
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemote rearrangement of the metal center in a (η6-C6Me6)Ru(II) complex†
Koichi
Takano
,
Yousuke
Ikeda
,
Shintaro
Kodama
and
Youichi
Ishii
*
Department of Applied Chemistry, Faculty of Science and Engineering, Chuo University, 1-13-27 Kasuga, Bunkyo-ku, Tokyo, Japan. E-mail: yo-ishii@kc.chuo-u.ac.jp; Fax: +81-3-3817-1895; Tel: +81-3-3817-1901
First published on 5th January 2015
Abstract
Reaction of [(η6-C6Me6)RuCl(Ph)(PMe3)] with internal alkynes in the presence of NaBArF4 gave rise to the 1,4-Ru migration to form the o-vinylaryl complex, providing the first example of 1,4-metal migration of a group 8 metal center; in one case further isomerization to an η3-allyl complex was observed.
C–H bond activation by intramolecular 1,4-metal migration has recently attracted considerable attention as the key step in the transition metal catalyzed synthetic reactions.1 In this process, a transition metal center is at first introduced into the carbon atom four bonds away from the ‘target’ C–H group by a standard organometallic method, and then exchanges its position with the hydrogen atom to complete the C–H activation. This chemistry was established by Miura, Larock, and others using Rh(I)2 and Pd(II)3 complexes, and since then 1,4-migration reactions of these metals have been investigated actively from synthetic as well as mechanistic points of view. Although 1,4-migration of other metals such as cobalt,4 iridium,5 nickel6 and platinum3f was also reported, only group 9 and 10 metal centers with a d8 electron configuration have served as the effective reaction sites throughout these studies, which limits generality of the 1,4-metal migration.
In the course of our investigation into the activation of internal alkynes by using group 9 d6 metal complexes, we have recently disclosed that the Rh(III) center in [Cp*Rh(CR1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR2Ph)(PR3)][BArF4] (R = Ph, Me; R1, R2 = alkyl, aryl; ArF = 3,5-(CF3)2C6H3), which is formed from the reactions of [Cp*RhCl(Ph)(PR3)] with R1C
CR2Ph)(PR3)][BArF4] (R = Ph, Me; R1, R2 = alkyl, aryl; ArF = 3,5-(CF3)2C6H3), which is formed from the reactions of [Cp*RhCl(Ph)(PR3)] with R1C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR2 in the presence of NaBArF4, takes part in the vinyl-to-aryl 1,4-migration to form the o-(vinyl)aryl complexes [Cp*Rh{o-C6H4CR2CHR1}(PR3)][BArF4] under very mild conditions.7a,b,8 Furthermore, similar reactions of the iridium analog complex [Cp*IrCl(Ph)(PMe3)] with internal alkynes result in not only 1,4- but also direct 1,3-Ir(III) migration, providing the first experimental evidence for the direct 1,3-metal migration accompanied by the C–H bond activation.7c,9 To broaden the scope of the 1,4-metal migration, we have now aimed to expand the reaction site to group 8 metal complexes. Herein we describe the first example of the 1,4-migration of a Ru(II) center.
CR2 in the presence of NaBArF4, takes part in the vinyl-to-aryl 1,4-migration to form the o-(vinyl)aryl complexes [Cp*Rh{o-C6H4CR2CHR1}(PR3)][BArF4] under very mild conditions.7a,b,8 Furthermore, similar reactions of the iridium analog complex [Cp*IrCl(Ph)(PMe3)] with internal alkynes result in not only 1,4- but also direct 1,3-Ir(III) migration, providing the first experimental evidence for the direct 1,3-metal migration accompanied by the C–H bond activation.7c,9 To broaden the scope of the 1,4-metal migration, we have now aimed to expand the reaction site to group 8 metal complexes. Herein we describe the first example of the 1,4-migration of a Ru(II) center.
When [(η6-C6Me6)RuCl(Ph)(PMe3)] (1), which was prepared by the reaction of [(η6-C6Me6)RuCl2(PMe3)] with PhMgCl, was allowed to react with PhCCPh in the presence of NaBArF4 in C2H4Cl2 at room temperature for a few min, the color of the reaction mixture turned red from orange, and the 31P{1H} NMR spectrum showed a new singlet at δ −0.58. After recrystallization, the o-(vinyl)arylruthenium complex [(η6-C6Me6)Ru{o-C6H4C(Ph)CHPh}(PMe3)][BArF4] (2a) with a Ru–(vinyl CH) agostic interaction was obtained in 89% yield as the sole product (Scheme 1). In the 1H NMR spectrum, the vinyl proton of 2a appears as a doublet at δ −4.47 (d, 2JPH = 13.7 Hz), confirming the presence of an effective agostic interaction. In fact, this vinyl signal exhibits considerably large high-field shift compared with those of closely related Rh(III) and Ir(III) complexes such as [Cp*M{o-C6H4C(Ph)CHPh}(PMe3)][BArF4] (M = Rh, δ 0.38; M = Ir, δ −0.30).7
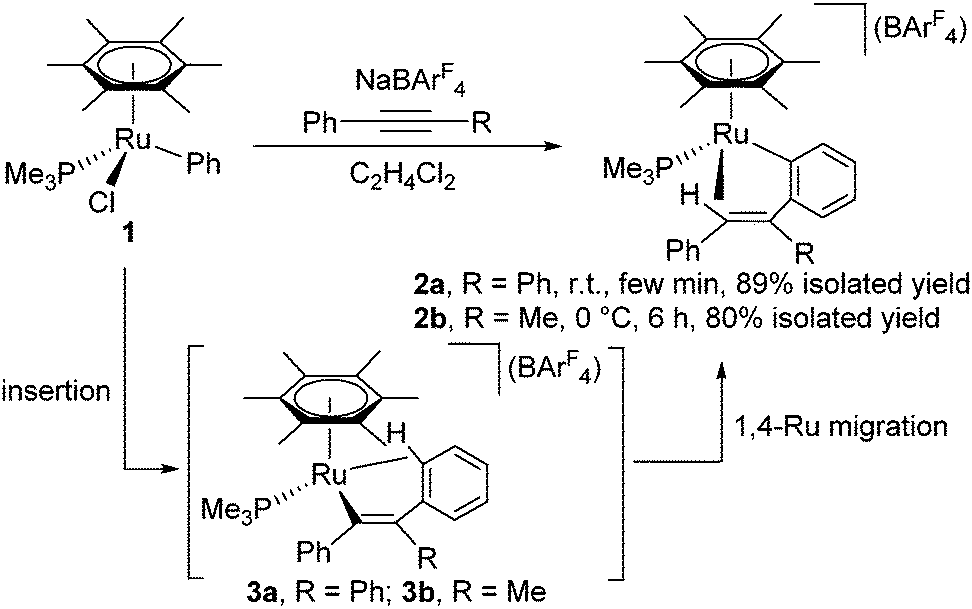 | ||
| Scheme 1 Reaction of 1 with PhCCR and NaBArF4. | ||
The molecular structure of 2a has been determined unambiguously by a single-crystal X-ray diffraction study (Fig. 1, left). Complex 2a displays a three-legged piano-stool structure with the η6-C6Me6, PMe3, and o-(vinyl)aryl ligands as well as the agostic vinyl C–H group. The Ru1–C1 distance is obviously longer than regular ruthenium–carbon σ-bond lengths but comparable with common agostic Ru–CH distances.10 This structure clearly indicates that 2a was formed by the vinyl-to-aryl 1,4-Ru migration from the vinylruthenium intermediate [(η6-C6Me6)Ru{C(Ph)CPh2}(PMe3)][BArF4] (3a) with a Ru–(o-CH of Ph) agostic interaction, which was too reactive to be characterized.11 Considering the isoelectronic nature of (η6-C6Me6)Ru(II) and Cp*Rh(III) fragments, the present 1,4-Ru(II) migration is presumed to proceed via the σ-complex assisted metathesis process (σ-CAM).7,12,13
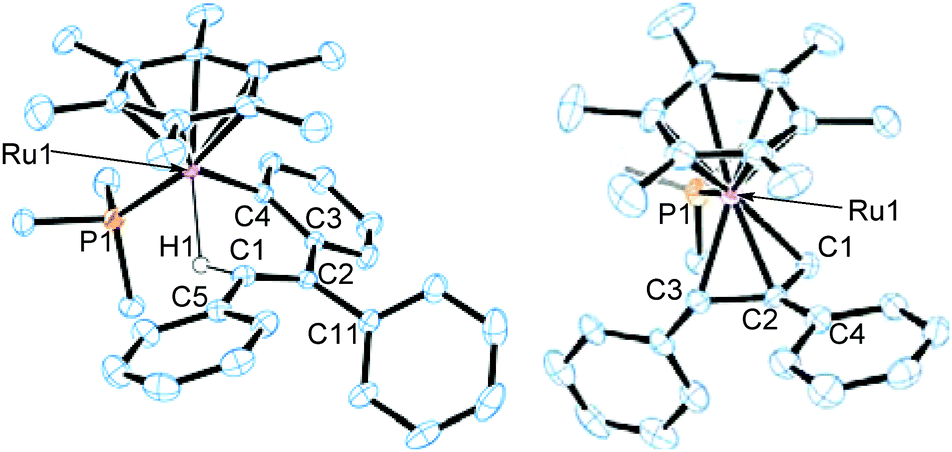 | ||
| Fig. 1 ORTEP drawings of 2a (left) and 4 (right). The anionic part and hydrogen atoms except for H1 of 2a are omitted for clarity. Selected bond lengths (Å): 2a, Ru1–P1, 2.3190(10); Ru1–C1, 2.426(4); Ru1–C4, 2.065(4); Ru1–H1, 1.87(4); C1–C2, 1.354(5). 4, Ru1–P1, 2.3517(16); Ru1–C1, 2.201(5); Ru1–C2, 2.165(5); Ru1–C3, 2.168(5); C1–C2, 1.452(6); C2–C3, 1.459(7). | ||
Similarly, the reaction of 1 with PhCCMe at 0 °C afforded [(η6-C6Me6)Ru{o-C6H4C(Me)CHPh}(PMe3)][BArF4] (2b) in 80% isolated yield,14 and its structure was also confirmed by a preliminary X-ray diffraction study. 2b exhibits a singlet at δ −0.26 in the 31P{1H} NMR and a high-field shifted vinyl signal at δ −4.24 (d, 2JPH = 10.3 Hz) in the 1H NMR, the latter of which is diagnostic of an agostic interaction.15
It would be interesting to note that a deuterium labeling experiment using [(η6-C6Me6)RuCl(C6D5)(PMe3)] (1-d5) and PhCCPh failed to give the product fully deuterated at the vinyl CH group; instead we observed the vinyl CH signal with 0.5 H intensity in the 1H NMR, suggesting the formation of [(η6-C6Me6)Ru{o-C6D4C(Ph)CDPh}(PMe3)][BArF4] (2a-d4/d1) and [(η6-C6Me6)Ru{o-C6H4C(C6D5)CHPh}(PMe3)][BArF4] (2a-d5/d0) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (see ESI†). This result clearly demonstrates that 3a-d5 is rapidly interconverting between 3a-d5 and 3a′-d5 by the CC bond rotation,16 and the subsequent 1,4-Ru migration can proceed from both structures to give 2a-d4/d1 and 2a-d5/d0 (Scheme 2). In addition, according to our previous study, vinyl-to-aryl 1,4-metal migration is a reversible process, and it is reasonable to consider that 2a-d4/d1 and 2a-d5/d0 are in equilibrium at room temperature.7b,c,17 It should also be mentioned that the CC bond rotation of 3a is even more facile than that of the rhodium system; only transfer of a D atom to the vinyl position was observed in the reaction of [Cp*Rh(C6D5)(PPh3)]+ with PhCCPh.7a
:1 ratio (see ESI†). This result clearly demonstrates that 3a-d5 is rapidly interconverting between 3a-d5 and 3a′-d5 by the CC bond rotation,16 and the subsequent 1,4-Ru migration can proceed from both structures to give 2a-d4/d1 and 2a-d5/d0 (Scheme 2). In addition, according to our previous study, vinyl-to-aryl 1,4-metal migration is a reversible process, and it is reasonable to consider that 2a-d4/d1 and 2a-d5/d0 are in equilibrium at room temperature.7b,c,17 It should also be mentioned that the CC bond rotation of 3a is even more facile than that of the rhodium system; only transfer of a D atom to the vinyl position was observed in the reaction of [Cp*Rh(C6D5)(PPh3)]+ with PhCCPh.7a
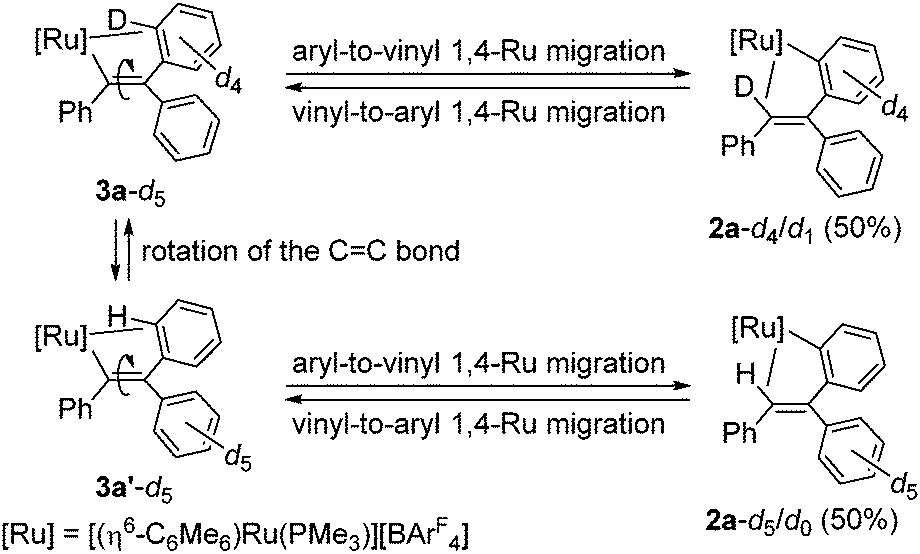 | ||
| Scheme 2 Interconversion between 2a-d4/d1 and 2a-d5/d0. | ||
Although a considerable number of C–H activation reactions utilizing the 1,4-metal migration have been described in the literature,1 they are exclusively performed at group 9 and 10 complexes. Very recently, Gunnoe reported that the reaction of [Cp*FePh(CO)(NCMe)] with MeCCMe ends in the formation of [Cp*Fe(η5-1-hydroxy-2,3-dimethylindenyl)], where a novel coupling reaction of the Ph, CO and MeCCMe ligands leads to an indenyl skeleton. For this transformation, 1,4-migration of the iron(II) center was suggested to be involved,18 while no experimental evidence for 1,4-migration of group 8 metals has been reported. The present reaction provides the first example of 1,4-metal migration in a structurally well-defined group 8 metal complex and reinforces Gunnoe's proposal.
Complex 2a is stable at 50 °C, and no further reaction was observed at this temperature. In contrast, 2b was further isomerized slowly at room temperature (Scheme 3). When an in situ generated C2H4Cl2 solution of 2b was stirred at room temperature for 48 h, the color of the reaction mixture turned orange from red, and formation of a new complex showing a 31P{1H} NMR signal at δ 3.03 was observed. Purification of this reaction mixture by column chromatography and recrystallization afforded the η3-allyl complex [(η6-C6Me6)Ru{η3-CH2C(Ph)CHPh}(PMe3)][BArF4] (4) in 59% yield as pale yellow crystals. The 1H NMR of 4 shows a set of CH signals of the η3-allyl ligand at δ 5.29 (br, CH2C(Ph)CHPh), 3.61 (d, 2JHH = 3.7 Hz, syn-CH2C(Ph)CHPh), and 1.91 (dd, 3JPH = 18.3 Hz, 2JHH = 3.7 Hz, anti-CH2C(Ph)CHPh), and the molecular structure of 4 has been determined unambiguously by a single-crystal X-ray diffraction study (Fig. 1, right). The bond lengths and angles of the η3-allyl ligand in 4 are comparable to those of known η3-allyl ruthenium complexes.19 It should be pointed out that the transformation of 2b into 4 can be viewed as a formal 1,3-metal migration accompanied by CH activation.
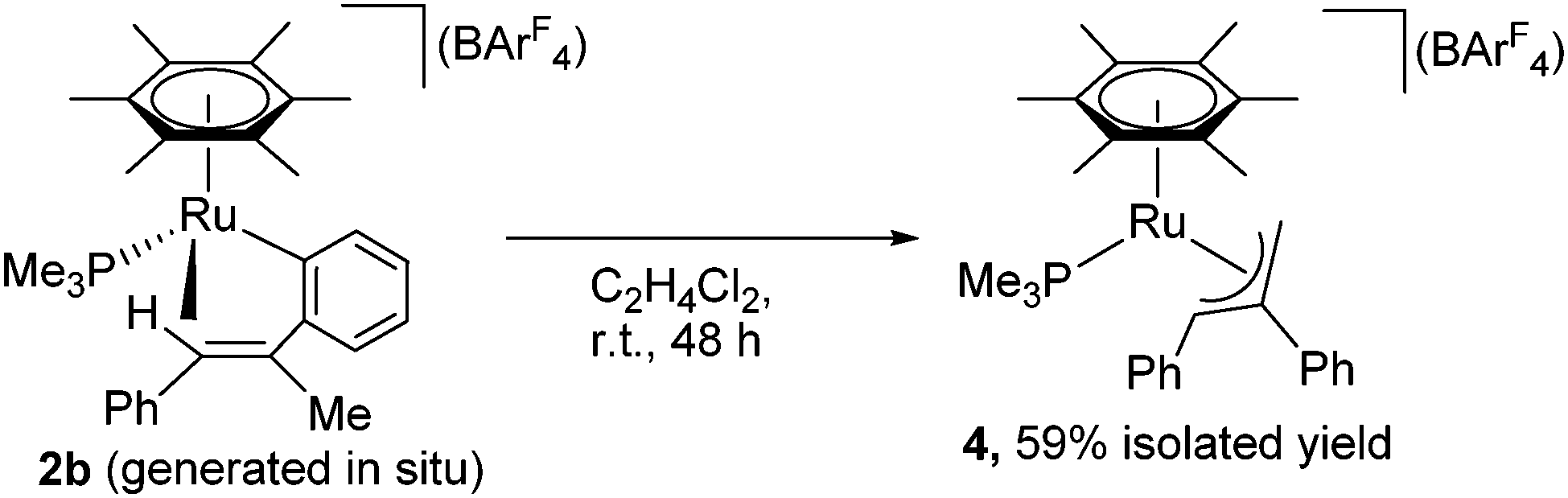 | ||
| Scheme 3 Isomerization of 2b to 4. | ||
To gain mechanistic information regarding this isomerization, we have performed deuterium labeling experiments by using 1-d5 and examined the destination of the deuterium atoms in 4 by means of 1H NMR. According to our previous study,7c we have hypothesized two mechanisms for the isomerization of 2b into 4 as illustrated in Scheme 4. Path A is constituted of the rotation of the Caryl–Cvinyl bond in 2b and the subsequent aryl-to-allyl 1,4-Ru migration, and this route would lead to 4-d1/d4 in which one deuterium atom is introduced into the C3 atom (see Fig. 1) of the η3-allyl ligand. On the other hand, path B includes the direct vinyl-to-allyl 1,3-Ru migration from 3b′ which is in turn generated from 2b through the aryl-to-vinyl 1,4-Ru migration (vide supra) followed by the rotation of the CC bond. In path B, 4-d0/d5, which has no deuterium atom in the η3-allyl moiety, is expected to be formed. The 1H NMR of the actual reaction product showed selective formation of 4-d1/d4, indicating that only path A is operative in the present reaction system, and the 1,3-Ru migration is not included (see ESI†).
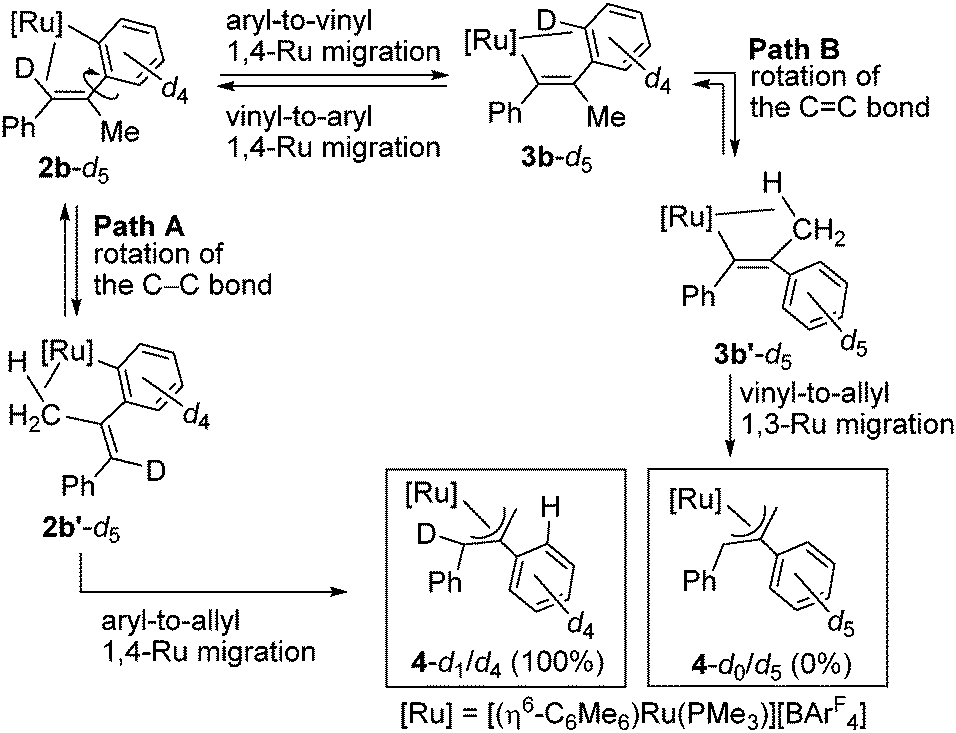 | ||
| Scheme 4 Plausible mechanisms of formation of 4. | ||
This result is in stark contrast to a related reaction with an iridium complex. Thus, we have recently reported the formation of a similar η3-allyliridium complex, [Cp*Ir{η3-CH2C(Ph)CHPh}(PMe3)][BArF4], by the reaction of [Cp*IrCl(Ph)(PMe3)] with PhCCMe and NaBArF4, and its deuterium labeling experiment with [Cp*IrCl(C6D5)(PMe3)] demonstrated that both path A and B are operative with the iridium system, where path B (direct 1,3-Ir migration) is the major reaction route (30:70).7c It is interesting to point out that the reaction mechanism for the present formal 1,3-metal migration is strongly dependent on the metal center involved, although we must await further investigation including DFT calculations to clarify the origin of such metal dependency.
In conclusion, we have revealed that the reaction of [(η6-C6Me6)Ru(Ph)(PMe3)]+ with PhCCR (R = Ph, Me) afforded the o-(vinylaryl) complex 2a,b by way of the alkyne insertion into the Ru–Ph bond to generate 3a,b followed by the vinyl-to-aryl 1,4-migration. Complex 2b was further isomerized to η3-allyl complex 4, and the deuterium labeling experiments disclosed that this isomerization proceeds through the rotation of the Caryl–Cvinyl bond in 2b and the second aryl-to-allyl 1,4-Ru migration. These reactions provide the first example of 1,4-migration of a Ru(II) center. A mechanistic study involving DFT calculations and synthetic applications of this reaction are now in progress.
This research was financially supported by JST ACT-C. Y. Ikeda thanks the Japan Society for the Promotion of Science (JSPS) Research Fellowships for Young Scientists.
Notes and references
- For recent reviews see: (a) S. Ma and Z. Gu, Angew. Chem., Int. Ed., 2005, 44, 7512–7517 CrossRef CAS PubMed; (b) F. Shi and R. C. Larock, Top. Curr. Chem., 2010, 292, 123–164 CrossRef CAS.
- For examples of 1,4-Rh(I) migration, see: (a) K. Oguma, M. Miura, T. Satoh and M. Nomura, J. Am. Chem. Soc., 2000, 122, 10464–10465 CrossRef CAS; (b) T. Hayashi, K. Inoue, N. Taniguchi and M. Ogasawara, J. Am. Chem. Soc., 2001, 123, 9918–9919 CrossRef CAS; (c) H. Yamabe, A. Mizuno, H. Kusama and N. Iwasawa, J. Am. Chem. Soc., 2005, 127, 3248–3249 CrossRef CAS PubMed; (d) M. Tobisu, I. Hyodo, M. Onoe and N. Chatani, Chem. Commun., 2008, 6013–6015 RSC; (e) K. Sasaki, T. Nishimura, R. Shintani, E. A. B. Kantchev and T. Hayashi, Chem. Sci., 2012, 3, 1278–1283 RSC; (f) J. Zhang, J.-F. Liu, A. Ugrinov, A. F. X. Pillai, Z.-M. Sun and P. Zhao, J. Am. Chem. Soc., 2013, 135, 17270–17273 CrossRef CAS PubMed.
- For examples of 1,4-Pd(II) migration, see: (a) R. F. Heck, J. Organomet. Chem., 1972, 37, 389–396 CrossRef CAS; (b) G. Bocelli, M. Catellani and G. P. Chiusoli, J. Organomet. Chem., 1984, 279, 225–232 CrossRef; (c) G. Dyker, Angew. Chem., Int. Ed. Engl., 1994, 33, 103–105 CrossRef; (d) Q. Tian and R. C. Larock, Org. Lett., 2000, 2, 3329–3332 CrossRef CAS PubMed; (e) M. A. Campo, Q. Huang, T. Yao, Q. Tian and R. C. Larock, J. Am. Chem. Soc., 2003, 125, 11506–11507 CrossRef CAS PubMed; (f) A. Singh and P. R. Sharp, J. Am. Chem. Soc., 2006, 128, 5998–5999 CrossRef CAS PubMed; (g) H. J. Lee, K. H. Kim, S. H. Kim and J. N. Kim, Tetrahedron Lett., 2013, 54, 170–175 CrossRef CAS PubMed; (h) T. Piou, A. Bunescu, Q. Wang, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 12385–12389 CrossRef CAS PubMed.
- (a) B.-H. Tan, J. Dong and N. Yoshikai, Angew. Chem., Int. Ed., 2012, 51, 9610–9614 CrossRef CAS PubMed; (b) B. Wu and N. Yoshikai, Angew. Chem., Int. Ed., 2013, 52, 10496–10499 CrossRef CAS PubMed.
- B. M. Partridge, J. S. González and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 6523–6527 CrossRef CAS PubMed.
- A. L. Keen, M. Doster and S. A. Johnson, J. Am. Chem. Soc., 2007, 129, 810–819 CrossRef CAS PubMed.
- (a) Y. Ikeda, K. Takano, S. Kodama and Y. Ishii, Chem. Commun., 2013, 49, 11104–11106 RSC; (b) Y. Ikeda, K. Takano, M. Waragai, S. Kodama, N. Tsuchida, K. Takano and Y. Ishii, Organometallics, 2014, 33, 2142–2145 CrossRef CAS; (c) Y. Ikeda, K. Takano, S. Kodama and Y. Ishii, Organometallics, 2014, 33, 3998–4004 CrossRef CAS.
- The 1,4-migration of Rh(III) center is rare, and only an example of the catalytic application of this process is reported, see: D. J. Burns and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 9931–9935 CrossRef CAS PubMed.
- Zhao reported a formal aryl-to-aryl 1,3-Rh(I) migration of a 2,6-dimethoxyphenyl complex, which occurred through consecutive two 1,4-migrations, see: ref. 2f.
- (a) R. Kuhlman, K. Folting and K. G. Caulton, Organometallics, 1995, 14, 3188–3195 CrossRef CAS; (b) D. Huang, K. Folting and K. G. Caulton, J. Am. Chem. Soc., 1999, 121, 10318–10322 CrossRef CAS; (c) W. Baratta, C. Mealli, E. Herdtweck, A. Ienco, S. A. Mason and P. Rigo, J. Am. Chem. Soc., 2004, 126, 5549–5562 CrossRef CAS PubMed; (d) W. Baratta, A. D. Zotto, G. Esposito, A. Sechi, M. Toniutti, E. Zangrando and P. Rigo, Organometallics, 2004, 23, 6264–6272 CrossRef CAS.
- With analogous Cp*RhIII complexes, a related vinylmetal intermediate was observed by NMR analysis, see; ref. 7a and b.
- For the Ru(II) mediated C–H activation reactions, the σ-bond metathesis or related mechanism has commonly been proposed rather than the oxidative addition/reductive elimination process, see: (a) I. Özdemir, S. Serpil, B. Çetinkaya, C. Gourlaouen, F. Maseras, C. Bruneau and P. H. Dixneuf, J. Am. Chem. Soc., 2008, 130, 1156–1157 CrossRef PubMed; (b) E. F. Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133, 10161–10170 CrossRef CAS PubMed.
- Y. Li, G. He and E. A. B. Kantchev, Phys. Chem. Chem. Phys., 2014, 16, 24250–24255 RSC.
- Complex 2b was not obtained in a pure form at room temperature because of the competing isomerization of 2b to 4.
- A carefully recrystallized sample of 2b shows a minor (∼10%) 31P{1H} NMR signal at δ 4.32 in addition to the major signal of 2b (see ESI†). This minor species has not been characterized but is tentatively assigned as a rotamer of 2b or 3b (see Scheme 4).
- For the mechanism of the CC bond rotation of vinyl complexes, see:
(a) R. S. Tanke and R. H. Crabtree, J. Am. Chem. Soc., 1990, 112, 7984–7989 CrossRef CAS;
(b) R. H. Crabtree, New J. Chem., 2003, 27, 771–772 RSC;
(c) B. C. Ranu, K. Chattopadhyay and S. Banerjee, J. Org. Chem., 2006, 71, 423–425 CrossRef CAS PubMed.
- As expected, the reaction of 1-d5 with PhCCMe and NaBArF4 leads to complete deuteration at the vinyl CH group of 2b.
- S. E. Kalman, T. B. Gunnoe and M. Sabat, Organometallics, 2014, 33, 5457–5463 CrossRef CAS.
- (a) G. E. Herberich and T. P. Spaniol, J. Chem. Soc., Dalton Trans., 1993, 2471–2476 RSC; (b) H. Nishiyama, M. Konno and K. Aoki, Organometallics, 2002, 21, 2536–2539 CrossRef CAS; (c) A. Ramirez-Monroy, M. A. Paz-Sandoval, M. J. Ferguson and J. M. Stryker, Organometallics, 2007, 26, 5010–5024 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Text, figures and CIF files giving experimental procedures and crystallographic data. CCDC 1036838 and 1036845. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc09699a |
| This journal is © The Royal Society of Chemistry 2015 |