
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergy of Mg2+ and poly(aspartic acid) in additive-controlled calcium carbonate precipitation†
Stefan L. P.
Wolf
,
Kathrin
Jähme
and
Denis
Gebauer
*
Department of Chemistry, Physical Chemistry, University of Konstanz, Universitätsstrasse 10, 78457 Konstanz, Germany. E-mail: denis.gebauer@uni-konstanz.de
First published on 10th June 2015
Abstract
Additive-controlled precipitation of calcium carbonate is central to various fields of research. Technically, scale formation is an important problem, where polycarboxylates are most commonly employed as inhibitors. Herein, we show that the combination of poly(aspartic acid) with magnesium ions leads to synergistic effects that bring about a dramatic increase in the efficiency towards inhibition of nucleation and growth of nanoscopic CaCO3 precursors. These effects can also be crucial in biomineralization processes, where polycarboxylates and magnesium ions are thought to play important roles.
Introduction
Calcium carbonate is a mineral of huge scientific importance due to its vast industrial,1 geological,2 and biological relevance.3 Not only from the point of view of industrial issues, such as the scaling (incrustation) of appliances from hard waters, the development of target-oriented means to direct and control the precipitation of calcium carbonate is highly desirable. While research on biomineralization can serve as an inspiration for the design of biomimetic additives,4,5 the mechanisms by which these are effective remain often unclear.The role of magnesium ions in calcium carbonate formation has been studied particularly well,6–8 not only thanks to its relevance in biomineralization.9–12 While their main function appears to relate to the kinetic stabilisation of amorphous calcium carbonate (ACC), magnesium ions are also required for the wetting of proteinaceous organic matrices by liquid precursors of CaCO3.13 Such polymer-induced liquid precursors (PILPs)14 are effectively stabilised by polycarboxylates, which have also been studied in combination with magnesium ions.15–17 More recent work suggests that liquid CaCO3 species are kinetically stabilised by the polymers rather than induced,18,19 suggesting that liquid precursors are inherent to indirect formation of crystalline CaCO3.20–24 In the case of magnesium-stabilised ACC, aspartic-acid rich molecules can trigger crystallisation, which can be understood by an enhanced desolvation of the hydrated precursors.16 On the other hand, carboxylated molecules regulate the Mg/Ca ratio in ACC based upon sequence-dependent binding affinities for the two ions.17 This illustrates that the combination of magnesium ions with carboxylated molecules can give rise to synergistic effects for controlling CaCO3 formation.
Quantitative studies utilising a so-called titration assay have shown that different classes of additives — ranging from simple ions and molecules like sugars and amino acids to complex macromolecules that can effectively influence CaCO3 precipitation — exhibit multiple roles,25–29 and interact with the nascent calcium carbonate before, during, and after nucleation. Here, we have used the established methodology where a calcium ion selective electrode is used to measure the calcium potential in a bi/carbonate buffer upon continuous addition of calcium solution, while the pH-value is kept constant by counter titration with sodium hydroxide solution. While magnesium ions30 and poly(aspartic acid)27 have already been investigated in this way individually, and exhibit distinct effects, we show that in combination, the efficiency is dramatically increased beyond the simple sum of their individual contributions. The highly efficient inhibition of particle nucleation brings about the effective stabilisation of mineral precursors. The pronounced synergistic effect may be especially interesting for novel formulations of antiscalant additives and could also be important in biomineralisation processes.
Experimental
The methodology and basic setup has been described in detail elsewhere.31 All experiments were carried out at 23 °C, chemicals were of analytical grade, and used as retrieved. Millipore water was employed for the preparation of aqueous solutions. All experiments were repeated at least in triplicate and generally showed a very good reproducibility.In brief, 10 mM calcium chloride solution was dosed at a constant rate of 100 μL min−1, or 10 μl min−1, as indicated, into 50 mL of 10 mM sodium bi/carbonate buffer at pH 9.75. The pH level was maintained constant by means of automatic counter-titration employing 100 mM NaOH, whereas the concentration of free calcium ions was monitored utilising an ion selective electrode. This experiment is the reference scenario. For experiments with additives, magnesium chloride and/or poly(aspartic acid) (PAsp, 1200–1800 g mol−1) were added to the calcium solution with concentrations as indicated (50 mM Mg2+; 25 mM Mg2+ + 5 μg mL−1 PAsp; 10 μg mL−1 PAsp). The calcium ion selective electrode was calibrated by dosing the respective solutions into pure water (with corresponding Ca/Mg concentrations where applicable, but without PAsp). The very low concentrations of the polyelectrolyte lead to insignificant complexation of calcium ions,25,27 whereas both polymer and ion concentrations are sufficiently low to prevent polymer salting out. We note that any measured ion concentrations reported herein are strictly speaking ion activities, however, owing to rather dilute solutions, any systematic errors resulting from our simplifying procedure only slightly exceed typical experimental errors, as explicitly demonstrated elsewhere.32
Mineral precursors were isolated from the titration assay experimentation at different points by a quench in excess ethanol (see ref. 33 for details) and the resulting powder was analysed by FT-IR, TGA and SEM. For TEM analyses, samples were drawn from the ethanolic solution and 10 μl droplets were blotted on the TEM grid and were allowed to dry in air.
FT-IR spectra were recorded utilizing a Perkin Elmer spectrometer 100 equipped with a diamond ATR crystal. TEM was carried out with an in-column OMEGA filter microscope (Zeiss Libra 120) operated at 120 kV. TGA measurements were performed under oxygen atmosphere with a heating rate of 10 K min−1 using an STA 449 F3 Jupiter instrument. The SEM pictures and EDS analyses were performed utilising a Hitachi TM 3000 microscope.
Results and discussion
The continuous addition of dilute calcium solution into carbonate buffer at a constant pH of 9.75 leads to a linear increase in the concentration of free calcium ions (Fig. 1). Once a critical stage is reached, nucleation occurs, and the concentration of free calcium ions drops to a level that correlates with the solubility of the most soluble phase (note the system is not yet in equilibrium according to Gibbs' phase rule). Typically, this is proto-vaterite amorphous calcium carbonate (pv-ACC) at this pH level (for the reference experiment without any additives, blue curve in Fig. 1; solubility data not shown).33,34 The measurements in presence of PAsp (10 μg mL−1 in the calcium solution; Fig. 1, green curve) and magnesium ions (50 mM in the calcium solution; Fig. 1, red curve) are consistent with previous investigations. Magnesium leads to a moderate inhibition of nucleation, and formation of thermodynamically less stable mixed calcium/magnesium carbonate pre-nucleation clusters30 (indicated by a steeper slope in the pre-nucleation stage, i.e. a less pronounced binding of calcium ions in the pre-nucleation ion associates, a correspondingly lower equilibrium constant of cluster formation, and, with it, a less negative change in free enthalpy) and of the initially precipitated phase exhibiting a distinctly increased solubility.30 PAsp, on the other hand, effectively inhibits nucleation already at very low concentrations (10 μg mL−1 in the calcium solution, which is furthermore considerably diluted upon addition into the carbonate buffer; note that in previous experiments, PAsp was added to the carbonate buffer and dilution effects were insignificant).27 After nucleation, a more soluble phase than pv-ACC occurs first (as evident from a higher concentration of free calcium ions) that over time transforms into a more stable form (in this case pv-ACC, solubility data not shown), according to Ostwald's rule of stages. Nevertheless, vaterite is already present at a certain time after nucleation (ESI,† Fig. S1), however, this does not contradict the higher level of free calcium ions, because the measured concentration of free calcium ions is governed by the most soluble species.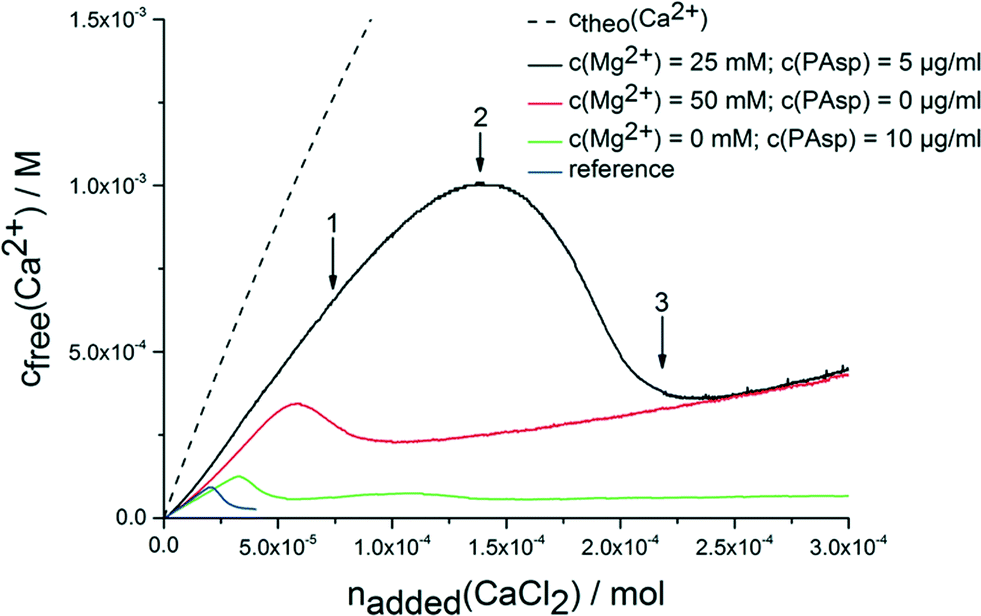 | ||
| Fig. 1 Development of free calcium concentration upon continuous addition of calcium solution with and without additives (as indicated) into 10 mM carbonate buffer (pH 9.75) at a rate of 100 μl min−1. Note that less calcium is detected than added (ctheo) already in the pre-nucleation stage, which can be ascribed to the binding of ions in pre-nucleation clusters.34 Arrows 1–3 indicate states, at which samples were quenched or drawn for additional analyses. For explanation see text. | ||
When magnesium and PAsp are combined in the calcium solution (25 mM Mg2+ and 5 μg mL−1 PAsp), the efficiency towards inhibition of nucleation is significantly increased (Fig. 1, black curve). Note that in this experiment with combined additives, the concentrations of PAsp and Mg2+ have been reduced in comparison to the individual measurements, to avoid a too high all-over additive concentration. Still, the inhibition of nucleation (apparent from the drop in the free calcium concentration) is far beyond the sum of the individual contributions. Moreover, a new effect arises; the pre-nucleation slope of the calcium concentration development is even steeper. This shows that the mixed calcium–magnesium carbonate pre-nucleation clusters are thermodynamically not as stable as in the experiment where only magnesium as additive is present (Fig. 1, red curvecf. also discussion above). Since electrode calibrations have been carried out with corresponding solutions containing Mg2+, any activity or electrode effects can be ruled out.
The free calcium concentration for the initially precipitated phase is distinctly higher than with PAsp alone, and agrees with the one of the phase formed in presence of Mg2+ alone, suggesting a thermodynamic de-stabilisation of the nucleated phase. However, since the composition of this phase may differ from that of the ones precipitated without magnesium (cf. below), this is merely a qualitative assessment. In any case, a link between pre- and post-nucleation speciation (i.e. thermodynamically de-stabilised clusters lead to less stable solid precipitates) is observed — similar as in the case of the pH-dependent switch between proto-calcite ACC and pv-ACC.33,34 This effect can also be found when reducing the addition rate of the calcium solution to 10 μl min−1 (ESI,† Fig. S2).
While the molecular explanation for the more pronounced thermodynamic de-stabilisation of pre-nucleation clusters in the presence of both Mg2+ and PAsp remains unknown, it has to be noted that it likely contributes to the synergistic increase in the kinetic efficiency towards inhibition of nucleation. Previous work has shown that an increase in ionic strength leads to similar effects, because the reduced activity of calcium and carbonate ions causes a lower amount of substance bound in pre-nucleation associates.32 Even though the nominal level of supersaturation at high ionic strengths can be significantly higher (cf.Fig. 1), nucleation is inhibited, highlighting that the bound calcium carbonate is central to the event of phase separation.20,32 Consequently, here, when mixed pre-nucleation clusters are thermodynamically destabilised, less calcium/magnesium carbonate is bound within pre-nucleation clusters and nucleation is inhibited to a greater extent than without the additives. However, the cluster destabilisation effect in presence of both Mg2+ and PAsp likely cannot explain the pronounced synergistic effect with respect to inhibition of nucleation alone.
To investigate the species occurring during the early stages in presence of both Mg2+ and PAsp, and during the eventual drop to the constant ion product characterizing the solubility of the precipitated phase, samples were drawn at different stages (indicated by arrows in Fig. 1), and analysed utilising TEM, SEM, EDS, FT-IR and TGA. TEM analyses illustrate that small amorphous particles (see ED pattern in Fig. 2, top) are forming larger aggregates (Fig. 2, top). At the second time point, the observed particles are still amorphous but have a higher contrast compared to the first sample (Fig. 2, middle). This points towards a densification of the amorphous particles due to the loss of water (note that unfortunately, the amount of sample obtained from time point 1 does not provide enough material for TGA measurements). The SEM micrographs (Fig. 2B and D) support this observation, as the amorphous material quenched from the pre-nucleation stage (Fig. 1, arrow 1) exhibits a rather loose structure compared to the one obtained at the later time point.
 | ||
| Fig. 2 TEM micrographs (A, C, E; scale bars 500 nm, 200 nm, 200 nm, respectively) with ED insets (scale bars; A 2/Å, C 2/Å, E 0.5/Å) and SEM (B, D, F; all scale bars 10 μm) images of specimens blotted from the quenched samples at different stages as indicated by arrows in Fig. 1; arrow 1: top; arrow 2: middle; arrow 3: bottom. | ||
After nucleation occurred, amorphous and crystalline particles are present in the sample (Fig. 2, bottom). This could be explained by a further densification of the particles and subsequent crystallisation yielding aragonite (ESI,† Fig. S5).
FT-IR spectra of specimens quenched at the three different time points (Fig. 3) show that the formed phase is amorphous, which is especially apparent from the ν4 spectral region, which does not show any clear bands related to crystalline calcium carbonates. The ν3 (1409/1468 cm−1), ν2 (861 cm−1) and ν1 (1083/1049 cm−1) bands are typical for ACC.7,33,35–39 However, considering band positions and shapes, there is no clear correlation between the ACC formed in presence of magnesium ions and PAsp and any ACC with distinct proto-structural features.40 While it is known that in presence of Mg2+, synthetic ACC can adopt aragonite-like short-range structural features,39 a more detailed assessment of the short-range structure of this phase, e.g. utilizing X-ray absorption and NMR spectroscopies, is beyond the scope of this work. It should be noted, though, that the ν1 band position coincides with that of aragonite,41 and indeed, aragonite is formed upon reaching the constant solubility product (Fig. 3, blue spectra).
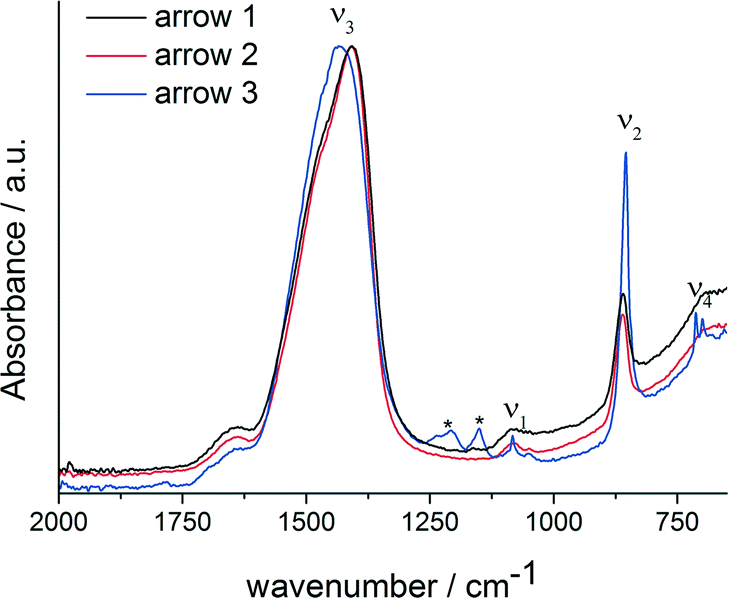 | ||
| Fig. 3 FT-IR spectra of the ACC samples quenched at different times in the titration assay, as indicated by arrows in Fig. 1; arrow 1: black; arrow 2: red; arrow 3: blue. Asterisks label bands that are due to polymer contaminations. | ||
In presence of PAsp, the first crystalline phase forming is vaterite (ESI,† Fig. S1), with Mg2+ alone, ACC is kinetically stabilised within experimental duration (ESI,† Fig. S3), whereas Rodriguez-Blanco et al.42 report the formation of monohydrocalcite after Mg–ACC. The reference experiment without any additives produces a mixture of calcite and vaterite.34 The ACCs formed in presence of Mg2+ alone and in presence of both Mg2+ and PAsp appear to be largely identical considering the IR spectra (ESI,† Fig. S3). Furthermore, the presence of aragonite only when magnesium and PAsp are combined, indicates that PAsp could drive the crystallisation of the amorphous precursors, in agreement with previous studies.16
SEM investigation of the phase obtained upon reaching a constant solubility level (Fig. 4) shows the presence of larger agglomerates, which are likely seen in TEM (cf. above) and pose similar aggregates of nanoscopic ACC entities. In addition, there are 2–3 μm sized aragonite crystals (flower-like structures, Fig. 2 bottom and 4B top). EDS analyses (Fig. 4, bottom) show that the amorphous precipitates contain significant amounts of magnesium ions whereas the aragonite crystals contain only a few percent of magnesium. This effect of magnesium depletion can also be observed in biogenic calcium carbonates.30
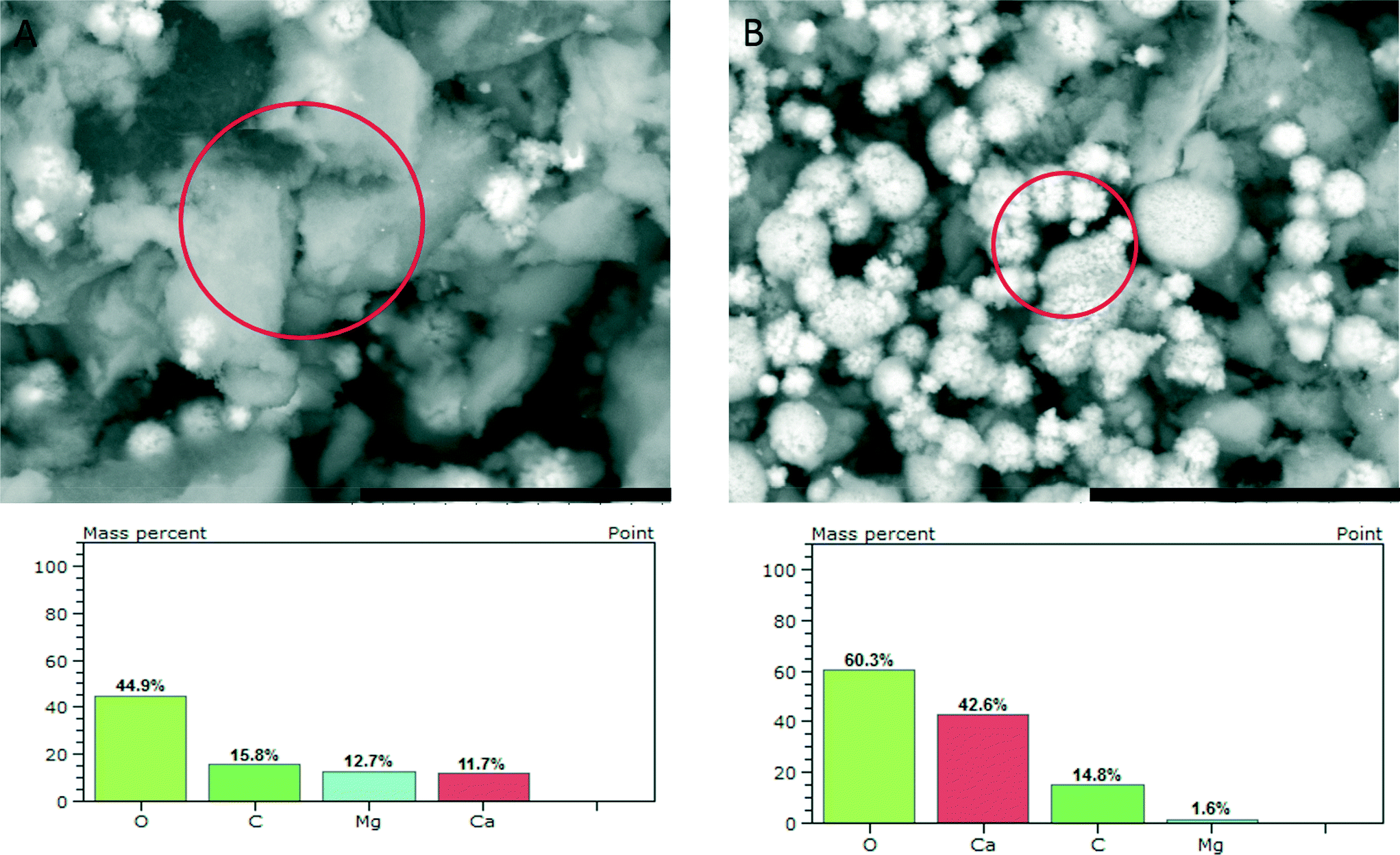 | ||
| Fig. 4 SEM micrographs (scale bars 20 μm) of ACC samples quenched upon reaching a constant solubility product, as indicated by arrow 3 in Fig. 1, showing agglomerates of nanoscopic ACC entities (A) and flower-like aragonite crystals (B), and the respective EDS results obtained from the areas marked by circles. | ||
However, owing to the presence of both amorphous (being more soluble and determining the solubility product, Fig. 1) and crystalline phases (aragonite typically contains insignificant amounts of magnesium), it is difficult to delineate the composition of the different species based on EDS alone. TGA (Fig. 5) of the fully amorphous precipitate quenched shortly before nucleation (arrow 2 in Fig. 1) was performed in order to determine the composition of the precipitates. The TGA trace (Fig. 5) shows three consecutive mass losses. The first one between 80–250 °C corresponds to the release of water, the second one between 300–500 °C to the calcination of magnesium carbonate,43 and the third one between 600–750 °C to the calcination of calcium carbonate. The corresponding relative weight losses are ~22%, ~18%, and ~18%, respectively, suggesting a MgCO3 to CaCO3 ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This also implies that the mass of PAsp within the precipitates is negligible, consistent with the minor PAsp concentration in the titration assay experiments. Thus, TGA suggests a molecular formula Ca0.5Mg0.5CO3·1.5 H2O, on average. Albeit EDS analyses indicate a slightly higher Mg content of the sample, it can be seen that magnesium and calcium ions are distributed evenly over the whole sample (ESI,† Fig. S4). This suggests that only about 1/5 of the Mg2+ ions present in the titration experiment is incorporated into the initially amorphous precipitate.
:1. This also implies that the mass of PAsp within the precipitates is negligible, consistent with the minor PAsp concentration in the titration assay experiments. Thus, TGA suggests a molecular formula Ca0.5Mg0.5CO3·1.5 H2O, on average. Albeit EDS analyses indicate a slightly higher Mg content of the sample, it can be seen that magnesium and calcium ions are distributed evenly over the whole sample (ESI,† Fig. S4). This suggests that only about 1/5 of the Mg2+ ions present in the titration experiment is incorporated into the initially amorphous precipitate.
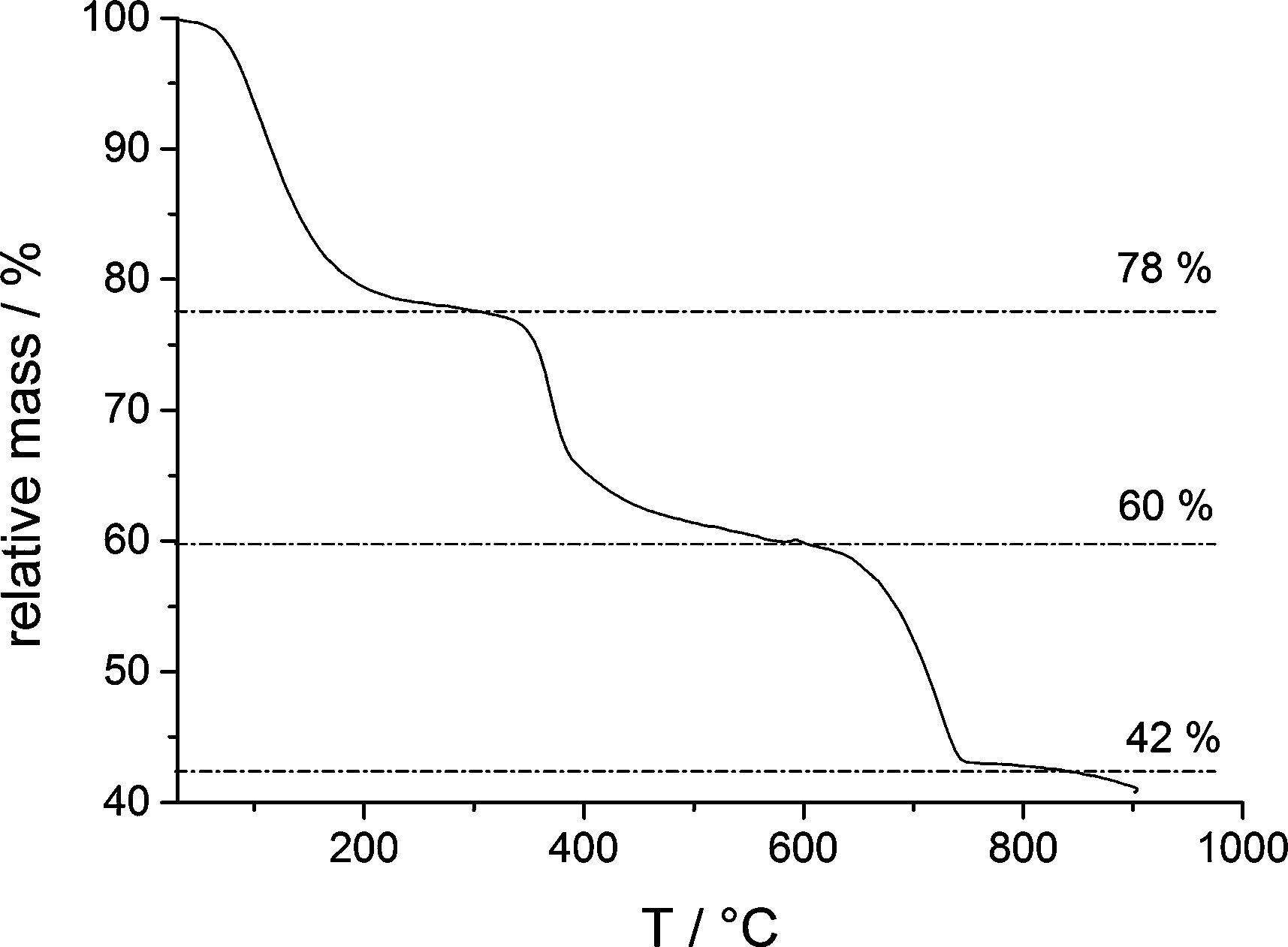 | ||
| Fig. 5 TGA of an ACC sample quenched shortly after the drop of the ion product, as indicated by arrow 2 in Fig. 1. | ||
Conclusions
Our analyses show that the combination of magnesium ions and poly(aspartic acid) (PAsp) brings about a pronounced synergistic effect leading to a significantly stronger inhibition of nucleation as compared to the individual contributions of the single additives. In part, this synergistic effect can be rationalised by the thermodynamic destabilisation of pre-nucleation species, and a lesser amount of calcium/magnesium carbonate bound in species central to phase separation. While a molecular explanation for this remains unknown, the stronger hydration of magnesium ions compared to calcium ions may lead to an even stronger hydration of the earliest precursors of calcium/magnesium carbonate in combination with PAsp. A stronger hydration of nucleation precursors can explain the inhibition of particle formation from the point of view of the notions of the so-called pre-nucleation cluster pathway,20 where the densification of precursors in combination with water loss are assumed to be the key steps underlying particle formation. From the point of view of classical nucleation theory, in contrast, the effect is difficult to understand, since the level of supersaturation is in fact distinctly increased when compared to the reference scenarios — already from the earliest stages of the experiment.Altogether, our findings strongly suggest that magnesium ions may also play a significant role for the effectiveness of biological proteins, which often contain acidic mineral binding domains based upon repeats of aspartic acid.44,45 The synergy of mixtures of additives may thus be central to the realisation of sophisticated crystallisation control, since it is likely tuneable by the relative concentrations of the active co-additives.
When it comes to the inhibition of unwanted precipitation in artificial settings (scaling from hard waters, e.g. in industrial heating and cooling circuits), the dramatic improvement of inhibition of nucleation by combined additives may be exploitable in advanced formulations of antiscalants.
Acknowledgements
DG is a Research Fellow of the Zukunftskolleg of the University of Konstanz and supported by the Fonds der Chemischen Industrie. We thank Masoud Farhadi Khouzani (University of Konstanz) for discussions and experimental support. We also thank In Flames for inspiration during data evaluation and writing of this manuscript.Notes and references
- J. Rieger, M. Kellermeier and L. Nicoleau, Angew. Chem., Int. Ed., 2014, 53, 12380–12396 CAS
.
-
J. Geyssant, E. Huwald and D. Strauch, Calcium carbonate: from the Cretaceous period into the 21st century, ed. F. Wolfgang Tegethoff, Birkhäuser Verlag, Basel; Boston, English edn., 2001 Search PubMed
-
H. A. Lowenstam, On Biomineralization, Oxford University Press, New York, 1989 Search PubMed
- L. B. Gower, Chem. Rev., 2008, 108, 4551–4627 CrossRef CAS PubMed
- C. Rodriguez-Navarro and L. G. Benning, Elements, 2013, 9, 203–209 CrossRef CAS
- R. A. Berner, Geochim. Cosmochim. Acta, 1975, 39, 489–504 CrossRef CAS
- E. Loste, R. M. Wilson, R. Seshadri and F. C. Meldrum, J. Cryst. Growth, 2003, 254, 206–218 CrossRef CAS
- Y. Politi, D. R. Batchelor, P. Zaslansky, B. F. Chmelka, J. C. Weaver, I. Sagi, S. Weiner and L. Addadi, Chem. Mater., 2010, 22, 161–166 CrossRef CAS
- S. Raz, S. Weiner and L. Addadi, Adv. Mater., 2000, 12, 38–42 CrossRef CAS
- S. Weiner, J. Mahamid, Y. Politi, Y. Ma and L. Addadi, Front. Mater. Sci. Chin., 2009, 3, 104–108 CrossRef
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959–970 CrossRef CAS PubMed
- S. Weiner and L. Addadi, Annu. Rev. Mater. Sci., 2011, 41, 21–40 CrossRef CAS
- J. K. Berg, T. Jordan, Y. Binder, H. G. Börner and D. Gebauer, J. Am. Chem. Soc., 2013, 135, 12512–12515 CrossRef CAS PubMed
- L. B. Gower and D. J. Odom, J. Cryst. Growth, 2000, 210, 719–734 CrossRef CAS
- G. Falini, M. Gazzano and A. Ripamonti, J. Cryst. Growth, 1994, 137, 577–584 CrossRef CAS
- J. Tao, D. Zhou, Z. Zhang, X. Xu and R. Tang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22096–22101 CrossRef CAS PubMed
- D. Wang, A. F. Wallace, J. J. De Yoreo and P. M. Dove, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21511–21516 CrossRef CAS PubMed
- M. A. Bewernitz, D. Gebauer, J. R. Long, H. Cölfen and L. B. Gower, Faraday Discuss., 2012, 159, 291–312 RSC
- S. E. Wolf, L. Müller, R. Barrea, C. J. Kampf, J. Leiterer, U. Panne, T. Hoffmann, F. Emmerling and W. Tremel, Nanoscale, 2011, 3, 1158–1165 RSC
- D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergström and H. Cölfen, Chem. Soc. Rev., 2014, 43, 2348–2371 RSC
- A. F. Wallace, L. O. Hedges, A. Fernandez-Martinez, P. Raiteri, J. D. Gale, G. A. Waychunas, S. Whitelam, J. F. Banfield and J. J. De Yoreo, Science, 2013, 341, 885–889 CrossRef CAS PubMed
- M. H. Nielsen, S. Aloni and J. J. De Yoreo, Science, 2014, 345, 1158–1162 CrossRef CAS PubMed
- S. E. Wolf, J. Leiterer, M. Kappl, F. Emmerling and W. Tremel, J. Am. Chem. Soc., 2008, 130, 12342–12347 CrossRef CAS PubMed
- S. E. Wolf, J. Leiterer, V. Pipich, R. Barrea, F. Emmerling and W. Tremel, J. Am. Chem. Soc., 2011, 133, 12642–12649 CrossRef CAS PubMed
- D. Gebauer, H. Cölfen, A. Verch and M. Antonietti, Adv. Mater., 2009, 21, 435–439 CrossRef CAS PubMed
- D. Gebauer, A. Verch, H. G. Börner and H. Cölfen, Cryst. Growth Des., 2009, 9, 2398–2403 CAS
- A. Verch, D. Gebauer, M. Antonietti and H. Cölfen, Phys. Chem. Chem. Phys., 2011, 13, 16811–16820 RSC
- A. Picker, M. Kellermeier, J. Seto, D. Gebauer and H. Cölfen, Z. Kristallogr., 2012, 227, 744–757 CAS
- A. Rao, J. K. Berg, M. Kellermeier and D. Gebauer, Eur. J. Mineral., 2014, 26, 537–552 CrossRef CAS PubMed
- A. Verch, M. Antonietti and H. Cölfen, Z. Kristallogr., 2012, 227, 718–722 CAS
-
M. Kellermeier, H. Cölfen and D. Gebauer, in Methods in Enzymology, Academic Press Inc, Burlington, 2013, vol. 532, pp. 45–69 Search PubMed
- M. Kellermeier, A. Picker, A. Kempter, H. Cölfen and D. Gebauer, Adv. Mater., 2014, 26, 752–757 CrossRef CAS PubMed
- D. Gebauer, P. N. Gunawidjaja, J. Y. P. Ko, Z. Bacsik, B. Aziz, L. J. Liu, Y. F. Hu, L. Bergström, C. W. Tai, T. K. Sham, M. Edén and N. Hedin, Angew. Chem., Int. Ed., 2010, 49, 8889–8891 CrossRef CAS PubMed
- D. Gebauer, A. Völkel and H. Cölfen, Science, 2008, 322, 1819–1822 CrossRef CAS PubMed
- L. Brečević and A. E. Nielsen, J. Cryst. Growth, 1989, 98, 504–510 CrossRef
- J. Aizenberg, G. Lambert, L. Addadi and S. Weiner, Adv. Mater., 1996, 8, 222–226 CrossRef CAS PubMed
- C. Günther, A. Becker, G. Wolf and M. Epple, Z. Anorg. Allg. Chem., 2005, 631, 2830–2835 CrossRef PubMed
- Y. Kojima, A. Kawanobe, T. Yasue and Y. Arai, J. Ceram. Soc. Jpn., 1993, 101, 1145–1152 CrossRef CAS
- R. S. K. Lam, J. M. Charnock, A. Lennie and F. C. Meldrum, CrystEngComm, 2007, 9, 1226–1236 RSC
- J. H. E. Cartwright, A. G. Checa, J. D. Gale, D. Gebauer and C. I. Sainz-Díaz, Angew. Chem., Int. Ed., 2012, 51, 11960–11970 CrossRef CAS PubMed
- N. Vagenas, A. Gatsouli and C. G. Kontoyannis, Talanta, 2003, 59, 831–836 CrossRef CAS
- J. D. Rodriguez-Blanco, S. Shaw, P. Bots, T. Roncal-Herrero and L. G. Benning, Geochim. Cosmochim. Acta, 2014, 127, 204–220 CrossRef CAS PubMed
- N. Khan, D. Dollimore, K. Alexander and F. W. Wilburn, Thermochim. Acta, 2001, 367–368, 321–333 CrossRef CAS
- B.-A. Gotliv, N. Kessler, J. L. Sumerel, D. E. Morse, N. Tuross, L. Addadi and S. Weiner, ChemBioChem, 2005, 6, 304–314 CrossRef CAS PubMed
- R. A. Metzler, G. A. Tribello, M. Parrinello and P. U. P. A. Gilbert, J. Am. Chem. Soc., 2010, 132, 11585–11591 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary IR and EDS spectra, and ED; Fig. S1–S5. See DOI: 10.1039/c5ce00452g |
| This journal is © The Royal Society of Chemistry 2015 |