
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A photoswitchable supramolecular complex with release-and-report capabilities†
Jesper R.
Nilsson
a,
Melanie C.
O’Sullivan
b,
S.
Li
a,
Harry L.
Anderson
b and
Joakim
Andréasson
*a
aDepartment of Chemical and Biological Engineering, Chalmers University of Technology, SE-412 96 Göteborg, Sweden. E-mail: a-son@chalmers.se; Fax: +46-31-772-3858; Tel: +46-31-772-2838
bDepartment of Chemistry, Oxford University, Oxford OX1 3TA, UK. E-mail: harry.anderson@chem.ox.ac.uk; Fax: +44-(0)1865-2-85002; Tel: +44-(0)1865-2-75704
First published on 26th November 2014
Abstract
A self-assembled supramolecular platform has been designed for reversibly controlling the concentration of a compound in solution, via a photochemical reaction. The system utilizes metal–ligand interactions between a Zn-porphyrin dimer and a pyridine-appended dithienylethene (DTE) photoswitch. In addition to reversible compound release, the spectral properties of the release scaffold provide a fluorescence-based reporting function.
Controlling when and where a substance is released and/or captured has great potential in a wide range of applications, e.g. the release of therapeutic compounds, sensing agents, or extraction of hazardous chemicals and pollutants. The unique properties of light sets it aside as a triggering stimulus for applications requiring spatiotemporally well-resolved, waste-free operation.1 A number of approaches have been investigated in pursuit of light-operated release systems and these efforts have consequently stimulated a rapidly progressing and inventive research field. A majority of the so far reported light-controlled release systems, however, are designed to exhibit irreversible release. This is typically achieved by light-induced cleavage of covalent bonds either directly using photolabile groups/linkers2 or second-hand by first generating for instance heat.3 Another well explored approach is the preparation of light-responsive drug loaded materials such as porous materials4 and/or micro/nanoparticles.5
An additional level of control is attained if the photo-release can be made reversible, as it allows for dynamic bidirectional control of the concentration profile in combination with precise timing of the dosage. Realization of such systems typically demands a photoswitchable component capable of interacting non-covalently with a host compound to form a supramolecular complex. There are several examples where this concept has been successfully implemented to manipulate for instance the release of small ions,6 as well as small molecules7 in a reversible manner. A particularly elegant example of photoreversible compound release was reported in the recent work by Clever and co-workers in which a photoswitchable coordination cage composed of Pd-coordinating dithienylethenes (DTEs) was shown to reversibly encapsulate inorganic guest molecules in response to light.7b
In this work, we report a conceptually different coordination-based approach for compound (capture and) release. Here, the differences in binding mode and binding strength between the two isomeric forms of a pyridine-appended DTE photoswitch (1, see Fig. 1) and a porphyrin dimer (P2) are combined into a self-assembled platform with release-and-report capabilities for lone pair-carrying guest molecules.
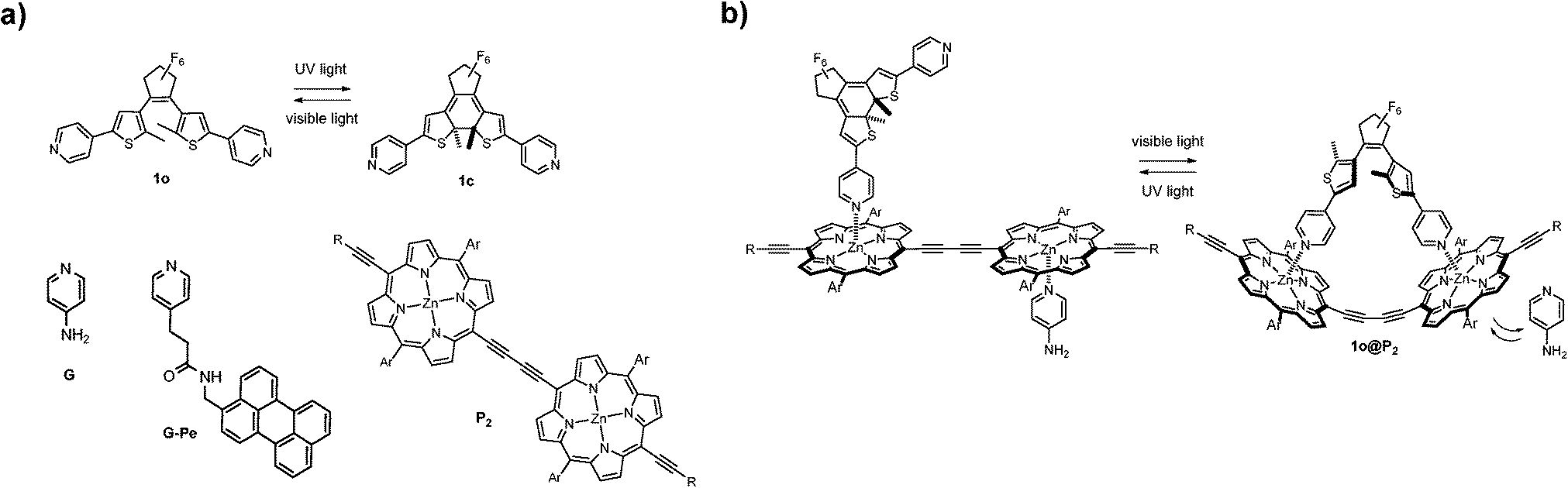 | ||
| Fig. 1 (a) Isomerization scheme of 1 and molecular structures of 4-aminopyridine (G), the perylene functionalized analog (G-Pe), and the porphyrin dimer (P2). (b) Photo-release principle. Ar = Si(C6H13)3, R = 3,5-di(tert-butyl)phenyl. | ||
Since its discovery, the DTE-backbone has found regular use in photoswitching applications due to its renowned high degree of photoconversion, thermal stability, and resistance to photofatigue.8 The photoinduced ring-closing (1o → 1c) is achieved with 302 nm UV-light (Φo→c = 0.57)9 and converts the sample to virtually 100% 1c. Subsequent visible light exposure (λ > 550 nm) completely opens the sample to 1o (Φc→o = 0.02). This switching cycle can be repeated several times without notable photodegradation10 (see Fig. S3 for absorption spectra of 1, and ESI† for details on isomerization quantum yield).
Porphyrin macrocycles have been included in numerous molecular and supramolecular constructs as building blocks with fluorescent, sensitizing, and/or energy/electron transfer capabilities.11 The two porphyrin units constituting the herein used P2 have a near-barrierless rotation around the central diethyne axis, thus allowing an even distribution of rotamers in the dimer.12 The aliphatic side chains (Ar and R in Fig. 1) effectively prevent dimer stacking, as no aggregation was detected up to mM concentrations. As for coordination to P2, the two DTE-isomers 1o and 1c are notably different. The ring-closed isomer (1c) coordinates axially to one Zn-center in P2 by donating a pyridine electron lone pair. This complexation occurs in a consecutive 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → 1:2 manner, with binding constants of Ka1 = 6.1 × 105 M−1 and Ka2 = 6.0 × 104 M−1, respectively as determined by UV/Vis titrations (see ESI† for titration details). For non-cooperative binding to a two-site host; Ka1 = 4 × Ka2, indicating that binding of the second 1c to P2 exhibits slightly negative cooperativity, possibly due to minor steric interactions. In sharp contrast, the structural flexibility of the ring-opened isomer (1o) allows it to stretch and instead form a 1:1 staple-like complex (1o@P2, Fig. 1). As a result of the double axial Zn-coordination, the latter binding is significantly stronger (Ka = 5.5 × 106 M−1), in effect causing initially bound compounds to be released into solution as a result of competitive binding. The 1o@P2 1:1 binding stoichiometry is strongly supported by the existence of no less than 8 isosbestic points throughout the 1o to P2 titration (Fig. S4, ESI†). There is also a clear resemblance to the spectral changes seen upon P2 planarization using static (non-photochromic) ligands.12 Furthermore, the 1o@P2 binding mode has been assessed by computational means.10 Binding of 1 to P2 has no significant effect on the rate of the photoinduced ring-opening reaction, while the corresponding closing rate is reduced by a factor of 6. This is likely due to coordination-induced restrictions in the conformational flexibility required for the isomerization process to occur. The usefulness of combining photoswitchable units and metalloporphyrins/porphyrinoids in supramolecular strategies is evidenced by the wide variety of processes brought under reversible photonic control using these building blocks. These include emission intensity,13 energy transfer,14 electron transfer,15 magnetic properties,16 and singlet oxygen generation.17
:1 → 1:2 manner, with binding constants of Ka1 = 6.1 × 105 M−1 and Ka2 = 6.0 × 104 M−1, respectively as determined by UV/Vis titrations (see ESI† for titration details). For non-cooperative binding to a two-site host; Ka1 = 4 × Ka2, indicating that binding of the second 1c to P2 exhibits slightly negative cooperativity, possibly due to minor steric interactions. In sharp contrast, the structural flexibility of the ring-opened isomer (1o) allows it to stretch and instead form a 1:1 staple-like complex (1o@P2, Fig. 1). As a result of the double axial Zn-coordination, the latter binding is significantly stronger (Ka = 5.5 × 106 M−1), in effect causing initially bound compounds to be released into solution as a result of competitive binding. The 1o@P2 1:1 binding stoichiometry is strongly supported by the existence of no less than 8 isosbestic points throughout the 1o to P2 titration (Fig. S4, ESI†). There is also a clear resemblance to the spectral changes seen upon P2 planarization using static (non-photochromic) ligands.12 Furthermore, the 1o@P2 binding mode has been assessed by computational means.10 Binding of 1 to P2 has no significant effect on the rate of the photoinduced ring-opening reaction, while the corresponding closing rate is reduced by a factor of 6. This is likely due to coordination-induced restrictions in the conformational flexibility required for the isomerization process to occur. The usefulness of combining photoswitchable units and metalloporphyrins/porphyrinoids in supramolecular strategies is evidenced by the wide variety of processes brought under reversible photonic control using these building blocks. These include emission intensity,13 energy transfer,14 electron transfer,15 magnetic properties,16 and singlet oxygen generation.17
Here, the drug chosen to illustrate the release event is the well known small molecule neurotransmitter 4-aminopyridine (G, see Fig. 1).18 In principle, any monodentate Lewis base can be used, the main prerequisite being adequate coordination capabilities (i.e. suitable binding strength) to Zn in P2. However, any change in the UV/Vis absorption of G upon coordination to P2 is obscured by the corresponding changes of the latter. Hence, a more straightforward means of monitoring binding/release of G is needed. Therefore, G-Pe was synthesized as a fluorescent model compound. G and G-Pe have identical binding modes to P2. The binding constants are: Ka1 = 3.2 × 105 M−1, Ka2 = 1.2 × 105 M−1 and Ka1 = 1.3 × 105 M−1, Ka2 = 3.6 × 104 M−1 for G and G-Pe respectively. The choice of the perylene fluorophore is motivated by the excellent spectral overlap between the emission spectra of G-Pe, and the P2 Soret band. Hence, binding to P2 efficiently quenches the G-Pe emission by excitation energy transfer (EET, R0 = 66 Å, see Fig. S12 for details, ESI†), possibly in combination with electron transfer (ET). Accordingly, the observed G-Pe emission originates exclusively from compound free in solution. It should be noted that G-Pe has nothing to do with the function of the release scaffold per se; it is used merely as a tool for monitoring the release.
The release of G-Pe is demonstrated in Fig. 2, where an initial cocktail of P2, 1o, and G-Pe gives rise to high emission, as the strongly coordinating 1o occupies both Zn binding-sites in P2. Subjecting the solution to UV-light causes a 1o → 1c isomerization, whereafter each DTE-unit cannot coordinate more than one Zn-center. In response, G-Pe binds to the liberated coordination site, and the emission is quenched. Subsequent visible light restores the high emission by reforming 1o and displacement of G-Pe from P2.
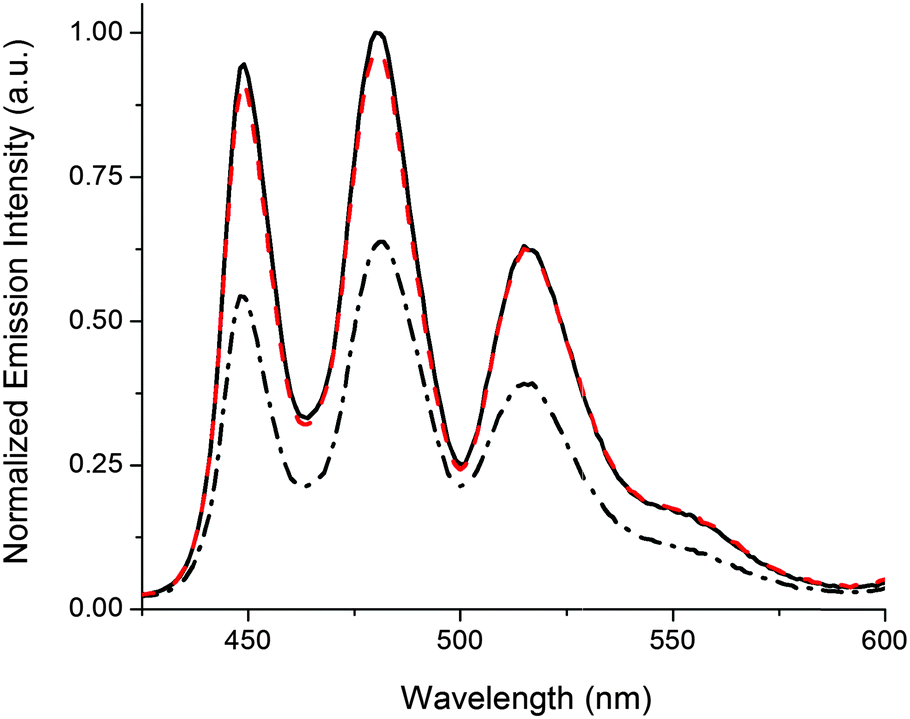 | ||
| Fig. 2 Reversible release of G-Pe in response to light monitored by the emission intensity of the uncomplexed G-Pe population. Applied concentrations; [P2] = 280 μM, [1] = 300 μM, [G-Pe] = 1 μM. Initially, 1 is in the open form 1o (solid black line). 2 min 302 nm UV-exposure triggers isomerization to 1c (dash-dotted black line). Subsequent visible light isomerizes the sample back to 1o (λ > 550 nm, 3 min, dashed red line). Please note that the experimental conditions/setup allows emission intensities unaffected by isomerization-induced inner filter effects.‡ | ||
In addition to triggering the compound release, formation of the doubly coordinated 1o@P2 complex forces the two porphyrin macrocycles to adopt a coplanar conformation. With this restriction in P2 rotameric distribution comes characteristic changes in the absorption spectrum (Fig. 3).
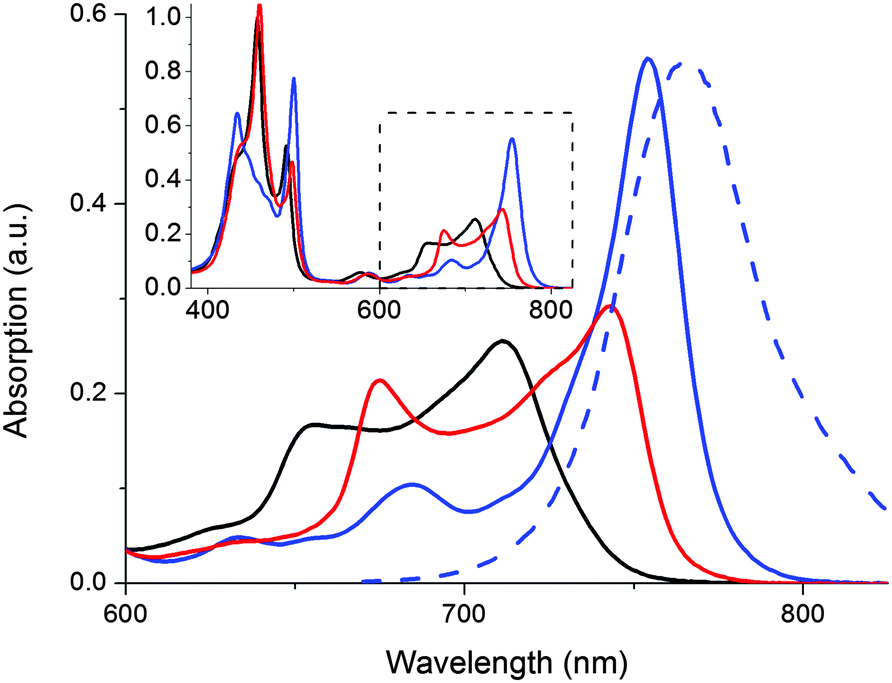 | ||
| Fig. 3 Absorption spectra of supramolecular P2-complexes in toluene: P2 (black line), 2G@P2 (red line), and the planarized 1o@P2 (blue line). The blue dashed line shows the 1o@P2 emission spectra (λexc = 510 nm). Inset: Absorption from 380–825 nm. | ||
In Fig. 3, the absorption spectral signatures of P2 and the two types of P2 complexes are shown. Monodentate species typically induce a red-shift in the P2 Q-band (711 nm → ∼745 nm). The planar complex (1o@P2) exhibits a further red shifted, and significantly hyperchromic absorption band centered at 754 nm. The spectral features inherent to the rotational distribution of diethyne-linked porphyrin dimers and oligomers have been used to control the rate of electron transfer19 as well as singlet oxygen generation,20 by allowing selective excitation of planar or randomly oriented rotamers. In our laboratory, we have devised a molecular memory capable of non-destructive readout based on photochromic planarization of a porphyrin dimer.10 Here, as the compound release proceeds concurrently with a marked increase in absorption of P2 around 750 nm, it is possible to read the state of the release scaffold to confirm the release event by probing the emission at 800 nm, following excitation at 790 nm. Hence, the inherent fluorescent properties of the scaffold are in line with the so-called release-and-report function.
The typical “release” (e.g. caged compounds2b) requires UV-light. For most light-controlled applications, this is not optimal, due to limited penetration depth and potential damage to surrounding tissue, materials, and/or the released compound itself. Here, a notable advantage is that both the release- and the report functions are triggered by low energy photons (λ up to ca. 700 nm and almost 800 nm, respectively). To illustrate the performance and stability of the release scaffold, we prepared a sample containing P2, 1o, and G, and subjected the solution to alternating irradiation of 302 nm and visible light (λ > 550 nm), probing the report output after each irradiation step (Fig. 4).
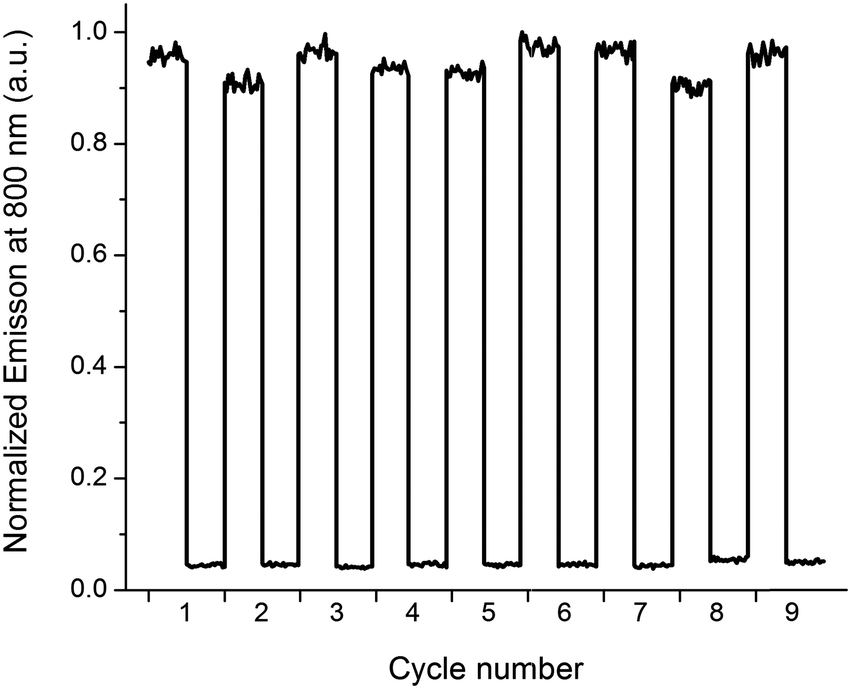 | ||
| Fig. 4 Photoswitching performance of the release-and-report system, as monitored via the report function, i.e. emission intensity at 800 nm (λexc = 790 nm). Applied concentrations; [P2] = 280 μM, [1] = 300 μM, [G] = 1 μM. Each cycle starts with 1 in the open form 1o (high intensity). 2 min 302 nm UV-exposure triggers isomerization to 1c (low intensity). Subsequent visible light (λ > 550 nm, 3 min) isomerizes the sample back to 1o. | ||
It is clear that 1c ⇌ 1o isomerization causes dramatic differences in emission intensity from P2, and that switching between the two types of complexes is fully reversible, and proceeds with no apparent photodegradation.
The authors appreciate the limited biological compatibility of the solvents used throughout this proof-of-principle study. Thus, for use in biological environments, solubilization of the release scaffold needs to be addressed. In this context it deserves mentioning that metal–ligand coordination approaches to photo-release (albeit irreversible) in living organisms have been reported,21 along with examples of porphyrin dimers22 and DTEs23 adapted for, and used in, biological applications.
A self-assembled system for reversible photo-control of compound release has been demonstrated. The unique spectral properties inherent to this system conveniently allows for fluorescence-based assessment of the state of the release scaffold, i.e. whether the compound is bound or not. This reporter ability has to our knowledge not been demonstrated in a reversible release system to date; thus this work represents a conceptually valuable addition to existing photo-operated release systems. The affinity of Zn-porphyrins for amine-based ligands implies that this reversible release system could be applied to a wide range of ligands, eliminating the need for guest-specific synthetic efforts.
This work was financed by the Swedish Research Council (Grant 622-2010-280) and the European Research Council (ERC FP7/2007–2013 Grant No. 203952). Professor Neil Branda is acknowledged for valuable discussions.
Notes and references
- R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982 RSC
.
-
(a) K. L. Ciesienski and K. J. Franz, Angew. Chem., Int. Ed., 2011, 50, 814 CrossRef CAS PubMed
-
(a) A. B. S. Bakhtiari, D. Hsiao, G. Jin, B. D. Gates and N. R. Branda, Angew. Chem., Int. Ed., 2009, 48, 4166 CrossRef CAS PubMed
-
(a) N. K. Mal, M. Fujiwara, Y. Tanaka, T. Taguchi and M. Matsukata, Chem. Mater., 2003, 15, 3385 CrossRef CAS
-
(a) H. Yan, C. Teh, S. Sreejith, L. Zhu, A. Kwok, W. Fang, X. Ma, K. T. Nguyen, V. Korzh and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 8373 CrossRef CAS PubMed
-
(a) M. Blank, L. Soo, H. Wassermann and B. Erlanger, Science, 1981, 214, 70 CAS
-
(a) H. Dube and J. Rebek, Angew. Chem., Int. Ed., 2012, 51, 3207 CrossRef CAS PubMed
- M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS PubMed
- Quantum yield for the ring-closing reaction was determined in methanol in; K. Matsuda, Y. Shinkai, T. Yamaguchi, K. Nomiyama, M. Isayama and M. Irie, Chem. Lett., 2003, 32, 1178 CrossRef CAS
- J. Kärnbratt, M. Hammarson, S. Li, H. L. Anderson, B. Albinsson and J. Andréasson, Angew. Chem., Int. Ed., 2010, 49, 1854 CrossRef PubMed
-
(a) I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659 CrossRef CAS PubMed
- M. U. Winters, J. Kärnbratt, M. Eng, C. J. Wilson, H. L. Anderson and B. Albinsson, J. Phys. Chem. C, 2007, 111, 7192 CAS
-
(a) T. B. Norsten and N. R. Branda, Adv. Mater., 2001, 13, 347 CrossRef CAS
- J. Otsuki, A. Yasuda and T. Takido, Chem. Commun., 2003, 608 RSC
- A. J. Myles and N. R. Branda, J. Am. Chem. Soc., 2000, 123, 177 CrossRef
- S. Thies, H. Sell, C. Bornholdt, C. Schütt, F. Köhler, F. Tuczek and R. Herges, Chem. – Eur. J., 2012, 18, 16358 CrossRef CAS PubMed
- L. Hou, X. Zhang, T. C. Pijper, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 910 CrossRef CAS PubMed
- M. Müller, P. W. Dierkes and W.-R. Schlue, Brain Res., 1999, 826, 63 CrossRef
- M. U. Winters, J. Kärnbratt, H. E. Blades, C. J. Wilson, M. J. Frampton, H. L. Anderson and B. Albinsson, Chem. – Eur. J., 2007, 13, 7385 CrossRef CAS PubMed
- M. K. Kuimova, M. Balaz, H. L. Anderson and P. R. Ogilby, J. Am. Chem. Soc., 2009, 131, 7948 CrossRef CAS PubMed
- L. Zayat, C. Calero, P. Alborés, L. Baraldo and R. Etchenique, J. Am. Chem. Soc., 2003, 125, 882 CrossRef CAS PubMed
- M. K. Kuimova, S. W. Botchway, A. W. Parker, M. Balaz, H. A. Collins, H. L. Anderson, K. Suhling and P. R. Ogilby, Nat. Chem., 2009, 1, 69 CrossRef CAS PubMed
- T. C. S. Pace, V. Müller, S. Li, P. Lincoln and J. Andréasson, Angew. Chem., Int. Ed., 2013, 52, 4393 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedure for G-Pe, spectroscopic details. See DOI: 10.1039/c4cc08513b |
| ‡ Under the selected conditions, [1] and [P2] give rise to a significant optical density in the solution. It should therefore be noted that the emission measurements were performed in a 1 mm cuvette with front-face detection and excitation at an isosbestic point, λexc = 410 nm. |
| This journal is © The Royal Society of Chemistry 2015 |