DOI:
10.1039/C4CS00507D
(Tutorial Review)
Chem. Soc. Rev., 2015,
44, 1740-1748
Progress in asymmetric biomimetic transamination of carbonyl compounds
Received
22nd December 2014
First published on 3rd February 2015
Abstract
Transamination of α-keto acids with transaminases and pyridoxamine phosphate is an important process to form optically active α-amino acids in biological systems. Various biomimetic transamination systems have been developed for carbonyl compounds including α-keto acid derivatives, fluoroalkyl ketones, and unactivated ketones with chiral vitamin B6 analogues, artificial transaminase mimics, chiral nitrogen sources, and chiral catalysts. This review provides a brief summary of this area.

Ying Xie
| Ying Xie was born in Hubei, China, in 1988. She received her BSc degree from Sichuan University in 2009 and PhD degree under the supervision of Professor Yian Shi at Institute of Chemistry, Chinese Academy of Sciences in 2014. |

Hongjie Pan
| Hongjie Pan was born in Anhui, China, in 1990. He received his BSc degree from Inner Mongolia University in 2011. He is currently a PhD graduate student with Professor Yian Shi at Institute of Chemistry, Chinese Academy of Sciences. |

Mao Liu
| Mao Liu was born in Chongqing, China, in 1986. He received his BSc degree from Tongji University in 2009 and PhD degree under the supervision of Professor Yian Shi at Institute of Chemistry, Chinese Academy of Sciences in 2014. He is currently working at Department of Chemistry, School of Chemistry and Chemical Engineering, Guizhou University. |

Xiao Xiao
| Xiao Xiao was born in Liaoning, China, in 1984. She received her BSc degree from Sichuan University in 2007 and PhD degree under the supervision of Professor Yian Shi at Institute of Chemistry, Chinese Academy of Sciences in 2012. She is currently a lecturer at School of Pharmaceutical Engineering, Shenyang Pharmaceutical University. |

Yian Shi
| Yian Shi was born in Jiangsu, China, in 1963. He obtained his BSc degree from Nanjing University in 1983, MSc degree from University of Toronto with Professor Ian W. J. Still in 1987, and PhD degree from Stanford University with Professor Barry M. Trost in 1992. After a postdoctoral study at Harvard Medical School with Professor Christopher Walsh, he joined Colorado State University as assistant professor in 1995 and was promoted to associate professor in 2000 and professor in 2003. He served as the director of CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences during 2009–2012. His current research interests include the development of new synthetic methods, asymmetric catalysis, and synthesis of natural products. |
Key learning points
(1) Introduction of biological transamination of α-keto acids.
(2) Design of chiral pyridoxamine analogues and artificial transaminase mimics.
(3) Development of catalytic asymmetric transamination of carbonyl compounds.
|
1. Introduction
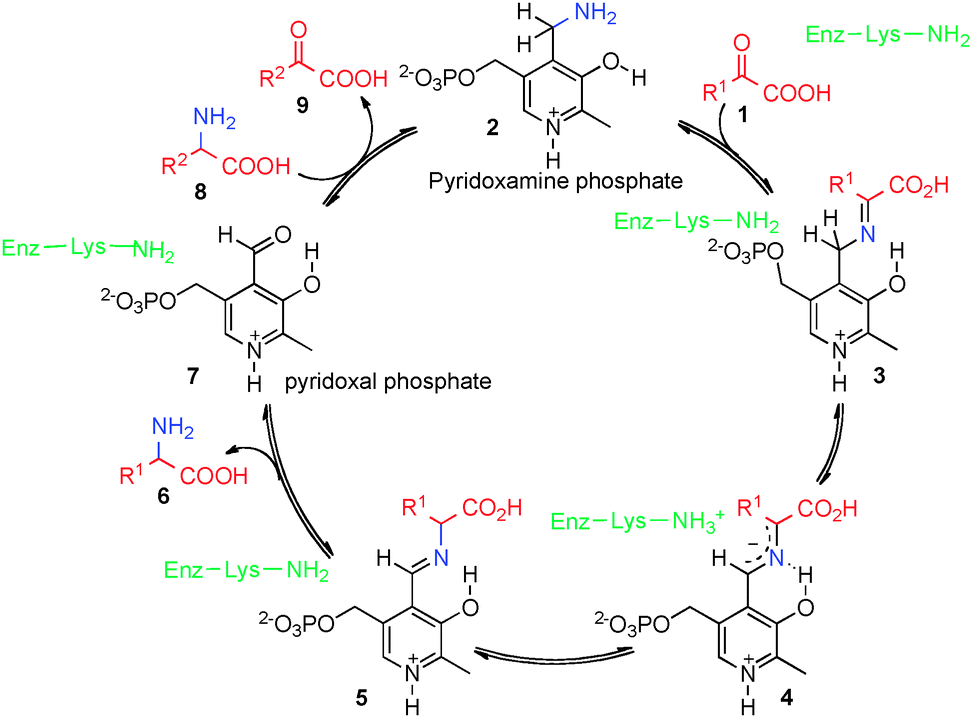
Optically active amino acids and their derivatives play a very important role in biological systems and chemical synthesis. Transamination of α-keto acids is an important biological process to generate α-amino acids (Scheme 1).1,2 In this process, α-keto acid 1 reacts with pyridoxamine phosphate (2) to form ketimine 3, which is converted into aldimine 5via asymmetric [1,3]-hydrogen shift mediated by a transaminase. Aldimine 5 is hydrolyzed to α-amino acid 6 and pyridoxal phosphate (7). Pyridoxamine phosphate 2 could be regenerated from pyridoxal phosphate (7) and another α-amino acid (8) via a reverse process. Great efforts have been made to develop asymmetric biomimetic transamination processes to generate optically active amino acids and related compounds via various strategies including chiral pyridoxamine derivatives, artificial transaminase mimics, chiral nitrogen sources, chiral catalysts, etc.3–7 This review will briefly highlight the development in this area.
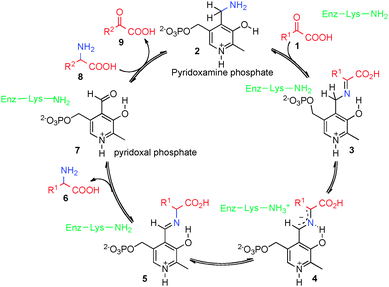 |
| | Scheme 1 Biological transamination of α-keto acids. | |
2. Vitamin B6 analogues
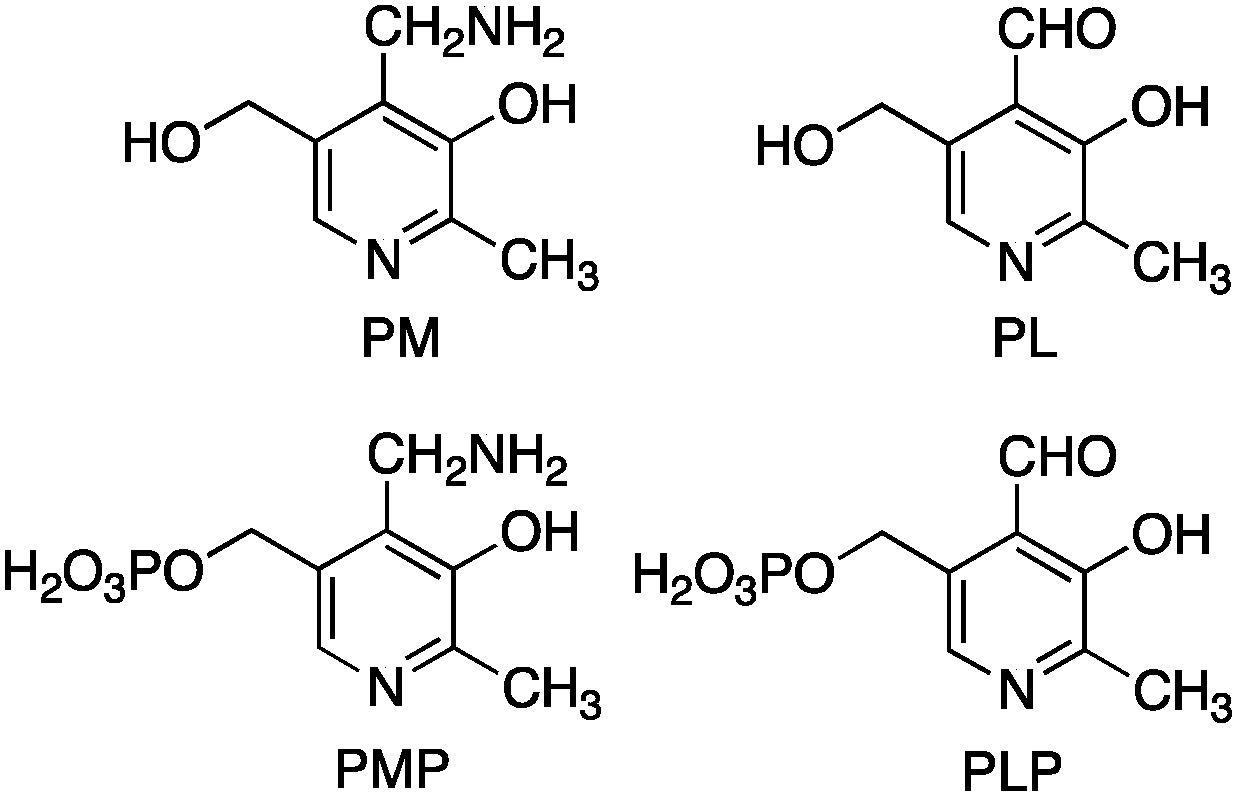
Pyridoxamine 5′-phosphate (PMP) and pyridoxal 5′-phosphate (PLP) belong to the vitamin B6 family (Scheme 2) and function as co-enzymes for the transamination in biological systems. A variety of chiral pyridoxamine analogues have been synthesized and investigated for the asymmetric biomimetic transamination.
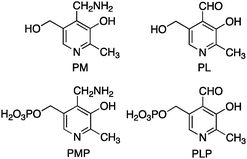 |
| | Scheme 2 Members of the vitamin B6 family. | |
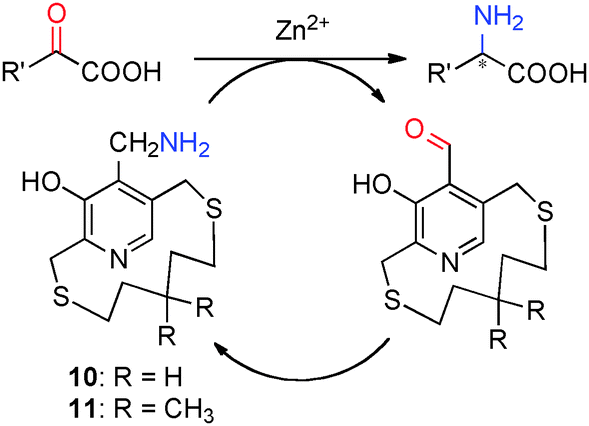
For example, in 1978, Kuzuhara and coworkers reported their studies on transamination of α-keto acids with chiral “ansa chain” based pyridoxamine analogues 10 (Scheme 3).8 Phenylalanine was obtained in 55–83% yield and 6–26% ee when the sodium salt of phenylpyruvic acid was treated with 10 in the presence of Zn2+ at room temperature.8 Further studies showed that higher ee was obtained with 11 than 10. The molar ratio of Zn2+/10 or 11 was important for the enantioselectivity. A number of amino acids were obtained in 60–96% ee when the reactions were carried out with sodium salts of α-keto acids (2 equiv.), pyridoxamine analogue 11 (1 equiv.), and Zn(ClO4)2·6H2O (0.5 equiv.) in MeOH at room temperature. In all cases, the corresponding aldehyde of 11 can be isolated in 75–85% yield.9–11 With chiral pyridoxamine analogue 11, several non-natural fluorophenylalanines were obtained in 33–66% ee from the corresponding α-keto acids.12
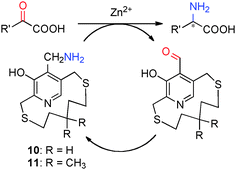 |
| | Scheme 3 Transamination of α-keto acids. | |
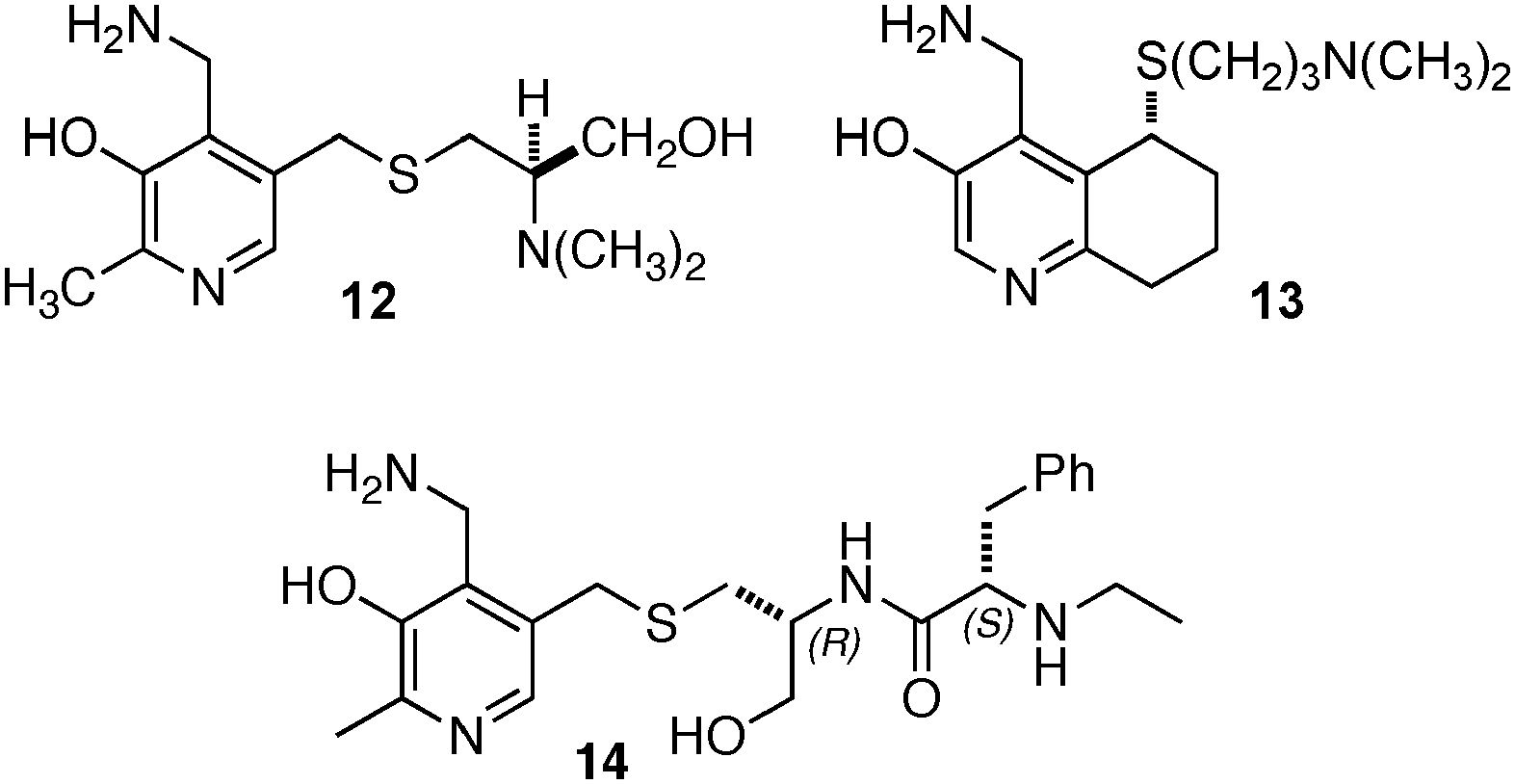
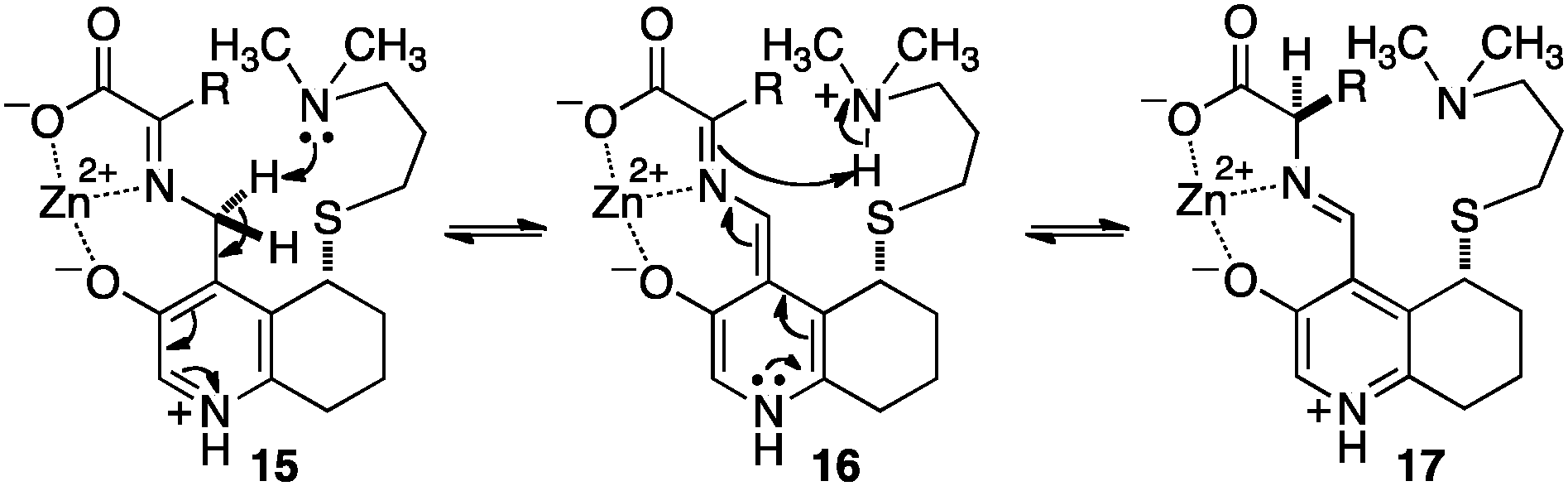
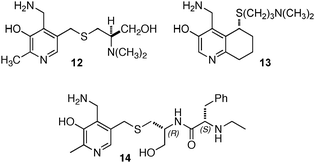
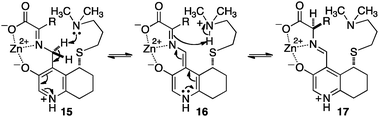
Breslow and coworkers investigated a series of chiral pyridoxamine analogues containing basic side chains such as 12,1313,14,15 and 1416 for the transamination of α-keto acids (Scheme 4). Up to 92% ee was obtained for α-amino acids when the reaction was carried out with α-keto acids and bicyclic compound 13 in the presence of Zn(OAc)2 in MeOH (pH = 4.0) at 30 °C.14,15 The high enantioselectivity obtained with 13 can be attributed to the rigidity of the basic side arm, which allows the proton transfer to occur predominately from one face (Scheme 5).14 Significant rate acceleration was also observed for the transamination as compared to the pyridoxamine analogue without the basic group in the side chain.13–15
 |
| | Scheme 4 Chiral pyridoxamine analogues with basic side chains. | |
 |
| | Scheme 5 Enantioselective deprotonation and protonation. | |
3. Artificial transaminase mimics
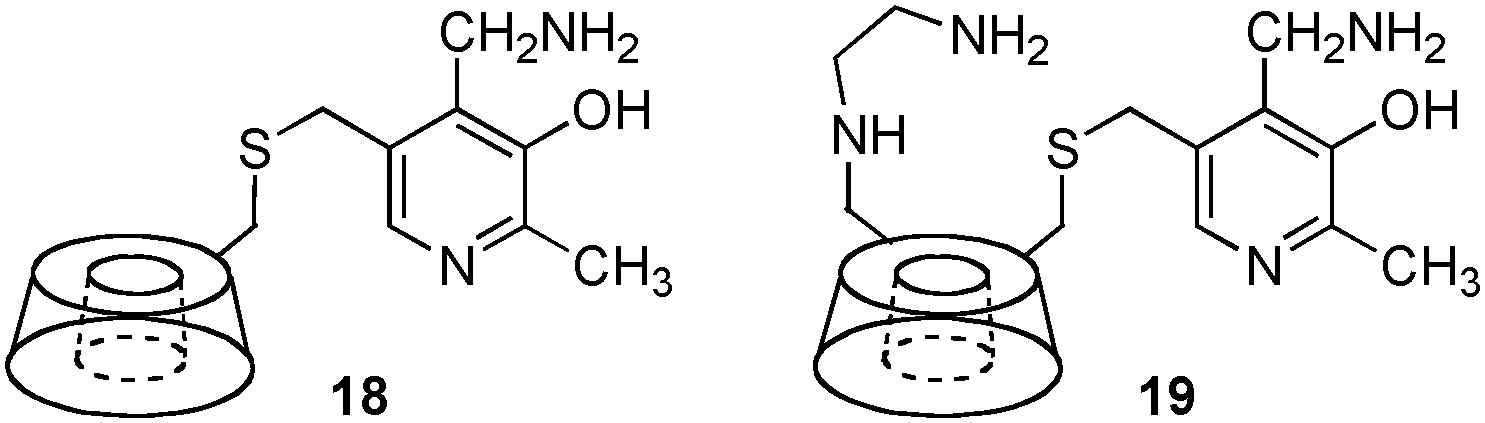
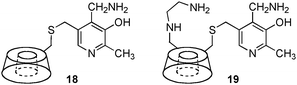
Transamination with a pyridoxamine linked to β-cyclodextrin (18) was reported by Breslow and coworkers in 1980 (Scheme 6).17 A 200-fold rate acceleration was observed with 18 as compared to pyridoxamine itself for the conversion of indolepyruvic acid to tryptophan. A 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of L-Phe to D-Phe was obtained for the transamination of phenylpyruvic acid with 18.15,18 In 1985, Tabushi and coworkers showed that β-cyclodextrin–pyridoxamine–ethylenediamine 19 was highly effective for the transamination, giving L-phenylalanine, L-tryptophan, and L-phenylglycine in 90–96% ee (Scheme 6).19,20
:1 ratio of L-Phe to D-Phe was obtained for the transamination of phenylpyruvic acid with 18.15,18 In 1985, Tabushi and coworkers showed that β-cyclodextrin–pyridoxamine–ethylenediamine 19 was highly effective for the transamination, giving L-phenylalanine, L-tryptophan, and L-phenylglycine in 90–96% ee (Scheme 6).19,20
 |
| | Scheme 6 β-Cyclodextrin–pyridoxamines. | |
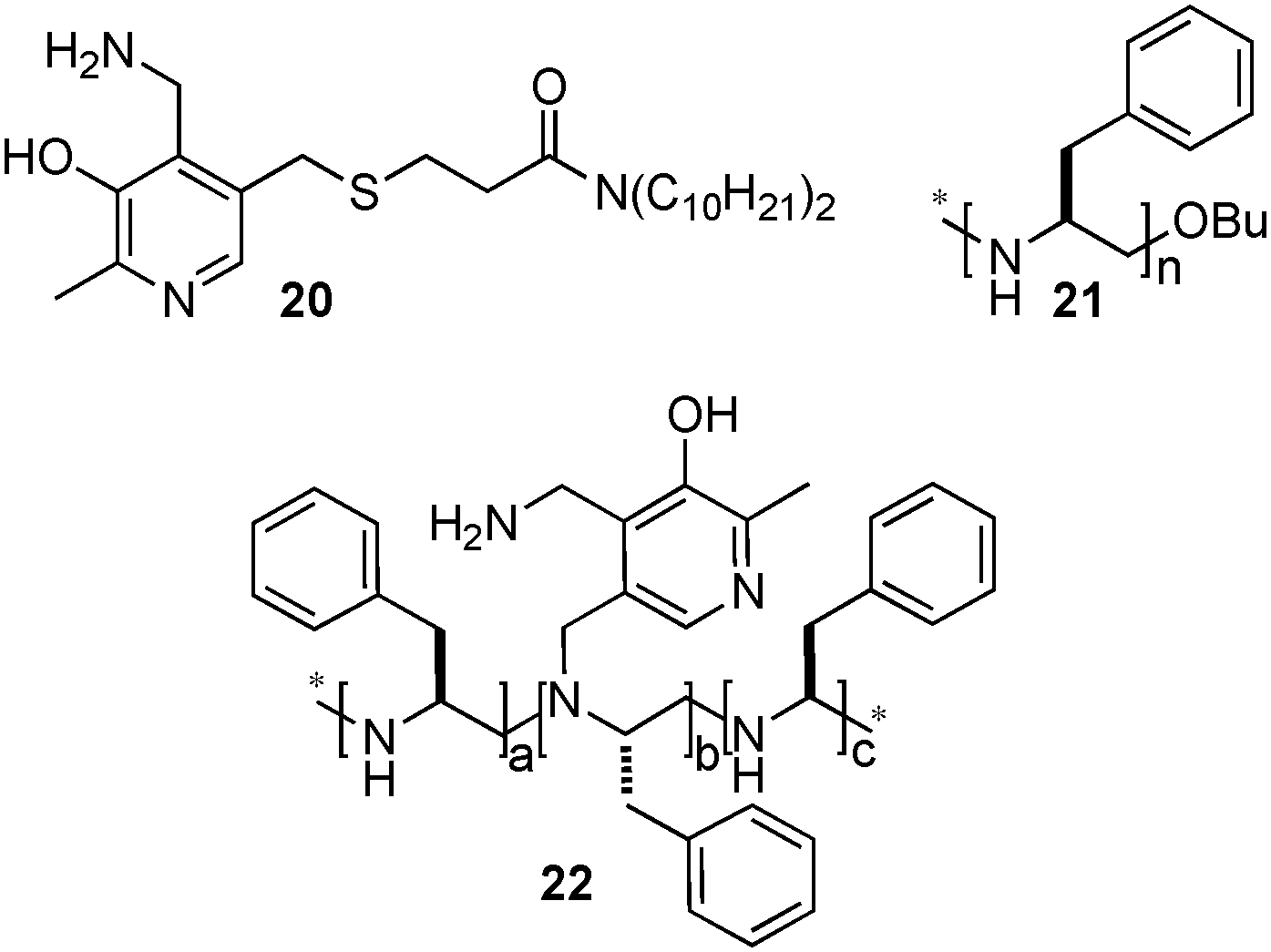
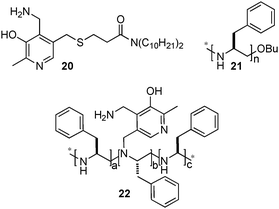
Breslow and coworkers also investigated the asymmetric transamination with pyridoxamine analogues bound to chiral dendrimers21,22 and polymers23 (Scheme 7). For example, up to 66% ee was obtained for L-Val at the initial stage of the reaction with pyridoxamine 20 bound to chiral PEI 21via hydrophobic interactions in 40% aqueous methanol at pH 7.3–7.8.23 Racemization of the amino acid was observed under the reaction conditions. With covalently bound chiral polymer-pyridoxamine 22, L-phenylalanine was obtained in 58% ee from phenylpyruvic acid.23 Little racemization of the amino acid occurred with this system. In 2004, Nicholls and coworkers reported that a transition state analogue-imprinted polymer could act as a transaminase mimic, giving phenylalanine in 32% ee.24
 |
| | Scheme 7 Chiral polymer-pyridoxamines. | |
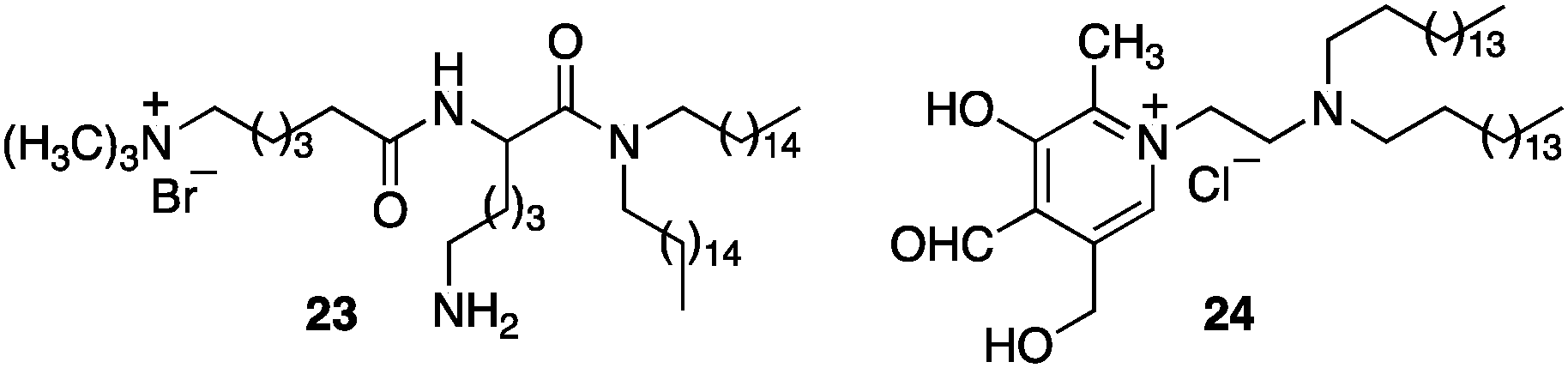
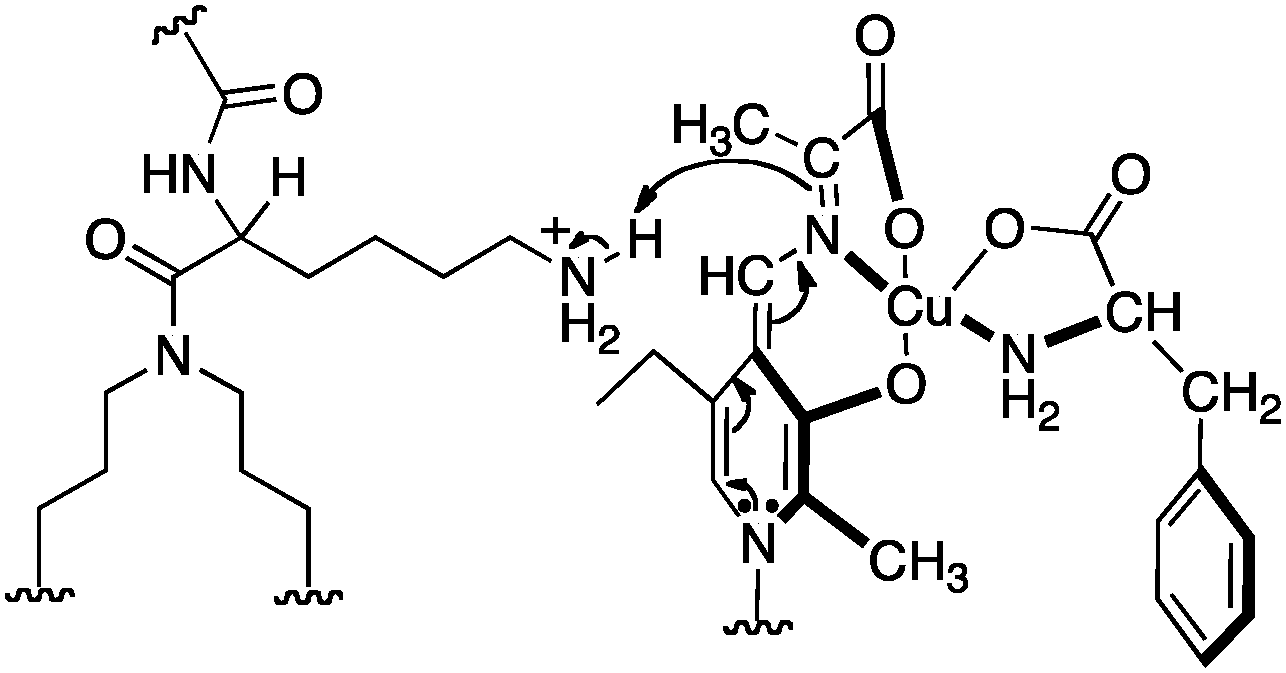
Murakami and coworkers reported the asymmetric transamination with supramolecular bilayer membrane based artificial aminotransferase.25–27 For example, up to 92% ee was obtained for D-alanine when the reaction was carried out with pyruvate (5 equiv.), L-Phe (5 equiv.), peptide lipid 23 (1 equiv.), hydrophobic pyridoxal analogue 24 (0.05 equiv.), and Cu(ClO4)2 (0.05 equiv.) in aqueous 2-[4-(2-hydroxyethyl)-1-piperazinyl]ethanesulfonic acid (HEPES) buffer (pH 7.0) at 30 °C (Scheme 8).27L-Phenylalanine not only acted as the nitrogen source for the conversion of pyruvate to D-alanine, but also played an important role in the enantioselectivity as a chiral ligand for the Cu(II) complex (Scheme 9).27 Distefano and coworkers illustrated that high enantioselectivity (up to 94% ee) can be achieved for the transamination with protein–pyridoxamine conjugates linked via the disulfide bond.28
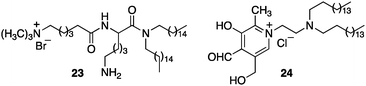 |
| | Scheme 8 Compounds for the supramolecular bilayer membrane. | |
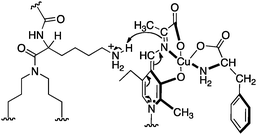 |
| | Scheme 9 Stereoselective protonation of the ketimine complex. | |
4. Catalytic asymmetric transamination
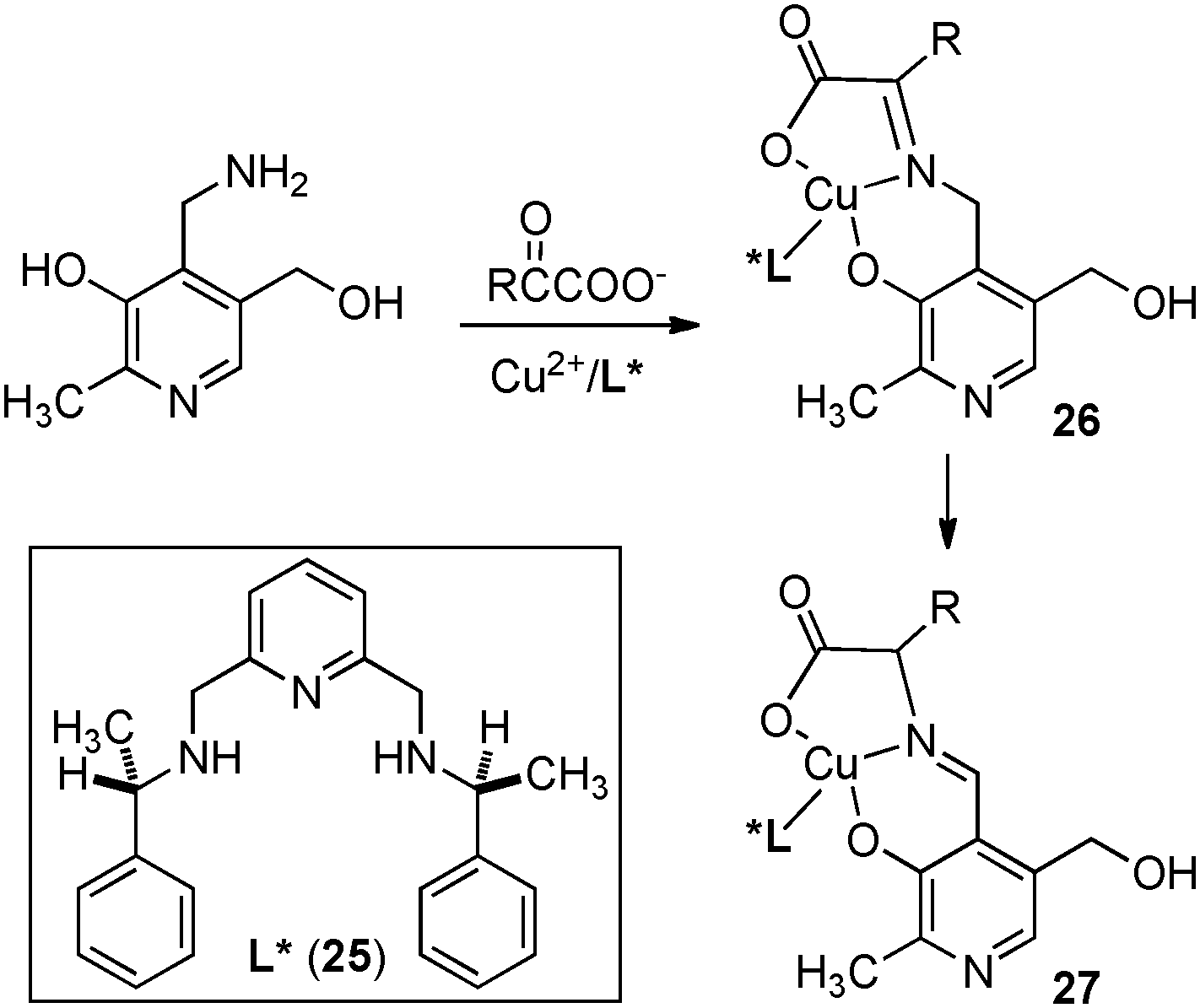
In 1983, Bernauer and coworkers reported that optically active phenylalanine was formed from phenylpyruvic acid with pyridoxamine and a chiral Cu(II) catalyst (Scheme 10).29,30 The Cu(II) catalyst promoted the ketimine–aldimine isomerization and induced the chirality for the reaction. It was found that the enantioselectivity decreased as the reaction proceeded likely due to in situ racemization of aldimine complex 27. The ee was estimated to be 80% ee at the beginning of the reaction.
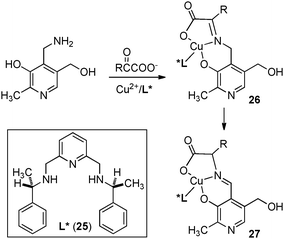 |
| | Scheme 10 Cu(II)-catalyzed asymmetric proton transfer. | |
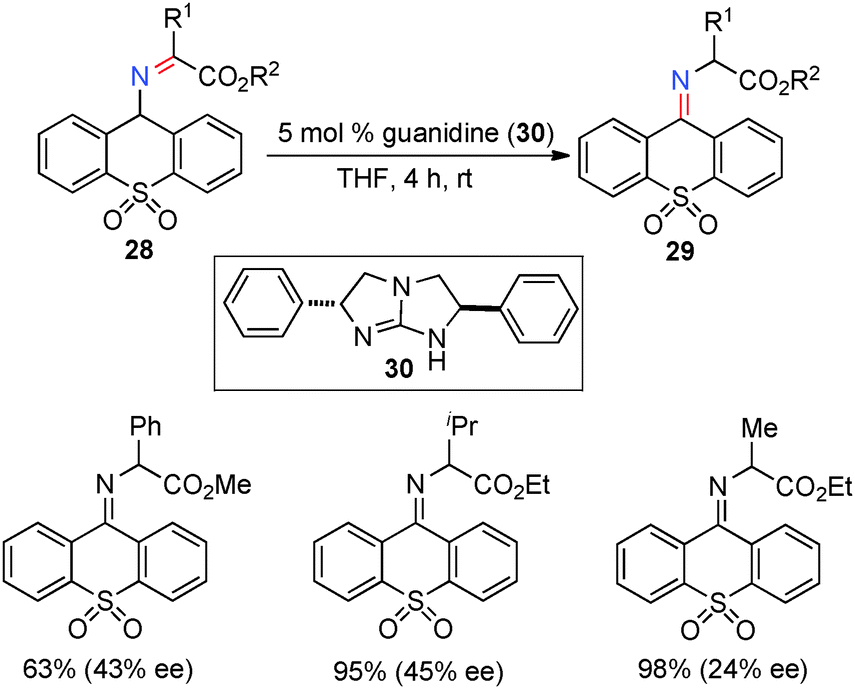
In 2002, Berg and coworkers showed that ketimines 28, derived from α-keto esters and 9-aminothioxanthene 10,10-dioxide, were efficiently isomerized to ketimines 29 in up to 45% ee with 5 mol% chiral guanidine catalyst 30 (Scheme 11).31 Studies suggest that the reaction may proceed via a stepwise, bifunctional mechanism, which provides valuable insight for the development of more effective systems.
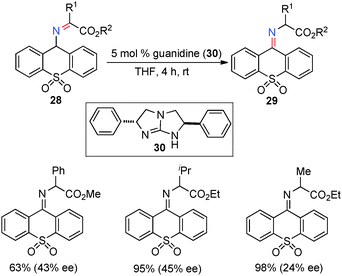 |
| | Scheme 11 Chiral guanidine-catalyzed ketimine–ketimine isomerization. | |
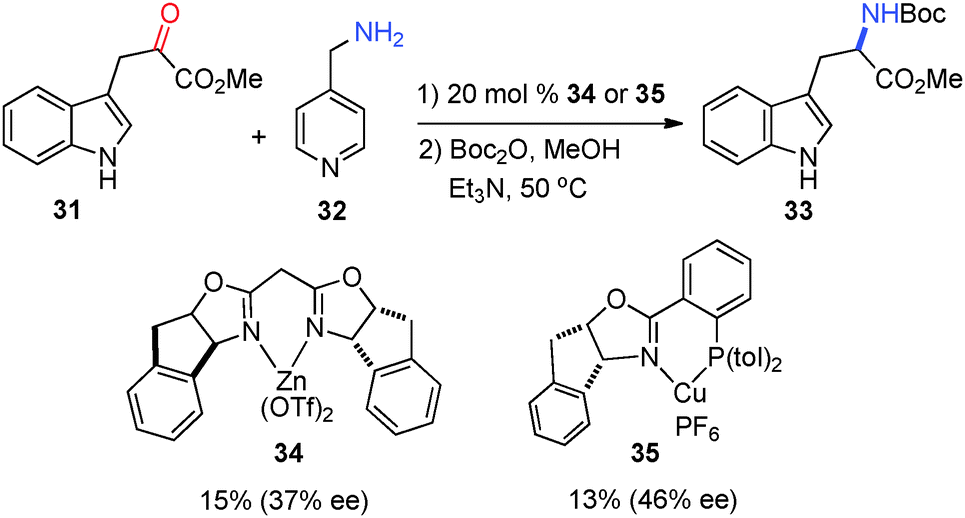
In 2003, Jørgensen and coworkers reported that asymmetric transamination was realized with chiral Lewis acids via in situ formation of the ketimine. Methyl-3-indole pyruvate (31) was converted into amino ester 33 in 37% and 46% ee, respectively, with catalysts 34 and 35 using 4-picolylamine (32) as the amine donor (Scheme 12).32,33 The pyridine of 32 was found to be important for the reactivity as benzylamine was shown to be ineffective for the reaction. The solvent had a significant impact on the enantioselectivity, with MeNO2 being the best.
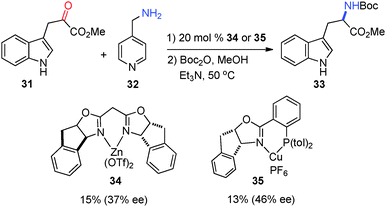 |
| | Scheme 12 Chiral Lewis acid-catalyzed transamination of α-keto ester. | |
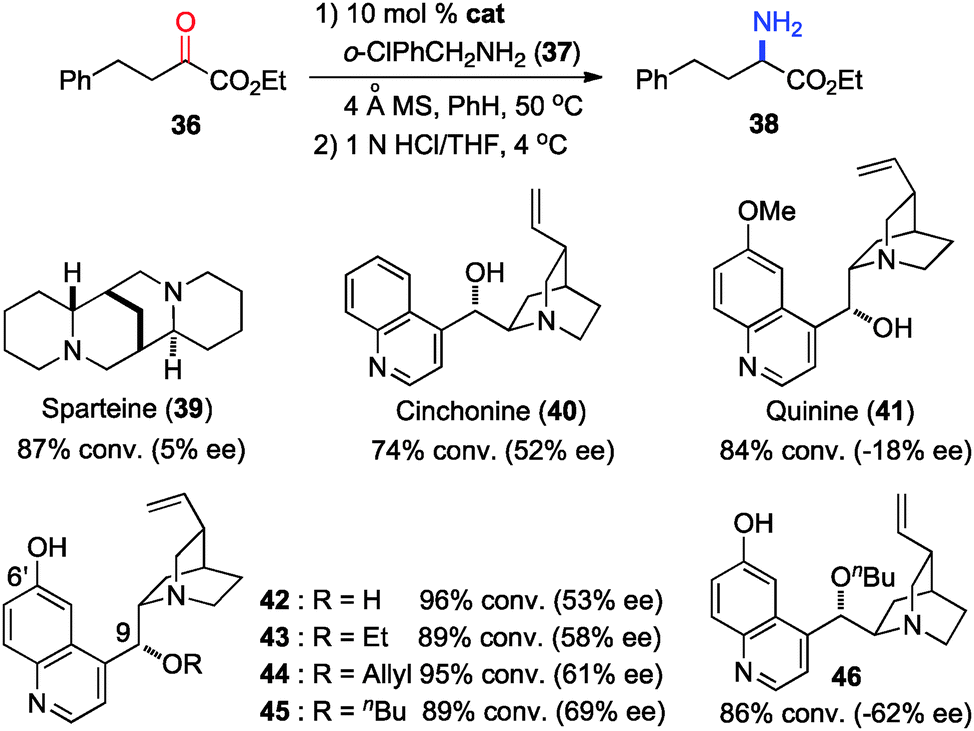
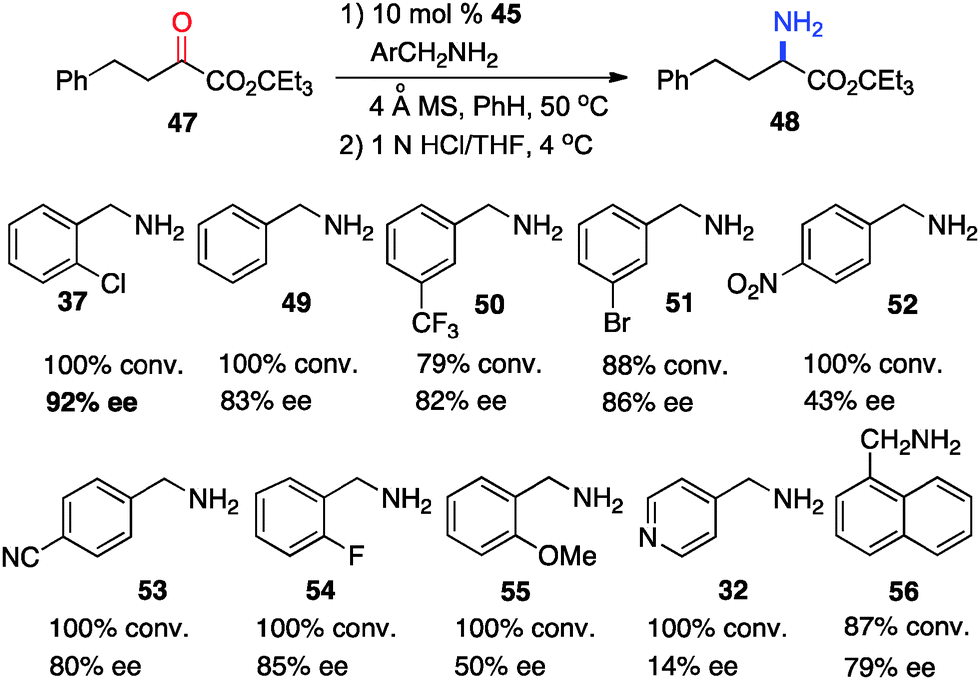
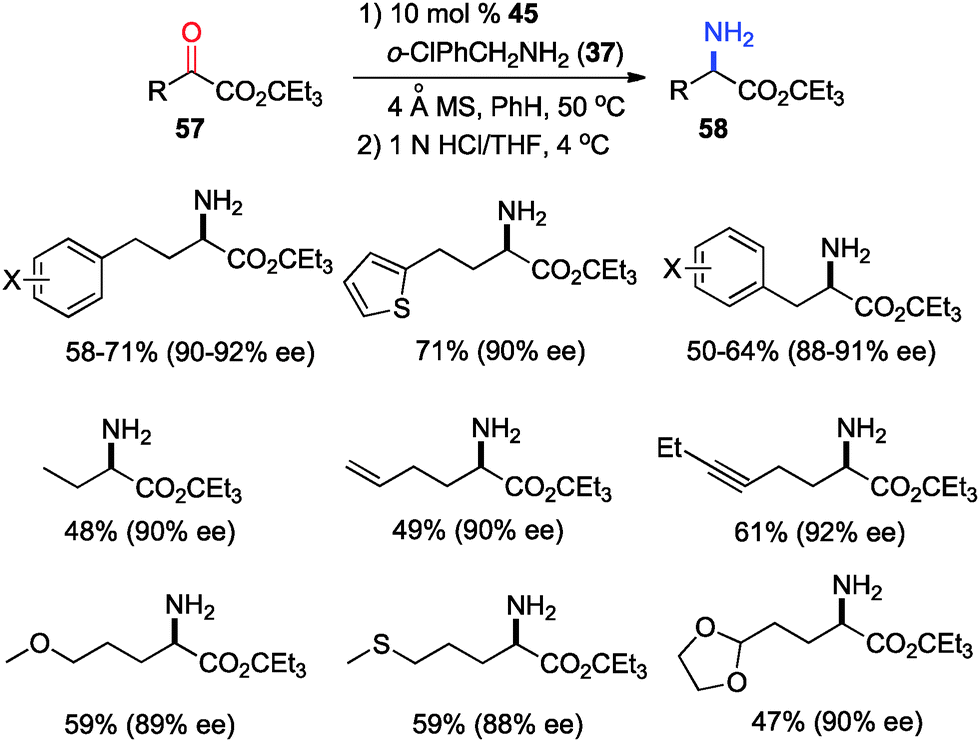
In 2011, Shi and coworkers reported an effective chiral base-catalyzed transamination of α-keto esters.34 Various chiral bases were examined with ethyl 2-oxo-4-phenylbutanoate (36) as the substrate and o-ClPhCH2NH2 (37) as the nitrogen source (Scheme 13). Up to 69% ee was obtained with quinine derived catalyst 45. Studies showed that the enantioselectivity was increased with a bigger ester group. For example, 92% ee was obtained with α-keto ester 47 using o-ClPhCH2NH2 (37) (Scheme 14). Enantioselectivity was found to be highly dependent on the structures of amine donors, with o-ClPhCH2NH2 (37) being the best in terms of both reactivity and enantioselectivity (Scheme 14). The transamination reaction with 45 and 37 was extended to a wide variety of α-keto esters, giving the corresponding α-amino esters in 88–92% ee (Scheme 15).
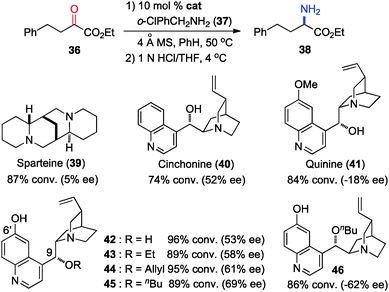 |
| | Scheme 13 Chiral base-catalyzed transamination. | |
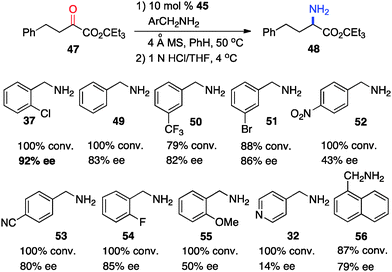 |
| | Scheme 14 The effect of benzylamine on transamination. | |
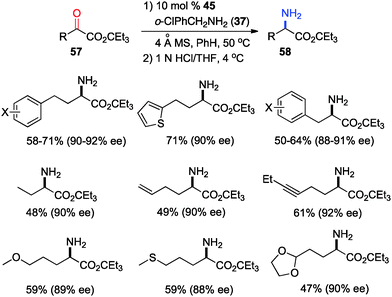 |
| | Scheme 15 Asymmetric transamination of α-keto esters. | |
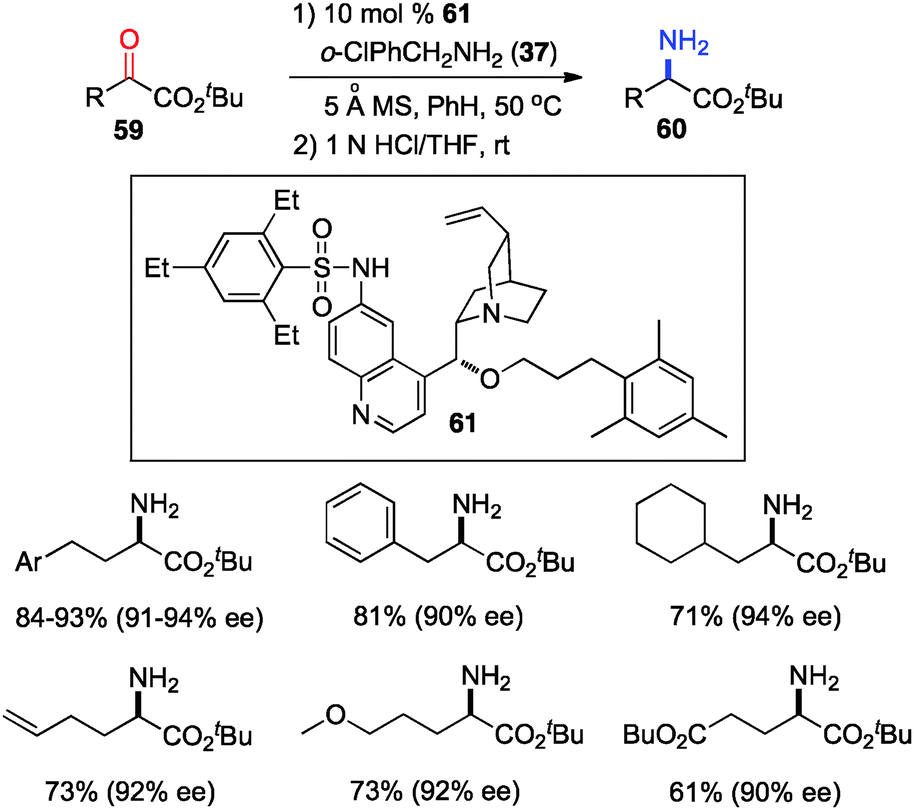
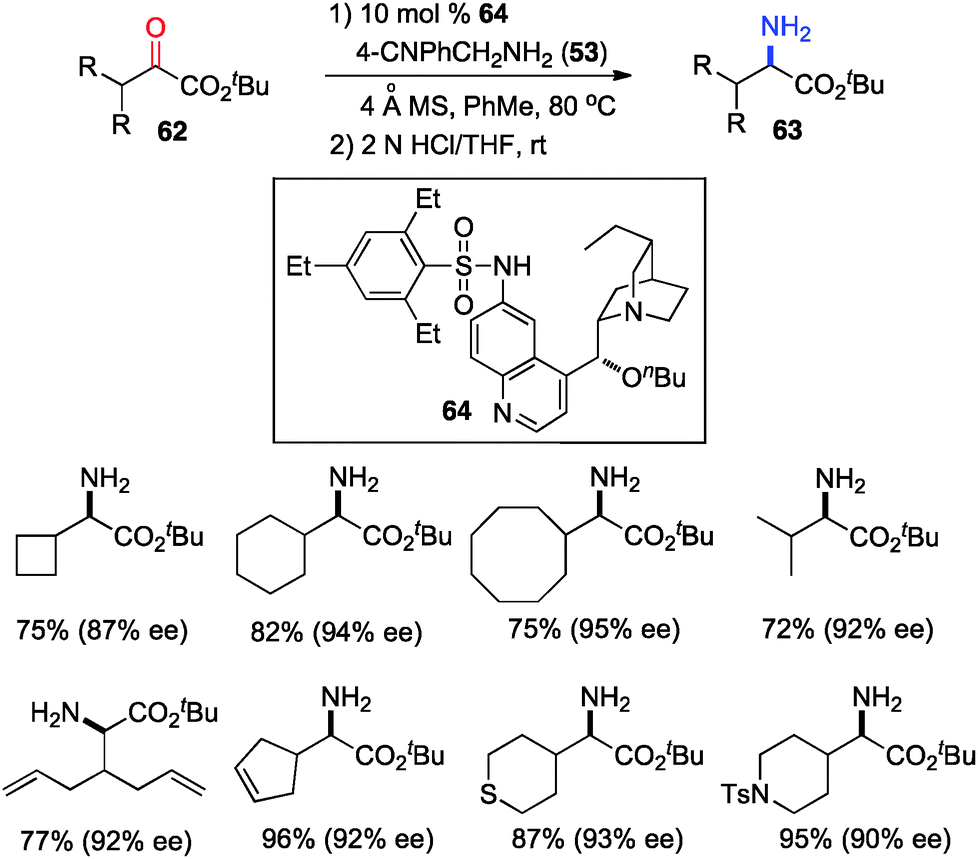
The 6′-OH in catalyst 45 played a very important role in the transamination for both reactivity and enantioselectivity, likely via a H-bond with the imine to facilitate the reaction and influence the enantioselectivity. To further understand the effect of H-bonding, catalysts with different H-bond donors at the 6′ position were investigated for the transamination.35 A quinine derivative containing 2,4,6-triethylbenzenesulfonamide (61) was found to be a highly effective catalyst (Scheme 16). A wide variety of α-amino esters were obtained from more readily available t-Bu keto esters in 61–93% yield and 90–94% ee with 61 and 37 in benzene at 50 °C. Transamination was amenable to the gram scale. With a related catalyst 64, various β-branched α-keto esters were transaminated to the corresponding amino esters in 50–96% yield and 87–95% ee with 4-CNPhCH2NH2 (53) as a nitrogen source (Scheme 17).36 The 4-CN group of the benzylamine likely enhanced the acidity of the ketimine and facilitated the proton transfer.
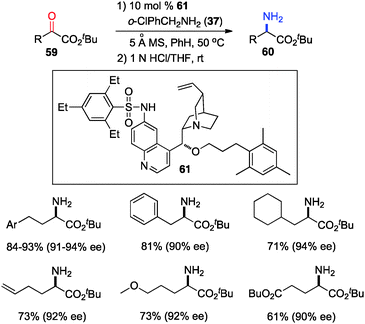 |
| | Scheme 16 Asymmetric transamination of α-keto esters. | |
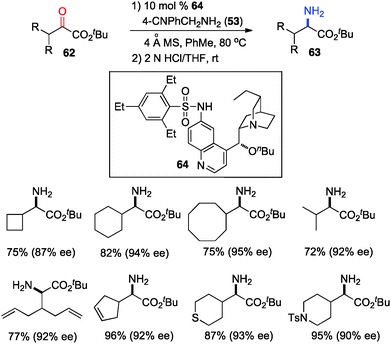 |
| | Scheme 17 Asymmetric transamination of β-branched α-keto esters. | |
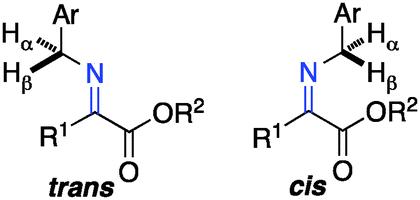
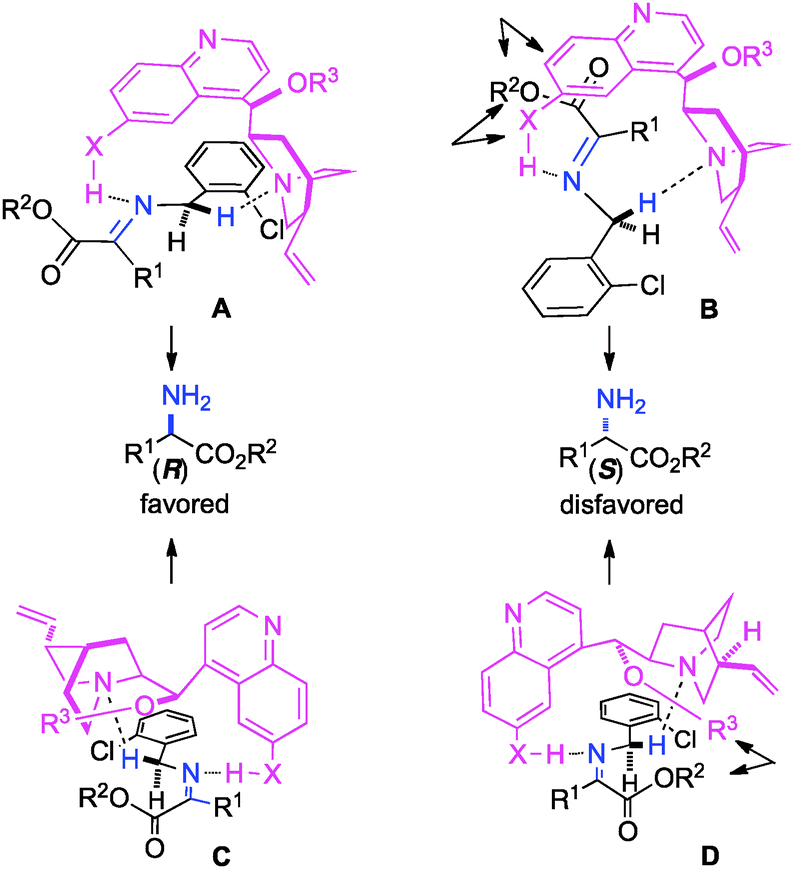
The ketimine can adopt two possible (trans and cis) configurations for the base catalyzed proton shift (Scheme 18). The relative content of the trans and cis configurations is likely dependent on the relative size of the side chain and the ester group of the keto ester. Two possible transition states for each configuration are outlined in Scheme 19. The (R)-amino ester is formed predominately via transition state A and/or C. The (S)-enantiomer is disfavored likely due to the steric interaction between the ester group of the substrate and the catalyst in transition state B and D. The enantioselectivity appears to be more influenced by the size of the ester group than the side chain, thus providing a broad scope for the keto ester substrate (Schemes 15–17).
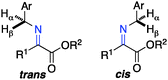 |
| | Scheme 18 The trans and cis configurations of the ketimine. | |
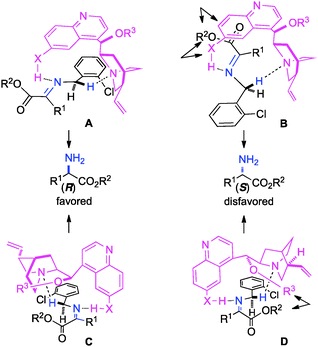 |
| | Scheme 19 Proposed transition state model for transamination. | |
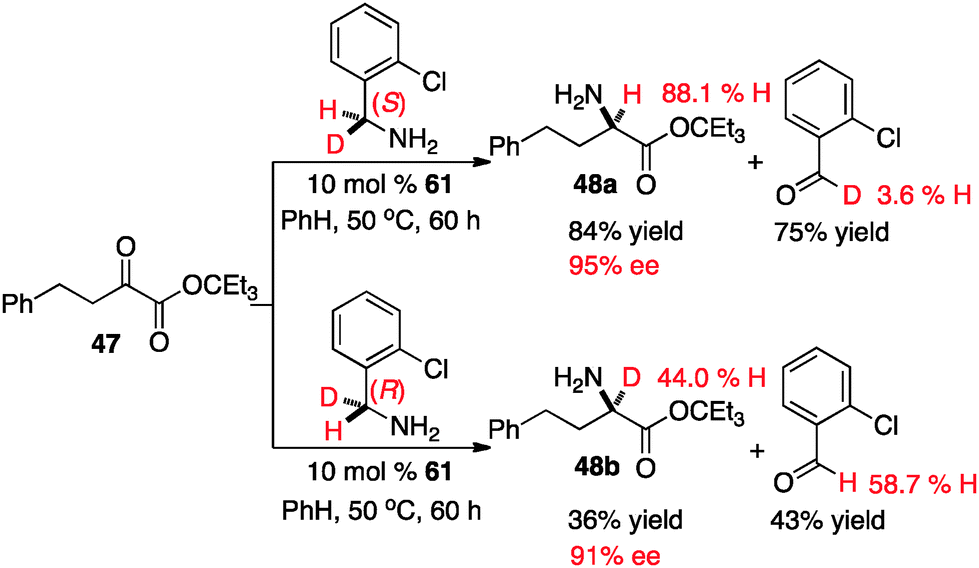
The extent of the involvement for each transition state likely depends on the structure of the keto ester. To further probe this issue, the transamination of keto ester 47 was carried out with catalyst 61 and optically active deuterated o-chlorobenzylamine37 (Scheme 20). It appears that the proton shift from the ketimine to the aldimine predominately proceeded via transition state A in this case, based on the yield and ee of the amino ester (48a & 48b) as well as the deuterium content of the amino ester and o-chlorobenzaldehyde.38
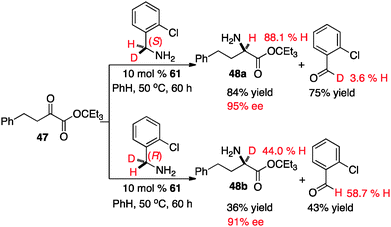 |
| | Scheme 20 Transamination with optically active deuterated benzylamines. | |
5. Transamination of fluoroalkyl ketones
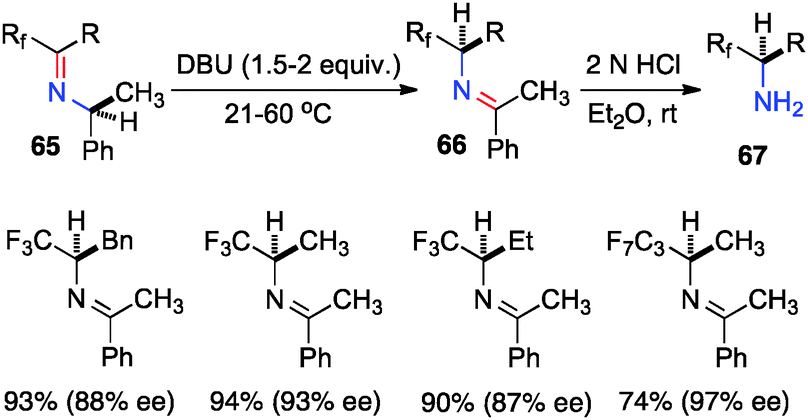
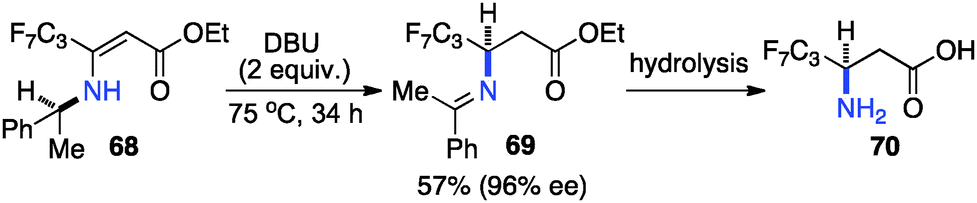
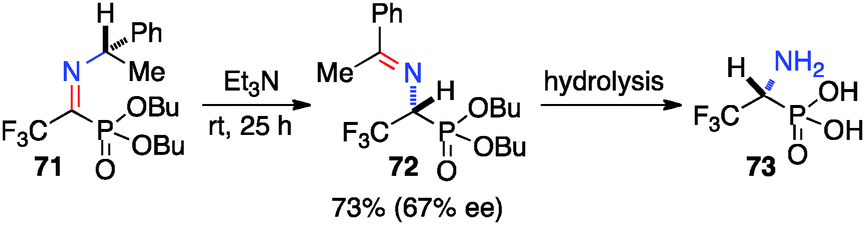
Optically active fluoroalkyl amines are very important functional moieties in various biologically and medicinally important molecules. Efforts have been made to synthesize fluoroalkyl amines from fluoroalkyl ketones via transamination using either chiral amine sources or chiral catalysts. In 1997, Soloshonok and coworkers showed that ketimines 65, prepared by the direct condensation of the corresponding fluoroalkyl ketones and (S)-α-phenylethylamine, were stereoselectively isomerized to ketimines 66 in up to 97% ee with DBU (Scheme 21).39 The use of DBU as both base and solvent was found to be crucial for the reaction. The isomerization process was used for the synthesis of optically active β-fluoroalkyl-β-amino acids.40 For example, 96% ee was obtained for aldimine 69 from the isomerization of 68 with DBU (Scheme 22). Yuan and coworkers reported that 1-amino-2,2,2-trifluoroethanephosphonic acid was synthesized from ketimine 71via a base-catalyzed transamination and subsequent hydrolysis (Scheme 23).41
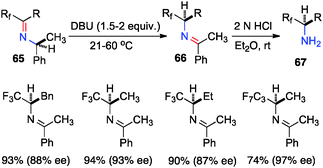 |
| | Scheme 21 Isomerization of fluoroalkyl ketimines. | |
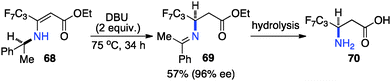 |
| | Scheme 22 Isomerization of fluoroalkyl enamines. | |
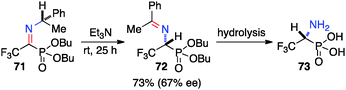 |
| | Scheme 23 Isomerization of 1-imino-2,2,2-trifluoroethanephosphonate. | |
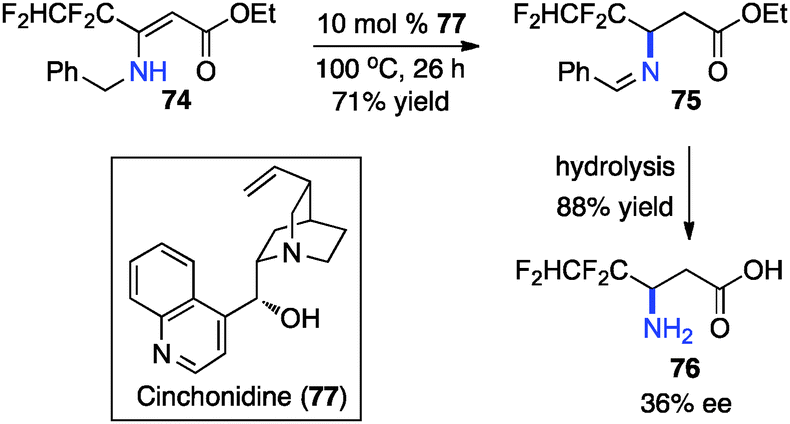
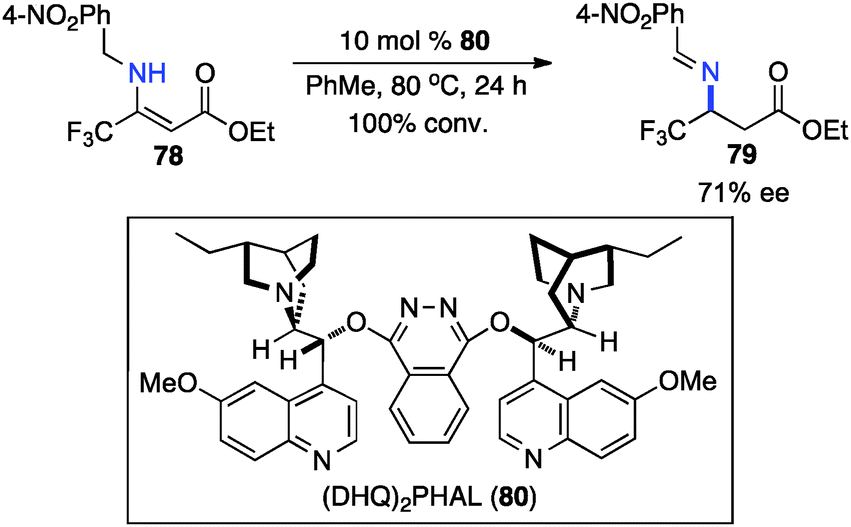
In 1994, Soloshonok and coworkers reported that β-fluoroalkyl-β-amino acids were obtained with up to 36% ee via chiral base-catalyzed isomerization of enamines under solvent-free conditions (Scheme 24).42 In 2007, Plaquevent and coworkers showed that trifluoromethyl enamines such as 78 were isomerized to the corresponding aldimines in up to 71% ee with a dimeric cinchona alkaloid (DHQ)2PHAL (Scheme 25).43
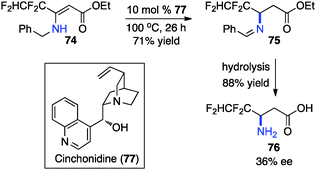 |
| | Scheme 24 Chiral base-catalyzed isomerization of enamines. | |
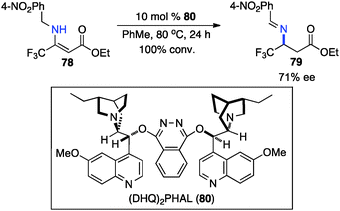 |
| | Scheme 25 Chiral base-catalyzed isomerization of trifluoromethyl enamines. | |
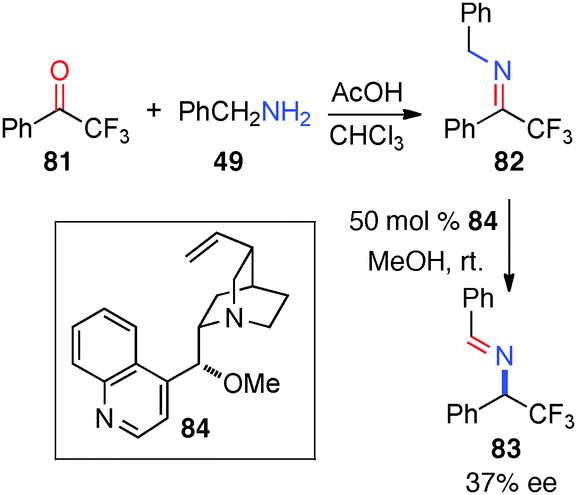
In 2007, Soloshonok and coworkers reported their studies on the chiral base-catalyzed isomerization of a trifluoromethyl ketimine (82).44 Aldimine 83 was obtained in 37% ee with cinchonidine-derived catalyst 84 in MeOH over an extended period of time (Scheme 26).
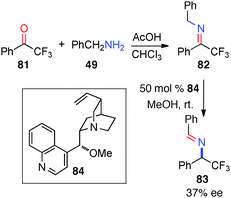 |
| | Scheme 26 Chiral base-catalyzed isomerization of trifluoromethyl ketimine. | |
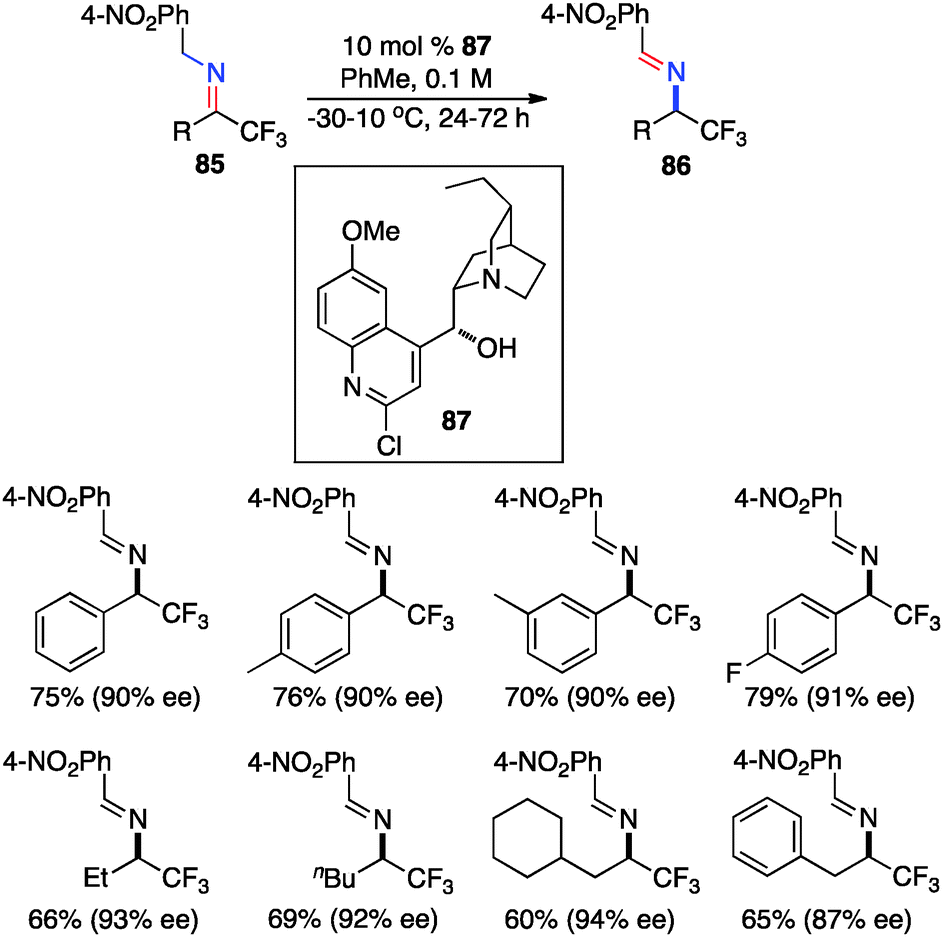
In 2012, Deng and coworkers reported the asymmetric isomerization of trifluoromethyl ketimines (85) derived from trifluoromethyl ketone and 4-NO2PhCH2NH2 (Scheme 27).45 The corresponding trifluoromethyl aldimines containing aryl or alkyl groups were obtained in up to 94% ee with cinchona alkaloid derivative 87 as the catalyst, and they were hydrolyzed to trifluoromethyl amines in high yields.
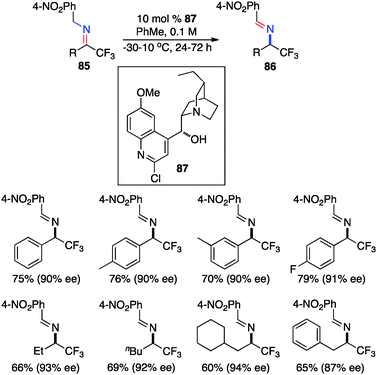 |
| | Scheme 27 Chiral base-catalyzed isomerization of trifluoromethyl ketimines. | |
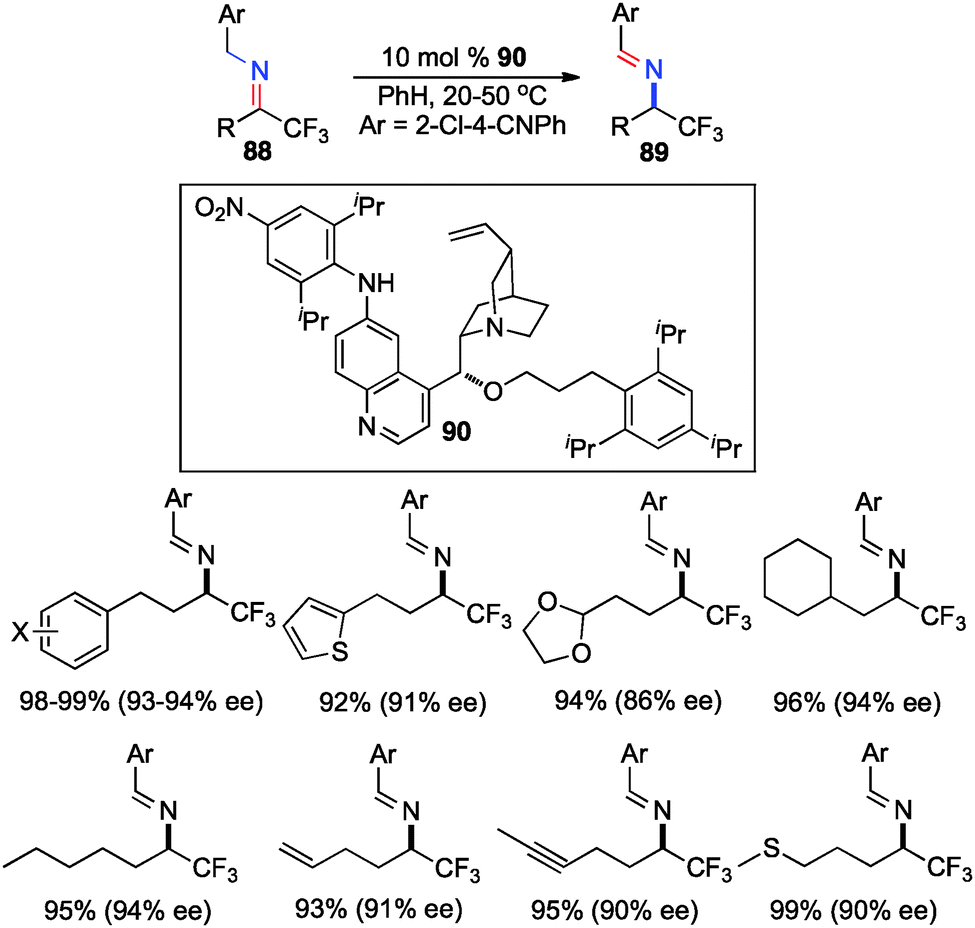
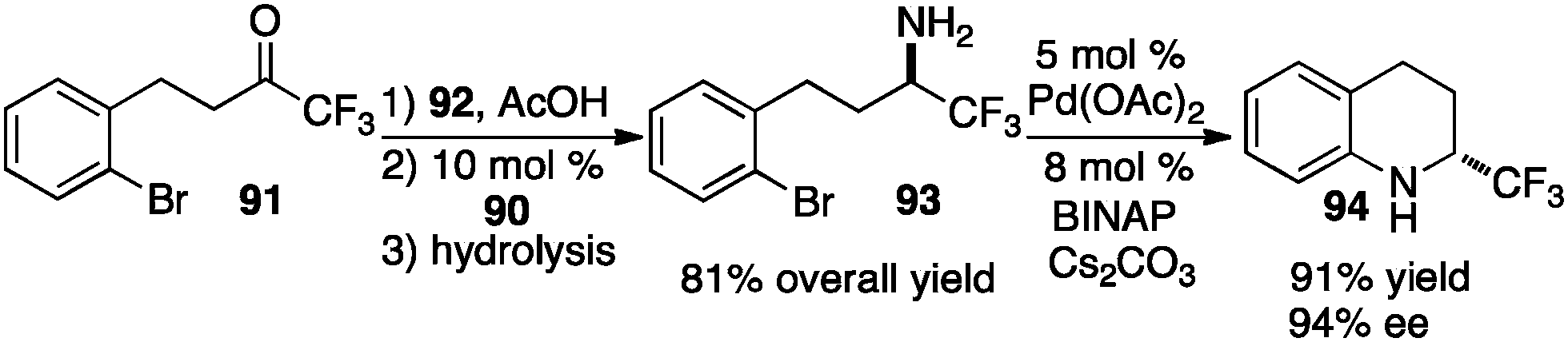
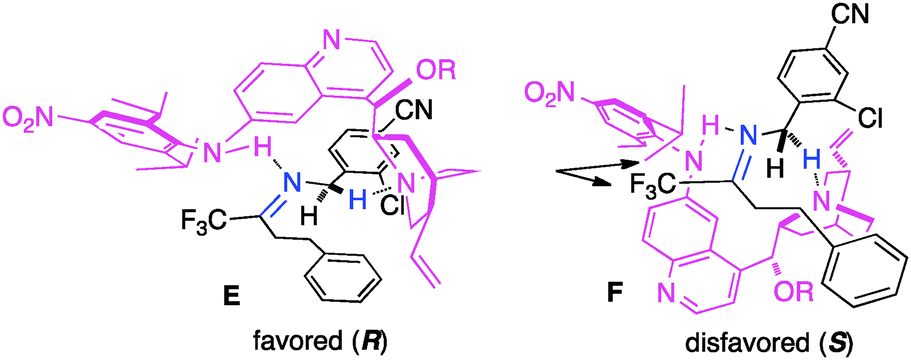
In their studies, Shi and coworkers showed that trifluoromethyl ketimines 88 were readily converted to aldimines 89 with catalyst 90 in up to 99% yield and up to 94% ee (Scheme 28).46 As illustrated in Scheme 29, trifluoromethyl amine 93 was prepared from the corresponding ketone (91) in 81% overall yield via condensation of 91 with 2-Cl-4-CNPhCH2NH2 (92), asymmetric proton shift, and subsequent hydrolysis. The Pd-catalyzed cyclization of amine 93 gave optically active tetrahydroquinoline 94 in 91% yield. The asymmetric isomerization of ketimine 88 to aldimine 89 was proposed to predominately proceed via transition state E, favoring the (R)-enantiomer (Scheme 30).
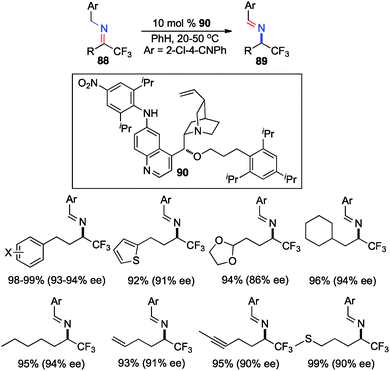 |
| | Scheme 28 Chiral base-catalyzed isomerization of trifluoromethyl ketimines. | |
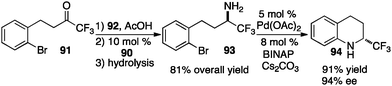 |
| | Scheme 29 Asymmetric transamination of trifluoromethyl ketone and cyclization. | |
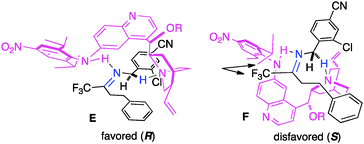 |
| | Scheme 30 The proposed transition state model for transamination. | |
6. Transamination of unactivated ketones
Significant progress has been made in the asymmetric transamination of α-keto esters and fluoroalkyl ketones. In these ketones, the electron-withdrawing ester and fluoroalkyl groups greatly facilitate the reactions. The transamination for ketones without these electron-withdrawing groups still remains challenging. Efforts have also been made in this area.
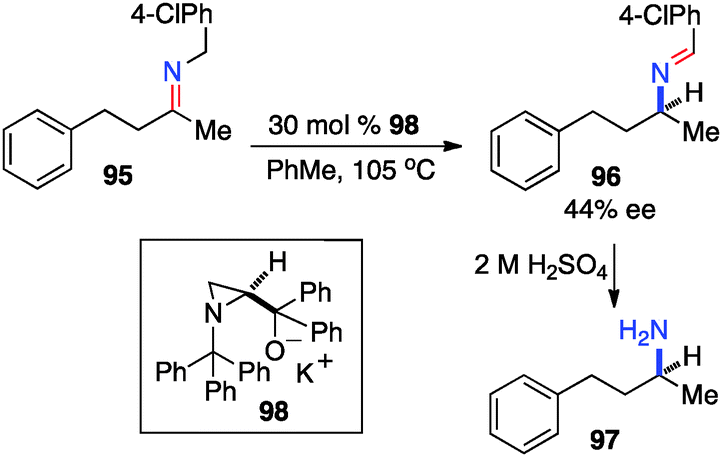
In 1995, Zwanenburg and coworkers reported the chiral base-catalyzed isomerization of ketimines to the corresponding aldimines.47 With aminoalcohol derived chiral base 98, aldimine 96 was obtained from ketimine 95 in up to 44% ee (Scheme 31).
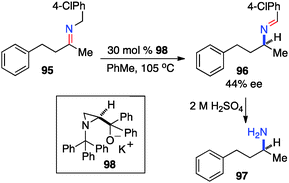 |
| | Scheme 31 Chiral base-catalyzed isomerization of ketimine. | |
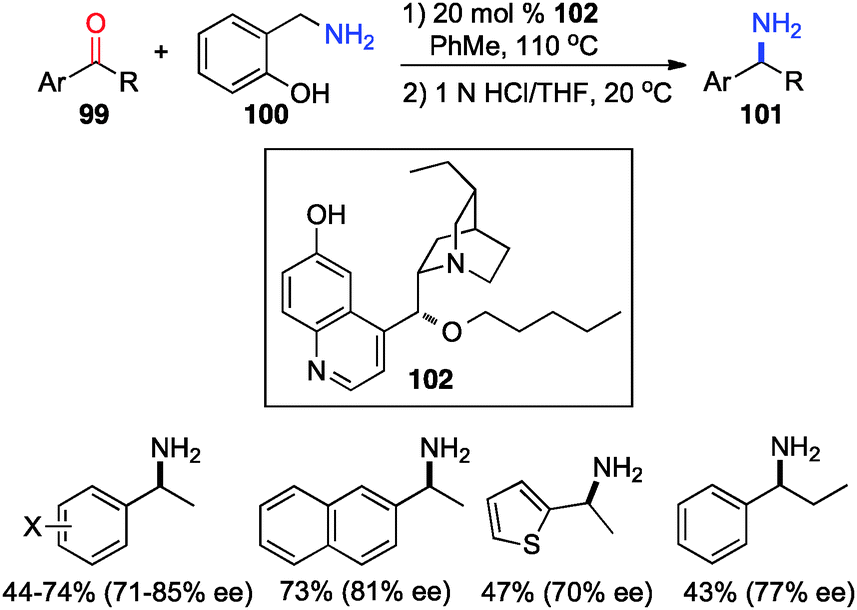
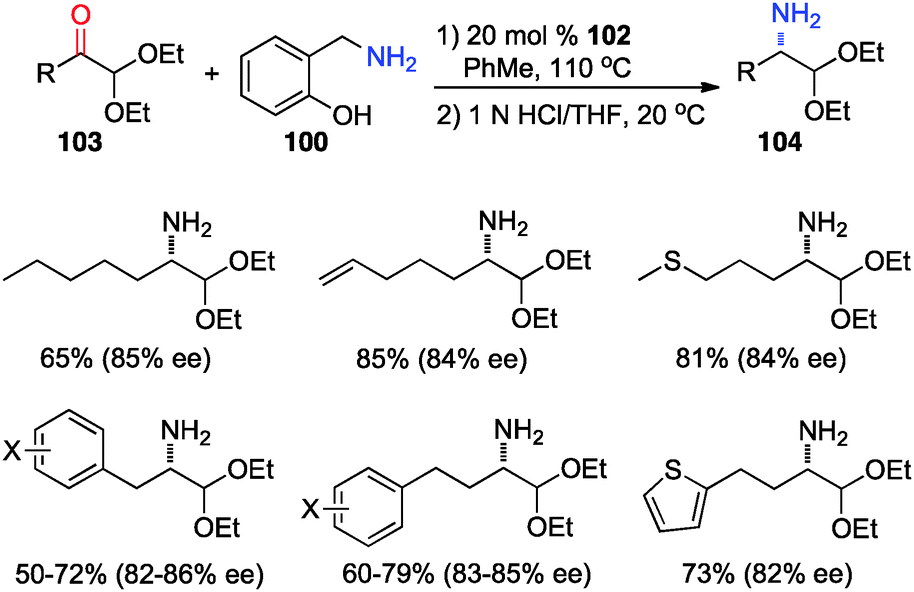
In 2012, Shi and coworkers showed that aromatic ketones 99 can be transaminated to the corresponding amines (101) in 70–85% ee with quinine-derived base 102 as the catalyst and o-HOPhCH2NH2 (100) as the nitrogen source in toluene at 110 °C (Scheme 32).48 Under similar reaction conditions, α-amino acetals were obtained from α-keto acetals in 82–86% ee (Scheme 33).49o-HOPhCH2NH2 was found to be crucial for the reaction. The o-OH group of the benzylamine likely formed a H-bond with the imine and facilitated the transamination.48,50
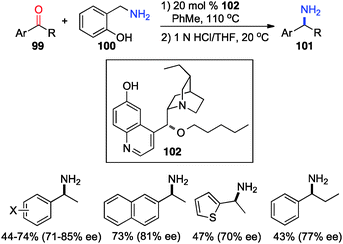 |
| | Scheme 32 Chiral base-catalyzed transamination of aromatic ketones. | |
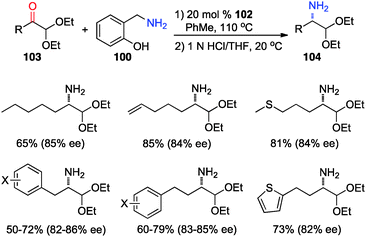 |
| | Scheme 33 Chiral base-catalyzed transamination of α-keto acetals. | |
7. Conclusions
Optically active α-amino acids can be efficiently generated from α-keto acids via transamination with vitamin B6 dependent transaminases in biological systems. Biomimetic asymmetric transamination of carbonyl compounds provides an attractive approach to optically active amine derivatives and has received considerable attention. Great progress has been made in the last few decades. Earlier studies focused on the development of chiral vitamin B6 analogues and artificial transaminase mimics. High enantioselectivity has been achieved in some cases. In recent years, highly enantioselective transamination processes have been developed for α-keto acid derivatives and fluoroalkyl ketones with chiral catalysts particularly chiral bases. Some mechanistic understanding of the enantioselectivity has also been gained. Asymmetric transamination of ketones without electron-withdrawing ester or fluoroalkyl groups has also been shown to be feasible. It can be expected that more effective transamination systems will emerge with the further understanding of the reaction mechanism and the development of new catalysts. It would be particularly useful if a simple ketone can be efficiently transaminated to an optically active amine under mild conditions. We hope that this review would stimulate new ideas in this area.
Acknowledgements
The authors gratefully acknowledge the National Basic Research Program of China (973 program, 2010CB833300) and the Chinese Academy of Sciences for the financial support.
Notes and references
- D. Zhu and L. Hua, Biotechnol. J., 2009, 4, 1420 CrossRef CAS PubMed.
- J. Ward and R. Wohlgemuth, Curr. Org. Chem., 2010, 14, 1914 CrossRef CAS.
- A. E. Martell, Acc. Chem. Res., 1989, 22, 115 CrossRef CAS.
- R. Breslow, Acc. Chem. Res., 1995, 28, 146 CrossRef CAS.
- Y. Murakami, J. Kikuchi, Y. Hisaeda and O. Hayashida, Chem. Rev., 1996, 96, 721 CrossRef CAS PubMed.
- J. Han, A. E. Sorochinsky, T. Ono and V. A. Soloshonok, Curr. Org. Synth., 2011, 8, 281 CrossRef CAS.
- E. Arceo and P. Melchiorre, ChemCatChem, 2012, 4, 459 CrossRef CAS.
- H. Kuzuhara, T. Komatsu and S. Emoto, Tetrahedron Lett., 1978, 3563 CrossRef CAS.
- Y. Tachibana, M. Ando and H. Kuzuhara, Chem. Lett., 1982, 1765 CrossRef CAS.
- Y. Tachibana, M. Ando and H. Kuzuhara, Chem. Lett., 1982, 1769 CrossRef CAS.
- M. Ando and H. Kuzuhara, Bull. Chem. Soc. Jpn., 1989, 62, 244 CrossRef CAS.
- M. Ando and H. Kuzuhara, Bull. Chem. Soc. Jpn., 1990, 63, 1925 CrossRef CAS.
- S. C. Zimmerman, A. W. Czarnik and R. Breslow, J. Am. Chem. Soc., 1983, 105, 1694 CrossRef CAS.
- S. C. Zimmerman and R. Breslow, J. Am. Chem. Soc., 1984, 106, 1490 CrossRef CAS.
- R. Breslow, A. W. Czarnik, M. Lauer, R. Leppkes, J. Winkler and S. Zimmerman, J. Am. Chem. Soc., 1986, 108, 1969 CrossRef CAS.
- W. Zhou, N. Yerkes, J. J. Chruma, L. Liu and R. Breslow, Bioorg. Med. Chem. Lett., 2005, 15, 1351 CrossRef CAS PubMed.
- R. Breslow, M. Hammond and M. Lauer, J. Am. Chem. Soc., 1980, 102, 421 CrossRef CAS.
- R. Breslow and A. W. Czarnik, J. Am. Chem. Soc., 1983, 105, 1390 CrossRef CAS.
- I. Tabushi, Y. Kuroda, M. Yamada, H. Higashimura and R. Breslow, J. Am. Chem. Soc., 1985, 107, 5545 CrossRef CAS.
- R. Breslow, J. Chmielewski, D. Foley, B. Johnson, N. Kumabe, M. Varney and R. Mehra, Tetrahedron, 1988, 44, 5515 CrossRef CAS.
- R. Breslow, S. Wei and C. Kenesky, Tetrahedron, 2007, 63, 6317 CrossRef CAS PubMed.
- S. Wei, J. Wang, S. Venhuizen, R. Skouta and R. Breslow, Bioorg. Med. Chem. Lett., 2009, 19, 5543 CrossRef CAS PubMed.
- S. Bandyopadhyay, W. Zhou and R. Breslow, Org. Lett., 2007, 9, 1009 CrossRef CAS PubMed.
- J. Svenson, N. Zheng and I. A. Nicholls, J. Am. Chem. Soc., 2004, 126, 8554 CrossRef CAS PubMed.
- J.-i. Kikuchi, Z.-Y. Zhang and Y. Murakami, Chem. Lett., 1994, 1559 CrossRef CAS.
- J.-i. Kikuchi, Z.-Y. Zhang, T. Miyajima and Y. Murakami, Chem. Lett., 1994, 1701 CrossRef CAS.
- J.-i. Kikuchi, Z.-Y. Zhang and Y. Murakami, J. Am. Chem. Soc., 1995, 117, 5383 CrossRef CAS.
- H. Kuang and M. D. Distefano, J. Am. Chem. Soc., 1998, 120, 1072 CrossRef CAS.
- K. Bernauer, R. Deschenaux and T. Taura, Helv. Chim. Acta, 1983, 66, 2049 CrossRef CAS.
- R. Deschenaux and K. Bernauer, Helv. Chim. Acta, 1984, 67, 373 CrossRef CAS.
- A. Hjelmencrantz and U. Berg, J. Org. Chem., 2002, 67, 3585 CrossRef CAS PubMed.
- K. R. Knudesn, S. Bachmann and K. A. Jørgensen, Chem. Commun., 2003, 2602 RSC.
- S. Bachmann, K. R. Knudsen and K. A. Jørgensen, Org. Biomol. Chem., 2004, 2, 2044 CAS.
- X. Xiao, Y. Xie, C. Su, M. Liu and Y. Shi, J. Am. Chem. Soc., 2011, 133, 12914 CrossRef CAS PubMed.
- X. Xiao, M. Liu, C. Rong, F. Xue, S. Li, Y. Xie and Y. Shi, Org. Lett., 2012, 14, 5270 CrossRef CAS PubMed.
- C. Su, Y. Xie, H. Pan, M. Liu, H. Tian and Y. Shi, Org. Biomol. Chem., 2014, 12, 5856 CAS.
- M. Liu, Y. Xie, J. Li, H. Pan, H. Tian and Y. Shi, J. Org. Chem., 2014, 79, 8417 CrossRef CAS PubMed.
-
M. Liu, Y. Xie, C. Su and Y. Shi, unpublished results.
- V. A. Soloshonok and T. Ono, J. Org. Chem., 1997, 62, 3030 CrossRef CAS.
- V. A. Soloshonok, T. Ono and I. V. Soloshonok, J. Org. Chem., 1997, 62, 7538 CrossRef CAS.
- J. Xiao, X. Zhang and C. Yuan, Heteroat. Chem., 2000, 11, 536 CrossRef CAS.
- V. A. Soloshonok, A. G. Kirilenko, S. V. Galushko and V. P. Kukhar, Tetrahedron Lett., 1994, 35, 5063 CrossRef CAS.
- V. Michaut, F. Metz, J.-M. Paris and J.-C. Plaquevent, J. Fluorine Chem., 2007, 128, 500 CrossRef CAS PubMed.
- V. A. Soloshonok and M. Yasumoto, J. Fluorine Chem., 2007, 128, 170 CrossRef CAS PubMed.
- Y. Wu and L. Deng, J. Am. Chem. Soc., 2012, 134, 14334 CrossRef CAS PubMed.
- M. Liu, J. Li, X. Xiao, Y. Xie and Y. Shi, Chem. Commun., 2013, 49, 1404 RSC.
- J. G. H. Willems, J. G. de Vries, R. J. M. Nolte and B. Zwanenburg, Tetrahedron Lett., 1995, 36, 3917 CrossRef CAS.
- Y. Xie, H. Pan, X. Xiao, S. Li and Y. Shi, Org. Biomol. Chem., 2012, 10, 8960 CAS.
- H. Pan, Y. Xie, M. Liu and Y. Shi, RSC Adv., 2014, 4, 2389 RSC.
- F. Xue, X. Xiao, H. Wang and Y. Shi, Tetrahedron, 2012, 68, 6862 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.