
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescent sensing of monofunctional platinum species†
Clara
Shen
a,
Benjamin D. W.
Harris
b,
Lucy J.
Dawson
a,
Kellie A.
Charles
b,
Trevor W.
Hambley
a and
Elizabeth J.
New
*a
aSchool of Chemistry, The University of Sydney, Building F11, NSW 2006, Australia. E-mail: elizabeth.new@sydney.edu.au; Tel: +61-2-9351-1993
bDepartment of Pharmacology, School of Medical Sciences, The University of Sydney, Building D06, NSW 2006, Australia
First published on 11th March 2015
Abstract
We report here FDCPt1, a novel selective fluorescent sensor for monofunctional platinum species. In the presence of such species, FDCPt1 exhibits a 70-fold increase in fluorescence emission, and can be used to monitor the metabolism of Pt(II)-based complexes in colorectal cancer cells. This probe is therefore expected to be valuable in studying changes in Pt coordination and distribution during chemotherapy.
The use of Pt(II)-based therapeutics has seen a significant improvement in the outcomes of tumour diagnoses, particularly for testicular cancer.1 It is generally accepted that Pt(II) drugs act through the formation of intrastrand adducts with DNA.2 However, it is also believed that only a small fraction of intracellular platinum will interact with DNA3 with the remainder ending up protein or peptide bound,4 forming adducts primarily with thiol-containing molecules such as glutathione, metallothienins,5 human serum albumin (HSA)6 and transferrin.7 There is therefore much interest in identifying other deactivating pathways and non-DNA-based contributions to cytotoxicity.
Pt(II) drugs typically bear two labile and two non-labile ligands, and in binding to DNA or protein will undergo two consecutive ligand replacement steps. An important intermediate in this process is the monofunctional Pt complex, bearing one labile and three non-labile ligands. A methodology to selectively identify monofunctional Pt species would therefore enable study of the rate and localisation of such species. Traditional methods for measuring Pt levels within cells, such as inductively-coupled plasma mass spectrometry (ICP-MS) and graphite-furnace atomic absorption spectrometry (GF-AAS) lack the spatial resolution and sensitivity to coordination environment required.8 We and others have previously used synchrotron radiation-induced X-ray emission to gain spatially-resolved information about Pt speciation within cells and tumour spheroids.9–12 However, these destructive techniques cannot be used to probe changes within living cells in response to stimuli.
Fluorescent-tagging of Pt(II) and Pt(IV) complexes has enabled study of Pt movement within cells,13–15 as well as dynamic imaging of intrinsically non-fluorescent Pt-drugs through the release of fluorescent ligands upon reduction of the Pt.16,17 A significant issue with this strategy is that the incorporation of organic dyes can considerably alter drug uptake and metabolism. An alternative strategy is to probe Pt through use of an exogenous fluorescent probe. Initial approaches have involved the use of reaction-based probes, which cannot be used for imaging changing Pt levels over time.18,19 A recent paper from Montagner et al.20 reports a reversible fluorescent probe for bifunctional Pt(II) complexes, such as cisplatin, and the utilisation of this probe to observe Pt(IV) reduction within cells. We report here FDCPt1, the first reversible fluorescent probe for monofunctional Pt(II).
FDCPt1 is a fluorescein-based compound bearing a dithiocarbamic acid moiety, commonly employed in chelators for platinum-group metals.21–24FDCPt1 was synthesised in two steps from fluorescein (Scheme 1), first with amidation in hydrazine to give the fluorescein hydrazide. This intermediate was heated in the presence of carbon disulfide and potassium hydroxide. Acidification gave the final dithiocarbamic acid, FDCPt1.
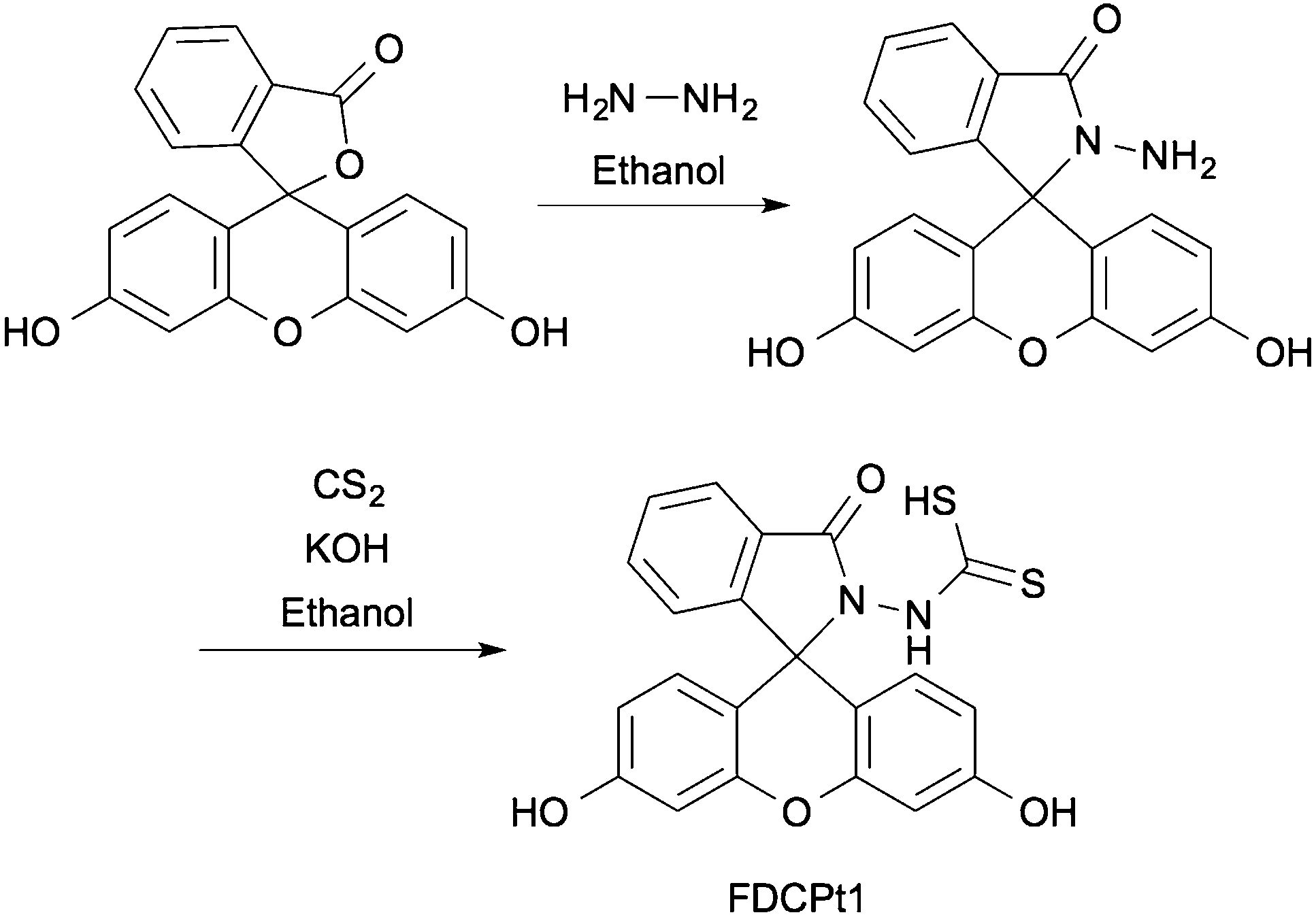 | ||
| Scheme 1 Synthesis of fluorescein dithiocarbamic acid (FDCPt1). | ||
The photophysical properties of FDCPt1 were studied in HEPES buffer (100 mM, pH 7.4, 50% DMF; Fig. 1). FDCPt1 exhibits a major absorption peak at 516 nm, and an emission maximum at 540 nm. Addition of one equivalent of (2-(aminoethyl)ethane-1,2-diamine)chlorido platinum(II) (1) resulted in a 7-fold change in absorbance (Fig. S1, ESI†) and a concomitant 70-fold change in fluorescence emission (Fig. 1a), and a 25 ppm shift in the 195Pt NMR spectrum (Fig. S2, ESI†). Fluorescence increased linearly with concentration of 1 between 0 and 1 equivalents (Fig. S3, ESI†). Binding analysis was carried out by the method of continuous variation, suggesting that 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 FDCPt1:Pt binding is responsible for the fluorescence increase (Fig. S4, ESI†). Interestingly, testing the fluorescence response of FDCPt1 to a range of Pt complexes of varied coordination environments revealed that the probe responds only to monofunctional Pt (1: Fig. 1b). This may be attributed to pi–pi stacking interactions upon the binding of two fluorophore molecules in the bifunctional case, quenching any resultant fluorescence. Furthermore, FDCPt1 is selective for Pt complexes over various transition metal ions (Fig. 1c). Since FDCPt1 responds selectively to monofunctional Pt, it is likely to have utility in distinguishing metabolites of platinum drugs within cells.
:1 FDCPt1:Pt binding is responsible for the fluorescence increase (Fig. S4, ESI†). Interestingly, testing the fluorescence response of FDCPt1 to a range of Pt complexes of varied coordination environments revealed that the probe responds only to monofunctional Pt (1: Fig. 1b). This may be attributed to pi–pi stacking interactions upon the binding of two fluorophore molecules in the bifunctional case, quenching any resultant fluorescence. Furthermore, FDCPt1 is selective for Pt complexes over various transition metal ions (Fig. 1c). Since FDCPt1 responds selectively to monofunctional Pt, it is likely to have utility in distinguishing metabolites of platinum drugs within cells.
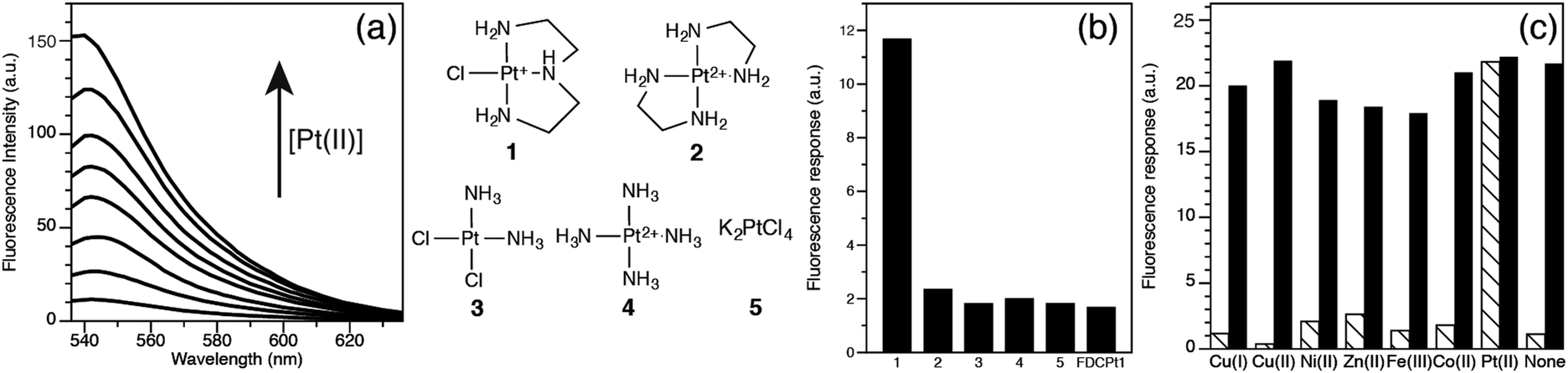 | ||
| Fig. 1 Photophysical behaviour of FDCPt1. (a) Fluorescence emission of FDCPt1 (λex = 516 nm) in the presence of increasing concentrations of 1 (from 0 to 5 equivalents). (b) Fluorescence response of FDCPt1 to Pt complexes. Bars represent integrated emission intensity (536 to 600 nm, λex = 516 nm) following addition of 200 μM Pt to 200 μM FDCPt1. (c) Fluorescence response of FDCPt1 to various metal ions. Bars represent integrated emission intensity (536 to 600 nm, λex = 516 nm). Hatched bars represent addition of an excess of the appropriate metal ion (concentrations) to a 200 μM solution of FDCPt1. Black bars represent subsequent addition of 200 μM 1 to the solution. | ||
The ability of FDCPt1 to respond exclusively to the presence of mono-functional Pt(II) suggests some useful applications in studying the cellular metabolism of Pt(II) drugs as they change coordination environment over time. Its stability over time was verified by absorption and emission, confirming suitability for such studies (Fig. S5, ESI†). Detecting intermediate species may reveal the timescale of nucleobase binding, in which we might expect an increase followed by decrease in fluorescence as Pt(II) becomes doubly bound. In addition, the effects of localisation of Pt(II) on the dynamics of drug deactivation can potentially be visualised with this probe.
In order to demonstrate the utility of FDCPt1 in cellular studies, we monitored the response of the probe to Pt treatment in Caco-2 human epithelial colorectal adenocarcinoma cells. Cells were treated with oxaliplatin (20 μM) for 1, 2, 6 or 24 h, followed by FDCPt1 (100 μM, 30 min), and were then visualised by fluorescence microscopy. Pt-treated cells clearly showed much higher fluorescence than cells treated with FDCPt1 alone (Fig. 2A and B). Furthermore, a time course with various incubation times showed an increase in fluorescence intensity after one and six hours, while cells treated with oxaliplatin for 24 h showed much lower FDCPt1 fluorescence. This is consistent with complete metabolism of oxaliplatin to the inactive form after this longer time period, while at early time-points there are appreciable amounts of partially metabolised, monofunctional platinum complexes. This effect was observed for a number of cell types (Fig. S6, ESI†) and for the other commercially available Pt drugs, cisplatin and carboplatin (Fig. S7 and S8, ESI†). Time course studies of HT29 cells treated with cisplatin or oxaliplatin and imaged with FDCPt1 demonstrate the ability of the probe to distinguish different cellular processing (Fig. 2 and Fig. S8, ESI†); the observed slower deactivation of cisplatin is consistent with findings that HT29 cells are more susceptible to oxaliplatin than cisplatin treatment.25 Furthermore, imaging of drug-treated cells alone, with no probe, confirmed that signal enhancement is due to the probe alone, not to background fluorescence (Fig. S9, ESI†).
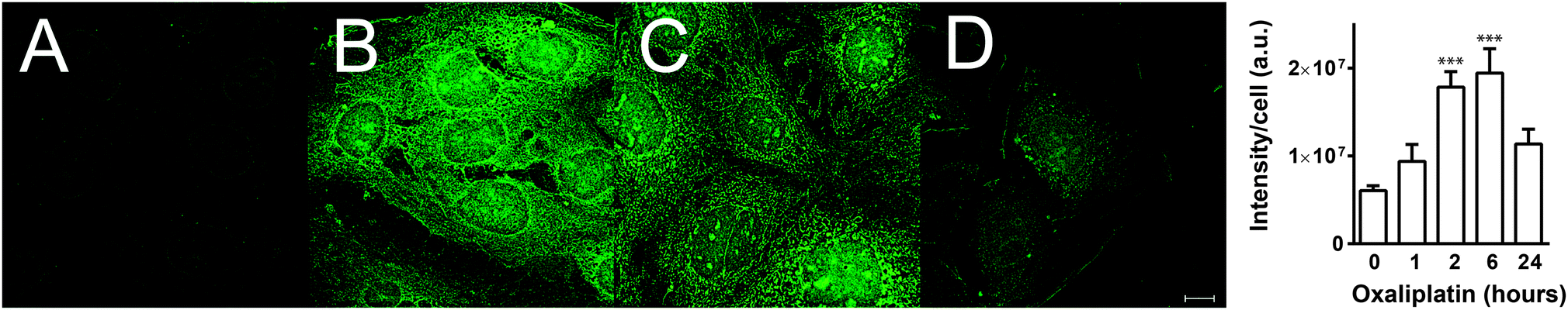 | ||
| Fig. 2 Deconvoluted fluorescence images of Caco-2 cells treated with FDCPt1 (20 μM) for 30 min alone (A) or with FDCPt1 after incubation with oxaliplatin (20 μM) for 1 (B), 6 (C) or 24 h (D). Scale bar represents 10 μm. | ||
In summary, here we have reported a sensitive probe FDCPt1 that is able to respond exclusively to reactive Pt(II) species within cells. Biological experiments were also performed using FDCPt1 in combination with Pt anti-cancer drugs. Results show that FDCPt1 is a 1:1 binder for monofunctional Pt species, and can reveal the presence of reactive Pt in colorectal cancer cells that have been treated with oxaliplatin. Some limitations of the probe still need to be overcome, such as its photostability, in future generations of Pt sensors. FDCPt1 will provide a useful scaffold for future probe design, as the ability to study the transformations of platinum's coordination environment is highly desirable for the future of platinum chemotherapy.
The authors acknowledge the technical assistance from Dr Louise Cole and Dr Yingying Su from the Advanced Microscopy Facility at the Bosch Institute, The University of Sydney.
Notes and references
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed.
- F. Yu, J. Megyesi and P. M. Price, Am. J. Physiol. Renal Physiol., 2008, 295, F44–F52 CrossRef CAS PubMed.
- D. C. Lemkuil, D. Nettesheim, C. F. Shaw and D. H. Petering, J. Biol. Chem., 1994, 269, 24792–24797 CAS.
- A. I. Ivanov, J. Christodoulou, J. A. Parkinson, K. J. Barnham, A. Tucker, J. Woodrow and P. J. Sadler, J. Biol. Chem., 1998, 273, 14721–14730 CrossRef CAS PubMed.
- C. S. Allardyce, P. J. Dyson, J. Coffey and N. Johnson, Rapid Commun. Mass Spectrom., 2002, 16, 933–935 CrossRef CAS PubMed.
- R. Stjernholm, F. W. Warner, J. W. Robinson, E. Ezekiel and N. Katayama, Bioinorg. Chem., 1978, 9, 277–280 CrossRef CAS.
- A. R. Timerbaev, S. S. Aleksenko, K. Polec-Pawlak, R. Ruzik, O. Semenova, C. G. Hartinger, S. Oszwaldowski, M. Galanski, M. Jarosz and B. K. Keppler, Electrophoresis, 2004, 25, 1988–1995 CrossRef CAS PubMed.
- E. L. Crossley, J. B. Aitken, S. Vogt, H. H. Harris and L. M. Rendina, Aust. J. Chem., 2011, 64, 253–257 CrossRef CAS.
- M. D. Hall, R. A. Alderden, M. Zhang, P. J. Beale, Z. Cai, B. Lai, A. P. J. Stampfl and T. W. Hambley, J. Struct. Biol., 2006, 155, 38–44 CrossRef CAS PubMed.
- M. D. Hall, G. J. Foran, M. Zhang, P. J. Beale and T. W. Hambley, J. Am. Chem. Soc., 2003, 125, 7524–7525 CrossRef CAS PubMed.
- J. Z. Zhang, N. S. Bryce, A. Lanzirotti, C. K. J. Chen, D. Paterson, M. D. de Jonge, D. L. Howard and T. W. Hambley, Metallomics, 2012, 4, 1209–1217 RSC.
- E. J. New, R. Duan, J. Z. Zhang and T. W. Hambley, Dalton Trans., 2009, 3092–3101 RSC.
- N. S. Bryce, J. Z. Zhang, R. M. Whan, N. Yamamoto and T. W. Hambley, Chem. Commun., 2009, 2673–2675 RSC.
- R. A. Alderden, H. R. Mellor, S. Modok, T. W. Hambley and R. Callaghan, Biochem. Pharmacol., 2006, 71, 1136–1145 CrossRef CAS PubMed.
- Y. Song, K. Suntharalingam, J. S. Yeung, M. Royzen and S. J. Lippard, Bioconjugate Chem., 2013, 24, 1733–1740 CrossRef CAS PubMed.
- Y. Yuan, Y. Chen, B. Z. Tang and B. Liu, Chem. Commun., 2014, 50, 3868–3870 RSC.
- K. Polec-Pawlak, J. K. Abramski, O. Semenova, C. G. Hartinger, A. R. Timerbaev, B. K. Keppler and M. Jarosz, Electrophoresis, 2006, 27, 1128–1135 CrossRef CAS PubMed.
- A. L. Garner and K. Koide, Chem. Commun., 2009, 83–85 RSC.
- D. Montagner, S. Q. Yap and W. H. Ang, Angew. Chem., 2013, 52, 11785–11789 CrossRef CAS PubMed.
- D. Montagner and P. J. Sanz Miguel, Dalton Trans., 2011, 40, 10809–10811 RSC.
- J. M. Bevilacqua and R. Eisenberg, Inorg. Chem., 1994, 33, 2913–2923 CrossRef CAS.
- G. Faraglia, D. Fregona, S. Sitran, L. Giovagnini, C. Marzano, F. Baccichetti, U. Casellato and R. Graziani, J. Inorg. Biochem., 2001, 83, 31–40 CrossRef CAS.
- W. M. Awni, J. V. Hoff, B. E. Shapiro and C. E. Halstenson, J. Clin. Pharmacol., 1994, 34, 1183–1190 CrossRef CAS.
- W. C. Reinhold, M. Sunshine, H. Liu, S. Varma, K. W. Kohn, J. Morris, J. Doroshow and Y. Pommier, Cancer Res., 2012, 72, 3499–3511 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc08077g |
| This journal is © The Royal Society of Chemistry 2015 |