
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in single chain technology
Marina
Gonzalez-Burgos
ab,
Alejandro
Latorre-Sanchez
ab and
José A.
Pomposo
*abc
aCentro de Física de Materiales (CSIC, UPV/EHU) – Materials Physics Center, Paseo Manuel de Lardizabal 5, E-20018 San Sebastián, Spain. E-mail: Josetxo.pomposo@ehu.eus
bDepartamento de Física de Materiales, Universidad del País Vasco (UPV/EHU), Apartado 1072, E-20800 San Sebastián, Spain
cIKERBASQUE – Basque Foundation for Science, María Díaz de Haro 3, E-48013 Bilbao, Spain
First published on 1st June 2015
Abstract
The recent ability to manipulate and visualize single atoms at atomic level has given rise to modern bottom-up nanotechnology. Similar exquisite degree of control at the individual polymeric chain level for producing functional soft nanoentities is expected to become a reality in the next few years through the full development of so-called “single chain technology”. Ultra-small unimolecular soft nano-objects endowed with useful, autonomous and smart functions are the expected, long-term valuable output of single chain technology. This review covers the recent advances in single chain technology for the construction of soft nano-objects via chain compaction, with an emphasis in dynamic, letter-shaped and compositionally unsymmetrical single rings, complex multi-ring systems, single chain nanoparticles, tadpoles, dumbbells and hairpins, as well as the potential end-use applications of individual soft nano-objects endowed with useful functions in catalysis, sensing, drug delivery and other uses.
 Marina Gonzalez-Burgos | Marina Gonzalez-Burgos received her bachelor's degree in industrial chemical engineering in 2013 and master's degree in applied chemistry and polymer materials in 2014 from the University of the Basque Country. She is currently a PhD student at Materials Physics Center under the supervision of Prof. José A. Pomposo. Her research focuses on exploring new synthesis routes and the applications of single chain polymer nanoparticles. |
 Alejandro Latorre-Sanchez | Alejandro Latorre-Sanchez obtained his degree in chemistry in 2011 from the University of Barcelona. He received his master's degree in applied chemistry and polymer materials in 2013 from the University of the Basque Country. Currently, he is a PhD student at Materials Physics Center under the supervision of Prof. José A. Pomposo. His research focuses on synthesis, characterization and interactions of single chain polymer nanoparticles with biomacromolecules. |
 José A. Pomposo | José A. Pomposo received his PhD from the University of the Basque Country in 1994. After more than 20 years research experience in Materials Science, including the heading of Cidetec New Materials Department for 12 years, currently, he is Ikerbasque Research Professor at Materials Physics Center, Polymers & Soft Matter Group, in San Sebastián. His research interests include the synthesis of uniform soft nano-objects, research in the structure, dynamics & self-assembly behaviour of complex single chain nano-objects, and construction of hybrid organic nanostructures. |
1. Introduction
The precise hierarchical structure found in many biomacromolecules (e.g., enzymes) to perform their specific functions is the result of millions years of natural evolution.1 It is well known that in nature, a diversity of functional nanoentities result from the self-structuring of dynamic individual biomacromolecules at multiscale levels. As a typical example, the precise monomer sequence in a protein (primary structure) encodes the information required for (i) the formation (or not) of α-helices or β-sheets (secondary structure), (ii) determining the global morphology of the protein (tertiary structure), and (iii) promoting the formation (or not) of well-defined multi-aggregates (quaternary structure).2,3Such a function-driven and precise level of hierarchical self-organization has remained elusive in the synthetic polymer field, even though some very promising examples of controlled chain compaction via folding/collapse have been demonstrated in recent years.4–9 Moreover, intriguing examples of soft nano-objects constructed through the compaction of linear single chains and endowed with useful enzyme-mimetic functions have been described.10–13 In this sense, taking inspiration from the topology and precise morphology of natural biomacromolecules continues to be a powerful driving force towards artificial functional soft nano-objects.14 By analogy to the development experienced in manipulating and visualizing single atoms at the atomic level, giving rise to modern bottom-up nanotechnology, a similar exquisite degree of control is expected to become a reality in the next few years at the individual synthetic polymeric chain level for producing functional soft nanoentities, through full development of what is called “single chain technology”.15 In the long term, unimolecular soft nano-objects endowed with useful, autonomous and smart functions are expected to be the main valuable output of single chain technology.
This review focuses in the recent advances in single chain technology for the construction of a variety of soft nano-objects via chain compaction.
Examples of single rings as primitive synthetic analogues of circular proteins16,17 found in nature are included. Circular proteins presumably evolved over ancestral linear proteins to confer advantages, such as reduced sensitivity to proteolytic cleavage and enhanced stability, while retaining their intrinsic biological functions. Particular emphasis is placed on dynamic (stimuli-responsive),19–23 letter-shaped24,25 and compositionally unsymmetrical single rings.26–31
A diversity of complex, high-precision multi-ring systems has recently been prepared by means of single chain technology via covalent and supramolecular interactions. Illustrative examples are also included in this review.32–42 Interestingly, some of these complex multi-ring nano-objects can be considered as topological analogues of natural macrocyclic peptides found in various organisms.
By means of single chain technology, individual copolymer chains of different natures, compositions and molar masses have been folded/collapsed to single chain nanoparticles (SCNPs) and tadpoles (monotailed SCNPs). As shown recently, even single chain dumbbells and hairpins can be constructed via single chain technology. Several review papers and chapter books on SCNPs have been published. Most focused on intra-chain cross-linking techniques for SCNP construction.4–9 Consequently, only the most recent and relevant results are considered here to clarify the current state-of-the-art and open up new avenues for research. In particular, the significant added value that is endowed to single chain nanoparticles by taking inspiration from the functions of both ordered and disordered proteins will be highlighted (see Fig. 1). Certainly, breakthrough research in the precision synthesis of precursors with predefined sequences, positionable reactive groups, tailored interactions and useful functions are needed to deploy the full possibilities of single chain technology.
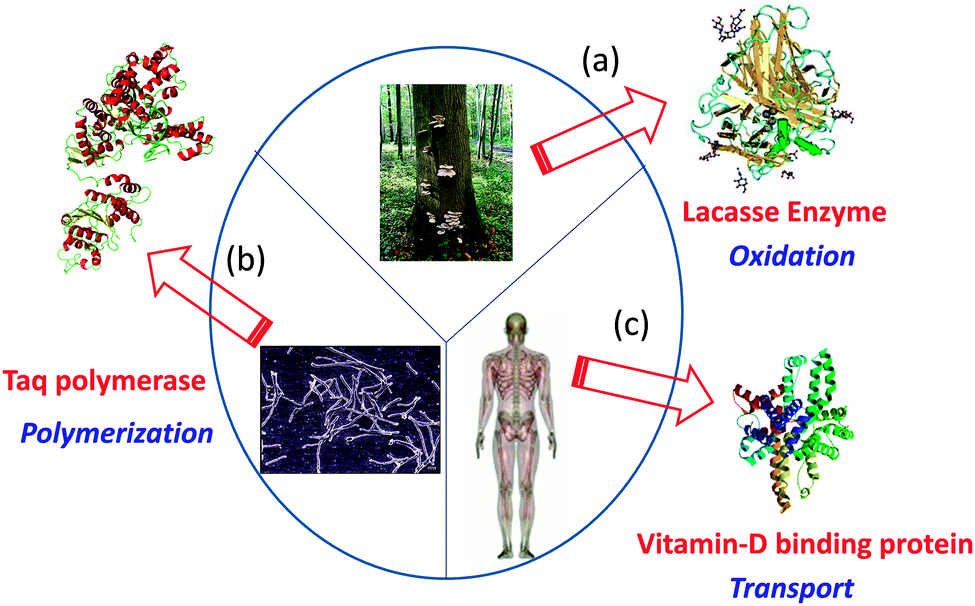 | ||
| Fig. 1 Several proteins with specific biological functions have been taken as inspiration source for the construction of functional biomimetic single chain nanoparticles (SCNPs): Laccase enzyme from Pleurotus ostreatus (a), Taq polymerase from Thermus aquaticus (b), and vitamin-D binding protein (DBP) from human serum (c). Reprinted with permission from ref. 14. | ||
Finally, potential end-use applications of these individual soft nano-objects in nanomedicine, catalysis, sensing and other uses are disclosed.
2. Soft nano-objects via chain compaction
2.1. Dynamic single rings
Many cyclic compounds have been isolated from living organisms, such as cyclotides (cyclo-peptides) (Fig. 2a) extracted from various organisms, including bacteria and plants.16,17 In nature, ring formation is employed to provide polypeptides with specific properties, such as improved stability against enzymatic degradation or acute toxicity via enhanced cellular membrane-disrupting activity.16 Often, cyclotides exhibit fast killing bactericidal activity. Interestingly, in addition to cyclization also precise folding through disulphide bonds is required for optimal cyclotide activity.17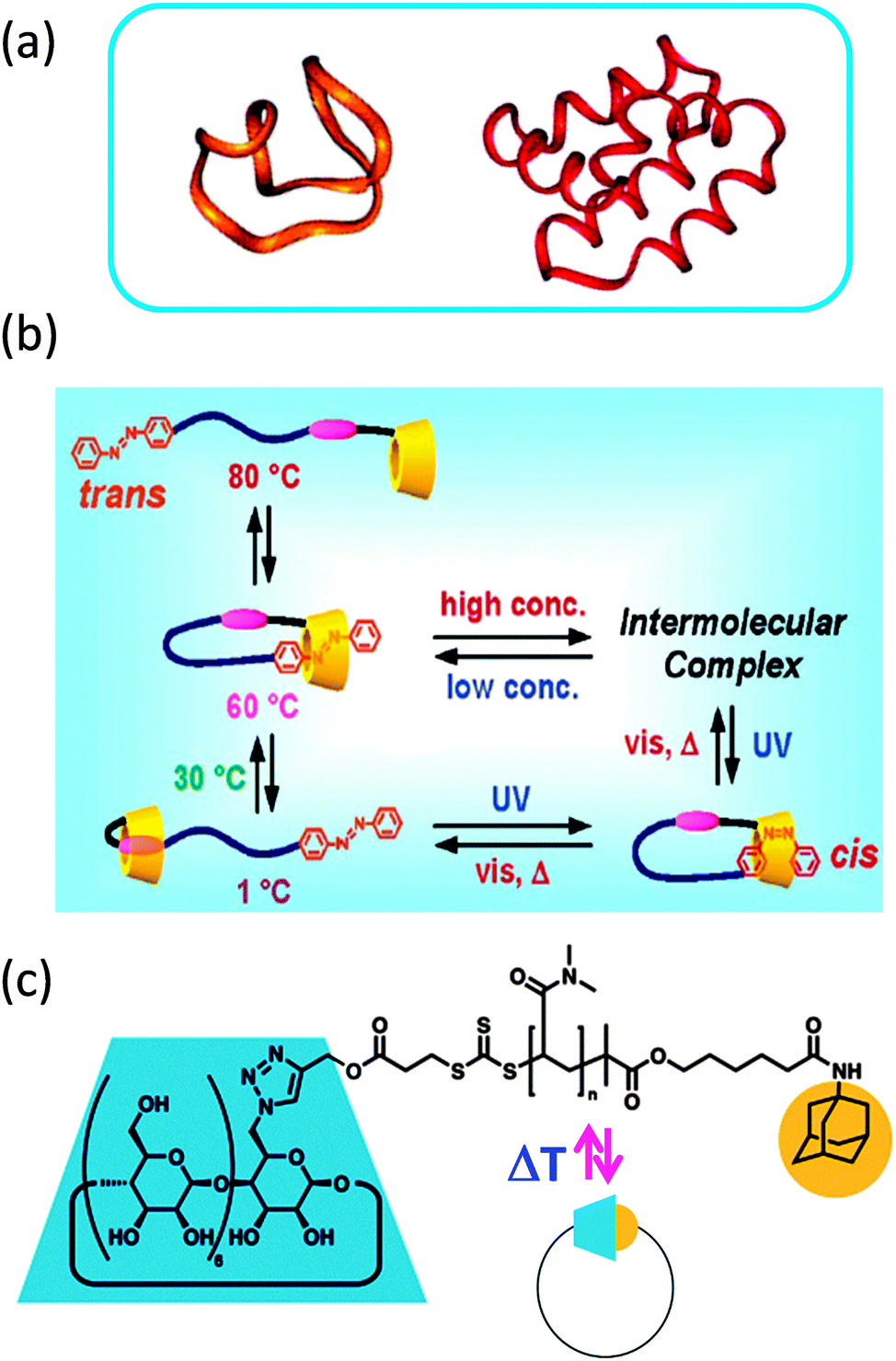 | ||
| Fig. 2 Ring formation endows cyclotides (cyclo-peptides) with specific properties, such as improved stability against enzymatic degradation or enhanced cellular membrane-disrupting activity (a). Dynamic single rings constructed via single chain technology that unfold back to linear chains triggered by external stimuli based on: β-cyclodextrin/azobenzene α,ω-functionalized polyethylene glycol (b), and β-cyclodextrin/adamantane α,ω-functionalized poly(N,N-dimethyl acrylamide) (c). Reprinted with permission from ref. 16, 19 and 20, respectively. | ||
Compaction of a linear synthetic polymer chain upon cyclization is well-known in the polymer chemistry field.18 As a consequence of ring formation, changes in many properties are observed when compared to those of the linear counterpart, such as intrinsic viscosity, glass transition temperature and order–disorder transition, to mention a few ones. Different strategies have been introduced for the cyclization of linear chains, some of them relying on the formation of a covalent, permanent bond between both chain ends. In recent years, however, several illustrative examples of the application of single chain technology to construct dynamic single rings that can unfold back to linear chains triggered by external stimuli have been reported.
In an illustrative work, Harada and colleagues19 showed thermal and photochemical switching of the conformation (from linear to ring structures) in aqueous solutions of polyethylene glycol (PEG)-substituted β-cyclodextrin (CD) with an azobenzene (AB) group at the chain end. The interplay between solution concentration, temperature and irradiation wavelength on the precise conformation and aggregation state of this CD–AB α,ω-functionalized PEG in aqueous solution is illustrated in Fig. 2b. Conformational changes were assessed by a combination of 2D rotating frame Overhauser effect spectroscopy (ROESY) and pulsed field gradient (PFG) 1H NMR experiments, circular dichroism and UV-vis spectra.
Reversible single chain selective point folding via β-cyclodextrin/adamantane (AD) host–guest chemistry in water has been reported by Barner-Kowollik and coworkers20 (Fig. 2c). Hence, a CD–AD α,ω-functionalized poly(N,N-dimethyl acrylamide) polymer was first synthesized by a combination of RAFT polymerization with a novel bifunctional RAFT agent and modular ligation chemistry. Subsequently, the CD–AD host–guest complexation-driven single chain cyclization and its reversion at elevated temperatures were monitored by dynamic light scattering (DLS) and nuclear Overhauser enhancement spectroscopy (NOESY). NOESY spectra recorded as a function of temperature revealed that chain unfolding takes place at around 50 °C.
The controlled self-folding of single polymer chains to individual rings induced by metal–ligand complexation has been recently demonstrated by the same group21 (Fig. 3a). Metal–ligand coordination belongs to the general class of non-covalent interactions and has proven to be one of the most prominent supramolecular motifs due to its ease of accessibility and strong binding constants. Upon metal-induced cyclization, a hydrodynamic diameter reduction of around 25% was observed as a consequence of the more compact topology of the cyclic polymer when compared to its linear precursor.
 | ||
| Fig. 3 Schematic illustration of the cyclization of single polymer chains to dynamic single rings by means of triphenylphosphine–palladium(II) complexation (a), cyanuric acid/Hamilton wedge hydrogen-bonding interactions (b), and thymine/diaminopyridine hydrogen-bonding interactions (c). Reprinted with permission from ref. 21, 22b and 23, respectively. | ||
The synthesis of dynamic single rings constructed via the single chain self-assembly of well-defined polymer precursors through α,ω-hydrogen-bonding between a cyanuric acid and a Hamilton wedge was also reported by Barner-Kowollik and colleagues22a (Fig. 3b). The existence of strong entropically driven hydrogen-bonding interactions between the α-donor and the ω-acceptor of the precursor at high dilution (<1 mM in CHCl3) leading to circular self-assembly was demonstrated by 1H NMR, DLS,22a and computer simulations.22b By increasing the concentration above 1 mM, the formation of inter-chain aggregates was observed by DLS. In a further work,23 dynamic single rings were synthesized using heterotelechelic polymer precursors containing thymine and diaminopyridine terminal groups (Fig. 3c).
These results show the increasing feasibility of generating dynamic single rings via single chain technology in which folding (or unfolding), which is accompanied by the corresponding change in material properties, is triggered by multiple external stimuli.
2.2. Letter-shaped single rings
Sequence control in synthetic macromolecules is currently a topic of significant interest for the construction of precise macromolecular origami via chain folding.15In a pioneering work by Lutz and coworkers,24 the synthesis of letter-shaped single rings from copolymers featuring functional groups at pre-selected position of the linear chain has recently been addressed. Controlled radical copolymerization of styrene with N-substituted maleimides (as ultra-reactive co-monomers) was used to prepare appropriate reactive precursors, some of them containing an azide functional group at the chain end. As a first example, alkyne functional groups were positioned in the polystyrene (PS) backbone using this procedure. The method does not lead to perfectly sequence-defined macromolecules but allows the inclusion of discrete functional patches in the PS chains. By performing intramolecular reactions from these precursors involving azide–alkyne cycloaddition and alkyne–alkyne coupling reactions, different macromolecular topologies were reached, such as P-, Q- and α-letter-shaped single rings (Fig. 4a). Chain compaction upon letter-shaped ring formation was clearly observed by size exclusion chromatography (SEC).
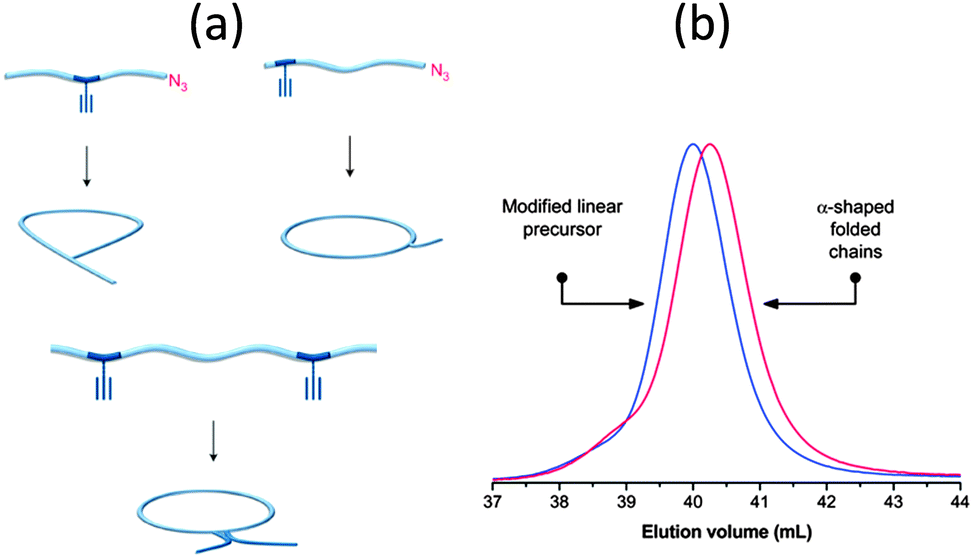 | ||
Fig. 4 Idealized image of the controlled compaction of linear polymer chains to P-, Q- and α-letter-shaped rings using positionable covalent bridges via azide(N3)–alkyne(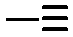 ) “click” cycloaddition and alkyne homocoupling reactions (a). Chain compaction upon letter-shaped ring formation was clearly observed by SEC (b). Reprinted with permission from ref. 24 and 25, respectively. ) “click” cycloaddition and alkyne homocoupling reactions (a). Chain compaction upon letter-shaped ring formation was clearly observed by SEC (b). Reprinted with permission from ref. 24 and 25, respectively. | ||
In a following work by the same group,25 two functional monomers were positioned at different locations in the PS chains and were then reacted with a short hetero-functional PEG spacer. The resulting α-letter-shaped rings obtained through intra-chain azide–alkyne “click” cycloaddition showed controllable loop and arm sizes. Chain compaction was also confirmed by SEC experiments (Fig. 4b).
The visualization of individual letter-shaped rings with atomic resolution by high resolution TEM or AFM, as well as an investigation of their presumably unique structure–property relationships and potential self-assembly behaviour in melt and solution are still pending issues.
2.3. Compositionally unsymmetrical single rings
Currently, the study of compositionally unsymmetrical polymer rings is receiving increasing attention, as a number of reports have suggested that cyclic block copolymers can play a unique role in a variety of different applications. Here we describe some illustrative examples of the unique properties of compositionally unsymmetrical single rings when compared with their linear counterparts.The highly efficient preparation of macrocyclic di-block copolymers via selective “click” reaction in micellar media taking advantage of the unimer-micelle exchange equilibrium has been described by Chen, Liu, and coworkers.26 Hence, by starting with heterodifunctional block copolymers in the form of micellar assemblies in which spatial separation between reactive groups took place, “click” cyclization was found to occur exclusively for unimers in solution that progressively self-assembled to flower-like micelles under the reaction conditions (Fig. 5). Interestingly, the compositionally unsymmetrical polymer rings prepared through this approach showed higher critical micelle concentration values, smaller hydrodynamic radii and lower aggregation numbers in self-assembled micelles, when compared to the linear precursors.
 | ||
| Fig. 5 Illustration of the highly efficient preparation of compositionally unsymmetrical polymer rings through the combination of supramolecular self-assembly (micellation) and selective intramolecular “click” ring closure at high polymer concentration (10 mg mL−1). Reprinted with permission from ref. 26. | ||
A remarkable topology effect showed by a cyclized block copolymer against its linear counterpart has been recently reported by Tezuka and colleagues.27 Flower-like micelles, approximately about 20 nm in size, were prepared from linear ABA block copolymers and their equivalent compositionally unsymmetrical AB rings, where A = polybutyl acrylate and B = polyethylene oxide, PEO. In spite of the similar morphology and size, the micelles formed from the cyclized block copolymer showed a drastic elevation (≥40 °C) of the cloud point (Tc). It was shown in this work that Tc can be tuned easily by co-assembly of the linear and cyclized block copolymers.
More recently, Grayson and colleagues28 prepared biocompatible amphiphilic PEG-b-polycaprolactone (PCL) macrocycles and investigated their stability and self-assembly behaviours. The resulting compositionally unsymmetrical AB rings were found to display reduced acid-catalysed degradation profiles and to form micelles in water of smaller sizes when compared to their equivalent linear block copolymers (Fig. 6a). The combination of reduced size and unique degradation behaviour suggest that they may be useful stimuli-responsive materials in the field of drug delivery carriers.
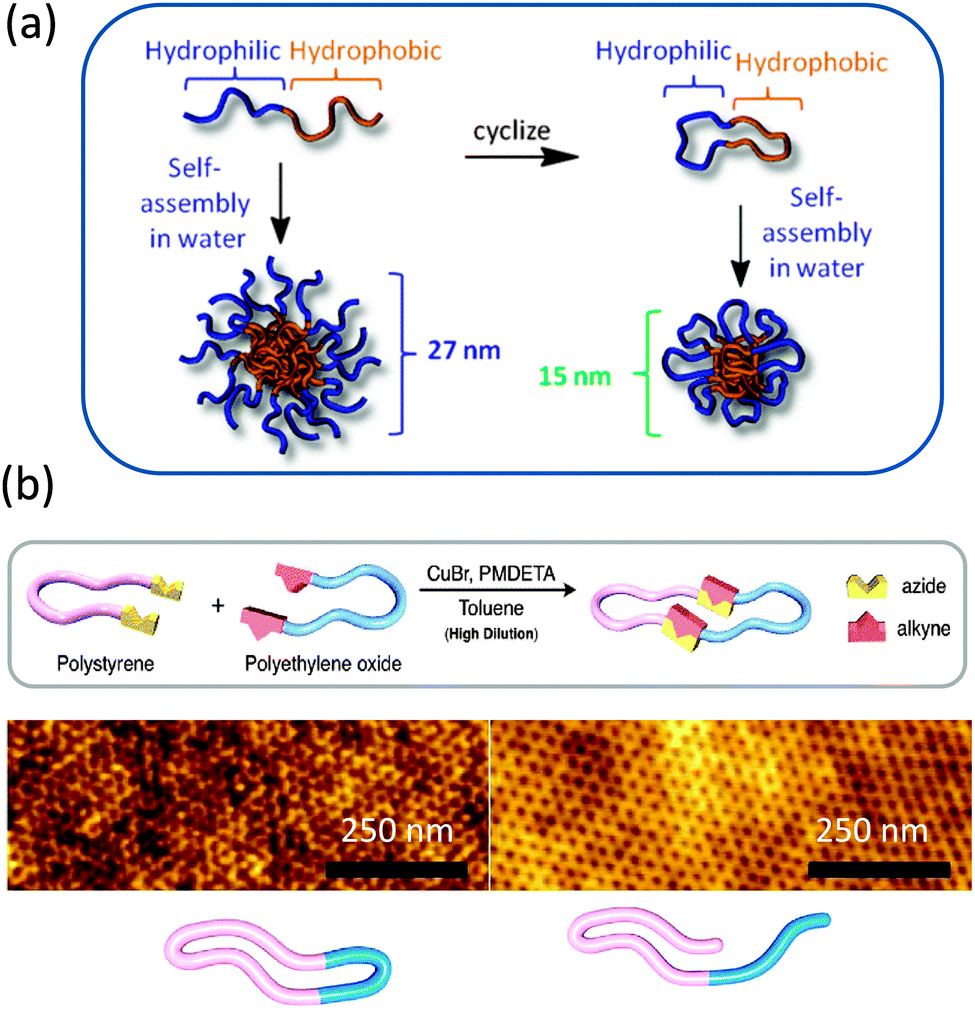 | ||
| Fig. 6 Amphiphilic polyethylene glycol-b-polycaprolactone macrocycles showed reduced acid-catalysed degradation profiles and formed micelles in water of smaller sizes when compared to their equivalent linear block copolymers (a). “Click” chemistry was used to obtain polystyrene-b-polyethylene oxide macrocycles that due to chain compaction showed a 30% decrease in domain spacing over their corresponding linear counterparts (b). Reprinted with permission from ref. 28 and 30, respectively. | ||
Linear and cyclic coil-crystalline di-block copolypeptoids that can efficiently form thermoreversible free-standing gels at moderate concentrations (5–10 wt%) in methanol at r.t. have been synthesized by Zhang and coworkers.29 These gels consist of a network of crystalline fibrils cross-linked by dynamic entanglement. Rheological studies showed that cyclic copolypeptoids produced stiffer gels than the linear counterparts. This was partially ascribed to the difference in the degree of crystalline packing of the solvophobic segments in the fibrils, resulting in enhanced rigidity for the fibrils and crystalline cross-linking sites for the cyclic gels than the linear counterparts.
The use of compositionally unsymmetrical AB rings (A = PS, B = PEO) for controlling the feature sizes in block copolymer lithography has recently been demonstrated by Hawker and coworkers.30 First, a modular approach to the synthesis of gram quantities of cyclic block copolymers by Cu(I)-catalysed azide–alkyne coupling of α,ω-azide-functionalized PS and α,ω-alkyne-functionalized PEO homopolymers was developed. Next, the film self-assembly of the compositionally unsymmetrical AB rings was compared to that of the corresponding linear analogues (Fig. 6b). PEO cylinders embedded in a PS matrix were observed for both nanophased separated thin films. The reduced hydrodynamic radii of the cyclic systems result in a decrease in domain spacing from 25.9 nm for the linear analogue to 19.5 nm for the cyclic block copolymer, as determined by grazing incidence X-ray scattering (GISAXS) experiments covering the entire thickness of the film. The corresponding domain spacing values obtained from 2D Fourier transform of AFM images were found to be 25 nm for the linear analogue and 20 nm for the cyclic block copolymer, which is in excellent agreement with the GISAXS data.
The facile access to cyclic PS-b-polyisoprene (PI) copolymers under high dilution conditions by combining anionic polymerization employing a protected acetylene-functionalized lithium initiator with azide–alkyne “click” chemistry has been reported by Touris and Hadjichristidis.31 The synthesis of macrocyclic copolymers with PS and PI brushes and their self-assembly into supramolecular tubes has been described by Schappacher and Deffieux.32
The above examples show how the unique properties of compositionally unsymmetrical AB rings, such as reduced hydrodynamic size, increased stability, tailored amphiphilic nature, and particular self-assembly behaviour, can be exploited for potentially relevant applications.
2.4. Complex multi-ring nano-objects
The extraordinary stability and bioactivity of natural cyclotides (cyclo-peptides) has been ascribed to their fused multi-cyclic structures stabilized through covalent folding via disulphide bonds (Fig. 7a).16,17 Several strategies have been followed in recent years to construct complex multi-ring nano-objects, which can be envisioned as topological analogues of natural multi-cyclic compounds.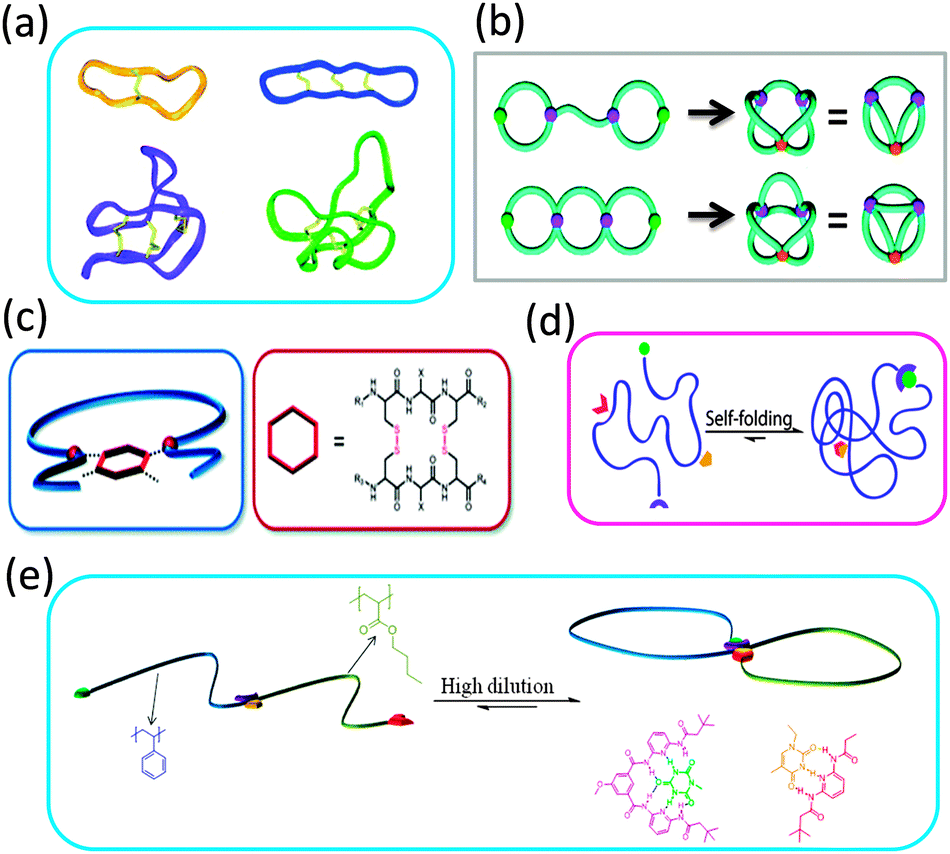 | ||
| Fig. 7 Some natural cyclotides show fused multi-cyclic structures formed through covalent folding via disulphide bonds (a). Complex multi-ring nano-objects prepared via cyclization of electrostatically template telechelic polymers (b). Bicyclic topology obtained through intramolecular twin disulphide bridge formation (c). Illustration of dual point single chain self-folding driven by two pairs of mutually orthogonal hydrogen bonding motifs placed at well-defined points within the polymer chain (d). Precision single chain folding of di-block copolymers via pairwise orthogonal multiple hydrogen bonding motifs and single chain selected point folding (e). Reprinted with permission from ref. 16, 36b, 37, 38 and 39, respectively. | ||
Different sequence controlled precursors have been used by Lutz and colleagues24,37 to construct permanent and stimuli-responsive 8-shaped polymer nano-objects, respectively. In a first report, azide groups introduced at both ends of a PS chain were coupled with alkyne moieties placed specifically at the middle of the chain by Cu(I)-catalysed azide–alkyne cycloaddition to yield a permanent 8-shaped nano-object.24 In a subsequent work by this group,37 an oligomer containing the cysteine–arginine–cysteine sequence was attached at specific locations on the PS backbones and used to give redox-responsive 8-shaped macrocycles via intramolecular twin disulphide bridge formation (Fig. 7c). This study paved the way for designing future water soluble multi-cyclic bio-hybrids, e.g., to enhance the therapeutic potential of peptide drugs.
Also, dynamic double ring-containing nano-objects have been constructed by Barner-Kowollik and coworkers,38 by employing pairwise orthogonal multiple hydrogen bonding motifs. Hence, dual point single chain self-folding driven by two pairs of mutually orthogonal hydrogen bonding motifs placed at well-defined points within the polymer chain was first demonstrated. Cyanuric acid/Hamilton wedge and thymine/diaminopyridine hydrogen bonding interactions were employed to construct such reversible complex-shaped nano-objects (Fig. 7d). In a very recent report by the same authors,39 well-defined 8-shaped cyclic block copolymers were synthesized via pairwise orthogonal multiple hydrogen bonding motifs (cyanuric acid/Hamilton wedge and thymine/diaminopyridine) and single chain selected point folding (Fig. 7e).
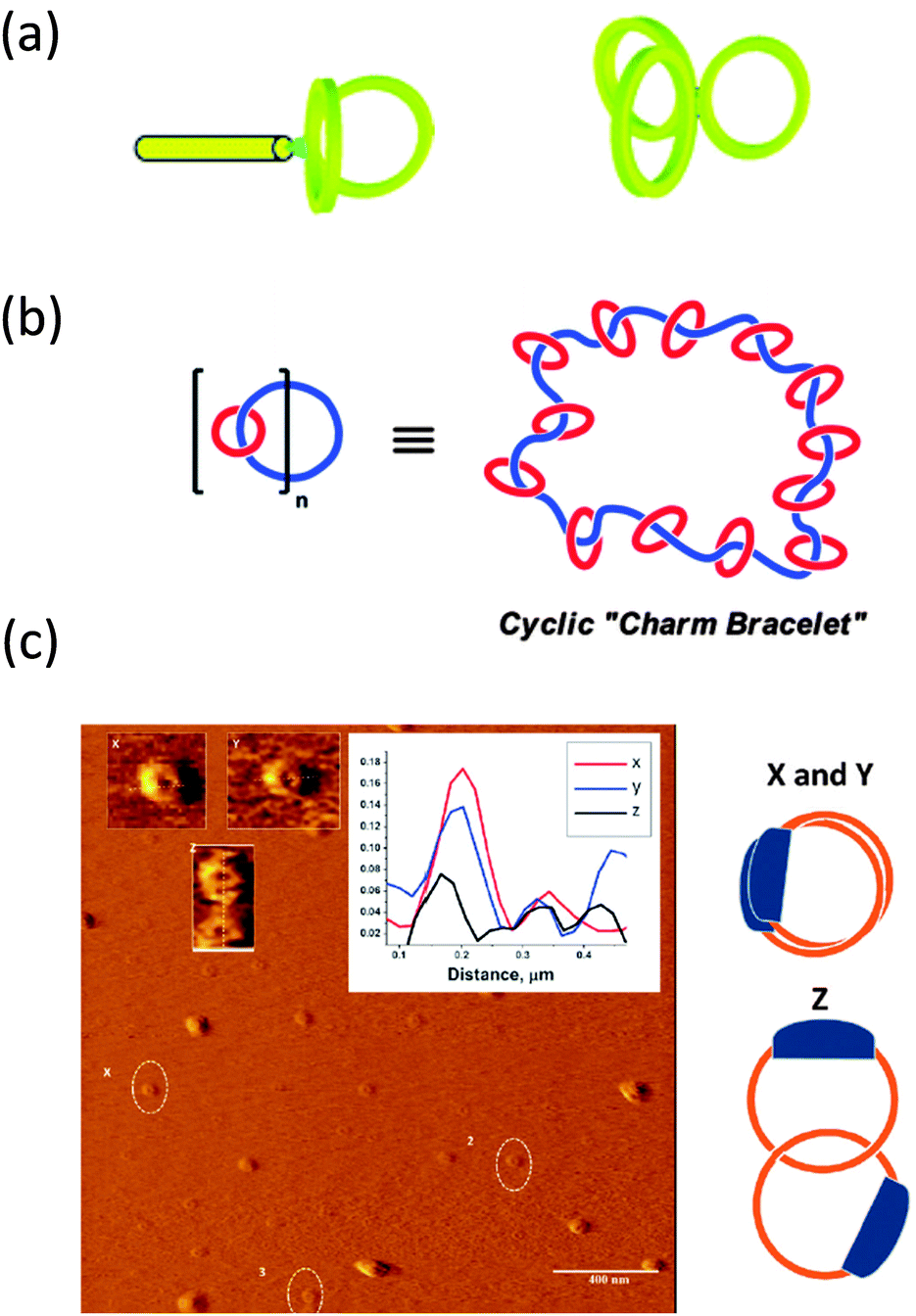 | ||
| Fig. 8 Illustration of complex multi-ring nano-objects synthesized from pre-formed rings: paddle-like structure and 3-arm cyclic star nano-object (a), polycatenated cyclic system (b), and different conformations of high molecular weight catenated polymers as visualized by AFM (c). Reprinted with permission from ref. 40, 41 and 42, respectively. | ||
The synthesis of a polycatenated cyclic polymer, a structure that resembles a molecular charm bracelet was described by Grubbs and coworkers.41 Ring-opening metathesis polymerization of an amine-containing cyclic olefin monomer in the presence of a chain transfer agent allowed for the synthesis of a linear polymer that was then functionalized and cyclized to the corresponding ring-shaped analogue as revealed by SEC, NMR and FTIR measurements and subjected to further functionalization reactions, affording a cyclic polyammonium scaffold. Diolefin polyether fragments were coordinated and clipped around the ammonium sites within the polymer backbone using ring-closing olefin metathesis, giving a molecular charm bracelet (Fig. 8b). Confirmation of the interlocked nature of the product was achieved via1H NMR and 2D DOSY measurements.
Synthesis of high molecular weight catenanes (i.e., molecules composed of two or more mechanically interlocked rings) in high yield is a formidable synthetic challenge. The efficient synthesis of a catenated polymer of Mn = 9.3 kDa via controlled ring expansion has been reported recently by Advincula and colleagues.42 The catenated polymer was isolated in 73% yield by dialysis and characterized by SEC, 1H NMR spectroscopy, and viscosimetry. Interestingly, different conformations of the catenated polymer were observed by AFM pointing to the relatively high conformational freedom of these nano-objects (Fig. 8c).
The above examples illustrate the significant advances in single chain technology applied to the construction of complex multi-ring nano-objects. An investigation in the near future of their presumably unique structure–property relationships and self-assembly behaviour should pave the way to the use of these multi-cyclic polymers in nanotechnology and nanomedicine, among other fields.
2.5. Single chain nanoparticles
The concept of single chain nanoparticles (SCNPs) constructed via intramolecular cross-linking of individual linear polymer chains was introduced 15 years ago by Mecerreyes, Miller and coworkers.43a This pioneering work established a new paradigm in polymer synthesis, i.e. manipulation of single polymer chains to construct ultra-small functional unimolecular nano-objects.15Recent reviews provide detailed information about the different techniques involved in SCNP construction (Fig. 9):4–9 (i) controlled polymerization for the synthesis of the SCNP precursor, (ii) polymer functionalization, if necessary, for the introduction of specific functional groups in the precursor, and (iii) intra-chain folding/collapse via covalent, non-covalent (supramolecular) or dynamic-covalent bonding interactions.
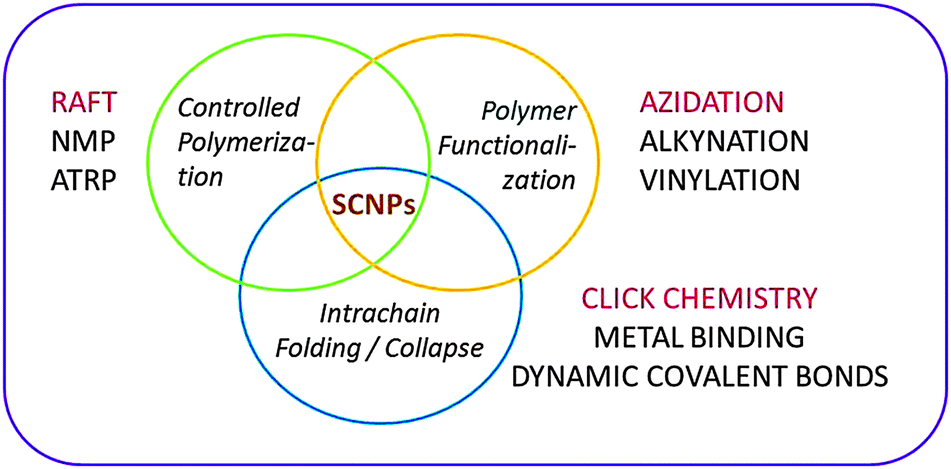 | ||
| Fig. 9 Different techniques involved in the construction of single chain nanoparticles (SCNPs). In red colour are indicated efficient procedures for permanent SCNP construction. Adapted from ref. 6a. | ||
A summary of the advances in single chain technology applied to the construction of permanent SCNPs via intra-chain folding/collapse by means of covalent interactions is provided in Fig. 10. The growing interest in this field is demonstrated by the constant introduction of new and refined intra-chain cross-linking procedures along time (in addition to alternative radical coupling procedures),43b,c such as Diels–Alder (DA) reaction44 in 2002, cross-metathesis45,46 and quinodimethane formation47–49 in 2007, copper(I)-catalysed azide–alkyne cycloaddition50–55 (CuAAC) and amide formation56,57 in 2008, urea formation58 in 2009, benzoxazine ring opening polymerization59,60 (ROP), Bergman cyclization61–64 and nitrene cross-linking65,66 in 2011, alkyne homocoupling67 in 2012, oxidative polymerization,68 thiol–ene coupling,69 Michael addition70–72 and epoxide ROP12 in 2013, and tetrazine–norbornene reaction,73 nitrile–imine ligation,74 thiol–yne coupling75 and lactone ROP76 in 2014. We will not enter into details here; instead, we refer the interested reader to previously published reviews focused on intra-chain cross-linking techniques for SCNP construction.4a,5,6a,c,7,8
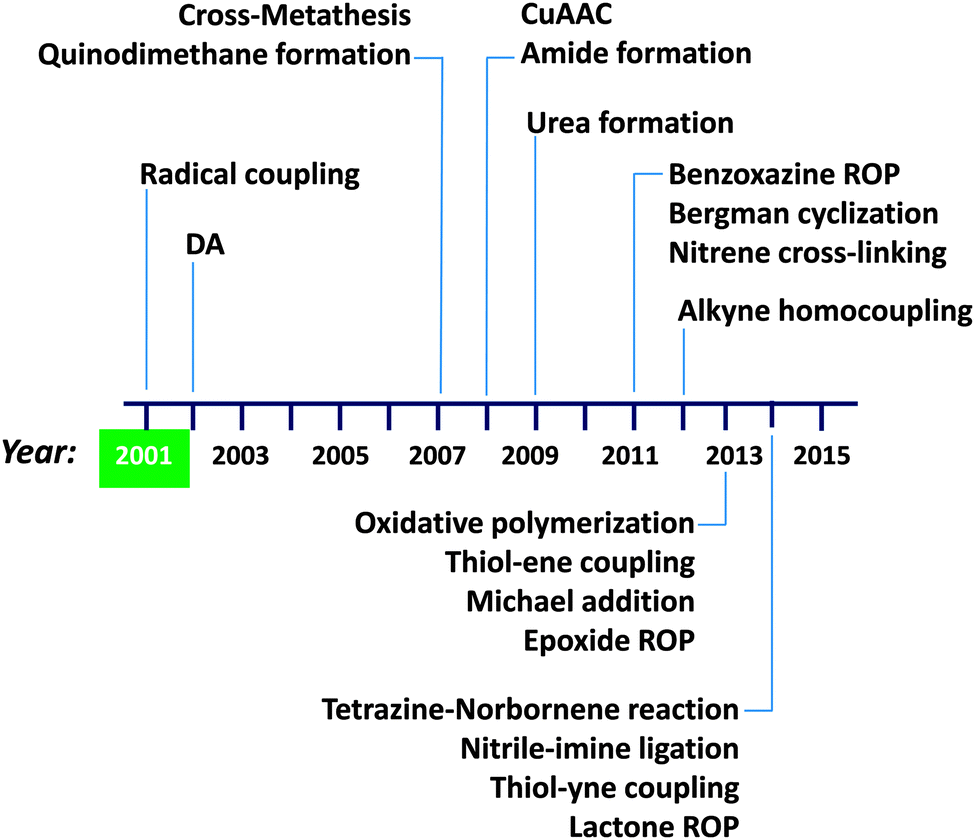 | ||
| Fig. 10 Summary of the advances in single chain technology applied to the construction of permanent SCNPs via intra-chain folding/collapse by means of covalent bonds (see text for references). First synthesis of irreversible SCNPs was carried out by Mecerreyes, Miller and coworkers in 2001. DA = Diels–Alder reaction, CuAAC = copper(I)-catalysed azide–alkyne cycloaddition, ROP = ring opening polymerization. | ||
Regarding the synthesis of structurally dynamic or reversible SCNPs via supramolecular interactions and dynamic-covalent bonds, a similar trend is observed, as illustrated in Fig. 11. Hence, different non-covalent bonding-based strategies have been introduced over recent years to afford the preparation of responsive SCNPs, such as benzamide dimerization77 developed in 2008, ureido-pyrimidinone (UPY) dimerization78–81 in 2009, benzene-tricarboxamide (BTA) helical stacking10,11,82–88 in 2011, cucurbit[n]uril complexation,89,90 hydrophobic L-phenylanaline (Phe)–Phe interactions91,92 and 3,3′-bis(acylamino)-2,2′-bipyridine (BiPy-BTA) self-assembly93 in 2012, Rh complexation,94,95 charged amphiphilic random copolymer self-assembly96–101 and Cu complexation in 2013,13,72 and finally, neutral amphiphilic random copolymer self-assembly102 in 2014. Complementary, successful approaches based on dynamic covalent bonds have been developed in recent years, such as those based on hydrazone bonds described in a pioneering work by Murray and Fulton,103,104 and coumarin dimerization,105,106 both reported in 2011, disulphide bonds107 in 2012, as well as enamine bonds108 and anthracene dimerization109 in 2014. For the interested reader, we suggest recent reviews of this field.5,6b,8,9
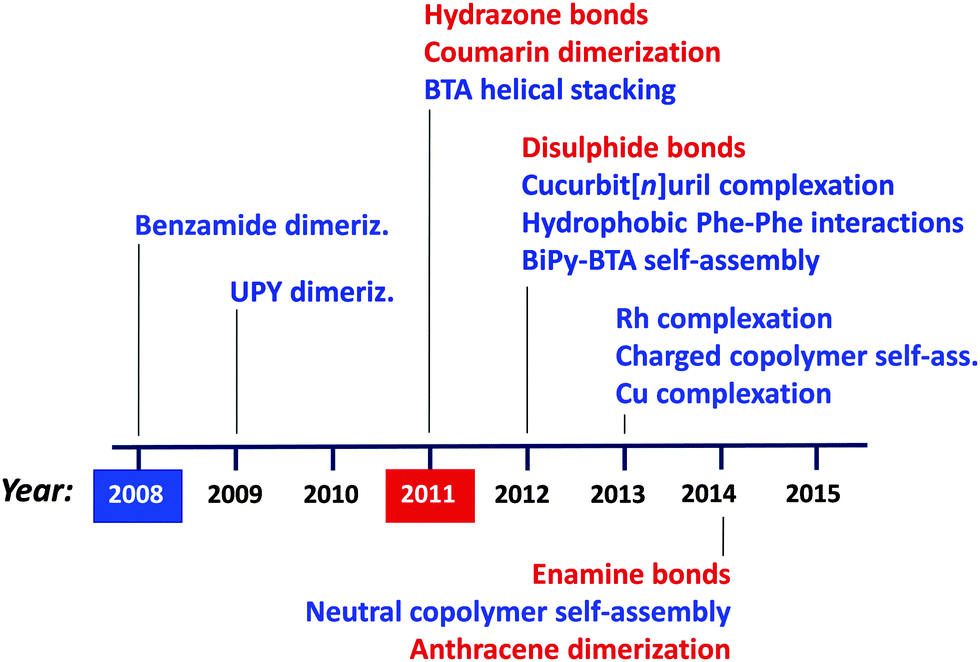 | ||
| Fig. 11 Evolution of the emerging field of structurally dynamic SCNPs synthesized via supramolecular, non-covalent interactions (blue) and dynamic covalent bonds (red) (see text for references). First synthesis of reversible SCNPs through non-covalent interactions was carried out by Hawker, Kim and colleagues in 2008. First preparation of dynamic covalent SCNPs was reported by Murray and Fulton in 2011. UPY = ureido-pyrimidinone, BTA = benzene-tricarboxamide, Phe = L-phenylanaline, BiPy-BTA = 3,3′-bis(acylamino)-2,2′-bipyridine substituted BTA. | ||
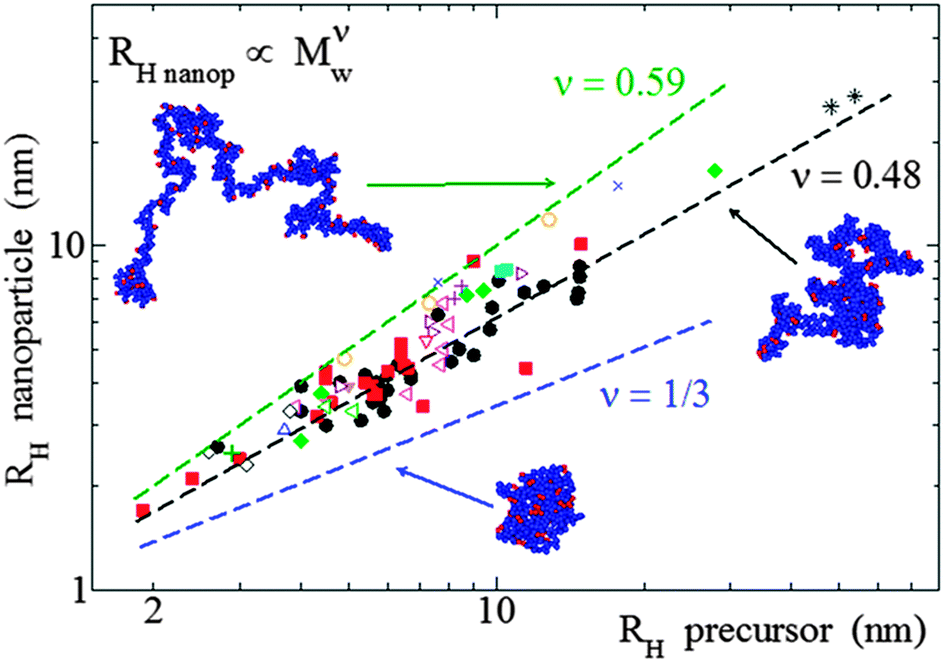 | ||
| Fig. 12 Analysis of size data reported in the literature for a large number of SCNPs in solution synthesized in good solvent, covering from covalent and dynamic-covalent to non-covalent bonded SCNPs, revealed that they adopt open, sparse morphologies (ν = 0.48) resembling those of intrinsically disordered proteins, instead of globular, compact conformations (ν = 0.33) (see text). RH = hydrodynamic radius, Mw = weight average molecular weight, ν = scaling law exponent. Reprinted with permission from ref. 110a. | ||
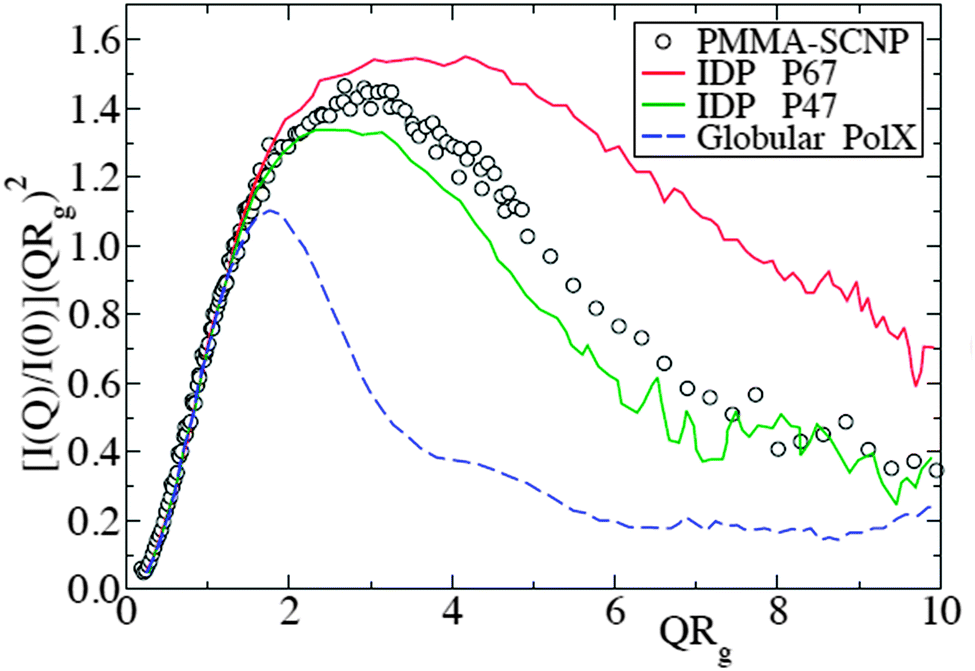 | ||
| Fig. 13 Kratky plot for the SANS form factor of representative polymethyl methacrylate SCNPs (symbols), compared with that of intrinsically disordered proteins – IDPs – (solid lines) and globular proteins (dashed line). I(Q), Q and Rg are the scattered intensity, wavevector and radius of gyration, respectively. Reprinted with permission from ref. 110a. | ||
The combination of SANS and SAXS measurements with MD simulations providing access to the SCNP form factor and the corresponding scaling law exponent, ν, (Rg ∝ Mνw) has been reported to be a powerful methodology to determine the actual conformation of SCNPs in solution.11,13,70–72,75 For linear polymers, the specific value of ν depends on the particular state of the chain, with a value ca. νF = 0.59 (Flory exponent) for the expanded coil state (i.e., chain in good solvent), 1/2 for the Θ-state and 1/3 for the most compact globule state.110 The values of ν reported for chemically denatured, intrinsically disordered, and folded proteins are 0.57, 0.51, and 0.29, respectively.110a
As illustrated in Fig. 12, the average value of ν for SCNPs prepared in good solvent is within statistics consistent with those of linear chains in the Θ-state, or intrinsically disordered proteins in solution (ν ≈ 0.5). On the contrary, SCNPs synthesized in good solvent and deposited on a surface often have a spherical, pancake-like shape, as observed in a large number of AFM and TEM images reported in the literature (Fig. 14). Interactions with the substrate upon solvent removal, dewetting effects or evaporative self-assembly could be responsible for the compact morphology, and often larger SCNP size observed by TEM and AFM when compared to their morphology and size in solution.8,117 In this sense, the morphology change suffered by SCNPs on passing from solution to the solid state is of paramount importance for the construction of efficient biosensors based on immobilized protein-mimic nano-objects and for the development of transient vitamin-binding systems, among other applications (see Section 3).
 | ||
| Fig. 14 Typical spherical morphology of polymethyl methacrylate SCNPs when deposited on a surface after solvent removal, as observed by TEM. Reprinted with permission from ref. 71. | ||
In order to increase the compaction degree of SCNPs, trying to mimic the conformation of functional globular proteins, several approaches have been investigated both experimentally and by means of computer simulations. Two independent experimental attempts were carried out to improve the folding degree of SCNPs by using heterofunctional polymers and orthogonal cross-linking chemistries. The first one by Palmans, Meijer and coworkers83 involved the sequential use of BTA helical stacking and UPY dimerization. The second one by Berda and colleagues employed a sequential combination of supramolecular folding and two different covalent coupling reactions, respectively.69 To draw a clear and general picture of the advantages of folding single polymer chains containing heterofunctional (A, B) reactive groups to soft nanoparticles via orthogonal intra-chain (A + B) cross-linking techniques, MD simulations of a generic bead-spring model for A + B cross-linked nanoparticles and their exactly equivalent homofunctional counterparts (i.e., A or B cross-linked nanoparticles) were carried out by our group.72 Moreover, the MD simulation results were compared with the experimental results by SEC/multi-angle laser light scattering (SEC/MALLS) and SAXS measurements on real SCNPs (Fig. 15). The MD simulation results, which is in good agreement with experimental data, showed the failure of this approach for obtaining SCNPs with a globular, compact morphology. Also SCNPs obtained through BTA helical stacking and UPY dimerization,83 even with internal left-handed helical column-structures, as revealed by circular dichroism (CD) experiments, showed a value of ν = 0.51, which is very far from ν = 0.33 of globular particles.
 | ||
| Fig. 15 Illustration of the advantages of folding single polymer chains to soft nanoparticles via orthogonal intra-chain (A + B) cross-linking techniques, as determined by SEC measurements. Increased retention time and, hence, reduced hydrodynamic size of the SCNPs was observed in the case of (A + B) cross-linking. Cross-linking techniques employed: A = Michael addition, B = copper complexation. Reprinted with permission from ref. 72. | ||
In a further extension of this work, MD simulations of the intramolecular cross-linking of individual polymer chains into SCNPs under good solvent conditions were carried out to explore the effect of the number x of different chemical species of linkers on the size and shape of the SCNPs.111 At a fixed backbone length and fraction of linkers, increasing x leads to nanoparticles that were, on average, smaller and more compact. The values of the scaling exponent ν where found to be 0.51, 0.47, 0.45, 0.44, and 0.43 for x = 2, 3, 4, 5, and 6, respectively. An analysis of the distributions of shape parameters revealed that cross-linking produces a significant fraction of highly non-spherical, sparse nanoparticles even for x = 6. The density profiles of the nanoparticles were smooth and no evidence of core–shell structures was found, neither particular spatial arrangement of the linkers, which were distributed randomly in the SCNP. The same conclusions were obtained for all the backbone lengths investigated. Consequently, multi-orthogonal protocols have a fundamental limitation for producing globular SCNPs (ν = 1/3), which is intimately connected to the inherent self-avoiding character of the polymer precursors under good solvent conditions. Even for x = 6, long-range looping was not sufficiently promoted to fully prevent the formation of sparse nanoparticles.111
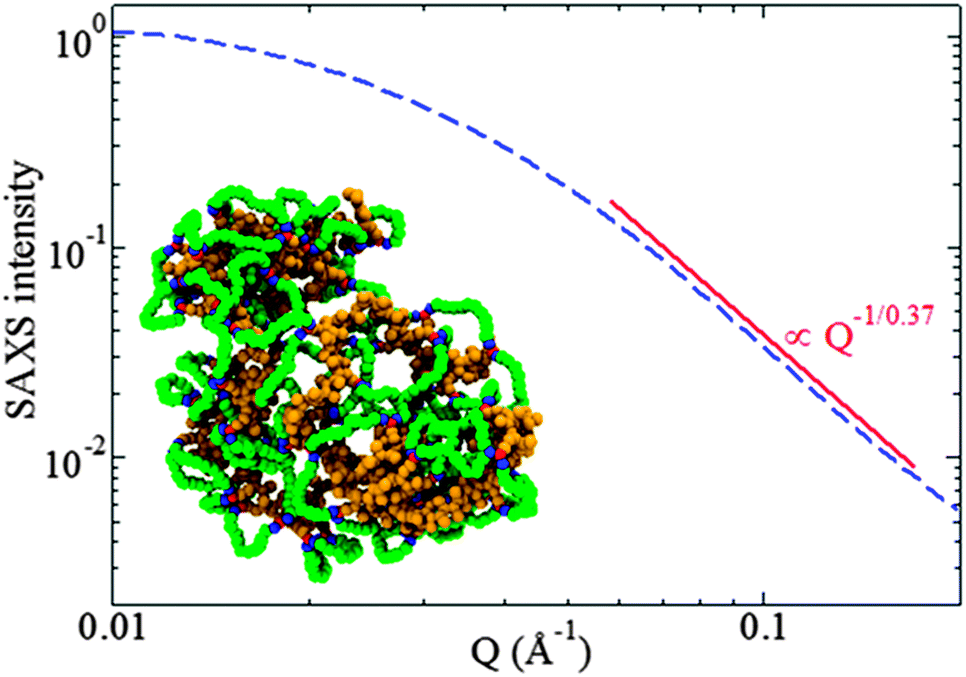 | ||
| Fig. 16 Rapid and efficient synthesis of SCNPs with a nearly globular morphology in solution by employing the photoactivated radical-mediated thiol–yne coupling (TYC) reaction as the driving force for chain folding/collapse and relatively long cross-linkers. Analysis of the scattering form factor from SAXS measurement revealed a scaling exponent ν ≈ 0.37 for the dependence of the SCNP size on its molecular weight, a value very close to that expected for globular objects, ν = 1/3. Reprinted with permission from ref. 75. | ||
Based on MD simulations, our group has proposed two new routes, which are experimentally realizable with state-of-the-art methods and do not require specific sequence control, for the design of permanent globular single chain nanoparticles by tuning solvent quality.112 In the first route (method I) cross-linking is performed under bad solvent conditions with the precursors anchored to a surface, at a density sufficiently low to prevent inter-chain contact. After completing the cross-linking, the nanoparticles are cleaved from the surface. In the second route (method II), a random copolymer with unreactive solvophilic and reactive solvophobic monomers is used, tuning the fraction of both monomers to form a single core–shell structure. The solvophilic shell prevents intermolecular aggregation, and the cross-linking of the solvophobic units is purely intramolecular. In both routes, after completing cross-linking and restoring good solvent conditions, the swollen single chain nanoparticles are globular objects, as confirmed by the analysis of their scaling behaviour and asphericity parameter (Fig. 17). It is worth mentioning that amphiphiles with a regular sequence are not useful for producing globular nanoparticles, unlike their (easier to synthesize) counterparts with random sequence.112 Although the cross-linking protocol based on amphiphiles with random composition is less successful than the route of the method I, from an experimental point of view, method II should be implemented more easily.
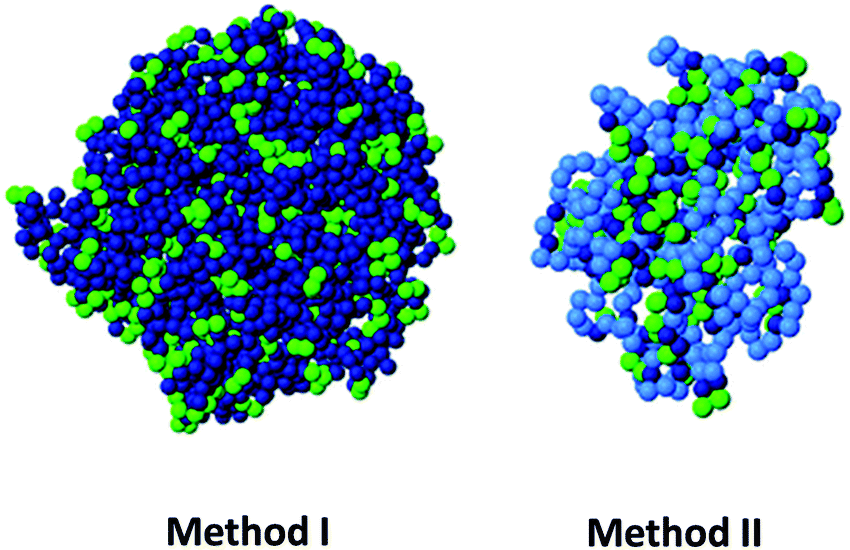 | ||
| Fig. 17 Based on MD simulations, two new routes (method I and method II) have been proposed, very recently, for the design of permanent globular single chain nanoparticles via tuning of solvent quality (see text for details). Reprinted with permission from ref. 112. | ||
Several approaches to structurally dynamic SCNPs displaying nearly globular morphology in solution have been recently reported based on neutral and charged amphiphilic random copolymer self-assembly, respectively. Hence, Akashi and coworkers91,92 reported unimer nanoparticles composed of hydrophobized poly(amino acid)s based on amphiphilic random copolymers of poly(γ-glutamic acid)-graft-L-phenylalanine (γ-PGA-Phe) self-assembled in aqueous media. The resulting γ-PGA-Phe SCNPs were characterized by DLS and static light scattering (SLS), SANS, as well as steady-state fluorescence measurements/quenching techniques. Using the molecular weights of γ-PGA higher than 140 kDa conjugated with Phe at 27–42%, spherical core–shell structures were observed by SANS combined with fluorescence quenching techniques. The number of hydrophobic domains per SCNP (NDomain) ranged from 3 to 7, whereas the number of Phe moieties per hydrophobic domain (NPhe) ranged from 100 to 200. NDomain was found to depend on the particle size, as well as the grafting degree of Phe. In addition, the stability of the SCNPs against pH revealed that the hydrophobic domains of the Phe groups were formed at pH values ranging from pH 5 to pH 10. A reduction of the NDomain was observed in alkaline conditions due to the cleavage of ester bonds. The differences in the inner structures of γ-PGA-Phe SCNPs offer great potential for applications as small drug carriers, because it has been demonstrated that γ-PGA-based nanoparticles are suitable for the intracellular delivery of protein-based drugs as well as tumour vaccines.92 However, the average SCNP sizes, as determined by SANS and DLS, are larger than those expected for compact globules of the corresponding actual molecular weights.
Multi-responsive SCNPs displaying spherical morphology in solution constructed via single chain folding of neutral amphiphilic random copolymers in water have been reported by Sawamoto and colleagues102 (Fig. 18). Hence, amphiphilic random methacrylate copolymers, consisting of PEG and alkyl side groups (20–40 mol% hydrophobic units with relatively long and/or large alkyl groups), were found to undergo reversible single chain self-folding in water to SCNPs comprising hydrophobic compartments, as revealed by solvatochromism experiments employing hydrophobic dyes. The self-assembled nanoparticles were dynamic, reversible and stimuli-responsive in water; unfolded via the addition of methanol and more mobile upon increasing temperature, as de-terminated by 1H NMR spectroscopy. Interestingly, these SCNPs were stable even at a high concentration in water (i.e., 60 mg mL−1) due to the PEG side-groups acting as efficient steric stabilizers. An estimation of the expected globule size based on its actual molecular weight showed that these SCNPs are not truly compact objects in water, but instead relatively swollen structures.
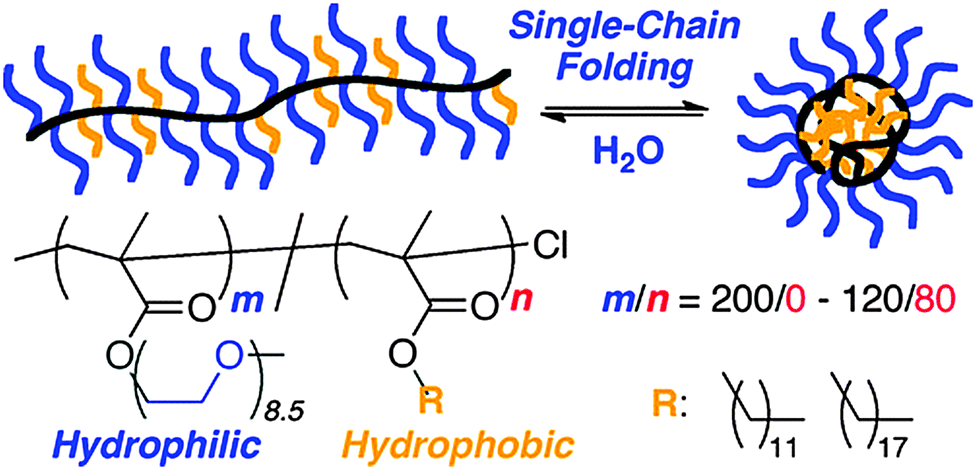 | ||
| Fig. 18 Illustration of the construction of structurally dynamic SCNPs displaying nearly globular morphology in solution via single chain folding of neutral amphiphilic random copolymers in water. Reprinted with permission from ref. 102. | ||
Reversible SCNPs displaying spheroidal morphology in solution have been synthesized by Van de Mark and colleagues96–101via single chain folding of charged amphiphilic random copolymers in water. The main steps involved in the preparation of these globular SCNPs are illustrated in Fig. 19. The new route is an innovative approach utilizing a small number of hydrophilic charged groups (ca. 10 mol%) along a hydrophobic polymer backbone which transitions from a random coil conformation in organic solvent to a hard sphere-like morphology in water through a slow gradient with subsequent organic solvent removal. The SCNPs showed diameters in agreement to those expected for globules of the same molecular weight, which ranged typically from 3 nm to 9 nm. The sodium or potassium salts of these SCNPs could be dried then redissolved in water with no aggregation. The diameters of the SCNPs synthesized in this way as measured by DLS correlated with the absolute molecular weight and distributions from SEC99 (Fig. 20), providing support of the globular conformation of these SCNPs stabilized in water by repulsive ionic forces.
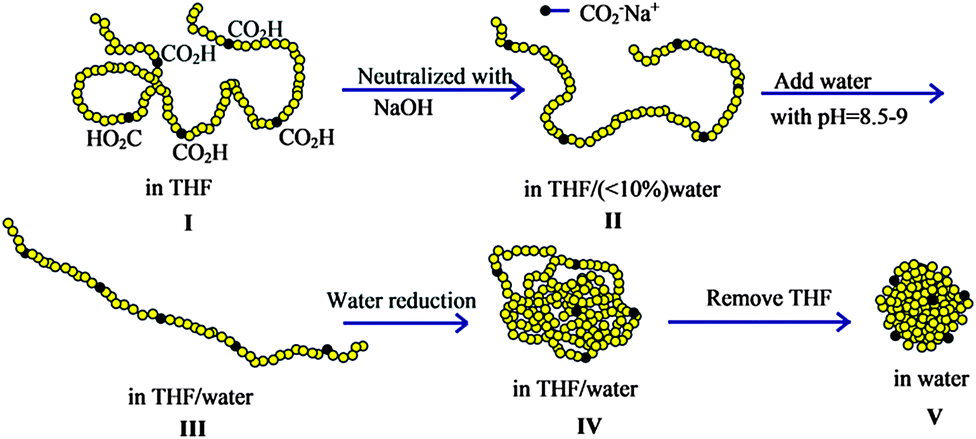 | ||
| Fig. 19 Illustration of the construction of structurally dynamic SCNPs displaying nearly globular morphology in solution via single chain folding of charged amphiphilic random copolymers in water. I = random coil configuration in THF solvent. II = random coil intimate ion pair, III = extended coil solvent separated ion pair, IV = collapsed coil, V = hard sphere. Reprinted with permission from ref. 98. | ||
 | ||
| Fig. 20 Particle size distribution as determined by DLS (points) and SEC (continuous line) for a SCNP of Mw = 169 kDa. The predicted size for a globule of identical molar mass is 7.6 nm, which is in good agreement with the experimental data. Reprinted with permission from ref. 99. | ||
The above results indicate that single chain technology allows, by proper selection of the synthesis route, the construction of SCNPs with a morphology resembling that of intrinsically disordered proteins (conventional synthesis methods in good solvent) or that showed by native, globular proteins (special synthesis methods, as illustrated above). Interestingly, as stated recently in a perspective article,14 “the potential applications of SCNPs broaden significantly by taking inspiration from the functions of both ordered and disordered proteins” (see Section 3).
2.6. Single chain tadpoles
Preparation (and isolation) of di-block copolymer tadpole molecules, i.e., single chain nano-objects that resemble a tadpole with a cross-linked block forming a globule and a soluble block assuming the random coil conformation, was described by Tao and Liu as early as 1997.113 Recent advances in single chain technology applied to the construction of permanent single chain tadpoles (SCTPs) are illustrated in Fig. 21. New intra-chain cross-linking procedures have been introduced over the years, such as a Diels–Alder (DA) reaction reported in 2002, pyridine quaternization in 2008, combined DA + CuAAC coupling, and alkylamine quaternization in 2013, as well as radical coupling and hydrolysis/polycondensation in 2014.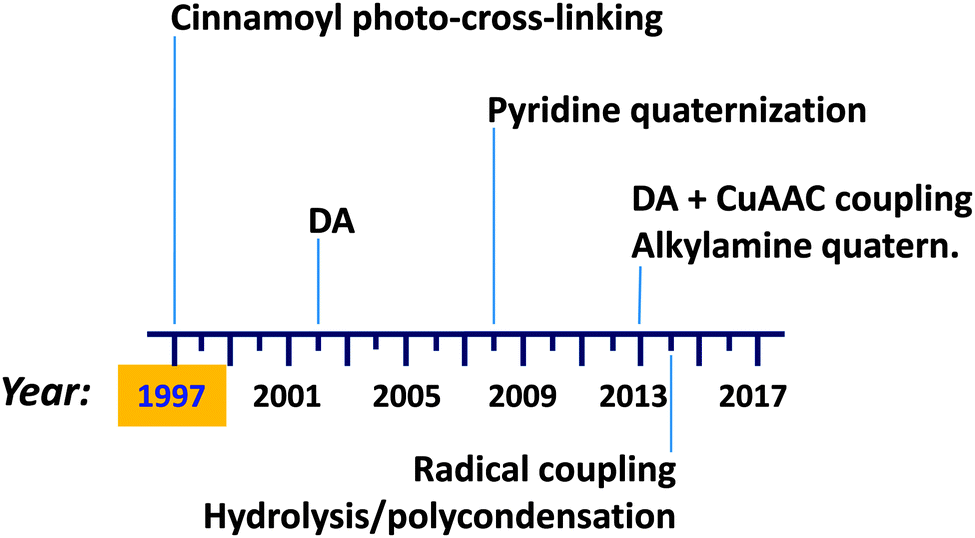 | ||
| Fig. 21 Illustration of the advances in single chain technology applied to the construction of permanent single chain tadpoles (SCTPs) (see text for references). First synthesis of SCTPs was carried out by Tao and Liu in 1997. DA = Diels–Alder reaction, CuAAC = copper(I)-catalysed azide–alkyne cycloaddition. | ||
Tao and Liu113 used cinnamoyl photo-cross-linking for the synthesis of SCTPs based on polystyrene-b-poly(2-cinnamoylethyl methacrylate) (PS-b-PCEMA) precursors that formed micelles in THF–cyclohexane mixtures with PCEMA as the core, in equilibrium with unimers. Photo-cross-linking the PCEMA block of the unimers allowed the preparation of permanent SCTPs in relatively low yield (5 to 25 wt%), which were isolated from cross-linked micelles by tedious SEC fractionation. PECMA double-bond conversion ranged from 26 and 38% based on the UV measurements. The mean size of the block copolymer reduced from 19.1 nm to 14.2 nm upon SCTP formation. In a further work by the same group,114 a new method for SCTP synthesis was examined based on the addition of the di-block copolymer solution at low pumping speed into a solvent mixture under constant UV irradiation and stirring. With this method, SCTPs were prepared free of micelles at a final tadpole concentration that was 27 times the critical micelle concentration (c.m.c.) of the copolymer. Such tadpoles showed a volume reduction of 33% relative to the corresponding copolymer precursor.
A Diels–Alder (DA) reaction and a special continuous addition technique were employed by Hawker and colleagues44 for the efficient synthesis of tadpoles based on AB block copolymers, in which A = PEG and B = PS containing DA-cross-linkable groups. A significant decrease in hydrodynamic size upon SCTP formation was observed clearly by SEC (Fig. 22). Moreover, dramatic morphological changes were observed in Langmuir–Blodgett (LB) film assemblies of these tadpoles compared to those of their linear counterparts.115 The linear block copolymer formed well-organized disk-like surface assemblies, as observed by AFM, while the SCTPs exhibited long (>10 μm) wormlike aggregates (Fig. 22). The driving force for the different morphologies observed was found to be a combination of the change in geometry of the block copolymer and the restricted degree of stretching in the nanoparticle block after intramolecular cross-linking.
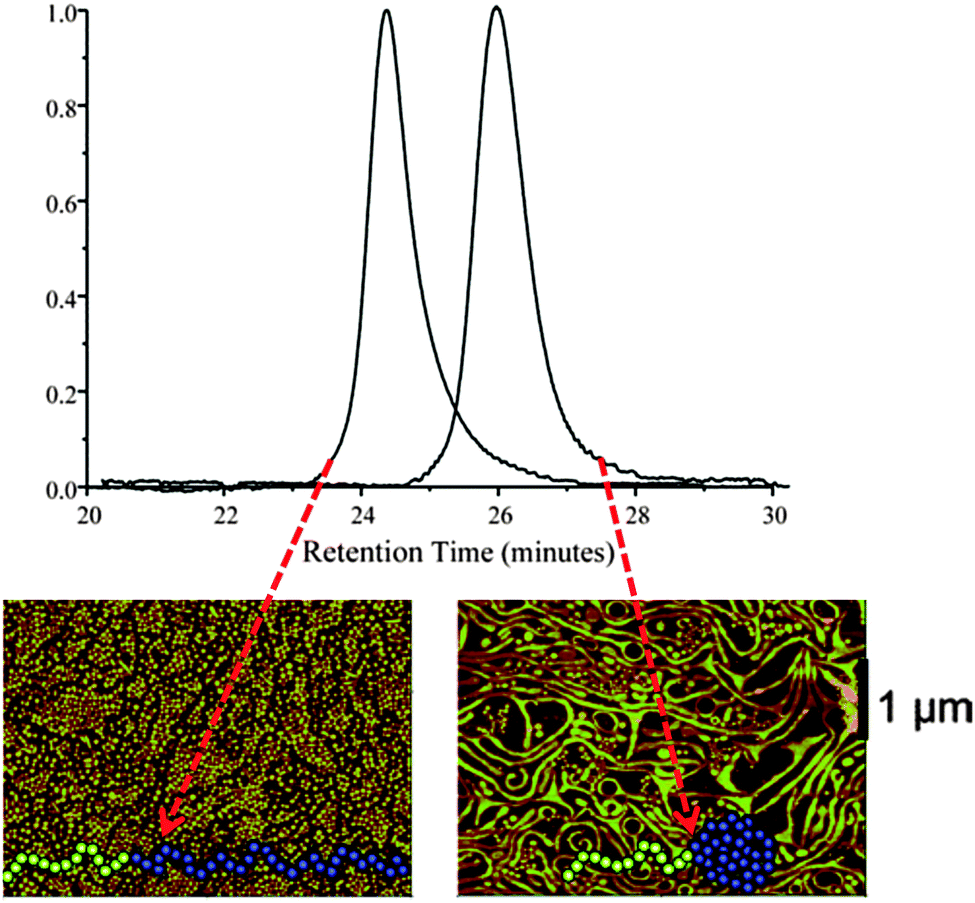 | ||
| Fig. 22 Hydrodynamic size reduction upon single chain tadpole (SCTP) formation was clearly observed by SEC, due to the longer retention time of the SCTPs when compared to their linear counterparts. In addition, the SCTPs exhibited long (>10 μm) wormlike aggregates, whereas the linear block copolymer formed well-organized disk-like surface assemblies, as observed by AFM. Reprinted with permission from ref. 44 and 115, respectively. | ||
Zhu and coworkers116 used pyridine quarterization with 1,4-dibromobutane (DBB) in dimethyl formamide (DMF) for the formation of unimolecular Janus tadpoles from ABC block copolymers, where A = PS, B = poly(2-vinyl pyridine) (P2VP) and C = poly(ethylene oxide) (PEO). Janus tadpoles result from the efficient intramolecular cross-linking of the middle P2VP block using DBB due to effective steric shielding of PS and PEO end blocks (Fig. 23). The SEC results indicated that intramolecular cross-linking of the middle P2VP block could take place when the polymer concentration was relatively high (20 mg mL−1) and/or the DBB-to-2VP molar ratio was high. This suggests that the relatively long PS and PEO end blocks effectively prohibited intermolecular cross-linking. Interestingly, concentration-dependent self-assembly in DMF was observed. At low concentrations (<2.0 mg mL−1), the majority of the Janus SCTPs existed in the unimolecular form.
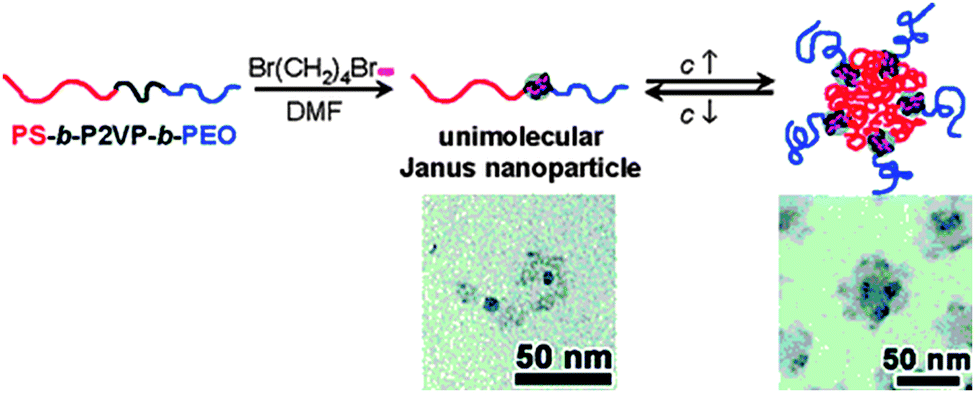 | ||
| Fig. 23 Unimolecular polymeric Janus tadpoles constructed via intramolecular cross-linking of poly(2-vinyl pyridine) (P2VP) blocks from PS-b-P2VP-b-PEO copolymers based on pyridine quaternization with 1,4-dibromobutane in N,N-dimethylformamide (DMF). Upon increasing solution concentration, Janus tadpoles were found to aggregate into super-micelles, as revealed by TEM. Reprinted with permission from ref. 116. | ||
When the concentration increased gradually, the Janus tadpoles began to aggregate into super-micelles (Rh = 50–100 nm), where PS formed a super-core and PEO formed the corona, with cross-linked P2VP nanoparticles in between. After intramolecular cross-linking of the middle P2VP block, DMF changed from a good solvent to a slightly poor solvent, as suggested by the second-virial coefficient (A2), as determined by SLS, changed from a positive value for the pure triblock copolymer to a negative value for the Janus tadpoles.116
The DA reaction and CuAAC coupling were recently combined by Barner-Kowollik and colleagues117 for the formation of amphiphilic PS-b-PEG-based tadpoles via intramolecular UV-light-triggered DA cross-linking. Tadpole precursors were synthesized in two main steps. First, a DA-cross-linkable PS was prepared containing one single alkyne functionality at one of the chain ends through combined NMP polymerization and polymer functionalization. Next, CuAAC “click” chemistry was used for the efficient coupling of an azide-terminated linear PEG to the single alkyne-containing PS. Upon tadpole formation, a significant reduction in hydrodynamic size was observed by SEC and DLS.
Alkylamine quaternization was recently used by Zhao and coworkers118 to construct amphiphilic PS-b-poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA)-based tadpoles. PS-b-PDMAEMA copolymers were synthesized by two-step RAFT polymerization and the PDMAEMA blocks were cross-linked intramolecularly by 1,4-diiodobutane, leading to amphiphilic SCTPs containing one hydrophilic nanoparticle head and one hydrophobic PS tail. Depending on the PDMAEMA block length and the size of the nanoparticle head, PS-b-PDMAEMA SCTPs were found to self-assemble into micellar or vesicular structures in aqueous and methanol solutions. In cyclohexane, they self-assembled into aggregates (bunchy micelles)118 with single chain nanoparticles in the cores and linear PS in the coronae (Fig. 24).
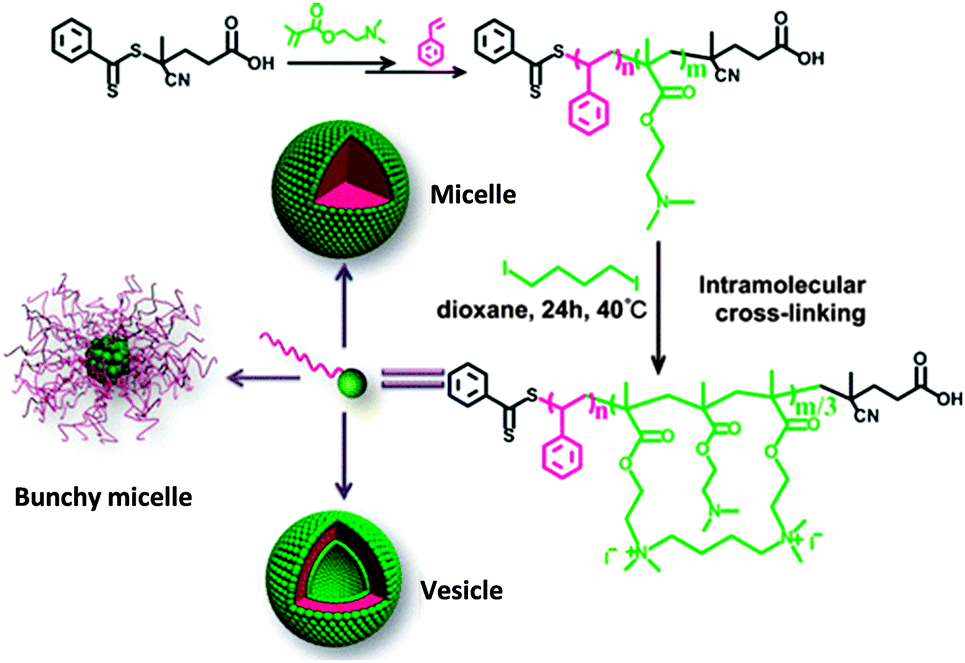 | ||
| Fig. 24 Illustration of the preparation of SCTPs from PS-b-poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) copolymers through intramolecular cross-linking via alkylamine quaternization, and self-assembled structures observed by TEM in water or methanol (micelles and vesicles) and in cyclohexane (bunchy micelles). Reprinted with permission from ref. 118. | ||
In a further study by the same group,119 the effect of charge density on self-assembly was investigated for PSx-b-PDMAEMAy tadpoles of different block length (x, y = number of PS and PDMAEMA repeat units, respectively). The morphology and the size of the aggregates changed significantly with the charge density. SCTPs made from PDMAEMA74-b-PS100, self-assembled into spherical micelles in aqueous solutions and the average size of the micelles increased with increasing charge density. Amphiphilic SCTPs with different charge densities were constructed from PDMAEMA30-b-PS169. With increasing charge density, the morphology of the aggregates in aqueous solutions changed from spherical micelles to vesicles, to form a mixed morphology of worm-like cylinders and vesicles. In a cyclohexane–THF mixture, with increasing charge density, the morphology of the aggregates made from PDMAEMA74-b-PS297 tadpoles was found to change from bunchy micelles to a mixture of vesicles and large compound vesicles.119
Radical coupling was used by Gao and colleagues120 to prepare PMMA-b-poly(4-vinyl pyridine) (P4VP) SCTPs by using propargyl bromide as cross-linker. Both SEC and DLS showed that the cross-linking of the P4VP block was an exclusively intramolecular reaction. The hydrodynamic size of the block copolymer reduced from 16 nm to 12 nm upon SCTP formation and the 1H NMR signals of the P4VP block disappeared completely due to the low mobility of the P4VP moieties in the tadpole head. It was estimated that during the cross-linking reaction, only about 10% of the PMMA block was wrapped in the cross-linked P4VP block.
Hydrolysis/polycondensation reactions have been employed by He and coworkers121 to construct hybrid SCTPs composed of silica-like heads and PEO tethers. Using di-block copolymers of PEO-b-[(PMMA-co-poly(3-(trimethoxysilyl)-propyl methacrylate))] (PEO-b-P(MMA-co-TMSPMA)), the intramolecular hydrolysis and polycondensation of silane moieties led to the formation of these hybrid SCTPs (Fig. 25). The tadpoles were characterized carefully by SEC, 1H NMR spectroscopy, TEM, and SLS/DLS. In a mixed solvent of THF–water, these hybrid SCTPs self-assembled into spherical micelles, vesicles, or large compound micelles, depending on the size of the silica heads and the initial concentrations.
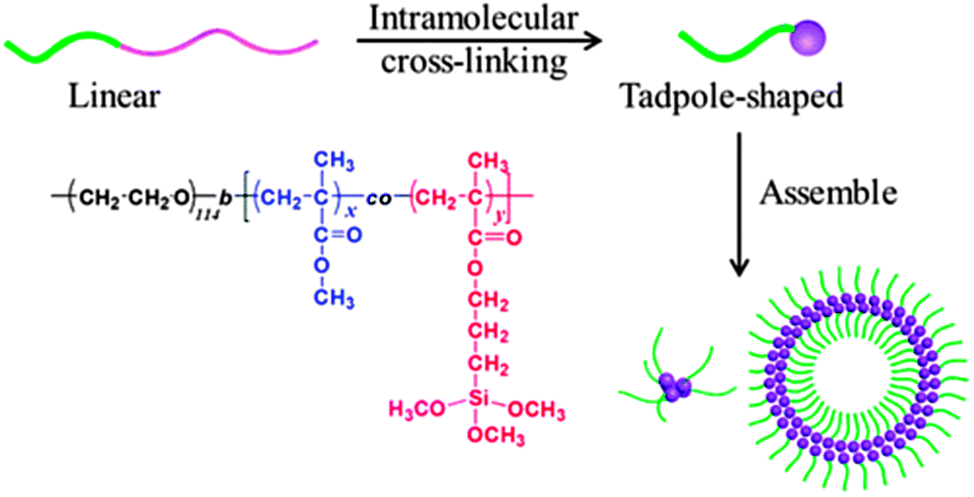 | ||
| Fig. 25 Schematic illustration of the formation of hybrid SCTPs through intramolecular cross-linking via hydrolysis/polycondensation reactions. The resulting SCTPs are composed of silica-like heads and PEO tethers, and can be self-assembled in to different structures (spherical micelles, vesicles, large compound micelles) depending on the size of the silica heads and the initial concentrations. Reprinted with permission from ref. 121. | ||
The above examples illustrate the interesting possibilities that single chain tadpoles offer to construct a range of responsive self-assembled structures. The potential applications of amphiphilic SCTPs are described in Section 3.
2.7. Single chain dumbbells
Recently, the construction of single chain dumbbells (SCDBs) by stepwise intramolecular cross-linking of sequence-controlled precursors has been reported by Roy and Lutz.122These compartmentalized single chain objects were prepared by performing successive cross-linking reactions in an orthogonal fashion. The foldable precursors were synthesized by sequence-controlled copolymerization of styrene with N-substituted maleimides, namely pentafluorophenyl 4-maleimidobenzoate (PFMI) and TIPS-protected N-propargyl maleimide (TIPS-PgMI). These two functional MIs allow intramolecular cross-linking. The activated ester pentafluorophenyl moieties of PFMI reacted with ethylenediamine, whereas the deprotected alkyne functions of TIPS-PgMI self-reacted via alkyne homocoupling.
The compaction of model copolymers containing only one cross-linkable zone (i.e., either PFMI or TIPS-PgMI) was first studied. 1H NMR and SEC analysis indicated that these structures could be compacted efficiently into single chain nano-objects. Therefore, more complex copolymers containing two individually addressable cross-linking zones were prepared and sequentially compacted to SCDBs (Fig. 26).
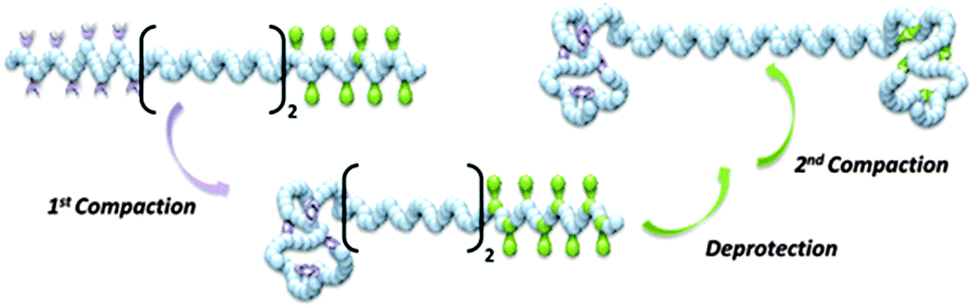 | ||
| Fig. 26 Schematic illustration of the formation of single chain dumbbells (SCDBs) via intramolecular double compaction of sequence-controlled linear macromolecules. Reprinted with permission from ref. 122. | ||
Characterization of the folding process by SEC indicated that double-compaction occurred and that the formed SCDBs contain, presumably, distinct cross-linked subdomains. Further characterization of the SCDBs by neutron and X-ray scattering techniques in solution, as well as TEM and AFM techniques in solid state is necessary to unravel the actual morphology of these complex soft nano-objects.
2.8. Single chain hairpins
The construction of responsive single chain hairpins (SCHPs) from ABC block copolymers, in which the external A and C blocks contain complementary side-chain recognition units, was pioneered by Weck and colleagues123,124 (Fig. 27).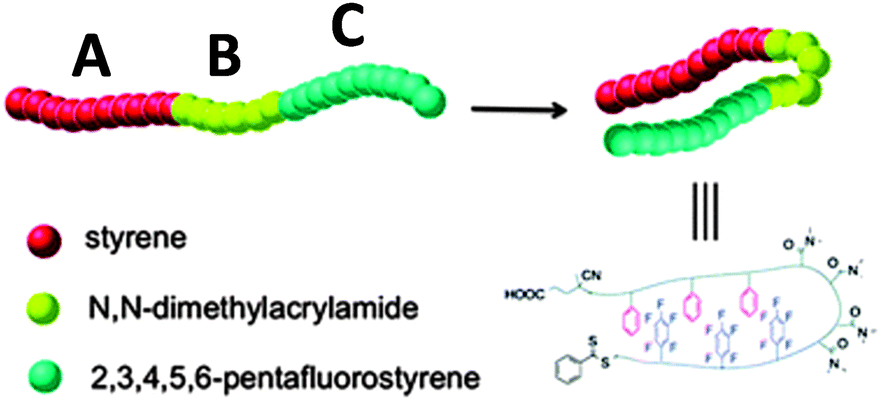 | ||
| Fig. 27 Illustration of the construction of responsive single chain hairpins (SCHPs) from ABC triblock copolymers via quadrupole interactions between polystyrene (A) and polypentafluorostyrene (C) blocks. Reprinted with permission from ref. 124. | ||
In pioneering work,123 ABC copolymers were synthesized by ROP of norbornenes, the A and C blocks contained protected ureidoguanosine (UG) and diaminonaphthyridine (DAN) pendant functional groups, respectively; B = poly(norbornene octyl ester). Removal of the protecting group was achieved by dialysis in dimethyl sulfoxide (DMSO). DMSO competed for the hydrogen bonding between the complementary UG–DAN pair allowing the release of the protecting group from the triblock copolymer. DLS and 1H NMR spectroscopy showed that the linear triblock copolymer chains self-assemble to SCHPs at high dilution, through intramolecular UG–DAN hydrogen bonding interactions.
In a subsequent work by the same group,124 SCHP formation was demonstrated from ABC triblock copolymers via quadrupole interactions between PS and polypentafluorostyrene blocks. The quadrupole interactions between the electron-rich and electron-deficient blocks were characterized in different solvents using 2D NMR spectroscopy and DLS. SCHP formation was observed in chloroform, whereas inter-chain aggregation was noted in DMF.
3. Endowing single chain nano-objects with function
Because this field is still in its infancy, only a limited number of proof-of-concept investigations have been carried out to endow single chain soft nano-objects with useful functions. In spite of this limitation, certain preliminary results illustrate the possibilities offered by these unimolecular nano-entities for a variety of potential applications. In general, the main advantages of folded/collapsed single chains over their linear counterparts are (i) presence of locally compact, but accessible, sites/cavities/zones, (ii) possibility to bound, temporally or permanently, active species, such as drugs or catalysts onto these local pockets, and (iii) reduced size and hydrodynamic volume. (i) and (ii) are useful for nanomedicine, catalysis and sensing applications, whereas (iii) is especially relevant for applications where a reduction in viscosity is required. Amphiphilic single chain nano-objects offer additional possibilities as emulsifiers and lithography agents, because of their particular self-assembly behaviour.3.1. Nanomedicine
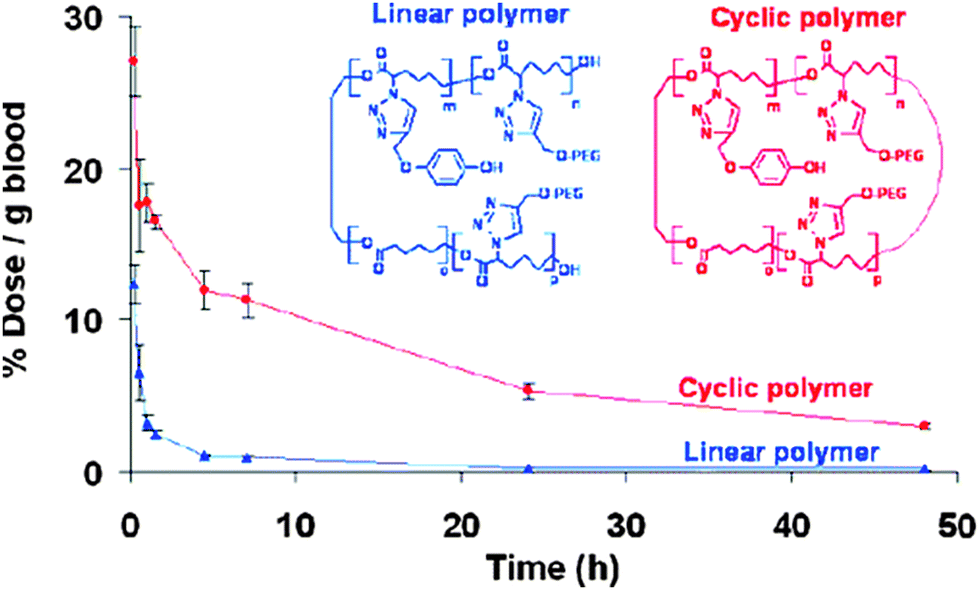 | ||
| Fig. 28 Cyclic polymers are attractive candidates as possible drug carriers, because they showed increased circulation half-lives compared to their linear counterparts. Reprinted with permission from ref. 125. | ||
Single chain nanoparticles are also promising nanocarriers for drug delivery applications. Biocompatible PCL-based SCNPs were recently prepared by Qiao and coworkers76 through organocatalysed ring opening polymerization. Cytotoxicity studies revealed that these SCNPs, which were covalently cross-linked by biodegradable polyester linkages, were nontoxic toward human embryonic kidney (HEK293T) cells. As demonstrated recently by Zhao and colleagues,106 preparation of <10 nm SCNPs through intra-chain photocrosslinking (under >320 nm UV) and their subsequent photodegradation via photoinduced chain scissions (254 nm UV) is possible by incorporating coumarin moieties into the chain backbone of a polyester precursor. Owing to the biocompatible and biodegradable nature of polyester-based SCNPs, they could be exploited for biomedical applications. Several examples showing the potential of SCNPs for the transport and delivery of peptides, natural amino acids, siRNAs and drug molecules have been published.
Hence, in a pioneering work by Hamilton and Harth,126 molecular dendritic transporter nanoparticle vectors based on hydrophilic SCNPs post-modified via amide coupling reactions with dendritic molecular transport units (Newkome dendrimers) were synthesized for the delivery of peptidic molecules into the cells. The rapid transport of multiple copies of peptide units per particle across the cellular barrier into the cytoplasm of NIT 3T3 mouse fibroblast cells using this novel nanoscopic delivery system (5–10 nm in size) was carried out, as observed by confocal microscopy with Alexa Fluor 568 dye-as the label of the nanoparticle backbone- and fluorescein as the marker of the peptide.
Ultra-fine (7.4 nm in size) L-phenylalanine anilide-imprinted SCNPs have been synthesized by Liu and coworkers.127 The selective binding of L-phenylalanine anilide (natural amino acid) vs.D-phenylalanine anilide (synthetic organic compound) by these SCNPs was investigated. Interestingly, the L-phenylalanine anilide-imprinted nanoparticles showed higher sorption capacity for L-phenylalanine anilide (238 μmol g−1) than for D-phenylalanine anilide (132 μmol g−1) and the rate constant for L-phenylalanine anilide release was found to be inversely proportional to the squared radius of the particles.
pH-sensitive polyamine nanogels containing PEG-tethered chains were able to form spontaneously a polyion complex with negative charged siRNA through electrostatic interactions under physiological pH conditions, as reported by Nagaski and colleagues.128 When combined with a siRNA that knocks down the firefly luciferase gene, the nanogel–siRNA complex showed a remarkable enhancement of gene-silencing activity against firefly luciferase gene expressed in HuH-7 cells. Within certain confidence limits, similar results could be expected for single chain nano-objects.
The facile hydrophobic guest (e.g., doxorubicin) encapsulation capabilities of biocompatible surface-functionalizable nanogels, showing a minimum diameter of 16 nm, approaching the size of the SCNPs, was demonstrated by Thayumanavan and coworkers,129 using disulphide bonds as the cross-linkers. The release of entrapped guest molecules induced by glutathione (GSH) was observed at high GSH concentrations (10 mM), corresponding to that found inside the cells, whereas at low GSH concentration (10 μM), as that outside the cell and within the blood plasma, no significant release was found. The cell viability investigated by treating 293T human kidney cell lines with these nanogels showed high cell viability and no concentration-dependent toxicity, suggesting the nontoxic characteristics of these nano-objects with potential applications in nanomedicine.
The preparation of nanoparticles with sizes between 30 and 200 nm for the development of vaccine and drug carriers by self-association of hydrophobically modified polyaminoacids was investigated by Akashi and colleagues,130 who found that the size of the nanoparticles plays a critical role in controlling the immune responses. These results motivated the synthesis of hydrophobically self-assembled SCNPs by these authors91,92 based on modified poly(γ-glutamic acid), a naturally occurring, water-soluble, biodegradable, edible, and non-toxic polyamide that is synthesized by several Bacillus strains.
Fulton and coworkers104 reported thermoresponsive water-soluble dynamic covalent SCNPs that reversibly transform into a hydrogel. Triggered gel formation required the simultaneous application of both low pH and temperature, which would be highly convenient for the development of improved drug delivery systems.
More recently, SCNPs showing form factors in solution similar to those of intrinsically disordered proteins (IDPs) have been synthesized by our group and assessed for controlled drug delivery applications.70,71 In the first example of bioinspired nanocarriers, SCNPs were synthesized from precursors containing β-ketoester functional groups via Michael addition-mediated multidirectional self-assembly. SANS measurements combined with MD simulations undoubtedly showed that the form factor of these SCNPs in solution resembles that of IDPs instead of that expected for globular, native proteins.70 Even without the precise sequence of proteins, the mimicking of the IDP morphology was a consequence of the intra-chain self-assembly process in a good solvent leading to the formation of local globules along the individual polymer chains. The resulting SCNPs were tested as novel transient-binding disordered nanocarriers (Fig. 29), from which the controlled delivery of both dermal protective (vitamin B9) and anticancer (hinokitiol) cargos was carried out.71
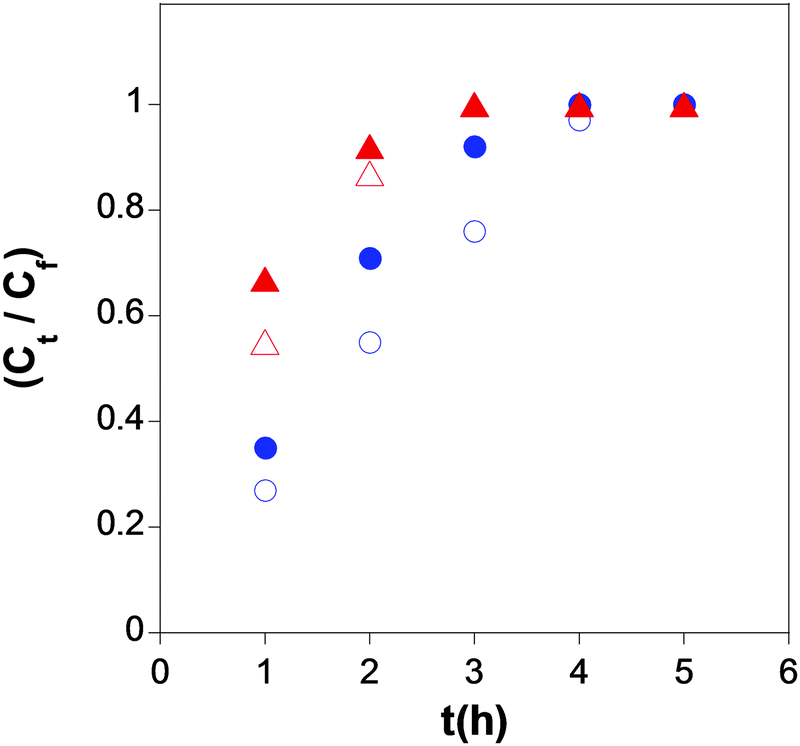 | ||
| Fig. 29 Simultaneous delivery data in water at pH = 6 (blue symbols) and pH = 8 (red symbols) of folic acid (open symbols) and hinokitiol (solid symbols) from the SCNPs synthesized via Michael addition-mediated multidirectional self-assembly. Reprinted with permission from ref. 71. | ||
Improved photoluminescence properties were reported by Harth and coworkers48 upon the site-isolation of semiconducting polymers from ABA block copolymers, in which A = functionalized PS and B = polyfluorene or poly(fluorene-co-thiophene) via SCNP formation through the intra-chain cross-linking of the A blocks. The photoluminescence measurements revealed the influence of the molecular weight of the A block to be crucial for the site isolation of the embedded conducting polymer block in the resulting SCNPs, with increased quantum efficiencies of 6% for the longer A blocks (3-fold increase compared to the linear ABA precursor).
Photoluminescent SCNPs containing ZnS nanocrystal (4.1 nm in size) were reported by Hu and coworkers.63 The fast in situ growth of ZnS crystalline nuclei, leading to ZnS quantum dot (QD) formation was performed by treating the Zn2+-containing SCNPs directly with a sodium sulphite solution. The maximum photoluminescence intensity of the SCNPs at 362 nm and the corresponding quantum yield were found to increase from 25 to 135 and from 2 to 17%, respectively, upon decreasing SCNP size. In addition, CdS QD-containing SCNPs were synthesized by these authors exhibiting bright fluorescence at 450 nm with a quantum yield of 45%. In subsequent work by the same group,64 the preparation of carbon nanodots from SCNPs and the theoretical investigation of their photoluminescence mechanism was carried out.
A high level of fluorescence was observed by our group for PS-based SCNPs, in which the intra-chain cross-linking points consisted in triazole–benzene–triazole segments.51 These SCNPs showed, after excitation at 350 nm, two maxima in the fluorescence spectrum located at 391 and 407 nm, respectively, as well as a small shoulder at 424 nm. Control experiments in which the triazole–benzene–triazole conjugation was disturbed or absent failed to show this fluorescence pattern.
Photochemical design of functional fluorescent SCNPs has been carried out by Barner-Kowollik and coworkers74 by employing intra-chain nitrile–imine ligation as the cross-linking technique for SCNP formation. This technique, in addition to finely controlling the size of the SCNP, allowed for adjusting the fluorescence properties of the nanoparticles by increasing the content of tetrazole units in the SCNPs.
Hydrophilic SCNPs exhibiting excellent fluorescence performance at 412 nm when irradiated under the light of 367 nm were synthesized by Pu's group60 based on SCNP precursors containing anthracene moieties.
More recently, fluorescent SCNPs containing fluorescein moieties were prepared Zimmerman and colleagues46via the intra-chain cross-linking of functional polynorbornene precursors. The protective effect of the SCNP against fluorescein photobleaching and their ability to enter live HeLa cells (human cervical cancers) (Fig. 30) was demonstrated. The excellent photostability and cellular uptake of these SCNPs suggest their use in future applications as long-term bioimaging and continuous tracking of living cells, or even as nanoscale delivery and imaging systems for potential intracellular theranostic applications.
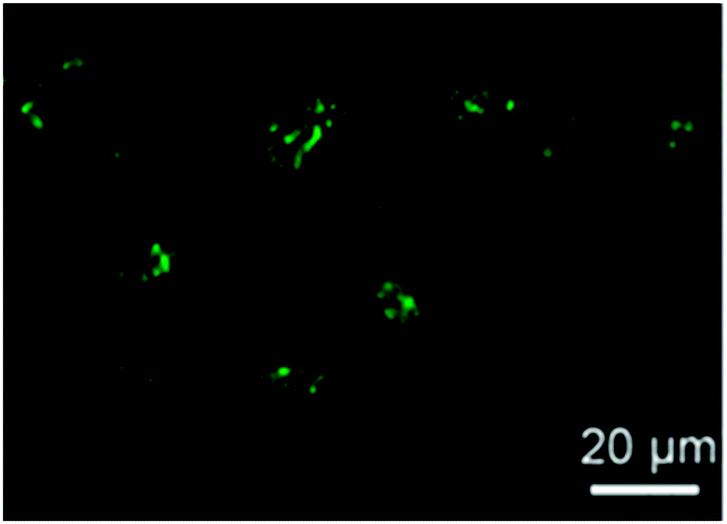 | ||
| Fig. 30 Confocal microscopy image of the HeLa cells treated with SCNPs containing fluorescein moieties exhibited bright green fluorescence upon SCNP internalization. Reprinted with permission from ref. 46. | ||
3.2. Catalysis
Wulff and colleagues published the first report of the use of catalytic single chain nanoparticles for carbonate hydrolysis.131 Soluble single molecule nanogels with molecular imprinted internal structure and containing just one active site per particle were reported by these authors. These SCNPs, 40 kDa in molecular weight, were soluble in water–acetonitrile mixtures and displayed Michaelis–Menten kinetics in close analogy to natural enzymes, but with a very low turnover frequency (TOF) of only 4.4 × 10−3 h−1.PMMA-based SCNPs synthesized by Zhao and coworkers105via coumarin (CM) photo-dimerization were evaluated as nanoreactors for the in situ synthesis of gold nanoparticles (AuNP). Interestingly, the intra-chain CM photo-dimerization degree was found to have a strong effect on the rate of AuNP formation. Hence, the relative rate of AuNP formation in a THF solution containing single chain nanoparticles with 64% and 27% degree of CM photo-dimerization was 4 and 2 times faster than that in a THF solution of the precursor. This provides a means to optically control the kinetics of AuNP formation. The whole reduction process of AuCl4 ions in water to 6–9 nm AuNPs was finished in 180 min at r.t., which was much faster than the reaction in THF solution due, presumably, to the more compact conformation of the SCNPs in aqueous media.
By taking inspiration from the hydrophobic pockets of globular enzymes, chiral nano-objects exhibiting catalytic activity towards carbonyl reductions in water without showing catalyst decomposition or hydrolysis were synthesized by Palmans, Meijer and coworkers.10 These catalytic SCNPs were prepared by folding through hydrogen-bonding interactions and helical self-assembly of a water-soluble amphiphilic precursor terpolymer containing both chiral and ruthenium-bonded units. The quantitative reduction of cyclohexanone to cyclohexanol in 18 h was demonstrated using 0.5 mol% of supported Ru catalyst, corresponding to a turnover frequency of TOF = 11 h−1. The catalytic activity was attributed to the formation of a ruthenium-protecting hydrophobic compartment inside the SCNPs. Following this compartmentalization concept, the same group reported12 a new family of SCNPs which catalyze the aldol reaction in water with a turnover frequency of TOF = 125 h−1 by using only 0.5 mol% of supported L-proline organocatalyst. Interestingly, these catalytic SCNPs can be easily recovered from the aqueous phase after separation of the aldol products by filtration and reused several times without additional purification. Moreover, a versatile, modular and efficient approach to introduce the L-proline organocatalyst moiety into benzene-1,3,5-tricarboxamide (BTA)-containing SCNPs was developed recently by Meijer, Palmans and coworkers.88 The catalytically active BTA moieties were introduced into the SCNPs via hydrophobic interactions and BTA self-recognition.
Recently, dual reductase/polymerase enzyme mimic SCNPs have been synthesized by our group11 through a new approach, which results in catalytic nano-objects allowing highly-efficient reductions to be performed (TOF > 5800 h−1), as well as the synthesis of high-molecular-weight polytetrahydrofuran in the presence of glycidyl phenyl ether (GPE) in an enzyme-like fashion. This new pathway to SCNPs endowed with enzyme-mimetic activity was based on the “concurrent” catalyst-assisted intramolecular cross-linking of linear precursors and concomitant binding of the catalyst to multiple SCNP intra-chain cross-linked sites (i.e., the “concurrent” approach). Hence, instead of possessing an isolated compact hydrophobic compartment, the resulting SCNPs in solution showed multiple, compartmentalized local catalytic sites, as well as a relatively open/sparse morphology in solution, as determined by the SANS experiments.11
As a first example of SCNPs displaying catalytic selectivity, oxidase enzyme-mimic SCNPs were recently synthesized in our laboratory via Cu2+-mediated intra-chain cross-linking, showing a disordered crumpled structure in solution, as revealed by SANS measurements.13 In spite of the lack of globular order, as found e.g. in Laccase enzymes, these SCNPs synthesized by the novel “concurrent” approach allow performing alkyne homocoupling reactions with unprecedented catalytic selectivity (Fig. 31).
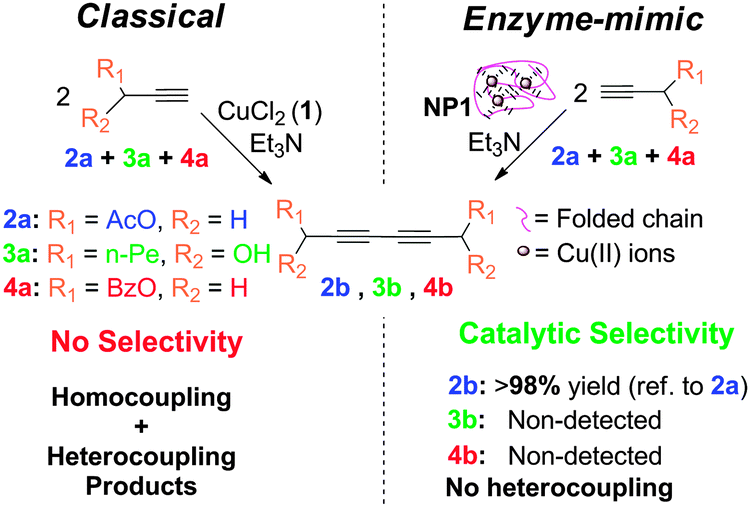 | ||
| Fig. 31 Schematic illustration of the catalytic selectivity showed by SCNPs synthesized via Cu2+-mediated intra-chain cross-linking during alkyne homocoupling experiments. Reprinted with permission from ref. 13. | ||
In addition, catalytic organo(bi)metallic SCNPs containing Rh(I), Ir(I) and Ni(0) have been reported by Lemcoff and coworkers.95 These SCNPs were found to be catalytically active in several reactions and the crowded environment provided by the folded/collapsed polymer precursor was suggested to provide novel catalytic performance. It was postulated by these authors that the close proximity of the catalytic centres led to distinctive reactivity compared to the isolated metallic complexes.
3.3. Sensing
Fluorescent polynorbornene-based SCNPs were reported by Palmans and coworkers93 based on the intra-chain self-assembly in a THF–methylcyclohexane (MCH) mixture of polynorbornene precursors containing bipyridine substituted BTA units (BiPy-BTAs). An increase in the green fluorescent intensity at 520 nm was observed with decreasing solvent polarity and increased degree of BiPy-BTA functionalization. An amount of BiPy-BTA of 12 mol% was found to be optimum to promote the intramolecular self-assembly and avoid the presence of a significant amount of multi-aggregates. Owing to the affinity of the bipyridine moieties towards metal ions such as Cu(II), these SCNPs were found to be efficient sensors for these metal ions, due to the strong quenching of nanoparticle fluorescence upon metal binding.3.4. Other uses
The reduced hydrodynamic size found upon soft nano-object construction via chain compaction often translates to a reduced intrinsic viscosity compared to the corresponding linear precursor (Fig. 32).132 The use of SCNPs as rheology-improving agents for the melts of thermoplastics,133 elastomeric polymers,51 nanocomposites134,135 and paints,55,86,96,100,101 or as polyelectrolytes with unconventional behaviour,52a has been proposed. Initially, SCNPs were evaluated as sacrificial porogens in microelectronics.43a,b The effect of SCNPs on the dynamics of all-polymer nanocomposites is currently the subject of intense interest.136,137 SCNPs are also promising materials for promoting miscibility in immiscible polymer blends, as revealed by several theoretical138–142 and experimental results.143,144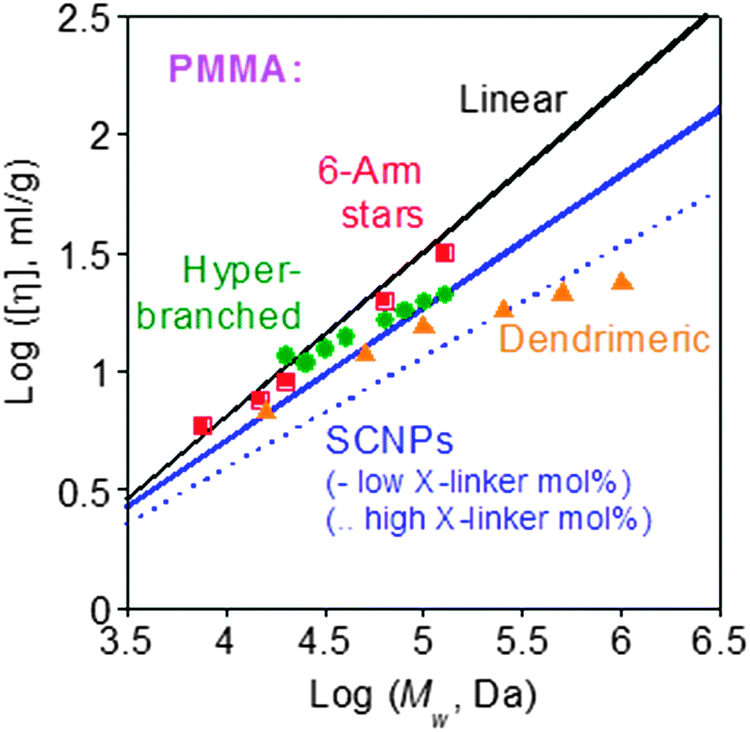 | ||
| Fig. 32 Intrinsic viscosity of tightly cross-linked SCNPs is lower than those of low-functionality stars, hyperbranched polymers and small dendrimers of identical nature and Mw. Reprinted with permission from ref. 132. | ||
Concerning the self-assembly possibilities of amphiphilic single chain nano-objects, different applications have been evaluated. Amphiphilic cyclic block copolymers have been used with success for controlling the feature sizes in block copolymer lithography.30 Single chain tadpoles have been applied as highly-efficient emulsifiers of water in oil emulsions, providing ideal media for the heterogeneous reactions and high long-term emulsion stability.120 In addition, super-particles composed of self-assembled Janus SCTPs showing ultrasonic sensitivity were reported with potential interest for controlled drug release.145
4. Future perspectives
Significant work has been devoted in recent years to enlarging the single chain technology toolbox for the construction of a variety of soft nano-objects via chain compaction (see Section 2). Further introduction of highly-efficient intra-chain cross-linking procedures, both covalent and supramolecular ones, is expected to continue over next years to produce permanent and stimulus responsive single chain nano-objects, respectively. For example, the construction of reversible cyclic block copolymers, supramolecular single chain tadpoles or other more complex unimolecular architectures are still pending. Bioinspired procedures will be presumably adopted as well. In addition the number of synthetic pathways towards both globular and non-globular (IDP-like) nano-object morphologies is expected to increase in next years. Precision synthesis of the corresponding linear precursors and fine-tuning of intra-chain interactions would contribute towards this end. New technological opportunities will be generated by controlling the chain positioning of functional elements in non-natural sequence-controlled polymers, because the solubility, phase transitions, biodegradability and optimal interactions with substrates have been found to be strongly dependent on subtle monomer sequence modifications.15b In addition, more complex nano-objects may require finer nanostructures, so sequence-controlled polymers will offer unique opportunities for preparing even more elaborated constructs. Further work in solving the scale up limitation often associated with the synthesis of gram quantities of well-defined single chain nano-objects is required.Visualization of single chain nano-objects constructed via chain compaction with atomic resolution continues to be a great challenge, both in solution and in the solid state. In the latter case, the interactions with the substrate and dewetting and evaporative self-assembly effects can modify, to a large extent, the conformation of these nano-objects compared to their true conformation in solution. In particular, the combination of SANS and SAXS measurements with MD simulations is expected to become a reference methodology for determining the actual morphology of soft nano-objects in solution with high level of confidence. The visualization and characterization of locally compact, but accessible, sites/cavities/zones of these nano-objects is a major target for establishing useful structure–properties relationships.
In addition, endowing soft nano-objects constructed via chain compaction with useful functions will be another field of intense activity. In addition to the potential uses indicated in Section 3, emerging fields, such as those of self-healing materials and complex hybrid biomaterials, could benefit from the advances in single chain technology towards the construction of unimolecular soft nano-objects endowed with useful, autonomous and smart functions.
A future full of possibilities is envisioned for single chain technology in next years.
Acknowledgements
Financial support from the Projects MAT2012-31088 (MINECO) and T-654-13 (GV) is acknowledged. M. G.-B. and A. L.-S. are grateful to the University of the Basque Country for their UPV/EHU pre-doctoral grants.References
- J. T. Goodwin, A. K. Mehta and D. G. Lynn, Acc. Chem. Res., 2012, 45, 2189 CrossRef CAS PubMed.
- K. A. Dill and J. L. MacCallum, Science, 2012, 338, 1042 CrossRef CAS PubMed.
- C. M. Dobson, Nature, 2003, 426, 884 CrossRef CAS PubMed.
- (a) M. K. Aiertza, I. Odriozola, G. Cabanero, H.-J. Grande and I. Loinaz, Cell. Mol. Life Sci., 2012, 69, 337 CrossRef CAS PubMed; (b) I. Odriozola, M. K. Aiertza, G. Cabanero, H. J. Grande and I. Loinaz, in Handbook of harnessing biomaterials in nanomedicine: preparation, toxicity, and applications, ed. D. Peer, Pan Standford Publishing, Singapore, 2012, ch. 2 Search PubMed.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2012, 33, 958 CrossRef CAS PubMed.
- (a) A. Sanchez-Sanchez, I. Perez-Baena and J. A. Pomposo, Molecules, 2013, 18, 3339 CrossRef CAS PubMed; (b) A. Sanchez-Sanchez and J. A. Pomposo, Part. Part. Syst. Charact., 2014, 31, 11 CrossRef CAS PubMed; (c) J. A. Pomposo, in Micelles: Structural biochemistry, formation and functions & usage, ed. D. Bradburn and T. Bittinger, Nova Science Publishers, New York, 2013, ch. 6 Search PubMed.
- L. Li, K. Raghupathi, C. Song, P. Prasad and S. Thayumanavan, Chem. Commun., 2014, 50, 13417 RSC.
- (a) C. K. Lyon, A. Prasher, A. M. Hanlon, B. T. Tuten, C. A. Tooley, P. G. Frank and E. B. Berda, Polym. Chem., 2015, 6, 181 RSC; (b) M. Huo, N. Wang, T. Fang, M. Sun, Y. Wei and J. Yuan, Polymer, 2015 DOI:10.1016/j.polymer.2015.04.011.
- M. Artar, E. Huerta, E. W. Meijer and A. R. A. Palmans, in Sequence-controlled polymers: synthesis, self-assembly, and properties, ed. J.-F. Lutz, T. Y. Meyer, M. Ouchi and M. Sawamoto, ACS Symp. Series, American Chemical Society, Washington, DC, 2014, ch. 21 Search PubMed.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742 CrossRef CAS PubMed.
- I. Perez-Baena, F. Barroso-Bujans, U. Gasser, A. Arbe, A. J. Moreno, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2013, 2, 775 CrossRef CAS.
- E. Huerta, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Angew. Chem., Int. Ed., 2013, 52, 2906 CrossRef CAS PubMed.
- A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2014, 3, 439 CrossRef CAS.
- J. A. Pomposo, Polym. Int., 2014, 63, 589 CrossRef CAS PubMed.
- (a) M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917 CrossRef CAS PubMed; (b) J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- M. Trabi and D. J. Craik, Trends Biochem. Sci., 2002, 27, 132 CrossRef CAS.
- R. Burman, S. Gunasekera, A. A. Strömstedt and U. Göransson, J. Nat. Prod., 2014, 77, 724 CrossRef CAS PubMed.
- Cyclic Polymers, ed. J. A. Semlyon, Kluwer Academic, Dordrecht, The Netherlands, 2nd edn, 2000 Search PubMed.
- Y. Inoue, P. Kuad, Y. Okumura, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2007, 129, 6396 CrossRef CAS PubMed.
- J. Willenbacher, B. V. K. J. Schmidt, D. Schulze-Suenninghausen, O. Altintas, B. Luy, G. Delaittre and C. Barner-Kowollik, Chem. Commun., 2014, 50, 7056 RSC.
- J. Willenbacher, O. Altintas, P. W. Roesky and C. Barner-Kowollik, Macromol. Rapid Commun., 2014, 35, 45 CrossRef CAS PubMed.
- (a) O. Altintas, P. Gerstel, N. Dingenouts and C. Barner-Kowollik, Chem. Commun., 2010, 46, 6291 RSC; (b) D. Danilov, C. Barner-Kowollik and W. Wenzel, Chem. Commun., 2015, 51 10.1039/C4CC10243F.
- O. Altintas, T. Rudolph and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2566 CrossRef CAS PubMed.
- B. V. K. J. Schmidt, N. Fechler, J. Falkenhagen and J.-F. Lutz, Nat. Chem., 2011, 3, 234 CrossRef CAS PubMed.
- M. Zamfir, P. Theato and J.-F. Lutz, Polym. Chem., 2012, 3, 1796 RSC.
- Z. Ge, Y. Zhou, J. Xu, H. Liu, D. Chen and S. Liu, J. Am. Chem. Soc., 2009, 131, 1628 CrossRef CAS PubMed.
- S. Honda, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 10251 CrossRef CAS PubMed.
- B. Zhang, H. Zhang, Y. Li, J. N. Hoskins and S. M. Grayson, ACS Macro Lett., 2013, 2, 845 CrossRef CAS.
- C.-U. Lee, L. Lu, J. Chen, J. C. Garno and D. Zhang, ACS Macro Lett., 2013, 2, 436 CrossRef CAS.
- J. E. Poelma, K. Ono, D. Miyajima, T. Aida, K. Satoh and C. J. Hawker, ACS Nano, 2012, 6, 10845 CAS.
- A. Touris and N. Hadjichristidis, Macromolecules, 2011, 44, 1969 CrossRef CAS.
- M. Schappacher and A. Deffieux, Science, 2008, 319, 1512 CrossRef CAS PubMed.
- H. Oike, H. Imaizumi, T. Mouri, Y. Yoshioka, A. Uchibori and Y. Tezuka, J. Am. Chem. Soc., 2000, 122, 9592 CrossRef CAS.
- Y. Tezuka, A. Tsuchitani, Y. Yoshioka and H. Oike, Macromolecules, 2003, 36, 65 CrossRef CAS.
- N. Sugai, H. Heguri, K. Ohta, Q. Meng, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 14790 CrossRef CAS PubMed.
- (a) Y. Tezuka and K. Fujiyama, J. Am. Chem. Soc., 2005, 127, 6266 CrossRef CAS PubMed; (b) N. Sugai, H. Heguri, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2011, 133, 19694 CrossRef CAS PubMed.
- O. Shishkan, M. Zamfir, M. A. Gauthier, H. G. Börner and J.-F. Lutz, Chem. Commun., 2014, 50, 1570 RSC.
- O. Altintas, E. Lejeune, P. Gerstel and C. Barner-Kowollik, Polym. Chem., 2012, 3, 640 RSC.
- O. Altintas, P. Krolla-Sidenstein, H. Gliemann and C. Barner-Kowollik, Macromolecules, 2014, 47, 5877 CrossRef CAS.
- D. E. Lonsdale and M. J. Monteiro, Chem. Commun., 2010, 46, 7945 RSC.
- P. G. Clark, E. N. Guidry, W. Yan Chan, W. E. Steinmetz and R. H. Grubbs, J. Am. Chem. Soc., 2010, 132, 3405 CrossRef CAS PubMed.
- P.-F. Cao, A. Bunha, J. Mangadlao, M. J. Felipe, K. I. Mongcopa and R. Advincula, Chem. Commun., 2012, 48, 12094 RSC.
- (a) D. Mecerreyes, V. Lee, C. J. Hawker, J. L. Hedrick, A. Wursch, W. Volksen, T. Magbitang, E. Huang and R. D. Miller, Adv. Mater., 2001, 13, 204 CrossRef CAS; (b) K. S. Park, D. Y. Kim, S. K. Choi and D. H. Suh, Jpn. J. Appl. Phys., 2003, 42, 3877 CrossRef CAS; (c) J. Jiang and S. Thayumanavan, Macromolecules, 2005, 38, 5886 CrossRef CAS.
- E. Harth, B. Van Horn, V. Y. Lee, D. S. Germack, C. P. Gonzales, R. D. Miller and C. J. Hawker, J. Am. Chem. Soc., 2002, 124, 8653 CrossRef CAS PubMed.
- A. E. Cherian, F. C. Sun, S. S. Sheiko and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11350 CrossRef CAS PubMed.
- Y. Bai, H. Xing, G. A. Vincil, J. Lee, E. J. Henderson, Y. Lu, N. G. Lemcoff and S. C. Zimmerman, Chem. Sci., 2014, 5, 2862 RSC.
- T. A. Croce, S. K. Hamilton, M. L. Chen, H. Muchalski and E. Harth, Macromolecules, 2007, 40, 6028 CrossRef CAS.
- C. T. Adkins, H. Muchalski and E. Harth, Macromolecules, 2009, 42, 5786 CrossRef CAS.
- J. N. Dobish, S. K. Hamilton and E. Harth, Polym. Chem., 2012, 3, 857 RSC.
- A. Ruiz de Luzuriaga, N. Ormategui, H. J. Grande, I. Odriozola, J. A. Pomposo and I. Loinaz, Macromol. Rapid Commun., 2008, 29, 1156 CrossRef CAS PubMed.
- L. Oria, R. Aguado, J. A. Pomposo and J. Colmenero, Adv. Mater., 2010, 22, 3038 CrossRef CAS PubMed.
- (a) A. Ruiz de Luzuriaga, I. Perez-Baena, S. Montes, I. Loinaz, I. Odriozola, I. García and J. A. Pomposo, Macromol. Symp., 2010, 296, 303 CrossRef CAS PubMed; (b) P. Khanjani, I. Pérez-Baena, L. Boruga and J. A. Pomposo, Macromol. Symp., 2012, 321–322, 145 CrossRef CAS PubMed.
- H. Cengiz, B. Aydogan, S. Ates, E. Acikalin and Y. Yagci, Des. Monomers Polym., 2011, 14, 68 Search PubMed.
- N. Ormategui, I. Garcia, D. Padro, G. Cabanero, H. J. Grande and I. Loinaz, Soft Matter, 2012, 8, 734 RSC.
- I. Perez-Baena, I. Loinaz, D. Padro, I. Garcia, H. J. Grande and I. Odriozola, J. Mater. Chem., 2010, 20, 6916 RSC.
- J. E. F. Radu, L. Novak, J. F. Hartmann, N. Beheshti, A.-L. Kjoniksen, B. Nyström and J. Borbély, Colloid Polym. Sci., 2008, 286, 365 CAS.
- A. Sanchez-Sanchez and J. A. Pomposo, J. Nanomater., 2015, 723492 Search PubMed.
- J. B. Beck, K. L. Killops, T. Kang, K. Sivanandan, A. Bayles, M. E. Mackay, K. Wooley and C. J. Hawker, Macromolecules, 2009, 42, 5629 CrossRef CAS PubMed.
- P. Wang, H. Pu and M. Jin, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5133 CrossRef CAS PubMed.
- P. Wang, H. Pu, J. Ge, M. Jin, H. Pan, Z. Chang and D. Wan, Mater. Lett., 2014, 132, 102 CrossRef CAS PubMed.
- B. Zhu, J. Ma, Z. Li, J. Hou, X. Cheng, G. Qian, P. Liu and A. Hu, J. Mater. Chem., 2011, 21, 2679 RSC.
- B. Zhu, G. Qian, Y. Xiao, S. Deng, M. Wang and A. Hu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5330 CrossRef CAS PubMed.
- G. Qian, B. Zhu, Y. Wang, S. Deng and A. Hu, Macromol. Rapid Commun., 2012, 33, 1393 CrossRef CAS PubMed.
- B. Zhu, S. Sun, Y. Wang, S. Deng, G. Qian, M. Wang and A. Hu, J. Mater. Chem. C, 2013, 1, 580 RSC.
- X. Jiang, H. Pu and P. Wang, Polymer, 2011, 52, 3597 CrossRef CAS PubMed.
- G. Li, F. Tao, L. Wang, Y. Li and R. Bai, Polymer, 2014, 55, 3696 CrossRef CAS PubMed.
- A. Sanchez-Sanchez, I. Asenjo-Sanz, L. Buruaga and J. A. Pomposo, Macromol. Rapid Commun., 2012, 33, 1262 CrossRef CAS PubMed.
- P. T. Dirlam, H. J. Kim, K. J. Arrington, W. J. Chung, R. Sahoo, L. J. Hill, P. J. Costanzo, P. Theato, K. Char and J. Pyun, Polym. Chem., 2013, 4, 3765 RSC.
- D. Chao, X. Jia, B. Tuten, C. Wang and E. B. Berda, Chem. Commun., 2013, 49, 4178 RSC.
- A. Sanchez-Sanchez, S. Akbari, A. Etxeberria, A. Arbe, U. Gasser, A. J. Moreno, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2013, 2, 491 CrossRef CAS.
- A. Sanchez-Sanchez, S. Akbari, A. J. Moreno, F. Lo Verso, A. Arbe, J. Colmenero and J. A. Pomposo, Macromol. Rapid Commun., 2013, 34, 1681 CrossRef CAS PubMed.
- A. J. Moreno, F. Lo Verso, A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, Macromolecules, 2013, 46, 9748 CrossRef CAS.
- C. F. Hansell, A. Lu, J. P. Patterson and R. K. O'Reilly, Nanoscale, 2014, 6, 4102 RSC.
- J. Willenbacher, K. N. R. Wuest, J. O. Mueller, M. Kaupp, H.-A. Wagenknecht and C. Barner-Kowollik, ACS Macro Lett., 2014, 3, 574 CrossRef CAS.
- I. Perez-Baena, I. Asenjo-Sanz, A. Arbe, A. J. Moreno, F. Lo Verso, J. Colmenero and J. A. Pomposo, Macromolecules, 2014, 47, 8270 CrossRef CAS.
- E. H. H. Wong, S. J. Lam, E. Nam and G. G. Qiao, ACS Macro Lett., 2014, 3, 524 CrossRef CAS.
- M. Seo, B. J. Beck, J. M. J. Paulusse, C. J. Hawker and S. Y. Kim, Macromolecules, 2008, 41, 6413 CrossRef CAS.
- E. J. Foster, E. B. Berda and E. W. Meijer, J. Am. Chem. Soc., 2009, 131, 6964 CrossRef CAS PubMed.
- E. B. Berda, E. J. Foster and E. W. Meijer, Macromolecules, 2010, 43, 1430 CrossRef CAS.
- E. J. Foster, E. B. Berda and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 118 CrossRef CAS PubMed.
- P. J. M. Stals, M. A. J. Gillissen, R. Nicolay, A. R. A. Palmans and E. W. Meijer, Polym. Chem., 2013, 4, 2584 RSC.
- T. Mes, R. van der Weegen, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 5085 CrossRef CAS PubMed.
- N. Hosono, M. A. J. Gillissen, Y. Li, S. S. Sheiko, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 501 CrossRef CAS PubMed.
- M. A. J. Gillissen, T. Terashima, E. W. Meijer, A. R. A. Palmans and I. K. Voets, Macromolecules, 2013, 46, 4120 CrossRef CAS.
- N. Hosono, P. J. M. Stals, A. R. A. Palmans and E. W. Meijer, Chem. – Asian J., 2014, 9, 1099 CrossRef CAS PubMed.
- P. J. M. Stals, M. A. J. Gillissen, T. F. E. Paffen, T. F. A. de Greef, P. Lindner, E. W. Meijer, A. R. A. Palmans and I. K. Voets, Macromolecules, 2014, 47, 2947 CrossRef CAS.
- N. Hosono, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2014, 50, 7990 RSC.
- E. Huerta, B. van Genabeek, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Macromol. Rapid Commun., 2014, 35, 1320 CrossRef CAS PubMed.
- E. A. Appel, J. Dyson, J. del Barrio, Z. Walsh and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 4185 CrossRef CAS PubMed.
- E. A. Appel, J. del Barrio, J. Dyson, L. Isaacs and O. A. Scherman, Chem. Sci., 2012, 3, 2278 RSC.
- T. Akagi, P. Piyapakorn and M. Akashi, Langmuir, 2012, 28, 5249 CrossRef CAS PubMed.
- P. Piyapakorn, T. Akagi, M. Hachisuka, H. Matsuoka and M. Akashi, Macromolecules, 2013, 46, 6187 CrossRef CAS.
- M. A. J. Gillissen, I. K. Voets, E. W. Meijer and A. R. A. Palmans, Polym. Chem., 2012, 3, 3166 RSC.
- S. Mavila, C. E. Diesendruck, S. Linde, L. Amir, R. Shikler and N. G. Lemcoff, Angew. Chem., Int. Ed., 2013, 52, 5767 CrossRef CAS PubMed.
- S. Mavila, I. Rozenberg and N. G. Lemcoff, Chem. Sci., 2014, 5, 4196 RSC.
- J. K. Mistry and M. R. Van De Mark, J. Coat. Technol. Res., 2013, 10, 453 CrossRef CAS.
- M. Chen, C. Riddles and M. R. Van De Mark, Colloid Polym. Sci., 2013, 291, 2893 CAS.
- M. Chen, C. J. Riddles and M. R. Van De Mark, Langmuir, 2013, 29, 14034 CrossRef CAS PubMed.
- C. J. Riddles, W. Zhao, H.-J. Hu, M. Chen and M. R. Van De Mark, Polymer, 2014, 55, 48 CrossRef CAS PubMed.
- A. M. Natu and M. R. Van De Mark, Prog. Org. Coat., 2015, 81, 35 CrossRef CAS PubMed.
- J. K. Mistry, A. M. Natu and M. R. Van De Mark, J. Appl. Polym. Sci. DOI:10.1002/app.40916.
- T. Terashima, T. Sugita, K. Fukae and M. Sawamoto, Macromolecules, 2014, 47, 589 CrossRef CAS.
- B. S. Murray and D. A. Fulton, Macromolecules, 2011, 44, 7242 CrossRef CAS.
- D. E. Whitaker, C. S. Mahon and D. A. Fulton, Angew. Chem., Int. Ed., 2013, 52, 956 CrossRef CAS PubMed.
- J. He, L. Tremblay, S. Lacelle and Y. Zhao, Soft Matter, 2011, 7, 2380 RSC.
- W. Fan, X. Tong, Q. Yan, S. Fu and Y. Zhao, Chem. Commun., 2014, 50, 13492 RSC.
- B. T. Tuten, D. Chao, C. K. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068 RSC.
- A. Sanchez-Sanchez, D. A. Fulton and J. A. Pomposo, Chem. Commun., 2014, 50, 1871 RSC.
- P. G. Frank, B. T. Tuten, A. Prasher, D. Chao and E. B. Berda, Macromol. Rapid Commun., 2014, 35, 249 CrossRef CAS PubMed.
- (a) J. A. Pomposo, I. Perez-Baena, F. Lo Verso, A. J. Moreno, A. Arbe and J. Colmenero, ACS Macro Lett., 2014, 3, 767 CrossRef CAS; (b) J. A. Pomposo, I. Perez-Baena, L. Buruaga, A. Alegría, A. J. Moreno and J. Colmenero, Macromolecules, 2011, 44, 8644 CrossRef CAS.
- F. Lo Verso, J. A. Pomposo, J. Colmenero and A. J. Moreno, Soft Matter, 2014, 10, 4813 RSC.
- F. Lo Verso, J. A. Pomposo, J. Colmenero and A. J. Moreno, Soft Matter, 2015, 11, 1369 RSC.
- J. Tao and G. Liu, Macromolecules, 1997, 30, 2408 CrossRef CAS.
- G. Njikang, G. Liu and S. A. Curda, Macromolecules, 2008, 41, 5697 CrossRef CAS.
- Y. Kim, J. Pyun, J. M. J. Fréchet, C. J. Hawker and C. W. Frank, Langmuir, 2005, 21, 10444 CrossRef CAS PubMed.
- L. Cheng, G. Hou, J. Miao, D. Chen, M. Jiang and L. Zhu, Macromolecules, 2008, 41, 8159 CrossRef CAS.
- O. Altintas, J. Willenbacher, K. N. R. Wuest, K. K. Oehlenschlaeger, P. Krolla-Sidenstein, H. Gliemann and C. Barner-Kowollik, Macromolecules, 2013, 46, 8092 CrossRef CAS.
- J. Wen, L. Yuan, Y. Yang, L. Liu and H. Zhao, ACS Macro Lett., 2013, 2, 100 CrossRef CAS.
- J. Wen, J. Zhang, Y. Zhang, Y. Yang and H. Zhao, Polym. Chem., 2014, 5, 4032 RSC.
- F. Xu, Z. Fang, D. Yang, Y. Gao, H. Li and D. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 6717 CAS.
- W. Li, C.-H. Kuo, I. Kanyo, S. Thanneeru and J. He, Macromolecules, 2014, 47, 5932 CrossRef CAS.
- K. Roy and J.-F. Lutz, J. Am. Chem. Soc., 2014, 136, 12888 CrossRef PubMed.
- J. Romulus and M. Weck, Macromol. Rapid Commun., 2013, 34, 1518 CrossRef CAS PubMed.
- J. Lu, N. ten Brummlhuis and M. Weck, Chem. Commun., 2014, 50, 6225 RSC.
- N. Nasongkla, B. Chen, N. Macaraeg, M. E. Fox, J. M. J. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842 CrossRef CAS PubMed.
- S. K. Hamilton and E. Harth, ACS Nano, 2009, 3, 402 CrossRef CAS PubMed.
- G. Njiang, G. Liu and L. Hong, Langmuir, 2012, 27, 7176 CrossRef PubMed.
- A. Tamura, M. Oishi and Y. Nagaski, Biomacromolecules, 2009, 10, 1818 CrossRef CAS PubMed.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227 CrossRef CAS PubMed.
- H. Kim, T. Uto, T. Akagi, M. Baba and M. Akashi, Adv. Funct. Mater., 2010, 20, 3925 CrossRef CAS PubMed.
- G. Wulff, B.-O. Chong and U. Kolb, Angew. Chem., Int. Ed., 2006, 45, 2955 CrossRef CAS PubMed.
- I. Perez-Baena, A. J. Moreno, J. Colmenero and J. A. Pomposo, Soft Matter, 2014, 10, 9454 RSC.
- M. E. Mackay, T. T. Dao, A. Tuteja, D. L. Ho, B. V. Horn, H.-C. Kim and C. J. Hawker, Nat. Mater., 2003, 2, 762 CrossRef CAS PubMed.
- M. E. Mackay, A. Tuteja, P. M. Duxbury, C. J. Hawker, B. V. Horn, Z. Guan, G. Chen and R. S. Krishnan, Science, 2006, 31, 1740 CrossRef PubMed.
- A. Tuteja, P. M. Duxbury and M. E. Mackay, Macromolecules, 2007, 40, 9427 CrossRef CAS.
- D. Bhowmik, J. A. Pomposo, F. Juranyi, V. García-Sakai, M. Zamponi, Y. Su, A. Arbe and J. Colmenero, Macromolecules, 2014, 47, 304 CrossRef CAS.
- D. Bhowmik, J. A. Pomposo, F. Juranyi, V. Garcia Sakai, M. Zamponi, A. Arbe and J. Colmenero, Macromolecules, 2014, 47, 3005 CrossRef CAS.
- J. A. Pomposo, A. Ruiz de Luzuriaga, A. Etxeberria and J. Rodriguez, Phys. Chem. Chem. Phys., 2008, 10, 650 RSC.
- A. Ruiz de Luzuriaga, A. Etxeberria, J. Rodriguez and J. A. Pomposo, Polym. Adv. Technol., 2008, 19, 756 CrossRef CAS PubMed.
- A. Ruiz de Luzuriaga, H. Grande and J. A. Pomposo, J. Nano Res., 2008, 2, 105 CrossRef CAS.
- S. Montes, H. Grande, A. Etxeberria and J. A. Pomposo, J. Nano Res., 2009, 6, 123 CrossRef CAS.
- A. Ruiz de Luzuriaga, H. J. Grande and J. A. Pomposo, J. Chem. Phys., 2009, 130, 084905 CrossRef PubMed.
- J. A. Pomposo, A. Ruiz de Luzuriaga, I. Garcia, A. Etxeberria and J. Colmenero, Macromol. Rapid Commun., 2011, 32, 573 CrossRef CAS PubMed.
- S. Y. Kim, K. S. Schweizer and C. F. Zukoski, Phys. Rev. Lett., 2011, 107, 225504 CrossRef.
- F. Zhou, M. Xie and D. Chen, Macromolecules, 2014, 47, 365 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |