
Electronic effects of ligand substitution on metal–organic framework photocatalysts: the case study of UiO-66†
Lijuan
Shen
,
Ruowen
Liang
,
Mingbu
Luo
,
Fenfen
Jing
and
Ling
Wu
*
State Key Laboratory of Photocatalysis on Energy and Environment, Fuzhou University, Fuzhou 350002, P. R. China. E-mail: wuling@fzu.edu.cn; Tel: +86 591 83779105
First published on 3rd November 2014
Abstract
UiO-66-X (X = H, NH2, NO2, Br) have been successfully synthesized and tested for their photocatalytic activity in water treatment. Results show that electronic effect of the ligand substituents greatly affects the photocatalytic activity of UiO-66. The rates obtained by different substituents are linearly correlated with their Hammett coefficients.
Heterogeneous photocatalysis has been considered as a promising route to address problems in chemical synthesis, environmental remediation and energy utilization.1–6 Although photocatalysis has been around for many years, continue efforts have been devoted to the development of highly efficient and active photocatalysts.7–10 Metal–organic frameworks (MOFs), which are a class of crystalline materials constructed from inorganic building units and organic linkers, have been garnering considerable research interest due to their distinctive physicochemical properties.11–20 Emerging research has demonstrated MOFs to be a new class of photocatalysts.21–25 For example, Garcia et al. have studied MOF-5 as a semiconductor for photodegradation of phenol in aqueous solutions.26 Then a water stable Zr-benzenedicarboxylate MOF has also been successfully applied to the photocatalytic hydrogen generation.27 In sharp contrast to the conventional photocatalysts, band gaps in MOFs can be tuned through simple modification of the organic linker or metal center, opening up the possibilities to facilely control the chemical and physical properties of photocatalysts at the molecular level.28–30 Moreover, the well-defined crystalline structures of MOFs are beneficial for the characterization and study of the structure–property relationship of these photocatalysts, which provides unprecedented opportunities to interrogate the basic mechanistic details of photocatalysis chemistry. In independent studies, Li et al.31 and Matsuoka et al.32 reported the successful tuning of the light absorption of a Ti-based MOF by the introduction of NH2 groups to the MIL-125(Ti), and its applications in the photocatalytic CO2 reduction and hydrogen production. Furthermore, a NH2-mediated zirconium MOF has also been obtained and used as an efficient visible-light-driven photocatalyst for selective oxidation of alcohols and reduction of aqueous Cr(VI).33,34 Despite the progress, work in this area is still in its early stages, particularly, we note that, engineering the optical response of the MOF-based photocatalysts by the functionalization of a ligand almost exclusively focuses on the NH2 substituent. For other substituents, such as NO2 and Br, although the corresponding functional MOFs have been developed, their applications mainly confined to the pore surface modification and adsorption properties tuning.35–38 The photocatalytic activity of functional MOFs except the NH2 substituent has not been experimentally uncovered. Moreover, a significant open issue is that the traditional photocatalytic mechanism has not been matched by exponential increases in publications on these novel photocatalysts. Research studies in open literature are inclined to attribute the enhanced photocatalytic activity of functional MOFs to more effective light harvest.31 But other affecting factors like electronic properties are yet to be thoroughly and systematically mapped. Understanding this step is envisioned to be essential in the future design of novel and more efficient photocatalysts.
Toward this goal, herein, we have embarked on a detailed study of the structure–photoactivity relation with an isoreticular series of MOFs UiO-66-X (X = H, NH2, NO2, Br) (Fig. S1, ESI†) and firstly showed that the electronic effect of the ligand substituents has a systematic and important impact on the photocatalytic activity of the MOFs. UiO-66 and its functionalised compounds are zirconium-based microporous MOFs, which built from Zr6O4(OH)4 oxoclusters related together by 1,4-benzene-dicarboxylate linkers (Fig. S2, ESI†). These materials stand out among the numerous MOFs due to their high stability.39,40 Moreover, recent studies have proved that the UiO-66 series behave as semiconductors when exposed to light, making them unique platforms for light harvesting and photoinduced catalysis.41–44
UiO-66 was prepared by the method reported earlier.34 UiO-66-X (X = NH2, NO2 and Br) were synthesized analogously by replacing terephthalic acid (H2BDC) with equivalent molar amounts of H2N–H2BDC, O2N–H2BDC and Br–H2BDC, respectively. X-ray powder diffraction (XRD) has been used to evaluate the crystallinity and structural topology of UiO-66-X (X = H, NH2, NO2 and Br). As shown in Fig. 1, all materials are crystalline, although no single crystals have been formed. The XRD pattern of the UiO-66 is in good agreement with the previous report.27 It is worth noting that in many cases, introduction of functional groups into the organic scaffold would alter the topology of the desired MOFs. Fortunately, comparison between the diffraction patterns of the parent UiO-66 and those of the functionalised samples demonstrates the formation of pure, topologically equivalent phases for UiO-66-X (X = NH2, NO2 and Br).
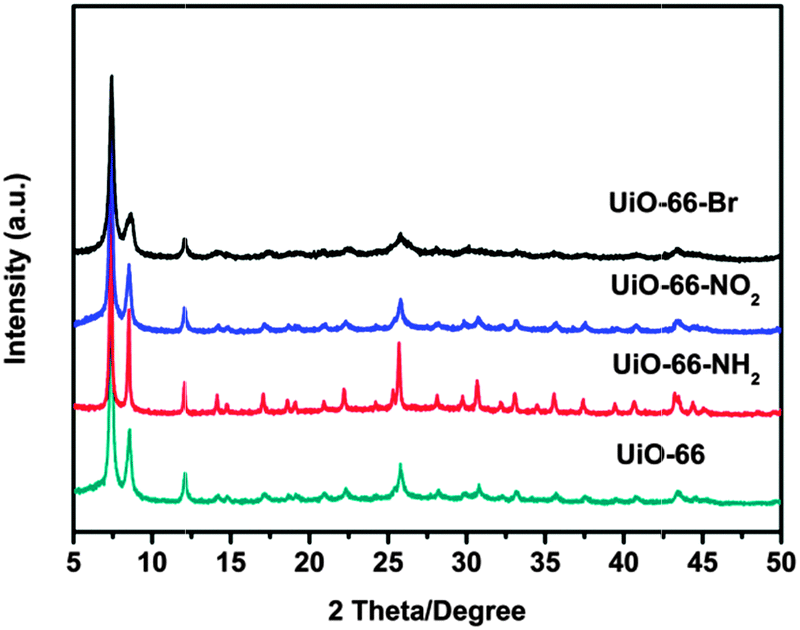 | ||
| Fig. 1 XRD patterns of the UiO-66-X (X = H, NH2, NO2 and Br). | ||
The presence of the introduced functional groups on the linkers has been further evidenced by characterizing the IR spectra on all the UiO-66-X samples, which also confirms that most of the free organic ligands and DMF molecules have been eliminated. Fig. S3 (ESI†) shows the comparison between the IR spectra of the parent UiO-66 and the UiO-66-X (X = NH2, NO2 and Br). The typical absorption modes of the NH2, NO2 and Br functional groups can be found, further confirming the effective functionalization of the linkers on each MOF, which is also consistent with previous reports.45,46 From these results coupled with the XRD analysis, it can be deduced that the functional groups (NH2, NO2 and Br) do not coordinate with metal ions but may be protruding into the empty space of the micropores.
Fig. S4 (ESI†) shows the N2 adsorption isotherm at 77 K measured on the synthesized UiO-66-X. All the samples are found to retain porosity despite the presence of different functional groups on the linker. The BET surface areas of UiO-66-X (X = H, NH2, NO2 and Br) are determined to be 1141.1, 732.2, 464.8 and 455.9 m2 g−1, respectively, suggesting the large surface areas of UiO-66-X. However, it is important to note that the surface area of the UiO-66 decreased after ligand substitution. The decrease in surface areas for the functionalised UiO-66 samples can be largely attributed to both the reduced free space available and an overall increased weight of the MOFs resulting from the introducing of the large and heavy functional groups.47
X-ray photoelectron spectroscopy (XPS) can be used as an exceptionally useful tool for providing information about the charged states of atoms in the molecule and probing the electronic effects of ligands.48,49 The curves in the Zr 3d region could be deconvoluted into two peaks for Zr 3d5/2 and Zr 3d3/2, respectively. Table S1 (ESI†) lists the Zr 3d5/2 binding energies for UiO-66 with different substituents. The XPS results for UiO-66(NH2) show a decrease in Zr 3d5/2 binding energy as compared to the UiO-66, demonstrating the electron-donating effect of the amino functional group. Comparison of UiO-66 with UiO-66-Br shows a 0.14 eV change due to the electron attracting ability of the bromine functional group. In particular, the shifts are pronounced for the UiO-66 bearing the nitro-group, as the electron-withdrawing ability of the para-ring substituents is increased, there is an increase in the Zr 3d5/2 and Zr 3d3/2 binding energies. These data are direct evidence for the effect of the substituents on the electronic nature at the metal centre.
Optical absorption of the UiO-66-X samples has been investigated using UV/vis spectroscopy (as shown in Fig. 2). The main absorption bands of UiO-66 are located in the UV region (ca. 320 nm), which could be attributed to ligand-to-metal charge transfer (LMCT). As expected, the presence of functional groups in the aromatic ring strongly affects the optical properties of the material. The absorption edges are around 450, 400 and 360 nm for UiO-66-X (X = NH2, NO2 and Br), respectively. For the sample of UiO-66-NH2, it shows an obvious red-shift in comparison with other samples. This is because when the amino-substituent is introduced to the ligand, it provides the lone pair of nitrogen for the interaction with the π*-orbitals of the benzene ring, donating electron density to the antibonding orbitals.50 This results in a higher HOMO level that brings absorption to the visible region. This characteristic absorption pattern suggests that the substitutions on the ligand narrow the band gap of UiO-66.
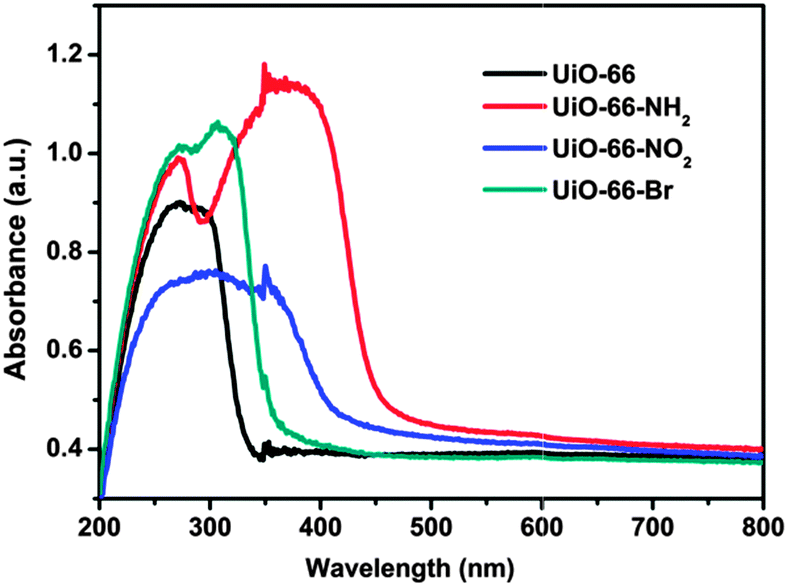 | ||
| Fig. 2 UV-vis absorption spectra of UiO-66-X (X = H, NH2, NO2 and Br). | ||
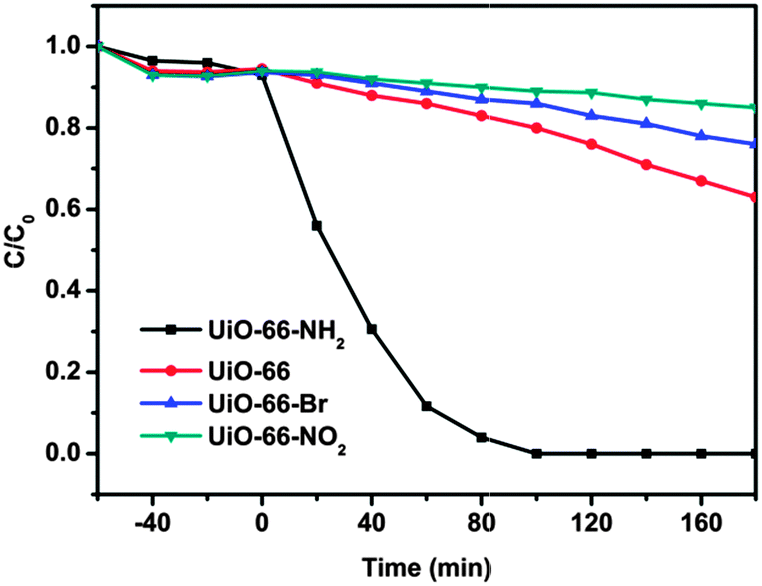
To get in-depth knowledge of the electronic effect of ligand substitution on the photocatalytic properties of MOFs, photocatalytic oxidation of As(III) over UiO-66-X was then carried out. Arsenic is believed to be one of the most toxic contaminants found in the environment and has attracted considerable attention from society and regulation authorities around the world.51–53 Long-term exposure to arsenic may cause cancer of the skin, lungs, urinary tract, kidneys and liver, and can also produce various other non-cancerous effects.54 Among various techniques, the semiconductor photocatalytic oxidizing transformation of As(III) to As(V) has been widely regarded as a promising technology.55 The photocatalytic oxidation of As(III) over UiO-66-X was investigated after a pre-setting of 60 min for achieving the adsorption–desorption equilibrium. Control experiments were first carried out to demonstrate the photocatalytic nature of the reactions. Taking photocatalytic oxidation of As(III) over UiO-66 as a testing reaction. As shown in Fig. S5 (ESI†), blank experiments in the absence of catalyst or light lead to no oxidation of As(III), which ensures that the reaction is driven by the photocatalytic process. Besides, experiment conducted in an inert N2-saturated atmosphere shows negligible photocatalytic activity, indicating that oxygen is the primary oxidant for the photocatalytic oxidation system. Fig. 3 displays the variations of As(III) concentrations (C/C0) in the photocatalytic process over UiO-66-X. It can be seen that all the samples show oxidation abilities toward As(III), the concentration of As(III) decreased with reaction time. Notably, the photocatalytic activity of UiO-66 can be drastically affected by the introduced substituents on the ligand. UiO-66-NH2 is by far the most active material, reaching full conversion after only 60 min. The reaction is first order oxidation abilities toward As(III) and the calculated rate constants are listed in Table 1. After the photocatalytic reaction, the UiO-66-X samples were recovered by filtration. As shown in Fig. S6 (ESI†), XRD patterns of the fresh and used UiO-66-X are almost the same, implying that the samples are stable during the reaction process.
| UiO-66-X | K X [h−1] | Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KX/KH KX/KH |
σ |
|---|---|---|---|
| –H | 0.0266 | 0 | 0 |
| –NH2 | 0.0417 | 0.3443 | −0.161 |
| –NO2 | 0.00369 | −0.6747 | 0.71 |
| –Br | 0.0128 | −0.3040 | 0.393 |
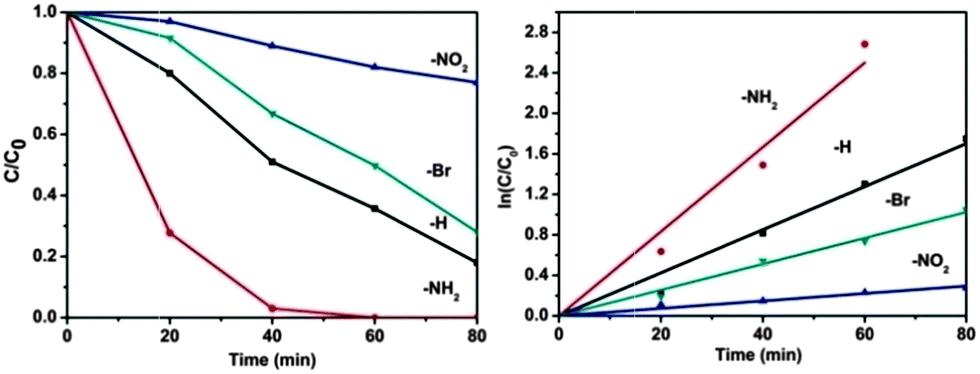 | ||
| Fig. 3 Photocatalytic oxidation of aqueous As(III) over UiO-66-X (X = H, NH2, NO2 and Br) under simulated sunlight (320–780 nm). Reaction solution: 40 mg photocatalyst, 40 mL 2 mg L−1 As(III) aqueous solution. | ||
As we know, the liquid–solid heterogeneous photocatalysis reactions are surface physical–chemical processes.56 Theoretically, a larger specific surface area of the photocatalyst can accommodate more surface active sites and facilitate the transport of charge carriers, which is expected to show positive effects on the reaction. However, in our case, the BET surface areas of the samples follow the order of UiO-66-H > UiO-66-NH2 > UiO-66-NO2 > UiO-66-Br. While the reaction rates are found to follow the order: UiO-66-NH2 > UiO-66 > UiO-66-Br > UiO-66-NO2. We further normalized the photocatalytic reaction rates of As(III) oxidation with the surface areas. As shown in Table S2 (ESI†), the order of the normalized rates is consistent with the original ones, indicating that the surface area could not be the decisive factor that determines the photocatalytic activity of the UiO-66-X. Moreover, in view of the significant difference in the optical spectra of UiO-66 and UiO-66-X, we envision that the extended absorption capability may be beneficial for improving the photocatalytic performance. Unexpectedly, the band gaps of the UiO-66-X still cannot correlate with their photocatalytic activities. These results are difficult to be explained by employing the conventional photocatalysis theory, suggesting that other explanations have to be sought. We attempt to correlate the activity to electronic effects of the linkers using Hammett's σm values. The Hammett constant represents the effect that different substituents have on the electronic character of a given aromatic system.57 A positive value of σ indicates an electron-withdrawing group and a negative value an electron-donating group. Apparently, an excellent correlation is found by plotting logKX values from Table 1versus the σm values (as shown in Fig. 4), confirming the influence of the electrophilic nature of the substituent on the photocatalytic oxidation of As(III) to As(V).
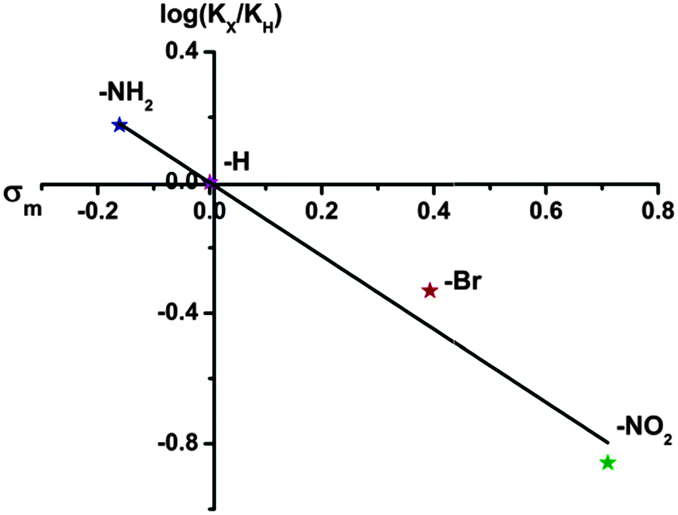 | ||
| Fig. 4 Hammett plot for oxidation of aqueous As(III) with UiO-66-X (X = H, NH2, NO2 and Br) catalysts. | ||
To check whether these trends are general, the reduction of Cr(VI) over UiO-66-X has also been studied. As can be observed from Fig. 5, similar to the case of photocatalytic oxidation of As(III), a large rate increase is realized when the electron-donating NH2 functional group is introduced, while the introduction of the Br and NO2 groups attenuates the reactivity of the UiO-66. This can be attributed to the fact that, zirconium–oxygen clusters within UiO-66-X have been identified as the active sites, which are spaced out by organic ligands. The introduction of functional groups into the organic scaffold affects the electron density around the metal centre. The higher electron density caused by the electron-donating functional group would promote the separation and transfer of photogenerated charge carriers, and hence improves the photocatalytic activity of the sample. These observations prove that the ruling factor affecting the photocatalytic activity of UiO-66-X is due to the electronic effect, rather than to the surface area and band gap of the sample.
 | ||
| Fig. 5 Photocatalytic reduction of aqueous Cr(VI) over UiO-66-X (X = H, NH2, NO2 and Br) under simulated sunlight (320–780 nm). Reaction conditions: 20 mg photocatalyst, 40 mL of 10 ppm Cr(VI), pH = 2. | ||
Conclusions
In conclusion, the photocatalytic activity of MOF catalysts can be strongly affected by using functionalized linkers. A clear structure–photoactivity relationship has been found between the electronic nature of the linker substituents and the reaction rate in the oxidation of As(III) over UiO-66-X (X = H, NH2, NO2 and Br), that is the UiO-66 bearing electron-donating substituent is more reactive, while the electron-withdrawing substituents attenuate the photoreactivity of UiO-66. The trend can be extended to other reactions like reduction of Cr(VI). This is an important finding, because it represents the electronic effect on photocatalysis over MOF catalysts that has not been widely studied. It is thus envisioned that our work would provide useful insights for future design of novel and more efficient photocatalysts.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21273036 and 21177024).Notes and references
- J. B. Joo, R. Dillon, I. Lee, Y. Yin, C. J. Bardeen and F. Zaera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7942–7947 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- L. Qi, J. Yu and M. Jaroniec, Phys. Chem. Chem. Phys., 2011, 13, 8915 RSC.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- L. Nie, J. Yu, X. Li, B. Cheng, G. Liu and M. Jaroniec, Environ. Sci. Technol., 2013, 47, 2777–2783 CrossRef CAS PubMed.
- Y. Wei, S. Han, D. A. Walker, S. C. Warren and B. A. Grzybowski, Chem. Sci., 2012, 3, 1090–1094 RSC.
- M.-Q. Yang and Y.-J. Xu, Phys. Chem. Chem. Phys., 2013, 15, 19102–19118 RSC.
- Y. Horiuchi, T. Toyao, M. Takeuchi, M. Matsuoka and M. Anpo, Phys. Chem. Chem. Phys., 2013, 15, 13243–13253 RSC.
- M.-Q. Yang, N. Zhang, M. Pagliaro and Y.-J. Xu, Chem. Soc. Rev., 2014 10.1039/c4cs00213j.
- J. Tang, Y. Liu, H. Li, Z. Tan and D. Li, Chem. Commun., 2013, 49, 5498–5500 RSC.
- Y.-S. Bae, O. K. Farha, J. T. Hupp and R. Q. Snurr, J. Mater. Chem., 2009, 19, 2131 RSC.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed.
- J. I. Feldblyum, A. G. Wong-Foy and A. J. Matzger, Chem. Commun., 2012, 48, 9828–9830 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- W. Li, M. R. Probert, M. Kosa, T. D. Bennett, A. Thirumurugan, R. P. Burwood, M. Parinello, J. A. Howard and A. K. Cheetham, J. Am. Chem. Soc., 2012, 134, 11940–11943 CrossRef CAS PubMed.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- H.-S. Lu, L. Bai, W.-W. Xiong, P. Li, J. Ding, G. Zhang, T. Wu, Y. Zhao, J.-M. Lee, Y. Yang, B. Geng and Q. Zhang, Inorg. Chem., 2014, 53, 8529–8537 CrossRef CAS PubMed.
- J. Gao, M. He, Z. Y. Lee, W. Cao, W.-W. Xiong, Y. Li, R. Ganguly, T. Wu and Q. Zhang, Dalton Trans., 2013, 42, 11367–11370 RSC.
- J. Gao, K. Ye, M. He, W.-W. Xiong, W. Cao, Z. Y. Lee, Y. Wang, T. Wu, F. Huo, X. Liu and Q. Zhang, J. Solid State Chem., 2013, 206, 27–31 CrossRef CAS PubMed.
- J. Gao, K. Ye, L. Yang, W.-W. Xiong, L. Ye, Y. Wang and Q. Zhang, Inorg. Chem., 2013, 53, 691–693 CrossRef PubMed.
- S. Han, Y. Wei and B. A. Grzybowski, Chem. – Eur. J., 2013, 19, 11194–11198 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- T. Zhou, Y. Du, A. Borgna, J. Hong, Y. Wang, J. Han, W. Zhang and R. Xu, Energy Environ. Sci., 2013, 6, 3229–3234 CAS.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed.
- J. Gao, J. Miao, P.-Z. Li, W. Y. Teng, L. Yang, Y. Zhao, B. Liu and Q. Zhang, Chem. Commun., 2014, 50, 3786–3788 RSC.
- M. Alvaro, E. Carbonell, B. Ferrer, F. X. Llabres i Xamena and H. Garcia, Chem. – Eur. J., 2007, 13, 5106–5112 CrossRef CAS PubMed.
- C. Gomes Silva, I. Luz, F. X. Llabres i Xamena, A. Corma and H. Garcia, Chem. – Eur. J., 2010, 16, 11133–11138 CrossRef PubMed.
- J. Gascon, M. D. Hernandez-Alonso, A. R. Almeida, G. P. van Klink, F. Kapteijn and G. Mul, ChemSusChem, 2008, 1, 981–983 CrossRef CAS PubMed.
- M. A. Nasalevich, M. G. Goesten, T. J. Savenije, F. Kapteijn and J. Gascon, Chem. Commun., 2013, 49, 10575–10577 RSC.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D'Arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 124, 1–9 CrossRef.
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848–20853 CAS.
- L. Shen, L. Huang, S. Liang, R. Liang, N. Qin and L. Wu, RSC Adv., 2014, 4, 2546–2549 RSC.
- L. Shen, S. Liang, W. Wu, R. Liang and L. Wu, Dalton Trans., 2013, 42, 13649–13657 RSC.
- T. Devic, P. Horcajada, C. Serre, F. Salles, G. Maurin, B. Moulin, D. Heurtaux, G. Clet, A. Vimont, J.-M. Grenèche, B. L. Ouay, F. Moreau, E. Magnier, Y. Filinchuk, J. Marrot, J.-C. Lavalley, M. Daturi and G. Férey, J. Am. Chem. Soc., 2009, 132, 1127–1136 CrossRef PubMed.
- F. Vermoortele, M. Vandichel, B. Van de Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Angew. Chem., Int. Ed., 2012, 51, 4887–4890 CrossRef CAS PubMed.
- S. T. Meek, S. L. Teich-McGoldrick, J. J. Perry, J. A. Greathouse and M. D. Allendorf, J. Phys. Chem. C, 2012, 116, 19765–19772 CAS.
- E. Flage–Larsen, A. Røyset, J. H. Cavka and K. Thorshaug, J. Phys. Chem. C, 2013, 117, 20610–20616 Search PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- H. Wu, T. Yildirim and W. Zhou, J. Phys. Chem. Lett., 2013, 4, 925–930 CrossRef CAS.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem. – Eur. J., 2013, 19, 14279–14285 CrossRef CAS PubMed.
- J. Long, S. Wang, Z. Ding, S. Wang, Y. Zhou, L. Huang and X. Wang, Chem. Commun., 2012, 48, 11656–11658 RSC.
- L. Shen, S. Liang, W. Wu, R. Liang and L. Wu, J. Mater. Chem. A, 2013, 1, 11473–11482 CAS.
- L. Shen, W. Wu, R. Liang, R. Lin and L. Wu, Nanoscale, 2013, 5, 9374–9382 RSC.
- Y. Lin, C. Kong and L. Chen, RSC Adv., 2012, 2, 6417–6419 RSC.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- V. Colombo, C. Montoro, A. Maspero, G. Palmisano, N. Masciocchi, S. Galli, E. Barea and J. A. Navarro, J. Am. Chem. Soc., 2012, 134, 12830–12843 CrossRef CAS PubMed.
- P. G. Gassman and C. H. Winter, J. Am. Chem. Soc., 1988, 110, 6130–6135 CrossRef CAS PubMed.
- L. Chiang, L. E. Allan, J. Alcantara, M. C. Wang, T. Storr and M. P. Shaver, Dalton Trans., 2014, 43, 4295–4304 RSC.
- M. A. Nasalevich, M. van der Veen, F. Kapteijn and J. Gascon, CrystEngComm, 2014, 16, 4919–4926 RSC.
- D. K. Nordstrom, Science, 2002, 296, 2143–2145 CrossRef CAS PubMed.
- J. Hu, S. Weng, Z. Zheng, Z. Pei, M. Huang and P. Liu, J. Hazard. Mater., 2014, 264, 293–302 CrossRef CAS PubMed.
- Y. Wang, J. Xu, J. Li and F. Wu, J. Hazard. Mater., 2013, 260, 255–262 CrossRef CAS PubMed.
- M. E. Pena, G. P. Korfiatis, M. Patel, L. Lippincott and X. Meng, Water Res., 2005, 39, 2327–2337 CrossRef CAS PubMed.
- J. Ryu, D. Monllor-Satoca, D. H. Kim, J. Yeo and W. Choi, Environ. Sci. Technol., 2013, 47, 9381–9387 CrossRef CAS PubMed.
- J. Zhang, J. Yu, M. Jaroniec and J. R. Gong, Nano Lett., 2012, 12, 4584–4589 CrossRef CAS PubMed.
- S. Parra, J. Olivero, L. Pacheco and C. Pulgarin, Appl. Catal., B, 2003, 43, 293–301 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental process and characterization. See DOI: 10.1039/c4cp04162c |
| This journal is © the Owner Societies 2015 |