
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transformation of rigid metal–organic frameworks into flexible gel networks and vice versa
José Juan
Marrero-Tellado
a and
David Díaz
Díaz
*bc
aDepartamento de Química Orgánica, IUBO, “Antonio González”, Universidad de La Laguna, Astrofísico Francisco Sánchez 2, 38206 La Laguna, Tenerife, Spain
bInstitut für Organische Chemie, Universität Regensburg, Universitätsstr. 31, 93040 Regensburg, Germany. E-mail: David.Diaz@chemie.uni-regensburg.de; Tel: + 49 941 9434373
cIQAC-CSIC, Jordi Girona 18-26, 08034 Barcelona, Spain
First published on 8th July 2015
Abstract
Understanding and controlling phase transformations is a timely subject of investigation because they are essential for the fabrication of high-performance materials with applications in energy, sensors, biomedical, and information-related technologies. Such transformations at the nanoscale arise from both diffusion kinetics and surface thermodynamics, whose reasoning represents a major intellectual challenge in multicomponent systems. In particular, the study of interconversion routes between stable and metastable states provides a useful foundation for the rational design of hard and soft materials. Here, we highlight some recent studies that have demonstrated the possibility of transforming rigid (hard) MOFs into flexible (soft) gel materials in quantitative (or nearly quantitative) yields, and vice versa albeit involving different mechanisms and starting materials. These works represent a new paradigm in the growing areas of crystal engineering and stimuli-responsive gels by building new bridges between advanced functional materials that have been traditionally studied in very different research fields.
Introduction
Viscoelastic gels1 exhibit solid-like rheological behavior under deformation2 and have been considered promising materials for bottom-up nanofabrication in numerous research fields.3 In contrast to chemical gels,4 which are based on covalent bonds (usually cross-linked polymers), physical (or supramolecular) gels5 are made of low-molecular-weight compounds or polymers so-called gelators- that self-assemble into 1D fibers by non-covalent interactions (e.g., hydrogen-bonding, π–π stacking, metal coordination, etc.). Subsequent entanglement of these fibers yields 3D matrices in whose interstices liquid molecules (major component) are entrapped via surface tension and capillary forces.6On the other hand, metal–organic frameworks (MOFs) are rigid and crystalline hybrid materials consisting of organic linkers with bridging organic ligands and metal ions. Owing to their tunable surface area and pore size, they have been of great interest as functional nanoporous materials for important applications such as gas storage, separation, sensors, catalysis, and drug delivery.7 Among other methods,8 MOFs are commonly synthesized by solvothermal reactions yielding uniform crystals with sharp edges and sizes ranging from nanometers to millimeters.9
In an attempt to connect both fields, the well-recognized balance between gelation and crystallization,10 has motivated numerous studies regarding the parameters that influence gel phase crystallization.11 Among these factors, solvent's nature plays a key role either during the crystallization of single gelator molecules,12 including coordination compounds as shown by Lloyd and co-workers,13 or during the assembly of various components (e.g., metal ions and organic ligands) in crystalline or amorphous phases.14 Indeed, among existing technologies to control crystal size and growth, the traditional sol–gel process has become a very important tool in which an inert gel matrix retains the crystal nuclei in its position of formation and growth.15 Yaghi and co-workers16 pioneered the use of sol–gel technique for growing MOF crystals. More recently, Steed and co-workers17 made also an important contribution to the field by employing low-molecular-weight organogels as inert gel niches to synthesize single crystals of a range of important organic molecules with controlled polymorphism, including active pharmaceutical ingredients (APIs). Afterward, other groups18 have also validated this approach.
Despite numerous advances in the preparation and formulation of these materials, prediction of either MOF structures or the gelation ability from molecular building blocks remains a great challenge. However, a number of reports have already revealed a powerful link between 3D crystal packing of small molecules and their ability to form gel networks.19
None of the above relationships are obvious if we consider that occurrence of crystals is predicted by equilibrium thermodynamics whereas gelation is a kinetics process.20 Herein, we highlight some recent and pioneering contributions that have demonstrated the feasibility of transforming directly (a) rigid MOFs into flexible gel particles or (b) supramolecular metallogels into MOFs. Nevertheless, the mechanisms involved in both transformations are different because the former involves covalent cross-linking of the starting material, whereas the latter involves an exchange process of the components by intermolecular interactions.
Transformation of metal–organic frameworks into polymer gels
In 2012, Sada and co-workers21 reported the first bottom-up approach for the fabrication of nano- and microsized cubic gel particles (CGPs) with well-defined edges and square faces using MOFs with cubic shape as templates. The strategy relies on the large reaction space of the 3D channels in the MOFs and consisted in three steps: 1) preparation of a hydroxyl-functionalized MOF, 2) subsequent cross-linking with a suitable bifunctional electrophile, and 3) removal of coordinated metal ions. Specifically, the authors used a γ-cyclodextrin (γ-CD) MOF (CD-MOF) that was easily prepared by reacting γ-CD with KOH in aqueous solution, followed by standard vapor diffusion of MeOH into the solution (Fig. 1). This method allowed the preparation of cubic crystals with 40–500 mm on a side, which were easily separated by decantation or filtration. Moreover, much smaller CD-MOF crystals could be obtained from the supernatant by adding a surfactant-containing mother liquor (i.e., cetyltrimethylammonium bromide, CTAB) and controlling the incubation time. Thus, uniform cubic crystals with sizes of ca. 10 mm (CD-MOF-Micro1), 1 mm (CD-MOF-Micro2) and 200–300 nm (CD-MOF-Nano) were obtained (Fig. 2a, c and d).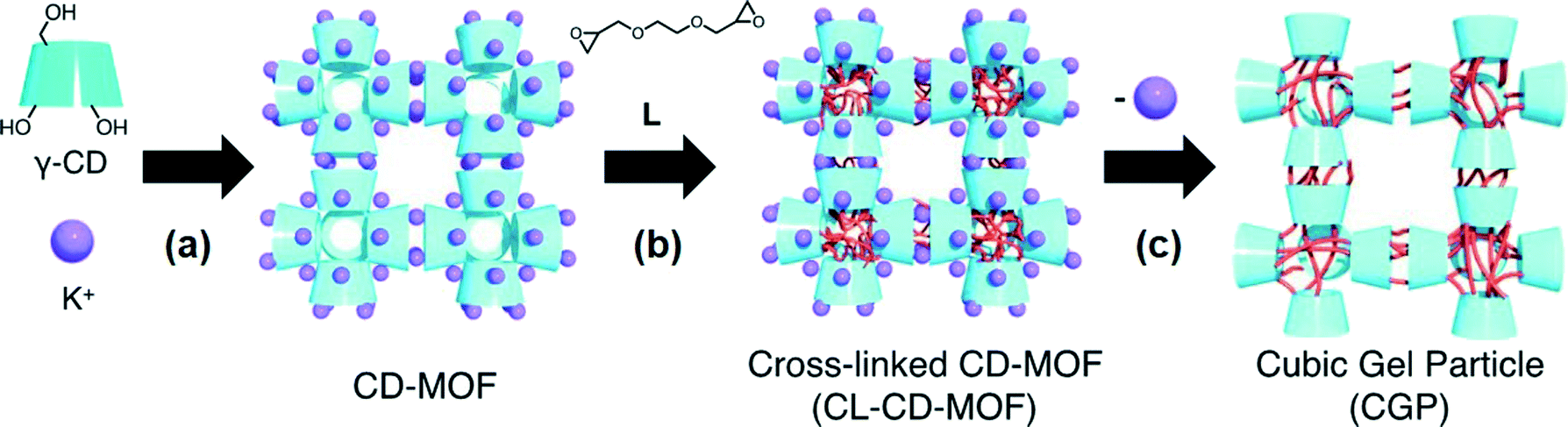 | ||
| Fig. 1 Schematic illustration of the synthesis of cubic gel particles (CGP) from MOF precursor (CL-CD-MOF): a) crystallization, b) cross-linking reaction, and c) removal of coordinated metal ions. Reprinted with permission from ref. 21. Copyright® Wiley-VCH. | ||
 | ||
| Fig. 2 SEM images of a) CL-CD-MOF-Micro1, b) CGP-Micro1, c) CLCD-MOF-Micro2, d) CGP-Micro2, e) CL-CD-MOF-Nano, and f) CGP-Nano. Adapted with permission from ref. 21. Copyright® Wiley-VCH. | ||
Subsequently, the cross-linking reaction of the γ-CDs in the CD-MOF pores was carried out by treatment with ethylene glycol diglycidyl ether (L) in EtOH at 65 °C for 3 days. Finally, removal of potassium ions and unreacted L was achieved by soaking the obtained cross-linked CD-MOF (CL-CD-MOF) in a mixed solvent (EtOH/H2O = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v)) and H2O, yielding CGPs with shape-memory of the starting crystalline materials (Fig. 2b, d and f).
:1 (v/v)) and H2O, yielding CGPs with shape-memory of the starting crystalline materials (Fig. 2b, d and f).
In sharp contrast to unmodified CD-MOFs, so prepared CL-CD-MOFs swelled in H2O. Although the original cubic shape of the CD-MOF was retained even after the cross-linking and degradation process, sizes of the cubes were expanded by 1.37 times the original CL-CD-MOF length (degree of swelling,22Q = 2.57) (Fig. 3). FT-IR, elemental analysis, PXRD, and TG analyses of the so prepared CGPs confirmed that the epoxy groups of L reacted with the hydroxy groups of each γ-CD, rising the thermal stability of the crystalline material and inducing the network formation. For CGP, no apparent diffraction peaks were observed after removing the solvent, indicating that CL-CD-MOF became amorphous after swelling, like a polymer gel.
 | ||
| Fig. 3 MOF-to-GP transition captured by SEM. Adapted with permission from ref. 21. Copyright® Wiley-VCH. | ||
This work demonstrated that cubic gel particles retained the shape and size of the original CD-MOF crystals. Therefore, it is expected that a fine control on the recrystallization conditions could be used to fabricate a wide range of sizes of polyhedral gel particles (micro- and nanosized) from a variety of MOF crystals as supramolecular templates. Therefore, the use of different cross-linkers and metals ions could be used to obtain gel particles with different shapes.
In order to expand the applicability of this strategy to other type of porous MOFs, the same group23 succeeded in transforming the rigid coordination networks of various “clickable” nanoporous azide-tagged MOFs to flexible organic PG networks by in situ cross-linking with a variety of polyvalent alkynes via copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC),24,25 followed by degradation of the metal coordination network (Fig. 4). In previous studies,26 the authors had demonstrated the feasibility of the Cu(I) catalyzed click reactions of azide-tagged MOFs (AzM) with alkynes without disrupting the crystal structures of the MOFs compounds. To demonstrate the conversion of different MOFs into gels, the former were easily prepared by treatment of a diazide-triphenyldicarboxylic acid ligand (AzTPDC) with Zn(NO3)2·6H2O in DEF at 80 °C for 3 days. After this time, simple decantation of the solution and washing of the crystal provided cubic AzM with sharp edges. Subsequent reaction of AzM with tetra-acetylene cross-linker (CL4) under the standard click reaction conditions at 80 °C in the presence of Cu+ as a catalyst in DEF yielded the corresponding cross-linked MOF (CLM) within 1 week. Finally, the coordination of carboxylate anion to Zn2+ ion was eliminated by acidification with concentrated HCl, providing a non-crystalline MOF-templated polymer (MTP) with rougher cubic shapes. Gradual size expansion of the MOFs by acid penetration could be observed under microscopy. In less than 10 min, the cubic crystals of CLM with 300–500 μm in length became cubic polymer gels. Even the cracks or scratches existing on the crystal surfaces were preserved on the gel surface after the hydrolysis reaction. Moreover, AzM treated with diacetylene cross-linker (CL2) instead of CL4 rapidly dissolved, and simultaneous click reaction during the crystallization process of AzM provided only amorphous powdery product. These results demonstrate the importance of both the degree of cross-linking and post-synthetic modification after formation of MOF crystal in order to facilitate the transition from an inorganic nanoporous brittle material to a viscoelastic organic framework.
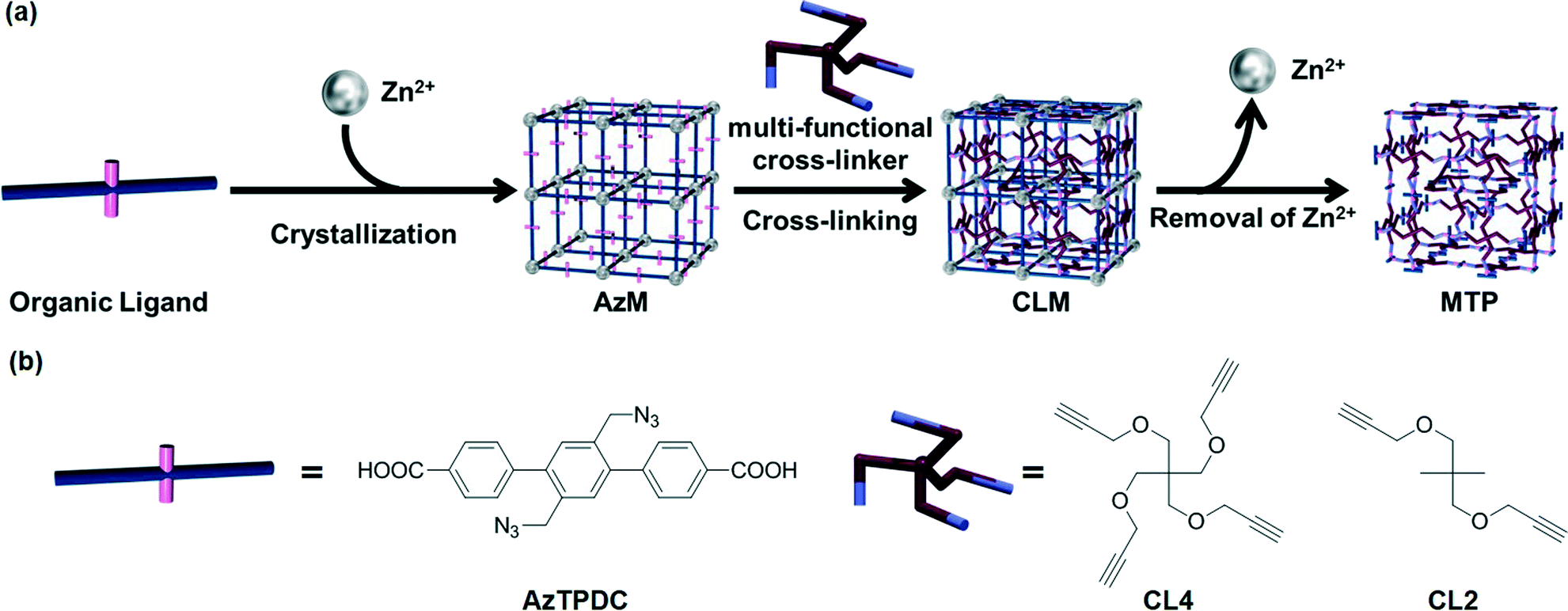 | ||
| Fig. 4 (a) Schematic illustration of cross-linking of organic linkers in MOF (AzM) and subsequent degradation to obtain polymer gel (PG). (b) Molecular structures of the organic ligand (AzTPDC) and the cross-linkers (CL4, CL2). Reprinted with permission from ref. 23. Copyright® American Chemical Society. | ||
FT-IR, XPS and ICP-AES measurements demonstrated that 1) all azide groups in the MOF crystals were consumed upon cross-linking, and 2) carboxylate anions were protonated and most Zn2+ ions (>90%) removed after acidification. On the other hand, TGA and XRD studies of the crystals before (AzM) and after (CLM) the click reaction indicated no impact of the cross-linking reaction on the thermal stability and inherent network structure.
The so prepared MTP swelled in aprotic polar solvents such as DMF, DEF and DMSO. The swelling without changes in shape and nanoporosity was mainly ascribed to suppression of hydrogen bonds among the carboxylic acid groups. Similarly to common polymer gels bearing carboxylic acids,27 the swelling degrees were nearly constant (ca. 1.2) in acidic solutions, but at higher pH (10.5), the swelling degrees increased (ca. 3.2) due to dissociation of the carboxylic groups, which induced repulsive interaction between anionic groups and generation of osmotic pressure between interior and exterior of MTP. Zinc ion was successfully entrapped in MTP by treatment of zinc nitrate. However, reformation of zinc carboxylates did not provide back crystallinity to the coordination network due to movement of coordination sites in length (ca. 10%) during the swelling process.
Very interestingly, Sada's group could also perform directional and partial hydrolysis of a single piece of CLM crystal to seamlessly fused hybrid material between PG and MOF, the first example of this class. The procedure consisted simply in contacting one crystal face of CLM on a wet membrane with 1 M NaOH solution for 5 min, followed by immersion in fresh DEF repeatedly to remove excess amount of the base.
The cross-linking of organic ligands in MOF crystals to form PG was successfully generalized by using other MOFs that consisted of biphenyl-type organic linkers with two azide groups (AzBPDC) and some other metal ions (Fig. 5). Treatment of AzBPDC with Zn2+ and Cu2+ provided colorless cubic and green truncated octahedral MOF crystal under standard solvothermal synthesis in DEF and DEF-DMSO, respectively. Moreover, solvothermal synthesis from AzTPDC and Zr4+ in DMF at 120 °C provided colorless octahedral crystal. XRD and FT-IR analyses of these MOF crystals demonstrated that they had the same crystal structures as their parents without azide groups. Moreover, they were cross-linked by CuAAC and followed by decomposition of metal coordination in acidic medium. After the acid treatment, all the crystals became insoluble and were successfully converted into the corresponding PG materials.
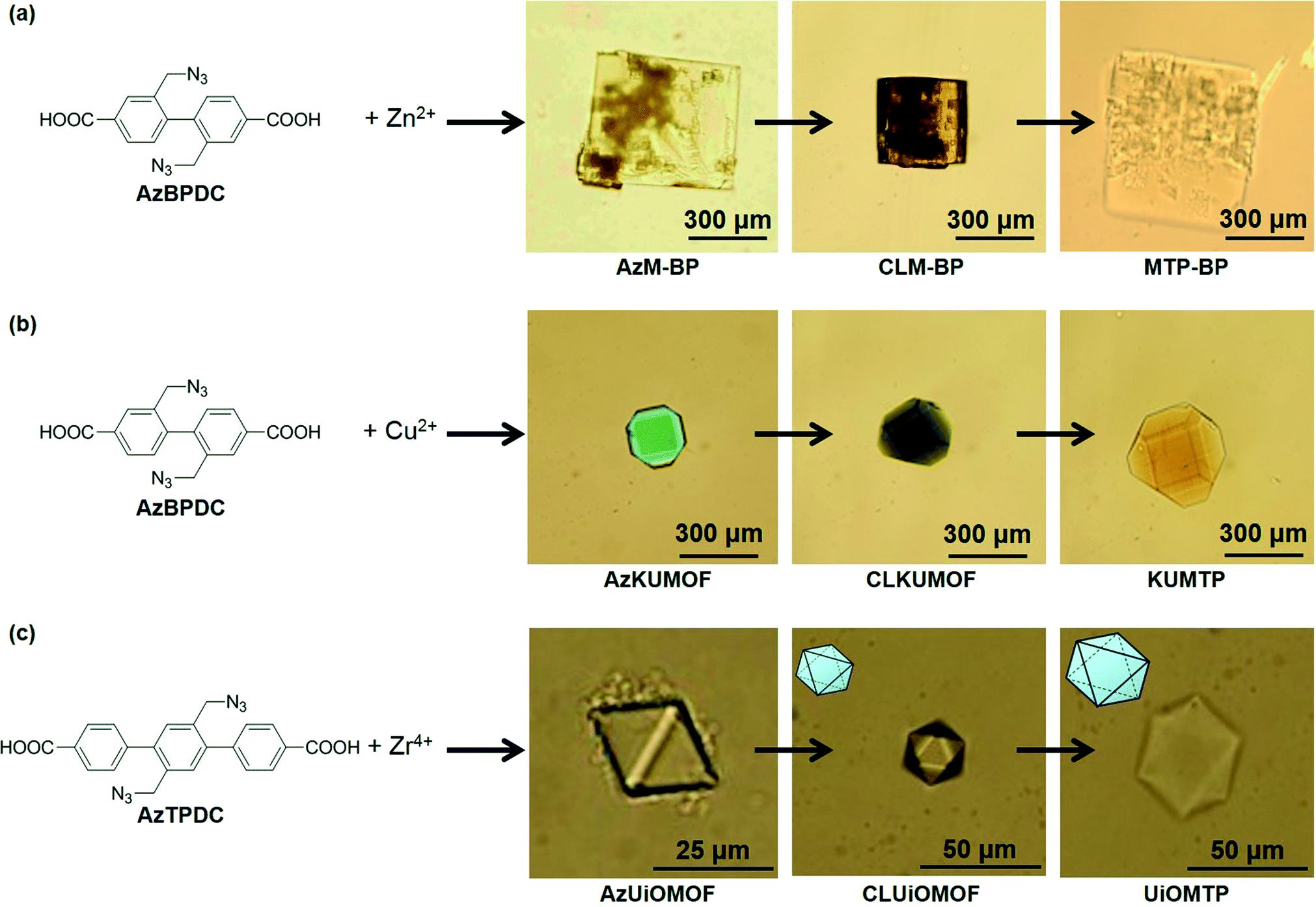 | ||
| Fig. 5 Optical microscopy images of (a) AzM-BP, (b) AzKUMOF, and (c) AzUiOMOF, and subsequent cross-linking and transformation into the corresponding MOF-templated polymers (MTPs), respectively. Reprinted with permission from ref. 23. Copyright® American Chemical Society. | ||
Transformation of metallogels into metal–organic frameworks
Banerjee and co-workers28 has recently closed the loop by developing a strategy to convert metallogels into MOFs. In sharp contrast to the previous examples involving covalent cross-linking, Banerjee's group exploits exchange processes of individual components by intermolecular interactions. As a proof of concept, Fe-based MOFs were in situ synthesized by PdCl2-mediated degradation of Fe-based metallogels. The gradual delivery of both the metal ions and the organic linker necessary for their re-assembly into a crystalline supramolecular structure is a challenging process due to high thermal and chemical stability of such metallogels.29The approach of Banerjee's team involves a very simple one-pot, two-step process. In the first step, MOF precursors Fe(NO3)3·9H2O and 1,3,5-benzene tricarboxylic acid (BTC) were dissolved in NMF, DMF or DEF (molar ratio Fe:CO2H = 1:3). Orange opaque metallogels were obtained in each solvent after heating the mixture for 6 h at 90 °C (Fig. 6). These gels were brittle in nature, as indicated by rheological measurements, and remained stable over weeks. ESEM revealed typical fibrillar morphologies of the supramolecular aggregates (Fig. 7 (a–c)). In the second step, a solution of PdCl2 (molar ratio Fe:Pd = 1:1) was added to the Fe-based metallogels and kept for 48 h at 90 °C to induce the degradation of the gel phase (after 24 h) and consequent thermodynamically controlled transformation into Fe-MOFs with well-defined faces as observed by SEM (Fig. 7 (d–f)). Traditionally, the solvents chosen in this approach are considered ideal for MOF synthesis due to their high boiling points and partial hydrolytic decomposition into corresponding amines and formic acid at higher temperature. The in situ formation of these amines has proven to deprotonate the ligand making the use of additional bases unnecessary.30 On the other hand, hydrogen-bond donors such as formic acid could also be involved in the stabilization of supramolecular gel phases.31
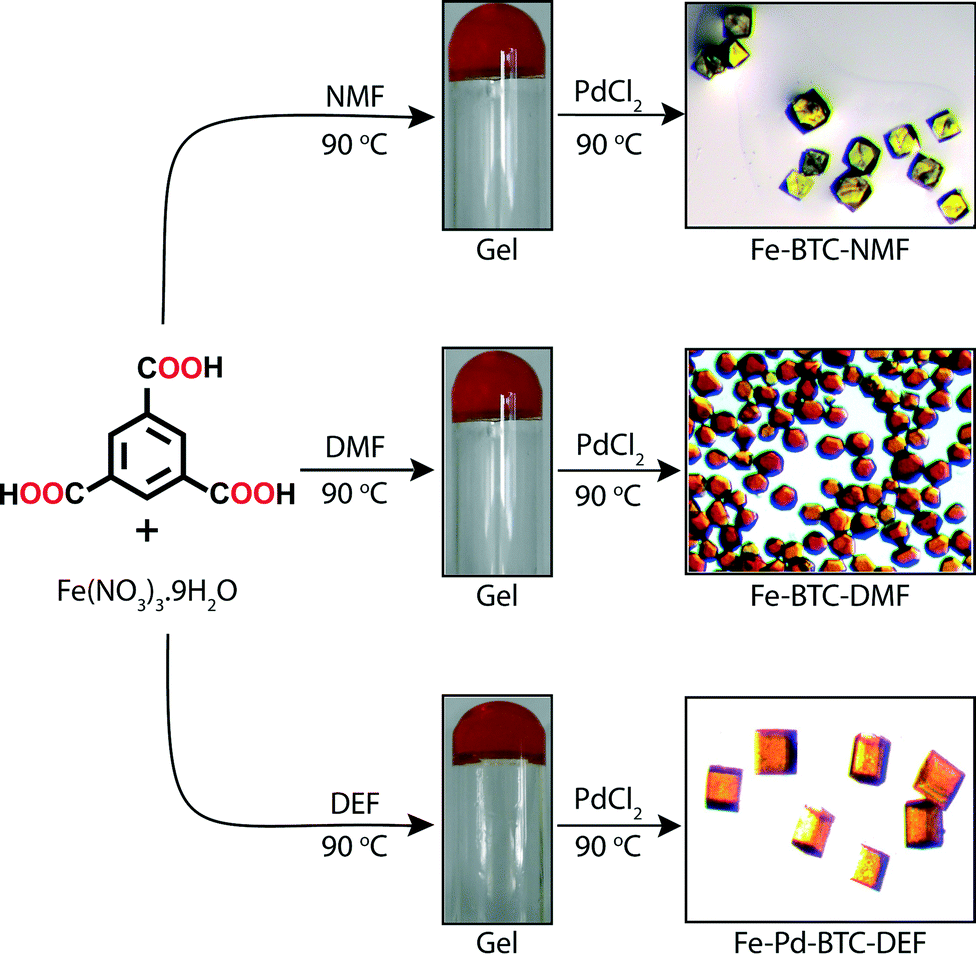 | ||
| Fig. 6 Formation of Fe-metallogels and their transformation into Fe-MOFs via PdCl2-mediated gel degradation. Adapted with permission from ref. 28. Copyright® American Chemical Society. | ||
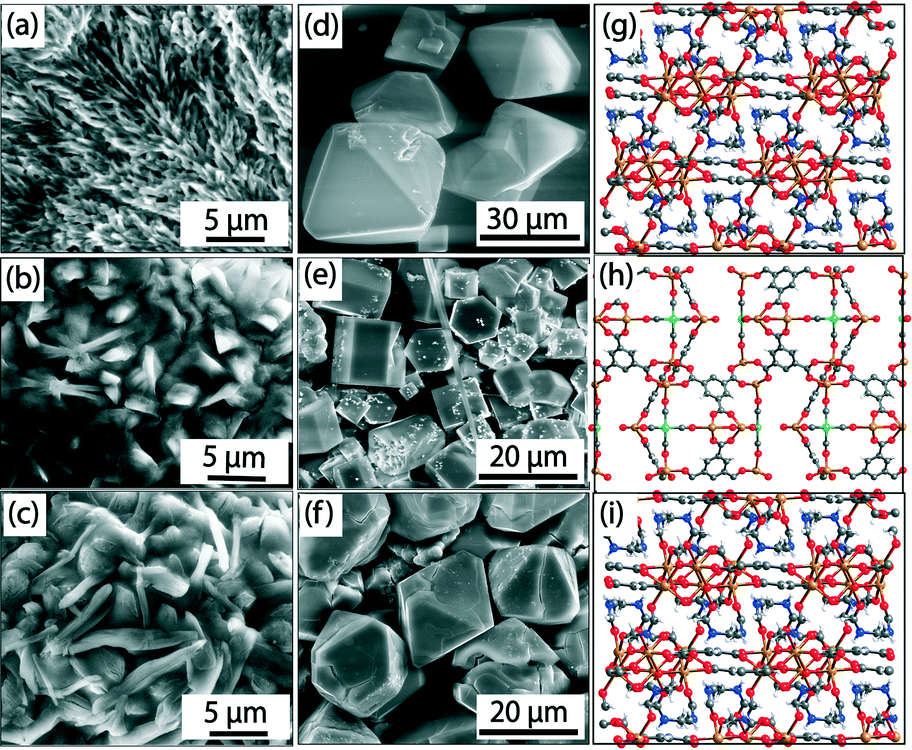 | ||
| Fig. 7 ESEM images of gels [(a), (b), (c)], SEM images [(d), (e), (f)], and single crystal structures [(g), (h), (i)] of Fe-BTC-NMR, Fe-Pd-BTC-DEF, and Fe-BTC-DMF MOFs, respectively. Color code: Fe = orange; O = red; C = grey; N = blue; Pd = green. Palladium residues on crystal faces are visible in Fe-Pd-BTC-DEF (e). Adapted with permission from ref. 28. Copyright® American Chemical Society. | ||
This facile protocol allowed the fabrication of a known MOF (i.e., Fe-BTC-DMF)17 and two new MOFs (i.e., Fe-BTC-NMF (2D) and Fe-Pd-BTC-DEF (3D)) (Fig. 7 (g–i)) in pure crystalline forms as indicated by PXRD analyses. Fe-BTC-NMF crystallized in trigonal space group R![[3 with combining overline]](https://www.rsc.org/images/entities/char_0033_0305.gif) . In the extended crystal structure, Fe3+ adopts an octahedral geometry coordinated to five oxygens from μ2-CO2− functionalities of BTC and one NMF oxygen atom. On the other hand, short-lived Fe-Pd-BTC-DEF crystallized in hexagonal space group P63/mmc and the secondary building unit contains two metal centers, Fe(II) and Pd(II). In the extended structure, one set of Fe centers is tetrahedrally coordinated to four O atoms, and another Fe is coordinated to five O atoms forming a square pyramidal geometry. The Pd(II) adopts a square planar geometry and is coordinated with four C atoms of CO molecules, which could be generated along the pathway of formic/formamide oxidation as suggested by continuous monitoring the gel-to-MOF transition by GC and a series of control experiments. In addition, formation of palladium black (confirmed from PXRD) was also observed indicating a close association between the reduction of PdCl2 and the onset of metallogel degradation. Therefore, a judicious balance between formulation components and hydrothermal stability of solvent molecules is required to drives the gel phase into MOFs. This work is especially relevant if we consider that standard bulk synthesis of Fe-based MOFs suffer from major limitations due to the easy oxidation of Fe2+ and ready hydrolysis of Fe3+ under hydrothermal conditions.32
. In the extended crystal structure, Fe3+ adopts an octahedral geometry coordinated to five oxygens from μ2-CO2− functionalities of BTC and one NMF oxygen atom. On the other hand, short-lived Fe-Pd-BTC-DEF crystallized in hexagonal space group P63/mmc and the secondary building unit contains two metal centers, Fe(II) and Pd(II). In the extended structure, one set of Fe centers is tetrahedrally coordinated to four O atoms, and another Fe is coordinated to five O atoms forming a square pyramidal geometry. The Pd(II) adopts a square planar geometry and is coordinated with four C atoms of CO molecules, which could be generated along the pathway of formic/formamide oxidation as suggested by continuous monitoring the gel-to-MOF transition by GC and a series of control experiments. In addition, formation of palladium black (confirmed from PXRD) was also observed indicating a close association between the reduction of PdCl2 and the onset of metallogel degradation. Therefore, a judicious balance between formulation components and hydrothermal stability of solvent molecules is required to drives the gel phase into MOFs. This work is especially relevant if we consider that standard bulk synthesis of Fe-based MOFs suffer from major limitations due to the easy oxidation of Fe2+ and ready hydrolysis of Fe3+ under hydrothermal conditions.32
Despite the attractiveness of the previous example, no quantitative conversion of the gels into the corresponding MOFs was achieved (approximately 80% yield was obtained with each gel). However, the same research group33 reported a few months later a one-pot synthesis of a metallohydrogel (ZAVA gel)34 and the in situ entrapment of uncapped CdS quantum dots to yield a luminescent metallohydrogel (CdS@ZAVA gel) (Fig. 8).
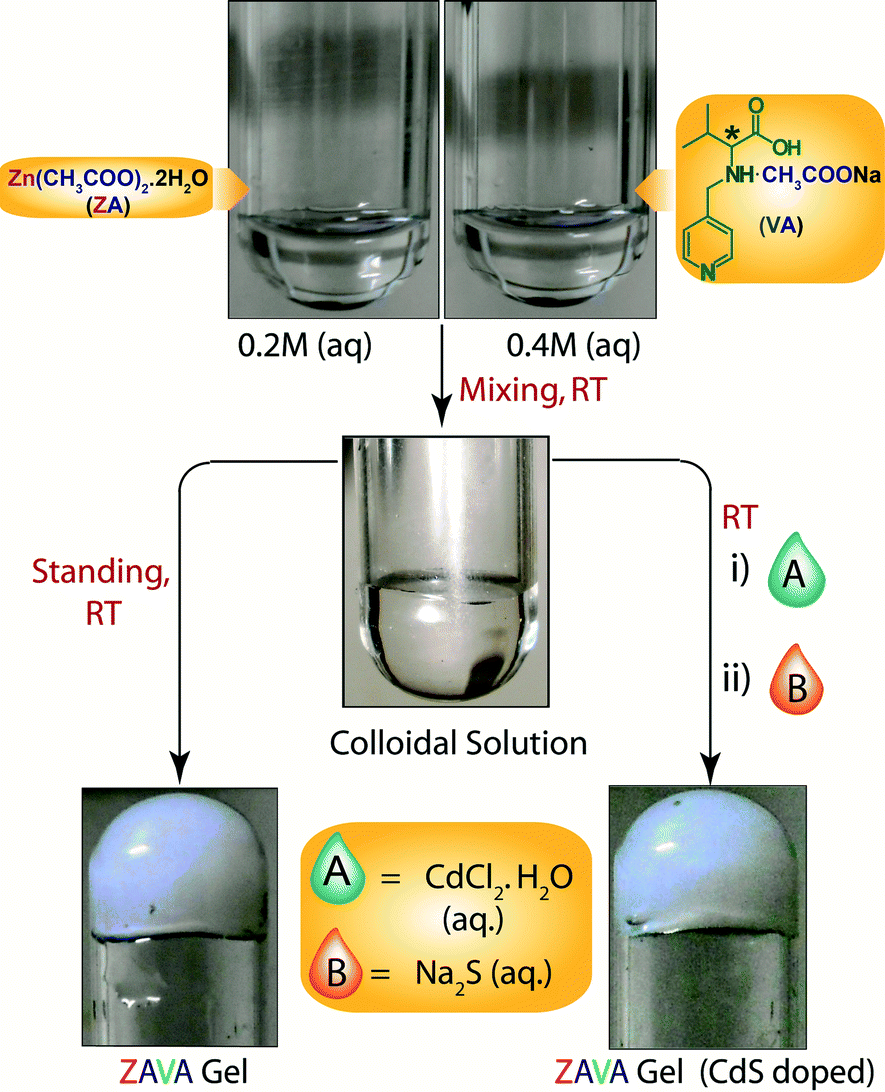 | ||
| Fig. 8 Room temperature synthesis of ZAVA gel and CdS-loaded ZAVA gel. Adapted with permission from ref. 33. Copyright® American Chemical Society. | ||
Briefly, ZAVA gel can be easily prepared after mixing stock solutions of zinc acetate dihydrate (ZA) and the acetate salt of L-3-methyl-2-(pyridin-4-ylmethylamino)butanoic acid (VA). Immobilization of quantum dots takes place efficiently upon addition of aqueous solutions of CdCl2·H2O and Na2S to the colloidal intermediate phase prior gelation. Very interestingly, CdS@ZAVA gel can be degraded and converted quantitatively into luminescent CdS-embedded MOF (CdS@ZAVCl MOF) via a simple and unique NaCl-mediated gradual degradation of the gel phase at room temperature (Fig. 9). The crystal structure of pure ZAVCl MOF (without CdS quantum dots) obtained by the same method revealed the presence of a chloride ion in the coordination sphere of Zn2+ along with two oxygens (from two carboxylate groups) and two nitrogens (one from pyridine and one from the amine of the ligand).35 On the other hand, PXRD analyses showed that immobilization of CdS particles did not cause alteration of the crystalline phase in comparison to pristine ZAVCl MOF. The ability of ZAVA gel to immobilize CdS particles was attributed to the presence of a pyridine moiety in the ligand (VA). In addition, the pyridinic nitrogen of VA is also involved in the formation of the hydrogen-bonded gel network,34 thus offering a remarkable dual stabilization role.
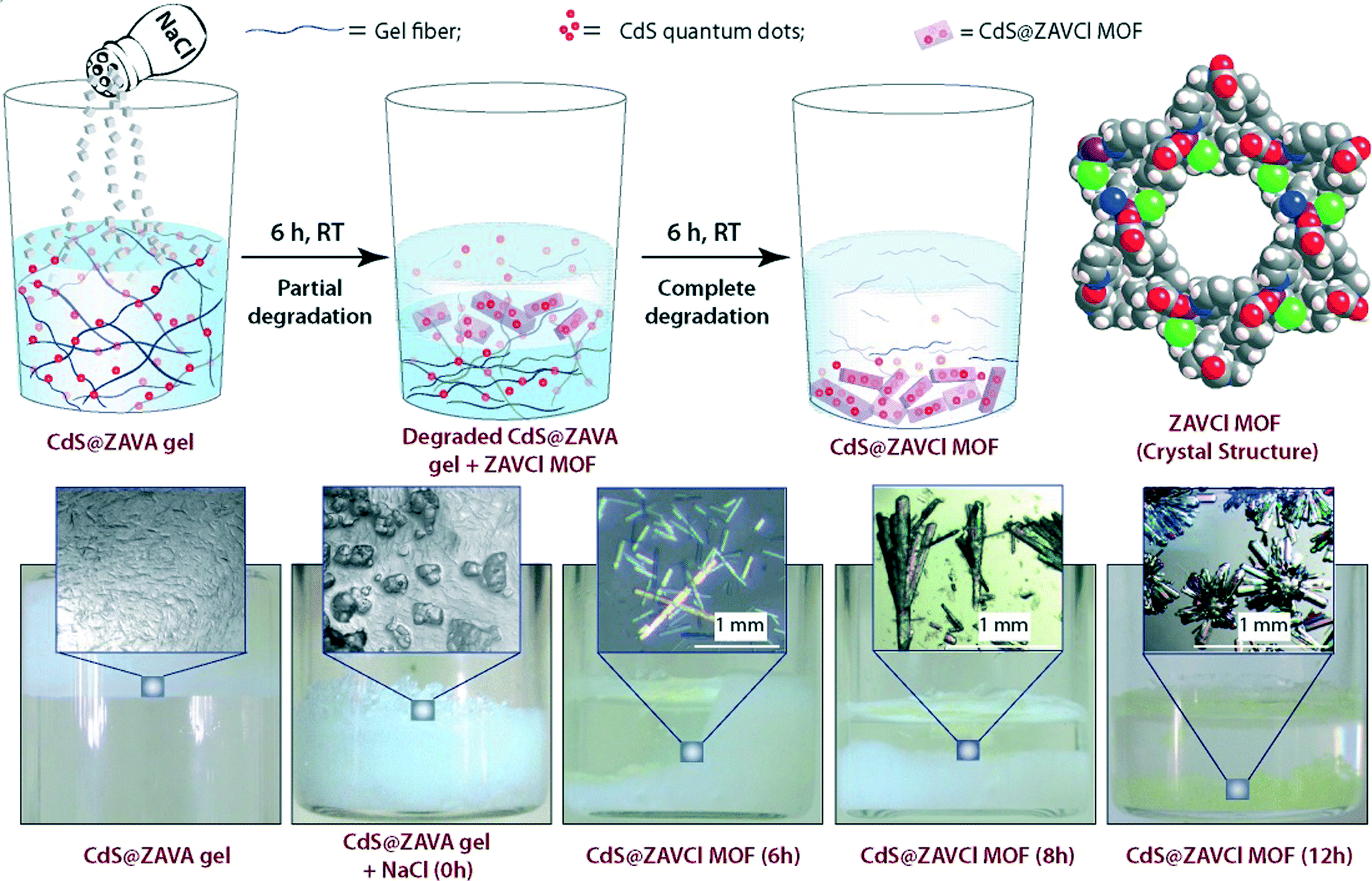 | ||
| Fig. 9 Top: Synthesis of CdS loaded ZAVCl MOF (CdS@ZAVCl MOF) from CdS loaded ZAVA gel (CdS@ZAVA gel), and space-filling representation of the crystal structure of ZAVCl (view through c axis. Color code: C = grey; H = white; Zn = purple; Cl = green; N = blue; O = red). Bottom: Real-time digital photographs of CdS@ZAVA gel to CdS@ZAVCl MOF conversion process. Adapted with permission from ref. 33. Copyright® American Chemical Society. | ||
It is worth mentioning that the conventional sovothermal method employs high-temperature conditions (90 °C) yielding small MOFs (ca. 50 μm).35 However, the room temperature gel-to-MOF approach affords millimeter sized rod-shaped, transparent ZAVCl MOF crystals (Fig. 9). Considering the constant photoluminescence of these MOFs after several washings in water, as well as their pore dimension (1.2 nm), CdS quantum dots (ca. 5–9 nm in diameter, as determined by HRTEM) are presumably sandwiched between crystallite surfaces of ZAVCl MOF.36
Last but not least, the so obtained CdS@ZAVCl MOF could also be used as an efficient photocatalyst for water-splitting under both UV and visible light irradiation (i.e., H2 evolution 500–510 μmol h−1 g−1) in spite of the low loading of the semiconductor in the material (ca. 1 wt%). Although there exist few reports on the utilization of CdS-MOF composites for water-splitting,37 their preparation require very high temperatures and much longer reaction times in comparison to CdS@ZAVCl MOF.
Summary and outlook
Until now, most research on MOFs and gel materials has remained as important topics albeit in very different fields. This has been mainly motivated by the opposite mechanical properties of both types of materials (i.e., hard vs. soft). However, Sada's group has demonstrated the possibility to transform rigid cubic MOF crystals into flexible polymer gel particles via internal cross-linking of the organic linkers in the void spaces, followed by loss of coordination metal ions. Remarkably, the resulting micro- or nanosized gel networks retained the shape and size of the original MOF crystals employed as templates in this approach. In addition, the first chimera-type hybrid material consisting of MOF and polymer gel was also obtained by directional and partial hydrolysis of resulting cross-linked MOF. On the other hand, Banerjee's group has recently advanced in the field by developing a simple and very efficient route for the synthesis of Fe-MOF crystals via PdCl2-mediated Fe-metallogel degradation. The bulk synthesis of such Fe-MOFs by standard hydrothermal methods is difficult due to the easy oxidation of Fe2+ and ready hydrolysis of Fe3+. Moreover, the same group also succeeded in preparing CdS quantum dot-loaded gel, which was then transformed quantitatively into luminescent MOF crystals by simple addition of chloride salt. The entire method works at room temperature and the so prepared MOFs were efficiently used as photocatalysts in visible light water-splitting reactions.Although fully reversible conversion between MOF and gels is still in demand to switch back and forth between different degrees of firmness, the works highlighted here represent a novel archetype in the growing areas of crystal engineering and stimuli-responsive gels. New bridges between crystalline (hard) and amorphous (soft) functional materials are now accessible, making possible to prepare one from the other in a simple way and with memory of specific features of the initial state. These results, together with envisioned future studies on the underlying physics and thermodynamics of these phase transitions, also open a new horizon toward the preparation of novel model networks and dynamic metal-biomolecule frameworks38 with precise properties.
Acknowledgements
Financial from DFG (PRJ 9209720) and Universität Regensburg are gratefully acknowledged. D.D.D. thanks Deutsche Forschungsgemeinschaft (DFG) for the Heisenberg Professorship Award.Notes and references
- S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649 RSC; R. G. Weiss and P. Terech, Molecular Gels: Materials with Self-Assembled Fibrillar Networks, Springer, New York, 2006 Search PubMed.
- E. Zaccarelli, J. Phys.: Condens. Matter, 2007, 19, 323101 CrossRef CAS; O. Gronwald, E. Snip and S. Shinkai, Curr. Opin. Colloid Interface Sci., 2002, 7, 148 CrossRef.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960 CrossRef CAS PubMed; R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664 RSC.
- Y. Osada and A. R. Khokhlov, Polymer Gels and Networks, Marcel Dekker, New York, 2002 Search PubMed.
- X. Yang, G. Zhang and D. Zhang, J. Mater. Chem., 2012, 22, 38 RSC; D. J. Adams, Macromol. Biosci., 2011, 11, 160 CrossRef CAS PubMed.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109 RSC.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015 10.1039/C4CS00395K; H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef CAS PubMed; G. K. H. Shimizu, J. M. Taylor and S. Kim, Science, 2013, 341, 354 CrossRef PubMed; S. Sen, N. N. Nair, T. Yamada, H. Kitagawa and P. K. Bharadwaj, J. Am. Chem. Soc., 2012, 134, 19432 CrossRef PubMed; L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC; A. U. Czaja, N. Trukhan and U. Mueller, Chem. Soc. Rev., 2009, 38, 1284 RSC; J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- H. Al-Kutubi, J. Gascon, E. J. R. Sudhoelter and L. Rassaei, ChemElectroChem, 2015, 2, 462 CrossRef CAS PubMed; N. A. Khan and S. H. Jhung, Coord. Chem. Rev., 2015, 285, 11 CrossRef PubMed; O. Karagiaridi, W. Bury, J. E. Mondloch, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2014, 53, 4530 CrossRef PubMed; J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3 CrossRef PubMed.
- E. A. Flügel, A. Ranft, F. Haase and B. V. Lotsch, J. Mater. Chem., 2012, 22, 10119 RSC.
- E.-M. Schön, S. Roelens and D. D. Díaz, CrystEngComm, 2015 10.1039/C5CE00397K; Y. Lan, M. G. Corradini, R. G. Weiss, S. R. Raghavan and M. A. Rogers, Chem. Soc. Rev., 2015 10.1039/C5CS00136F; Y. Yin, Z. Gao, Y. Bao, B. Hou, H. Hao, D. Liu and Y. Wang, Ind. Eng. Chem. Res., 2014, 53, 1286 CrossRef CAS; I. Kapoor, E.-M. Schön, J. Bachl, D. Kühbeck, C. Cativiela, S. Saha, R. Banerjee, S. Roelens, J. J. Marrero-Tellado and D. D. Díaz, Soft Matter, 2012, 8, 3446 RSC; D. J. Adams, K. Morris, L. Chen, L. C. Serpell, J. Bacsa and G. M. Day, Soft Matter, 2010, 6, 4144 RSC; Y. Wang, L. Tang and J. Yu, Cryst. Growth Des., 2008, 8, 884 Search PubMed.
- D. Krishna Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080 RSC.
- Y. Xu, C. Khang, Y. Chen, Z. Bian, X. Qiu, L. Gao and Q. Meng, Chem. – Eur. J., 2012, 18, 16955 CrossRef CAS PubMed; P. Zhu, X. Yan, Y. Su, Y. Yang and J. Li, Chem. – Eur. J., 2010, 16, 3176 CrossRef PubMed.
- C. D. Jones, J. C. Tan and G. O. Lloyd, Chem. Commun., 2012, 48, 2110 RSC.
- G. Nandi, H. M. Titi, R. Thakuria and I. Goldberg, Cryst. Growth Des., 2014, 14, 2714 Search PubMed; A. Mallick, E.-M. Schön, T. Panda, K. Sreenivas, D. D. Díaz and R. Banerjee, J. Mater. Chem., 2012, 22, 14951 RSC.
- N. Ahmad, M. M. Ahmad and P. N. Kotru, J. Cryst. Growth, 2015, 412, 72 CrossRef CAS PubMed; L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42 CrossRef PubMed; R. Daly, O. Kotova, M. Boese, T. Gunnlaugsson and J. L. Boland, ACS Nano, 2013, 7, 4838 CrossRef PubMed; V. R. Thalladi, S. Brasselet, H.-C. Weiss, D. Bläser, A. K. Katz, H. L. Carrell, R. Boese, J. Zyss, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1998, 120, 2563 CrossRef; G. R. Desiraju, D. Y. Curtin and I. C. Paul, J. Am. Chem. Soc., 1977, 99, 6148 CrossRef.
- O. M. Yaghi, G. Li and H. Li, Chem. Mater., 1997, 9, 1074 CrossRef CAS.
- J. A. Foster, M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke, J. A. K. Howard and J. W. Steed, Nat. Chem., 2010, 2, 1037 CrossRef CAS PubMed.
- C. Sudha, P. Parimaladevi and K. Srinivasan, Mater. Sci. Eng., C, 2015, 47, 150 CrossRef CAS PubMed; F. Aparicio, E. Matesanz and L. Sánchez, Chem. Commun., 2012, 5757 RSC; D. Braga, S. d'Agostino, E. D'Amen, F. Grepioni, D. Genovese, L. Prodi and M. Sgarzi, Dalton Trans., 2013, 42, 16949 RSC; D. Braga, S. d'Agostino, E. D'Amen and F. Grepioni, Chem. Commun., 2011, 5154 RSC.
- T. K. Adalder and P. Dastidar, Cryst. Growth Des., 2014, 14, 2254 Search PubMed; T. K. Adalder, U. K. Das, J. Majumder, R. Roy and P. Dastidar, J. Indian Inst. Sci., 2014, 94, 9 Search PubMed; T. K. Adalder, D. P. Kumar and P. Dastidar, Cryst. Growth Des., 2014, 14, 11 Search PubMed; S. Banerjee, N. N. Adarsh and P. Dastidar, Soft Matter, 2012, 8, 7623 RSC; N. N. Adarsh and P. Dastidar, Cryst. Growth Des., 2011, 11, 328 Search PubMed; N. N. Adarsh, P. Sahoo and P. Dastidar, Cryst. Growth Des., 2010, 10, 4976 Search PubMed.
- N. M. Dixit and C. F. Zukoski, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys, 2003, 67, 061501 CrossRef.
- Y. Furukawa, T. Ishiwata, K. Sugikawa, K. Kokado and K. Sada, Angew. Chem., Int. Ed., 2012, 51, 10566 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS PubMed.
- T. Ishiwata, Y. Furukawa, K. Sugikawa, K. Kokado and K. Sada, J. Am. Chem. Soc., 2013, 135, 5427 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- C. W. Torøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef PubMed.
- Y. Goto, H. Sato, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2008, 130, 14354 CrossRef CAS PubMed.
- J. Ricka and T. Tanaka, Macromolecules, 1984, 17, 2916 CrossRef CAS.
- H. B. Aiyappa, S. Saha, B. Garai, J. Thote, S. Kurungot and R. Banerjee, Cryst. Growth Des., 2014, 14, 3434 CAS.
- S. K. Nune, P. K. Thallapally and B. P. McGrail, J. Mater. Chem., 2010, 20, 7623 RSC; M. R. Lohe, M. Rose and S. Kaskel, Chem. Commun., 2009, 6056 RSC.
- L. Rajput, D. Kim and M. S. Lah, CrystEngComm, 2013, 15, 259 RSC; A. D. Burrows, K. Cassar, R. M. W. Friend, M. F. Mahon, S. P. Rigbyb and J. E. Warren, CrystEngComm, 2005, 7, 548 RSC.
- D. K. Smith, Self-assembling fibrillar networks-Supramolecular gels, in, Supramolecular Chemistry: From Molecules to Nanomaterials, ed. J. W. Steed and A. P. Gale, Wiley & Sons Ltd., Chichester, UK, 1st edn, 2012, vol. 7, pp. 3355–3376 RSC; D. D. Díaz, D. Kühbeck and R. J. Koopmans, Chem. Soc. Rev., 2011, 40, 427 RSC; T. Yu, R. Cristiano and R. G. Weiss, Chem. Soc. Rev., 2010, 39, 1435 RSC; Y. Gao, F. Zhao, Q. G. Wang, Y. Zhang and B. Xu, Chem. Soc. Rev., 2010, 39, 3425 RSC.
- K. Kongpatpanich, S. Horike, M. Sugimoto, S. Kitao, M. Setoc and S. Kitagawa, Chem. Commun., 2014, 50, 2292 RSC; L. Xie, S. Liu, C. Gao, R. Cao, J. Cao, C. Sun and Z. Su, Inorg. Chem., 2007, 46, 7782 CrossRef CAS PubMed; P. Horcajada, S. Surble, C. Serre, D. Y. Hong, Y. K. Seo, J. S. Chang, J. M. Greneche, I. Margiolaki and G. Ferey, Chem. Commun., 2007, 2820 RSC; T. R. Whitfield, X. Wang, L. Liu and A. Jacobson, ECS J. Solid State Sci. Technol., 2005, 7, 1096 CrossRef PubMed; C. Serre, F. Millange, S. Surble and G. Ferey, Angew. Chem., Int. Ed., 2004, 43, 6286 CrossRef PubMed; C. Serre, F. Millange, S. Surble, J.-M. Greneche and G. Ferey, Chem. Mater., 2004, 16, 2706 CrossRef.
- S. Saha, G. Das, J. Thote and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 14845 CrossRef CAS PubMed.
- S. Saha, J. Bachl, T. Kundu, D. D. Díaz and R. Banerjee, Chem. Commun., 2014, 3004 RSC; S. Saha, J. Bachl, T. Kundu, D. D. Díaz and R. Banerjee, Chem. Commun., 2014, 7032 RSC.
- S. C. Sahoo, T. Kundu and R. Banerjee, J. Am. Chem. Soc., 2011, 133, 17950 CrossRef CAS PubMed.
- D. Buso, J. Jasieniak, M. D. H. Lay, P. Schiavuta, P. Scopece, J. Laird, H. Amenitsch, A. J. Hill and P. Falcaro, Small, 2012, 8, 80 CrossRef CAS PubMed.
- R. Lin, L. Shen, Z. Ren, W. Wu, Y. Tan, H. Fu, J. Zhang and L. Wu, Chem. Commun., 2014, 50, 8533 RSC; J. He, Z. Yan, J. Wang, J. Xie, L. Jiang, Y. Shi, F. Yuan, F. Yu and Y. Sun, Chem. Commun., 2013, 49, 6761 RSC.
- I. Imaz, M. Rubio-Martínez, J. An, I. Solé-Font, N. L. Rosi and D. Maspoch, Chem. Commun., 2011, 7287 RSC.
| This journal is © The Royal Society of Chemistry 2015 |