
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmine-catalyzed tunable reactions of allenoates with dithioesters: formal [4+2] and [2+2] cycloadditions for the synthesis of 2,3-dihydro-1,4-oxathiines and enantioenriched thietanes†
Hai-Bin
Yang
,
Yu-Chao
Yuan
,
Yin
Wei
* and
Min
Shi
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: weiyin@mail.sioc.ac.cn; mshi@mail.sioc.ac.cn
First published on 5th March 2015
Abstract
The chemoselective [4+2] vs. [2+2] cycloaddition between allenoates and dithioesters can be controlled by switching the nucleophilic amine catalyst. The two modes of cyclizations represent the first example of controllable and chemoselective annulations between allenoates and dienophiles catalyzed by amine. These cyclizations are useful in offering a divergent synthesis of sulfur-containing heterocycles. On the basis of this investigation, it can be realized that dithioesters with a vicinal electron-withdrawing group can react not only like a Michael acceptor but also as a ketone or imine.
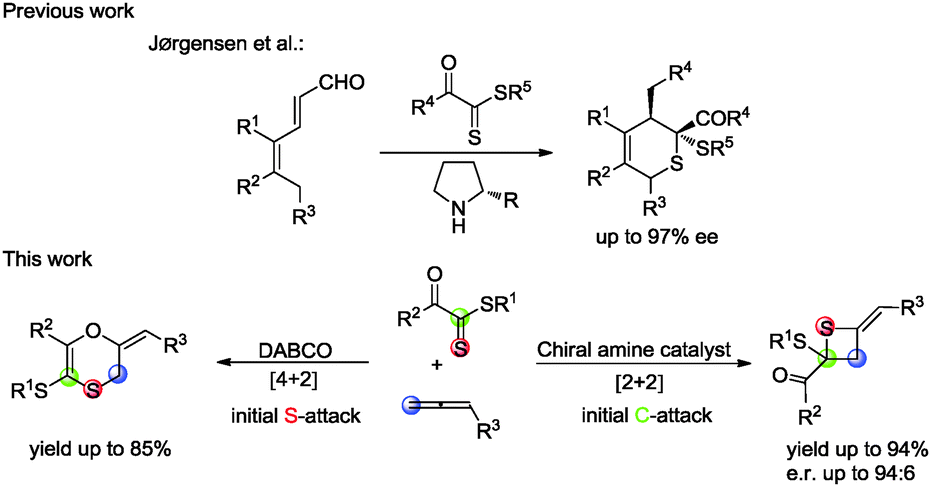
Lewis base catalysis, often classified as nucleophilic catalysis, remains an active and dynamic area of interest for synthetic chemists. Allenoates as a class of attractive substrates are often used in Lewis base catalyzed reactions due to their facile preparation and diverse reactivity.1 The addition of a Lewis base to the electrophilic, sp-hybridized, β-carbon of an α-allenic ester results in the generation of a zwitterionic enolate-like intermediate which subsequently takes part in divergent annulation reaction modes with alkene, ketone and imine, including [3+2],2 [4+2],3 [3+3]4 and [2+2]5 annulations. However, to the best of our knowledge, thiocarbonyls have not been employed in the cycloaddition with allenoates catalyzed by phosphines or amines. In 2013, Jørgensen reported an asymmetric organocatalytic thio-Diels–Alder reaction between dienals and dithioesters via trienamine catalysis.6 Based on theoretical investigations, they suggested that this thio-Diels–Alder reaction was a stepwise process rather than a concerted [4+2] cycloaddition and dithioesters with a vicinal electron-withdrawing group acted like a Michael acceptor in their reaction (Scheme 1). With these precedents in mind and in connection with our ongoing efforts on developing novel reactions using nitrogen-containing Lewis bases as nucleophilic catalysts,7 we envisaged that treating allenoates with dithioesters under the catalysis of the nucleophilic amine might afford 2,3-dihydro-1,4-oxathiine derivatives (Scheme 1).
 | ||
| Scheme 1 Amine catalyzed cycloaddition based on thiocarbonyls. | ||
Gratifyingly, we obtained the expected 1,4-oxathiine derivatives which were generated by a formal [4+2] cycloaddition between allenoates and dithioesters under the catalysis of DABCO. 1,4-Oxathiine represents an important structural motif featured in biologically active compounds (Fig. 1).8 For example, carboxin (Vitavaxa®) and its 4,4-dioxide analogue, oxycarboxin (Plantavaxa®), are well known as systemic fungicides and both are the active components of many effective commercially available pesticides used worldwide to control crop smuts and rust diseases.9 Motivated by these significances, intensive investigations have been conducted to develop practically useful and step-economic methodologies to the 1,4-oxathiine architecture.10 Accordingly, the discovery of novel strategies for the synthesis of 1,4-oxathiine with good functional-group tolerance through simple operation is highly desirable. Besides the expected [4+2] cycloaddition products in our experiment, we could also obtain enantioenriched [2+2] cycloaddition products by choosing different chiral catalysts. It remains a challenge to selectively generate different products from identical substrates, utilizing catalyst rather than substrate control. Thietanes also commonly present in a variety of natural products as well as biologically active compounds.11,12 Although thietanes can be synthesized via a lot of known methods, one-step asymmetric catalysis methodology to construct this scaffold has not been reported.13 Herein, we wish to report the amine-catalyzed tunable cycloadditions between allenoates and dithioesters.
 | ||
| Fig. 1 Biologically active molecules containing 1,4-oxathiine or thietane. | ||
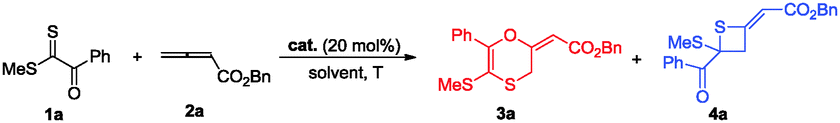
Initial studies using dithioester 1a and allenoate 2a as the substrate were aimed at determining the reaction outcome and subsequently optimizing the reaction conditions. The results are summarized in Table 1. We found that [4+2] cycloaddition product 3a was obtained in 20% yield as a major product with concomitant formation of [2+2] cycloaddition compound 4a in 10% yield when the reaction was carried out in CH2Cl2 under the catalysis of DABCO (20 mol%) at room temperature for 24 h (Table 1, entry 1). Instead of DABCO, other commonly used nitrogen-containing catalysts such as DMAP, DBU and Et3N were also tested; however, they afforded a complex product mixture under the same reaction conditions. The examination of solvent effects using DABCO (20 mol%) as the catalyst revealed that toluene was the solvent of choice (Table 1, entries 1–5). After extensive screening of the reaction temperature, we found that this [4+2] cycloaddition was sensitive to the reaction temperature and the reaction provided compound 3a in 85% yield along with 4a in 9% yield at −40 °C. Further attempts to switch the regioselectivity to produce 4a were carried out with 1a and 2a using various cinchona alkaloid-derived catalysts. Using Quinine, (DHQD)2PHAL or C1 as a catalyst, almost all starting materials 1a were recovered (Table 1, entries 10–12). When β-ICD was employed as a catalyst, this reaction could afford 4a in 30% yield with 54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :46 e.r. value (Table 1, entry 13). We rationalized that the cyclic ether motif of β-ICD is critical to promote the reaction between allenoates and dithioesters because of the reduced steric hindrance around the nucleophilic nitrogen of β-ICD by restraining the conformational freedom of the bulky aromatic moiety.14 To improve the stereoselectivity, further studies were focused on the effect of the hydrogen bonding donor motif of the catalyst on this annulation reaction. Under the catalysis of C2 which was designed and prepared by Deng, cycloadduct 4a was obtained in similar results as that of β-ICD (Table 1, entry 14).15 The reaction could give better results in terms of yield and chemoselectivity in the presence of C3 containing a sterically hindered thiourea group (Table 1, entry 15). The examination of solvent effects using C3 (20 mol%) as the catalyst also revealed that toluene was the solvent of choice, affording 4a in 71% yield with 83:17 e.r. value (Table 1, entries 16 and 17). Lowering the temperature to 0 °C did not improve the enantioselectivity of this [2+2] cycloaddition (Table 1, entry 18). Reducing the catalyst loading to 10 mol% had no significant influence on the reaction outcome (Table 1, entry 19).
:46 e.r. value (Table 1, entry 13). We rationalized that the cyclic ether motif of β-ICD is critical to promote the reaction between allenoates and dithioesters because of the reduced steric hindrance around the nucleophilic nitrogen of β-ICD by restraining the conformational freedom of the bulky aromatic moiety.14 To improve the stereoselectivity, further studies were focused on the effect of the hydrogen bonding donor motif of the catalyst on this annulation reaction. Under the catalysis of C2 which was designed and prepared by Deng, cycloadduct 4a was obtained in similar results as that of β-ICD (Table 1, entry 14).15 The reaction could give better results in terms of yield and chemoselectivity in the presence of C3 containing a sterically hindered thiourea group (Table 1, entry 15). The examination of solvent effects using C3 (20 mol%) as the catalyst also revealed that toluene was the solvent of choice, affording 4a in 71% yield with 83:17 e.r. value (Table 1, entries 16 and 17). Lowering the temperature to 0 °C did not improve the enantioselectivity of this [2+2] cycloaddition (Table 1, entry 18). Reducing the catalyst loading to 10 mol% had no significant influence on the reaction outcome (Table 1, entry 19).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Cat. | Solvent | T (°C) | Yield of 3ab (%) | Yield of 4ab (%) |
3a:4ac |
e.r. of 4ad (%) |
a Dithioester 1a (0.1 mmol), allenoate 2a (0.1 mmol), and the catalyst (0.02 mmol) were stirred in 1 mL of solvent.
b Isolated yields after chromatography are shown.
c Determined by 1H NMR spectroscopy.
d Determined by HPLC analysis.
e 10 mol% of catalyst loading.
|
|||||||
| 1 | DABCO | CH2Cl2 | rt | 20 | 10 | 2.0:1 |
— |
| 2 | DABCO | Toluene | rt | 37 | 7 | 5.3:1 |
— |
| 3 | DABCO | THF | rt | 36 | 4 | 9.0:1 |
— |
| 4 | DABCO | DMF | rt | 32 | Trace | — | — |
| 5 | DABCO | CH3CN | rt | 33 | Trace | — | — |
| 6 | DABCO | Toluene | 0 | 60 | 12 | 5.0:1 |
— |
| 7 | DABCO | Toluene | −20 | 75 | 9 | 8.3:1 |
— |
| 8 | DABCO | Toluene | −40 | 82 | 9 | 9.1:1 |
— |
| 9 | DABCO | Toluene | −40 | 85 | 9 | 9.4:1 |
— |
| 10 | Quinine | Toluene | rt | — | — | — | — |
| 11 | (DHQD)2PHAL | Toluene | rt | — | — | — | — |
| 12 | C1 | Toluene | rt | — | — | — | — |
| 13 | β-ICD | THF | rt | 30 | 30 | 1:1.0 |
54:46 |
| 14 | C2 | THF | rt | 30 | 30 | 1:1.0 |
53:47 |
| 15 | C3 | THF | rt | 47 | 53 | 1:1.1 |
68:32 |
| 16 | C3 | Toluene | rt | 27 | 71 | 1:2.6 |
83:17 |
| 17 | C3 | CH2Cl2 | rt | 28 | 68 | 1:2.4 |
64:36 |
| 18 | C3 | Toluene | 0 | 33 | 65 | 1:2.0 |
83:17 |
| 19e | C3 | CH2Cl2 | rt | 27 | 70 | 1:2.6 |
68:32 |
With the optimized reaction conditions in hand, we next investigated the generality of this [4+2] cyclization reaction (Table 2). As for dithioester 1, both electron-deficient (1b–1c and 1e) and electron-rich (1d and 1f) aromatic substituents at the α-position were tolerated in this [4+2] cyclization reaction although 1d and 1f afforded the desired products with lower chemoselectivity (Table 2, entries 1–5). Even for dithioester 1g having a naphthalen-2-yl group, the corresponding product 3g was furnished in 81% yield (Table 2, entry 6). We were pleased to find that heteroaromatic group-substituted dithioester 1h was also suitable for this reaction, affording 3h in moderate yield (Table 2, entry 7). The structure of compound 3h was confirmed by X-ray diffraction.16 Furthermore, the optimized reaction conditions were also applicable to dithioester 1i bearing an alkyl group at the α-position (Table 2, entry 8). Using dithioester 1j containing a benzylthio group in this [4+2] cyclization reaction afforded 3j in 72% yield under standard conditions (Table 2, entry 9). Notably, besides allenoate 2a, 1-phenylbuta-2,3-dien-1-one 2b was also applicable to this [4+2] cyclization reaction without the formation of a [2+2] cycloadduct (Table 2, entries 10–15). We reasoned that the better chemoselectivity may be caused by the stronger electron withdrawing ability of the carbonyl group in 2b.
|
|
||||
|---|---|---|---|---|
| Entrya | R1, R2 | R3 | Yield of 3b (%) |
3:4c |
| a Dithioester 1 (0.2 mmol), allene 2 (0.2 mmol) and DABCO (0.04 mmol) were stirred in 2 mL of toluene at −40 °C. b Isolated yields after chromatography are shown. c Determined by 1H NMR spectroscopy. | ||||
| 1 | 1b, Me, 2-chlorophenyl | 2a, CO2Bn | 3b, 73 | >99:1 |
| 2 | 1c, Me, 3-bromophenyl | 2a, CO2Bn | 3c, 71 | 17.0:1 |
| 3 | 1d, Me, 3-methoxyphenyl | 2a, CO2Bn | 3d, 81 | 11.0:1 |
| 4 | 1e, Me, 4-bromophenyl | 2a, CO2Bn | 3e, 76 | >99:1 |
| 5 | 1f, Me, p-tolyl | 2a, CO2Bn | 3f, 78 | 6.0:1 |
| 6 | 1g, Me, 2-naphthalenyl | 2a, CO2Bn | 3g, 81 | 10.0:1 |
| 7 | 1h, Me, 2-thienyl | 2a, CO2Bn | 3h, 52 | 4.4:1 |
| 8 | 1i, Me, tBu | 2a, CO2Bn | 3i, 44 | 1.3:1 |
| 9 | 1j, Bn, Ph | 2a, CO2Bn | 3j, 72 | 16.0:1 |
| 10 | 1j, Bn, Ph | 2b, COPh | 3k, 68 | — |
| 11 | 1b, Me, 2-chlorophenyl | 2b, COPh | 3l, 67 | — |
| 12 | 1c, Me, 3-bromophenyl | 2b, COPh | 3m, 68 | — |
| 13 | 1e, Me, 4-bromophenyl | 2b, COPh | 3n, 57 | — |
| 14 | 1g, Me, 2-naphthalenyl | 2b, COPh | 3o, 45 | — |
| 15 | 1h, Me, 2-thienyl | 2b, COPh | 3p, 43 | — |

Using catalyst C3, we were able to suppress the [4+2] cycloaddition of allenoates with dithioesters and selectively access thietane products. Under the optimized reaction conditions, we next investigated the generality of this [2+2] cyclization reaction and the results are summarized in Table 3. In the case of dithioester 1g, the corresponding product 4b was obtained in 62% yield with 84:16 e.r. value (Table 3). As for 1h, the reaction afforded thietane 4c in 53% yield along with 61:39 e.r. value (Table 3). However, using dithioester 1i as a substrate, the reaction became sluggish so that the conversion of dithioester 1i was only 50% after 2 days, affording thietane 4d in 34% yield with 84:16 e.r. value (Table 3).
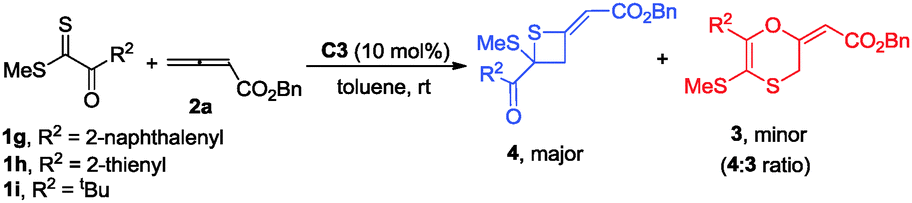

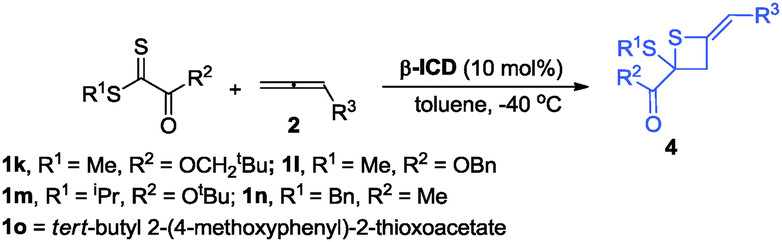
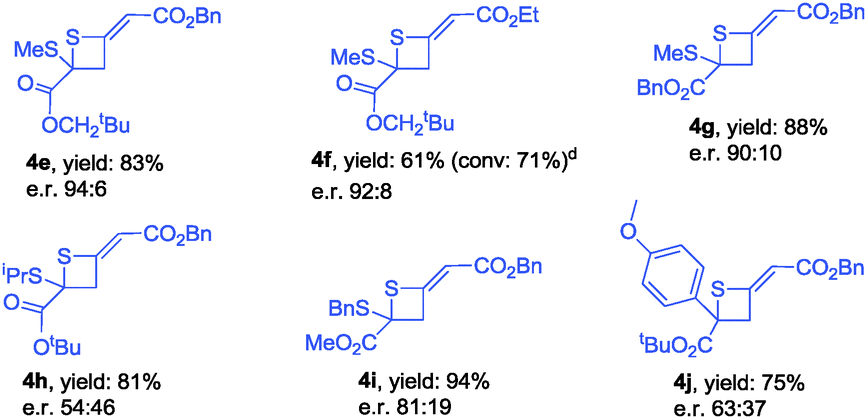
During further exploration, we found that this [2+2] cycloaddition was not only catalyst-dependent but also substrate-dependent. When 2-thioxoacetates were employed as substrates, thietanes were the exclusive products no matter what kinds of nucleophilic amine catalysts were used. In terms of controlling the enantioselectivity in this [2+2] cyclization reaction, β-ICD was better than C3 in this case. After simple examination, the optimal reaction conditions had been identified to carry out the reaction in toluene at −40 °C for 24 h using 10 mol% of β-ICD as the catalyst. As for the reaction of neopentyl 2-(methylthio)-2-thioxoacetate 1l with benzyl buta-2,3-dienoate and ethyl buta-2,3-dienoate, the reactions proceeded smoothly to afford the corresponding products 4e and 4f in good e.r. values (Table 4). The enantioselectivity of this [2+2] cyclization was sensitive to the substituent linked to the sulfur atom and product 4h was obtained in 54:46 e.r. value. Changing the neopentyl ester group to benzyl, and methyl ester group, the corresponding products 4g and 4i were obtained in good yields and good e.r. value. Notably, thioketone was also applicable to this novel [2+2] cyclization reaction, affording the desired product 4j in good yield albeit with moderate e.r. value (Table 4).
To illustrate the synthetic utility of these obtained products, we further developed some transformations of [2+2] cycloadducts (Scheme 2). Treating 4j with MCPBA in CH2Cl2 gave sulfone 5a in 39% yield. Ozonation of 4j afforded β-thiolactone 6a at −78 °C in 34% yield, which underwent ring-opening with phenylmethanamine to generate product 7a. The structure of 7a has been identified by X-ray diffraction and the CIF data are presented in the ESI.†17
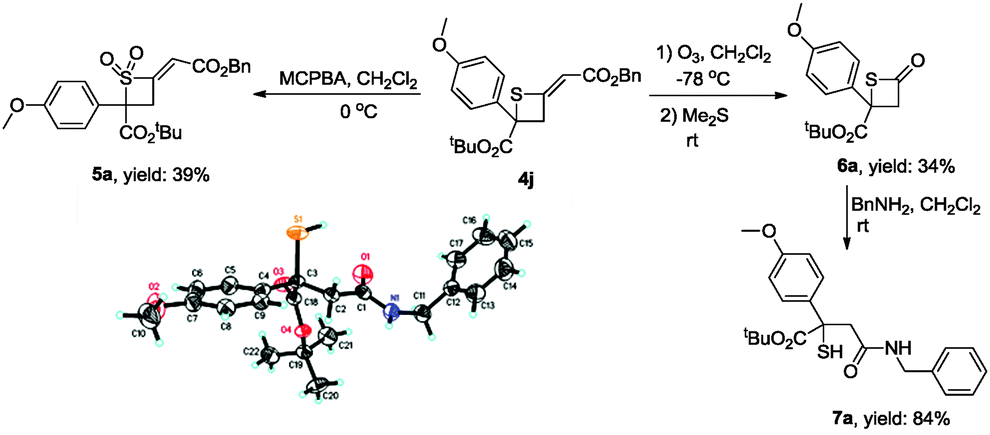 | ||
| Scheme 2 The transformations of thietane 4j. | ||
A plausible mechanism is depicted in Scheme 3 to account for the selective control. The [4+2] and [2+2] cyclization reactions are initiated by the formation of zwitterionic intermediate Avia the nucleophilic addition of amine to allenoate. When amine is DABCO, the thiophilic attack of A on the sulfur atom of the thiocarbonyl group in 1 generates intermediate B. The subsequent cyclization delivers product 3a along with the liberation of the catalyst. Based on this mechanism, the reaction of dithioesters bearing electron-deficient R2 group with allenoate is favored because the negative charge in intermediate B can be stabilized by delocalization. This is why they have better chemoselectivity (Table 2, entries 1 and 4). When the amine catalyst is C3 or β-ICD, the nucleophilic attack of zwitterionic intermediate A on the carbon atom of the thiocarbonyl group in 1 is preferred, perhaps due to the observation that the hydrogen bonding interaction between the catalyst with its hydrogen bonding donor and the substrate leads to the chemoselective [2+2] exceeding over [4+2] cycloaddition (Scheme 4).18 Thus, the C–S bond is formed and the catalyst is released to give product 4a.
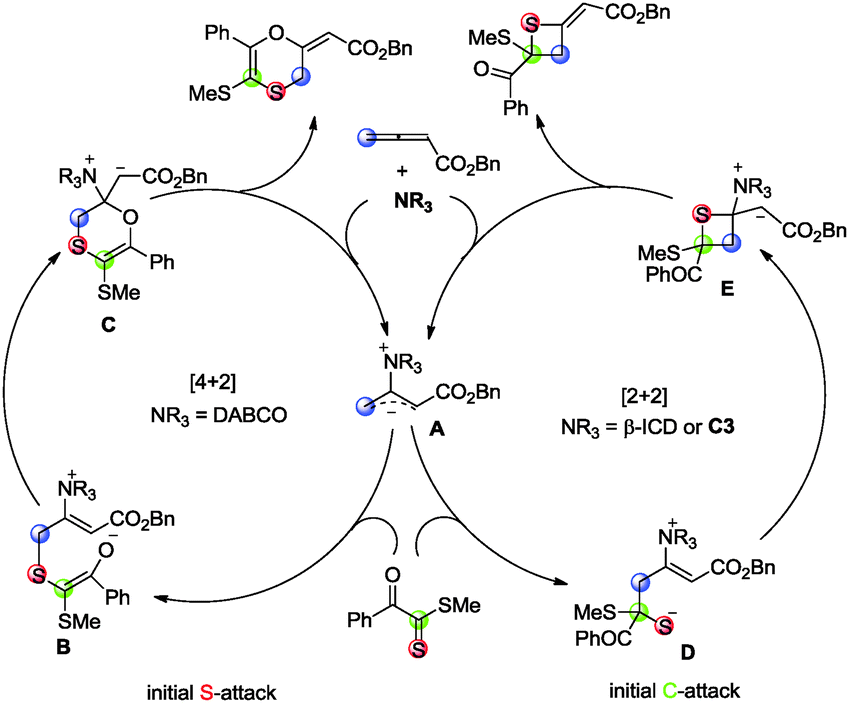 | ||
| Scheme 3 Proposed mechanism. | ||
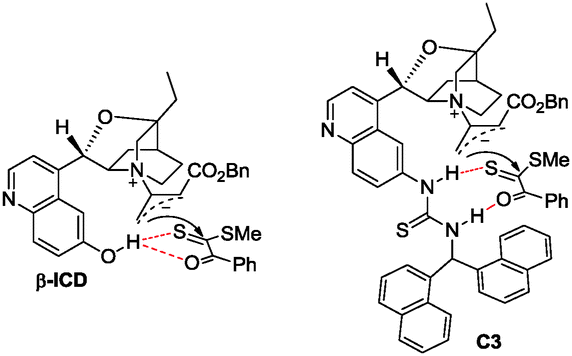 | ||
| Scheme 4 Proposed hydrogen bonding interaction mode. | ||
In summary, we have developed novel amine-catalyzed tunable cycloadditions between allenoates and dithioesters, providing a divergent synthesis of 2,3-dihydro-1,4-oxathiines and enantioenriched thietanes. Through this finding, we can realize that dithioesters with a vicinal electron-withdrawing group can react not only like a Michael acceptor but also as a ketone or imine. The exploration of novel catalysts to further improve the enantioselectivity of [2+2] cycloaddition between allenoates and dithioesters is underway in our laboratory.
We thank the National Basic Research Program of China (973)-2015CB856603, and the National Natural Science Foundation of China (20472096, 21372241, 21361140350, 20672127, 21421091, 21302203, 21102166, 21372250 and 20732008).
Notes and references
- (a) M. Schmittel and C. Wohrle, J. Org. Chem., 1995, 60, 8223 CrossRef CAS; (b) K. Nakatani, A. Okamoto and I. Saito, Angew. Chem., Int. Ed., 1999, 38, 3378 CrossRef CAS.
- (a) C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906 CrossRef CAS; (b) G. Zhu, Z. Chen, Q. Jiang, D. Xiao, P. Cao and X. Zhang, J. Am. Chem. Soc., 1997, 119, 3836 CrossRef CAS; (c) J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1426 CrossRef CAS PubMed; (d) A. Voituriez, A. Panossian, N. Fleury-Brégeot, P. Retailleau and A. Marinetti, J. Am. Chem. Soc., 2008, 130, 14030 CrossRef CAS PubMed; (e) H. Xiao, Z. Chai, C.-W. Zheng, Y.-Q. Yang, W. Liu, J.-K. Zhang and G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 4467 CrossRef CAS PubMed; (f) Y. Fujiwara and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 12293 CrossRef CAS PubMed; (g) F. Zhong, X. Han, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 7837 CrossRef CAS PubMed; (h) C. E. Henry, Q. Xu, Y. C. Fan, T. J. Martin, L. Belding, T. Dudding and O. Kwon, J. Am. Chem. Soc., 2014, 136, 11890 CrossRef CAS PubMed.
- (a) X. F. Zhu, J. Lan and O. Kwon, J. Am. Chem. Soc., 2003, 125, 4716 CrossRef CAS PubMed; (b) R. P. Wurz and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 12234 CrossRef CAS PubMed; (c) Y. S. Tran and O. Kwon, J. Am. Chem. Soc., 2007, 129, 12632 CrossRef CAS PubMed; (d) T. Wang and S. Ye, Org. Lett., 2010, 12, 4168 CrossRef CAS PubMed; (e) X. Wang, T. Fang and X. Tong, Angew. Chem., Int. Ed., 2011, 50, 5361 CrossRef CAS PubMed; (f) K. D. Ashtekar, R. J. Staples and B. Borhan, Org. Lett., 2011, 13, 5732 CrossRef CAS PubMed; (g) X.-Y. Chen, M.-W. Wen, S. Ye and Z.-X. Wang, Org. Lett., 2011, 13, 1138 CrossRef CAS PubMed; (h) Z. G. Shi and T.-P. Loh, Angew. Chem., Int. Ed., 2013, 52, 8584 CrossRef CAS PubMed.
- C. Li, Q. Zhang and X. Tong, Chem. Commun., 2010, 46, 7828 RSC.
- (a) J. B. Denis, G. Masson, P. Retailleau and J.-P. Zhu, Angew. Chem., Int. Ed., 2011, 50, 5356 CrossRef CAS PubMed; (b) L. B. Saunders and S. J. Miller, ACS Catal., 2011, 1, 1347 CrossRef CAS; (c) T. Wang, X.-Y. Chen and S. Ye, Tetrahedron Lett., 2011, 52, 5488 CrossRef CAS PubMed; (d) S. Takizawa, F. A. Arteaga, Y. Yoshida, M. Suzuki and H. Sasai, Org. Lett., 2013, 15, 4142 CrossRef CAS PubMed; (e) P. Selig, A. Turočkin and W. Raven, Chem. Commun., 2013, 49, 2930 RSC.
- H. Jiang, D. C. Cruz, Y. Li, V. H. Lauridsen and K. A. Jørgensen, J. Am. Chem. Soc., 2013, 135, 5200 CrossRef CAS PubMed.
- (a) C.-K. Pei, Y. Jiang, Y. Wei and M. Shi, Angew. Chem., Int. Ed., 2012, 51, 11328 CrossRef CAS PubMed; (b) H.-B. Yang, Y.-Z. Zhao, R. Sang and M. Shi, J. Org. Chem., 2014, 79, 3519 CrossRef CAS PubMed; (c) H.-B. Yang, Y.-Z. Zhao, R. Sang, Y. Wei and M. Shi, Adv. Synth. Catal., 2014, 356, 3799 CrossRef CAS.
- (a) C. J. Paget, E. M. Dennis, J. Nelson and D. C. DeLong, J. Med. Chem., 1970, 13, 620 CrossRef CAS; (b) M. Eltze, Eur. J. Pharmacol., 1996, 311, 187 CrossRef CAS; (c) M. Harfenist, D. M. Joseph, S. C. Spence, D. P. C. Mcgee, M. D. Reeves and H. L. White, J. Med. Chem., 1997, 40, 2466 CrossRef CAS PubMed; (d) J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719 CrossRef CAS PubMed.
- (a) B. von Schmeling and M. Kulka, Science, 1966, 152, 659 CAS; (b) B. von Schmeling, M. Kulka, D. S. Thiara and W. A. Harrison, US Pat., 3249499, 1966Chem. Abstr., 1966, 65, 7190 Search PubMed; (c) M. Kulka, Can. J. Chem., 1980, 58, 2044 CrossRef CAS PubMed.
- (a) J. Mattay and C. Dittmer, J. Org. Chem., 1986, 51, 1894 CrossRef CAS; (b) W. S. Lee, H. G. Hahn and and K. D. Nam, J. Org. Chem., 1986, 51, 2789 CrossRef CAS; (c) W. S. Lee, O. S. Park, J. K. Choi and K. D. Nam, J. Org. Chem., 1987, 52, 5374 CrossRef CAS; (d) W. S. Lee, H. G. Hahn and K. H. Chang, J. Org. Chem., 1989, 54, 2455 CrossRef CAS; (e) S. Kim and C. M. Cho, Heterocycles, 1994, 38, 1971 CrossRef CAS; (f) G. Capozzi, R. G. W. Franck, M. Mattioli, S. Menichetti, C. Nativi and G. Valle, J. Org. Chem., 1995, 60, 6416 CrossRef CAS; (g) V.-H. Nguyen, H. Nishino, S. Kajikawa and K. Kurosawa, Tetrahedron, 1998, 54, 11445 CrossRef CAS; (h) S. Watanabe, E. Mori, H. Nagai, T. Iwamura, T. Iwama and T. Kataoka, J. Org. Chem., 2000, 65, 8893 CrossRef CAS PubMed.
- (a) W. C. Lo, J. E. Hunter, G. B. Watson, A. Patny, P. S. Iyer and J. Boruwa, US Pat. 171308 A1, 2014 Search PubMed; (b) C. J. Yves, R. Peter, P. Thomas and E. Q. Myriem, WO Pat. 26931 A1, 2013 Search PubMed; (c) S. K. Chunga, S. H. Banb, S. H. Kimb, B. E. Kimb, S. H. Woob, J. B. Summers and R. G. Conway, Bioorg. Med. Chem. Lett., 1995, 5, 1091 CrossRef; (d) C. H. Tilford, J. Med. Chem., 1971, 14, 1020 CrossRef CAS.
- S. Aubry, K. Sasaki, L. Eloy, G. Aubert, P. Retailleau, T. Cresteil and D. Crich, Org. Biomol. Chem., 2011, 9, 7134 CAS.
- (a) H. Kohn, P. Charumilind and Y. Gopichand, J. Org. Chem., 1978, 43, 4961 CrossRef CAS; (b) K. Nagasawa and A. Yoneta, Chem. Pharm. Bull., 1985, 33, 5048 CrossRef CAS; (c) K. Muthuramu, B. Sundari and V. Ramamurthy, J. Org. Chem., 1983, 48, 4482 CrossRef CAS; (d) M. Machida, K. Oda and E. Yoshida, J. Org. Chem., 1985, 50, 1681 CrossRef CAS; (e) J. D. Coyle, P. A. Rapley, J. Kamphuis and H. J. Bos, J. Chem. Soc., Perkin Trans. 1, 1985, 1957 RSC.
- Y. Iwabuchi, M. Nakatani, N. Yokoyama and S. Hatakeyama, J. Am. Chem. Soc., 1999, 121, 10219 CrossRef CAS.
- J. Song, Y. Wang and L. Deng, J. Am. Chem. Soc., 2006, 128, 6048 CrossRef CAS PubMed.
- The crystal data of 3h have been deposited in CCDC with number 992536.
- The crystal data of 7a have been deposited in CCDC with number 1029698.
- (a) K. Martinez-Mayorga, E. Juaristi and G. Cuevas, J. Org. Chem., 2004, 69, 7266 CrossRef CAS PubMed; (b) J. T. Lenthall, J. A. Foster, K. M. Anderson, M. R. Probert, J. A. K. Howard and J. W. Steed, CrystEngComm, 2011, 13, 3202 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds. CCDC 992536 and 1029698. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc01313e |
| This journal is © The Royal Society of Chemistry 2015 |