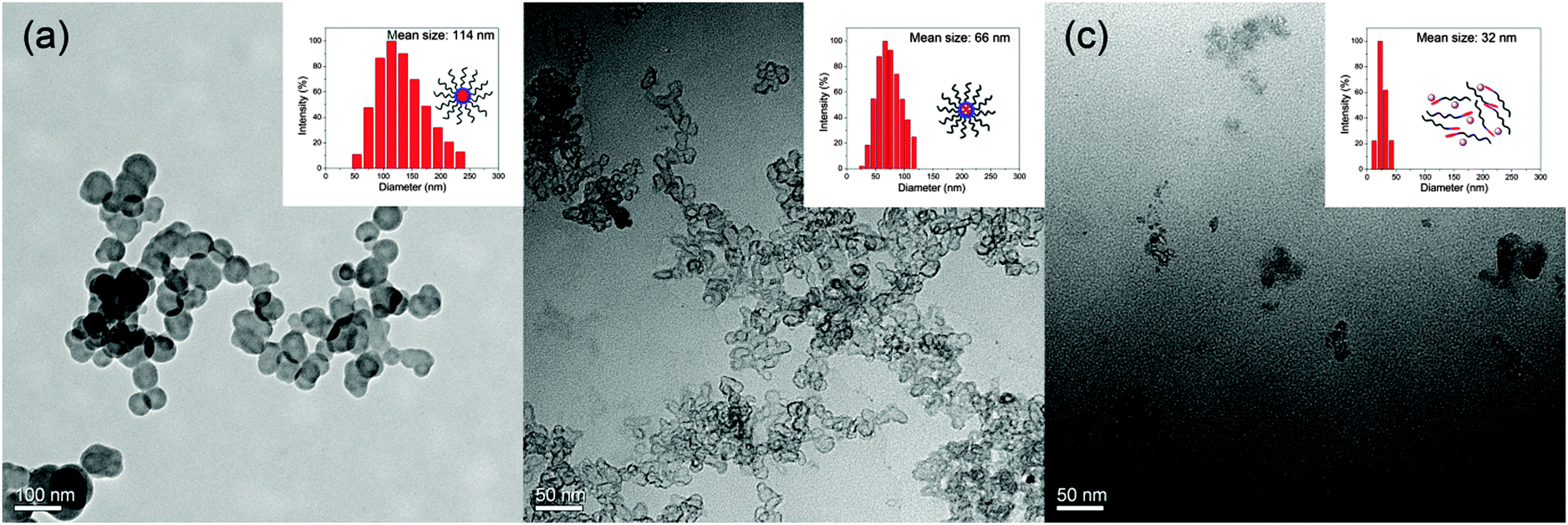 | ||
| Fig. 2 TEM images and hydrodynamic diameters of (a) PEG-SS-DOX nanoparticles at pH 7.4, and PEG-SS-DOX-Cu nanoparticles at (b) pH 7.4 and (c) pH 5.0. | ||
For a superb therapeutic effect, a rapid lysosomal drug release should be achieved. However, compared with a fast entry of free DOX into cell nuclei after cell uptake, transportation efficiency of DOX loaded in the polymeric carriers is much lower.15 Therefore, we next focused on whether core-cross-linking affected the cell internalization of prodrug nanoparticles. As illustrated in Fig. S6a (ESI†), free DOX displayed a quick accumulation toward nuclei after only 4 h of incubation. However, for PEG-SS-DOX, very low DOX fluorescence intensity was determined in the cytoplasm around nuclei and no fluorescence signal was found in nuclei even after 8 h of incubation (Fig. S6b, ESI†), and most of the DOX fluorescence was still outside of cell nuclei even after 24 h of incubation (Fig. S6c, ESI†), which can be due to the poor cell internalization resulting from the stealth effect of PEG shells.5a,16 It is worth noting that the DOX location of PEG-SS-DOX-Cu was detected in both the cytoplasm and nuclei after 8 h of incubation and was mainly concentrated in nuclei after 24 h of incubation, revealing that core-cross-linking was an effective strategy beneficial for the endocytosis of PEG-SS-DOX and the transportation of DOX into the cell nucleus. In addition, the presence of Cu2+ helped DOX to escape from endosomes as well as enter into nuclei. Although the underlying mechanism was unclear, the role of Cu2+ in DOX delivery should not be neglected, which will be investigated in future work. To understand the influence of the overexpressed intracellular reductive signal on DOX transportation,17 the GSH level in HeLa cells was enhanced by pretreatment with GSH according to a general method.18 A stronger fluorescence intensity of DOX in HeLa cells was observed for both PEG-SS-DOX and PEG-SS-DOX-Cu upon GSH treatment (Fig. 3a), compared with the non-pretreated samples after the same incubation time (Fig. S6a, ESI†), which was due to the accelerated DOX release resulting from the fast cleavage of disulfide bonds and dissociation of aggregated nanoparticles in the presence of GSH. It was also exciting that the red fluorescence was mainly focused in the nuclei of HeLa cells for the core-cross-linked prodrug after only 4 h of incubation, indicating the efficient delivery of DOX into the cell nucleus which was quite difficult to be achieved previously and can be explained by the excellent internalization of cross-linked nanoparticles and the cleavage of the disulfide bond linking DOX with PEG, demonstrating a particularly combined pH- and reduction-responsive characteristic. As designed, the smart prodrug nanoparticle system can collect biological stimuli to transport hydrophobic anticancer drugs (Fig. 3b).
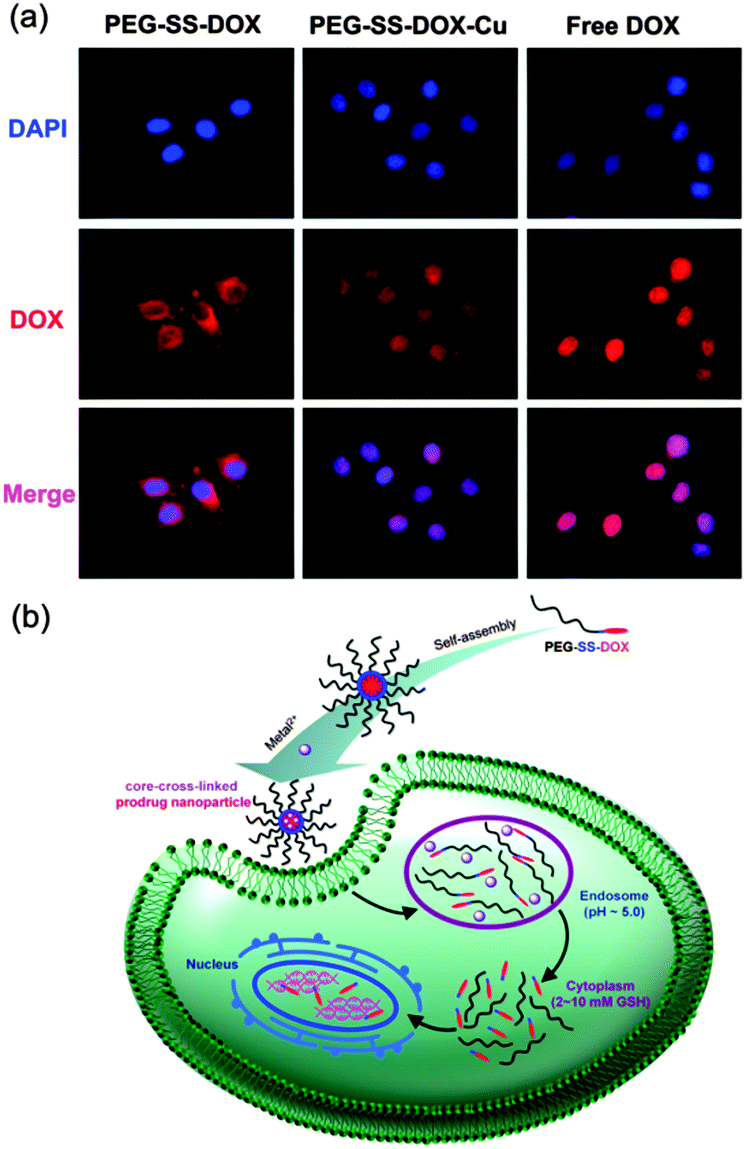 | ||
| Fig. 3 (a) Fluorescence microscopy images of intracellular tracking of free DOX, PEG-SS-DOX and PEG-SS-DOX-Cu nanoparticles after 4 h of incubation. The HeLa cells were previously treated with 5 mmol L−1 GSH. Nuclei were labeled with DAPI. Images were taken from the DAPI channel (blue), the DOX channel (red), and their overlapped images. (b) Schematic illustrations of intracellular pH-triggered disassembly of Cu2+-cross-linked nanoparticles and reduction-triggered release of DOX from a PEG-SS-DOX prodrug. | ||
The pH and reduction dual-responsive drug release behaviors were then investigated for the prodrug system. For the non-cross-linked prodrug (Fig. S7, ESI†), the cumulative release of DOX did not vary obviously between pH 7.4 and 5.0 except at the early stage of 10 h. After cross-linking with Cu2+ (Fig. 4a), the prodrug nanoparticles showed a much lower release rate and the cumulative DOX release was less than 10% within 48 h at pH 7.4 in the presence of 2 µM DTT, due to the highly inhibited premature drug release at the OFF state of the AND logic gate. Moreover, although increasing the DTT concentration to 2 or 5 mM could increase the release rate of DOX, most of the drug (>60%) was still not released and would not release further after 48 h. In addition, the cleavage of the coordination bond between Cu2+ and DOX under acidic conditions obviously enhanced the drug release rate of PEG-SS-DOX-Cu, which was close to that of the non-cross-linked prodrug, but the cumulative DOX release was still less than 40%. The results demonstrated that the sole existence of either input signal could not completely turn the logic gate ON. Upon increasing the intensity of the reduction signal, DOX was remarkably released from the PEG skeleton with the cumulative release close to 100%, acting as the ON state of the AND logic gate at which the nanoparticles were disintegrated into small fragments to trigger drug release.
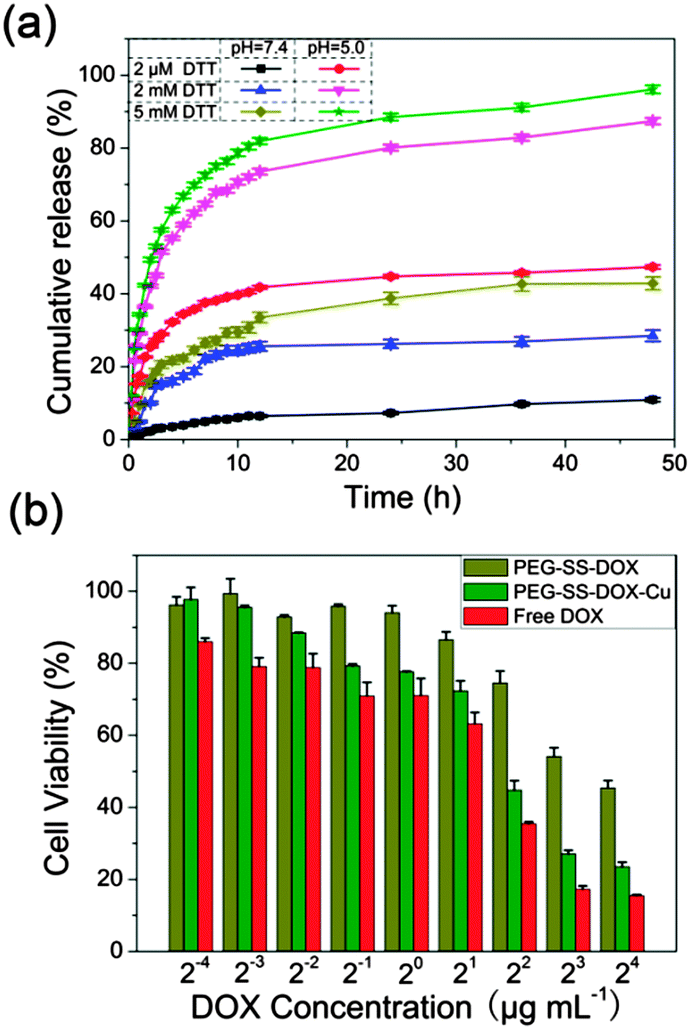 | ||
| Fig. 4 (a) DOX release profiles of PEG-SS-DOX-Cu nanoparticles (mean ± standard deviation (SD), n = 3); (b) cytotoxicity of PEG-SS-DOX nanoparticles, PEG-SS-DOX-Cu nanoparticles and free DOX against HeLa cells at different DOX concentrations for 24 h. | ||
These results indicated that the Cu2+-cross-linked approach was favorable for improving the physiological circulation stability as well as realizing a high efficient intracellular drug delivery. In order to evaluate in vitro therapeutic efficiency of the prodrug system, the cytotoxicity toward HeLa cells was studied after 24 h of incubation (Fig. 4b). PEG-SS-DOX-Cu effectively inhibited the proliferation of HeLa cells with the half maximal inhibitory concentration (IC50) of 3.6 µg DOX equiv. mL−1, which was lower than that of PEG-SS-DOX (11.2 µg DOX equiv. mL−1). In particular, the prodrug at a high dosage possessed an excellent therapeutic effect with the cell viability lower than 25%, which was close to that of free DOX, further confirming that the reversible core-cross-linking strategy was favorable for improving the anti-tumor activity of the prodrug system. Notably, the cell viability of HeLa cells incubated with Cu2+ still remained more than 95% even after 72 h of incubation (Fig. S8, ESI†), indicating that the cytotoxicity toward HeLa cells was generated from anticancer drug rather than Cu2+. These results demonstrated that the core-cross-linked prodrug system could efficiently improve the endocytosis of DOX and transport the drug to cell nuclei, which was not realized in previous studies. Characteristics such as excellent cell internalization, nuclear drug delivery and an in vitro therapeutic effect, which are comparable to the recently reported advanced drug carriers,19 suggest that this smart prodrug system is a new platform for chemotherapy.
In summary, we present a metal-mediated strategy to develop a novel core-cross-linked prodrug nanoparticle for efficient intracellular drug delivery. Premature drug release can be inhibited under normal tissue conditions, while the drug release is activated upon exposure to an acidic and reductive environment. Compared with the poor drug transportation of traditional carriers, the prodrug system can carry DOX to cell nuclei within a short period. The results of cell viability investigation confirm that this AND logic gate regulated dual-responsive core-cross-linked prodrug system is a promising platform for cancer therapy.
This work was supported by the Research Fund for the Doctoral Program of Higher Education of China (20130181120067), the National Natural Science Foundation of China (51403136, 51273123 and 51121001), and the National 863 Program of China (2012AA062904). The authors are grateful to Prof. J.L. Yang and Ms. L. Yu (State Key Laboratory of Biotherapy and Cancer Center, Sichuan University) for their help in cell experiments.
Notes and references
- (a) L. Bildstein, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2011, 63, 3 CrossRef CAS PubMed; (b) Y. Shen, E. Jin, B. Zhang, C. J. Murphy, M. Sui, J. Zhao, J. Wang, J. Tang, M. Fan and E. Van Kirk, J. Am. Chem. Soc., 2010, 132, 4259 CrossRef CAS PubMed; (c) A. Nori and J. Kopeček, Adv. Drug Delivery Rev., 2005, 57, 609 CrossRef CAS PubMed; (d) D. Trung Bui, A. Maksimenko, D. Desmaële, S. Harrisson, C. Vauthier, P. Couvreur and J. Nicolas, Biomacromolecules, 2013, 14, 2837 CrossRef CAS PubMed.
- (a) Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640 CrossRef CAS PubMed; (b) S. Aryal, C.-M. J. Hu and L. Zhang, ACS Nano, 2009, 4, 251 CrossRef PubMed.
- Z. Jia, L. Wong, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 3106 CrossRef CAS PubMed.
- (a) P. Liu, B. Shi, C. Yue, G. Gao, P. Li, H. Yi, M. Li, B. Wang, Y. Ma and L. Cai, Polym. Chem., 2013, 4, 5793 RSC; (b) M. T. Basel, S. Balivada, T. B. Shrestha, G. M. Seo, M. M. Pyle, M. Tamura, S. H. Bossmann and D. L. Troyer, Small, 2012, 8, 913 CrossRef CAS PubMed.
- (a) F. Zhan, W. Chen, Z. Wang, W. Lu, R. Cheng, C. Deng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2011, 12, 3612 CrossRef CAS PubMed; (b) P. Gou, W. Liu, W. Mao, J. Tang, Y. Shen and M. Sui, J. Mater. Chem. B, 2013, 1, 284 RSC.
- (a) J. Wang, M. A. Cohen Stuart, A. T. M. Marcelis, M. Colomb-Delsuc, S. Otto and J. van der Gucht, Macromolecules, 2012, 45, 7179 CrossRef CAS; (b) R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068 RSC; (c) R. Wei, L. Cheng, M. Zheng, R. Cheng, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2012, 13, 2429 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 118, 6120 CrossRef.
- (a) I. Imaz, J. Hernando, D. Ruiz-Molina and D. Maspoch, Angew. Chem., Int. Ed., 2009, 48, 2325 CrossRef CAS PubMed; (b) I. Imaz, M. Rubio-Martínez, L. García-Fernández, F. García, D. Ruiz-Molina, J. Hernando, V. Puntes and D. Maspoch, Chem. Commun., 2010, 46, 4737 RSC.
- (a) L. Xing, H. Zheng and S. Che, Chem. – Eur. J., 2011, 17, 7271 CrossRef CAS PubMed; (b) P. F. Gao, L. L. Zheng, L. J. Liang, X. X. Yang, Y. F. Li and C. Z. Huang, J. Mater. Chem. B, 2013, 1, 3202 RSC; (c) G. H. Hwang, K. H. Min, H. J. Lee, H. Y. Nam, G. H. Choi, B. J. Kim, S. Y. Jeong and S. C. Lee, Chem. Commun., 2014, 50, 4351 RSC.
- (a) S. Angelos, Y. W. Yang, N. M. Khashab, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11344 CrossRef CAS PubMed; (b) Y. W. Yang, Y. L. Sun and N. Song, Acc. Chem. Res., 2014, 47, 1950 CrossRef CAS PubMed.
- S. Santra, C. Kaittanis, O. J. Santiesteban and J. M. Perez, J. Am. Chem. Soc., 2011, 133, 16680 CrossRef CAS PubMed.
- Y. L. Colson and M. W. Grinstaff, Adv. Mater., 2012, 24, 3878 CrossRef CAS.
- (a) F. Greenaway and J. Dabrowiak, J. Inorg. Biochem., 1982, 16, 91 CrossRef CAS; (b) H. Beraldo, A. Garniersuillerot and L. Tosi, Inorg. Chem., 1983, 22, 4117 CrossRef CAS; (c) M. Fiallo and A. Garnier-Suillerot, J. Inorg. Biochem., 1987, 31, 43 CrossRef CAS.
- S. A. Abraham, K. Edwards, G. Karlsson, S. MacIntosh, L. D. Mayer, C. McKenzie and M. B. Bally, Biochim. Biophys. Acta, 2002, 1565, 41 CrossRef CAS.
- (a) X. Wang, G. Wu, C. Lu, W. Zhao, Y. Wang, Y. Fan, H. Gao and J. Ma, Eur. J. Pharm. Sci., 2012, 47, 256 CrossRef CAS PubMed; (b) Y. Jin, L. Song, Y. Su, L. Zhu, Y. Pang, F. Qiu, G. Tong, D. Yan, B. Zhu and X. Zhu, Biomacromolecules, 2011, 12, 3460 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS PubMed.
- (a) M. H. Lee, J. H. Han, J.-H. Lee, H. G. Choi, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 17314 CrossRef CAS PubMed; (b) F. Meng, R. Cheng, C. Deng and Z. Zhong, Mater. Today, 2012, 15, 436 CrossRef CAS.
- (a) Y. Zhang, C. Xiao, M. Li, J. Ding, C. He, X. Zhuang and X. Chen, Polym. Chem., 2013, 5, 2801 RSC; (b) S. Yu, C. He, J. Ding, Y. Cheng, W. Song, X. Zhuang and X. Chen, Soft Matter, 2013, 9, 2637 RSC.
- (a) X. Xu, Y. Li, H. Li, R. Liu, M. Sheng, B. He and Z. Gu, Small, 2013, 6, 1133 Search PubMed; (b) M. Li, Z. Tang, S. Lv, W. Song, H. Hong, X. Jing, Y. Zhang and X. Chen, Biomaterials, 2014, 35, 3851 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc07012g |
| This journal is © The Royal Society of Chemistry 2015 |