
Low-temperature aqueous-phase reforming of ethanol on bimetallic PdZn catalysts†
Haifeng
Xiong
a,
Andrew
DeLaRiva
a,
Yong
Wang
bc and
Abhaya K.
Datye
*a
aDepartment of Chemical & Biological Engineering and Center for Micro-engineered Materials, University of New Mexico, Albuquerque, New Mexico 87131, USA. E-mail: datye@unm.edu; Fax: +1 505 277 1024; Tel: +1 505 277 0477
bInstitute for Integrated Catalysis, Pacific Northwest National Laboratory, Richland, WA 99352, USA
cThe Gene & Linda Voiland School of Chemical Engineering and Bioengineering, Washington State University, Pullman, WA 99164-2710, USA
First published on 28th August 2014
Abstract
Bimetallic PdZn catalysts supported on carbon black (CB) and carbon nanotubes (CNTs) were found to be selective for CO-free H2 production from ethanol at low temperature (250 °C). On Pd, the H2 yield was low (~0.3 mol H2/mol ethanol reacted) and the CH4/CO2 ratio was high (~1.7). Addition of Zn to Pd formed the intermetallic PdZnβ phase (atomic ratio of Zn to Pd is 1) with increased H2 yield (~1.9 mol H2/mol ethanol reacted) and CH4/CO2 ratio of <1. The higher H2 yield and low CH4 formation was related to the improved dehydrogenation activity of the L10 PdZnβ phase. The TOF increased with particle size and the CNTs provided the most active and selective catalysts, which may be ascribed to pore-confinement effects. Furthermore, no significant changes in either the supports or the PdZnβ particles was found after aqueous-phase reforming (APR) indicating that the metal nanoparticles and the carbon support are hydrothermally stable in the aqueous phase at elevated temperatures and pressures (>200 °C, 65 bar). No CO was detected for all the catalysts performed in aqueous-phase reaction, indicating that both monometallic Pd and bimetallic PdZn catalysts have high water-gas shift activity during APR. However, the yield of H2 is considerably lower than the theoretical value of 6 H2 per mole ethanol which is due to the presence of oxygenated products and methane on the PdZn catalysts.
1. Introduction
H2 is a clean energy carrier for future energy systems, especially for vehicles with fuel cells.1 The conventional approach to produce H2 is steam reforming (SR) which is energy intensive since it requires vaporization of water and the reaction is carried out at high temperatures (>400 °C).2 The high temperature SR process can also lead to the formation of coke on the catalyst, which results in catalyst deactivation.3,4 Aqueous-phase reforming (APR), on the other hand, can be carried out at low temperatures (<300 °C).5 Compared to SR in the gas phase, APR exhibits several advantages such as reduced energy consumption and low amounts of CO due to the water gas shift (WGS) reaction.6In this study we focus on bioethanol which is a renewable resource that can be obtained from biomass. There have been numerous studies on gas phase ethanol steam reforming (SR), showing that a high yield of hydrogen can be obtained, but only at elevated temperatures (>400 °C).7 APR is conducted at lower temperatures, and the conversion of oxygenated hydrocarbons such as methanol, ethylene glycol and glycerol has been reported, but there is little reported on the APR of bioethanol.8 A major consideration for APR reactions is the limited hydrothermal stability of conventional supports in liquid water at elevated temperatures (>200 °C).9 Roy et al. found by XPS that a nickel oxide layer had formed on the nickel/alumina catalyst and the γ-Al2O3 support had transformed to boehmite during ethanol APR.10 Likewise, Ravenelle and coworkers found that the transformation of γ-Al2O3 into boehmite caused the sintering of the supported nickel and Pt particles during aqueous phase reactions.11 The development of stable catalytic materials for APR is therefore of great interest.12
Carbon supports have been shown to be hydrothermally stable under conditions used for APR.13,14 A recent study using a carbon-supported ruthenium catalyst showed that the Ru metal phase remained in the reduced state in sub- and supercritical water and the catalyst was stable over long term operation.15 The reaction products included CH4, CO2 and H2 but the ratio of CH4/CO2 was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with a resulting low yield of H2. To achieve a high yield of H2 it is necessary to suppress the formation of CH4 and enhance CO2 formation. Among the noble metal catalysts tested for APR of oxygenates, it was found that Pt shows the highest reactivity, but Pd shows the best selectivity to CO2 formation.8,16 The activity and selectivity of these catalysts can be further improved by alloying the noble metal with components that enhance water gas shift activity. ZnO was found to be a selective support for SR of ethanol on cobalt catalysts,7 but it was not clear whether an alloy was formed during reaction. In the PdZn system it has been shown that the formation of the intermetallic PdZnβ phase (Zn:Pd atomic ratio of 1) was critical for obtaining a high activity and selectivity for methanol steam reforming to produce H2.17 But the stability of the PdZn intermetallic has not been tested under aqueous phase conditions, hence the main objective of this study was to investigate the behavior of the intermetallic PdZn phase for H2 production during ethanol APR.
:1 with a resulting low yield of H2. To achieve a high yield of H2 it is necessary to suppress the formation of CH4 and enhance CO2 formation. Among the noble metal catalysts tested for APR of oxygenates, it was found that Pt shows the highest reactivity, but Pd shows the best selectivity to CO2 formation.8,16 The activity and selectivity of these catalysts can be further improved by alloying the noble metal with components that enhance water gas shift activity. ZnO was found to be a selective support for SR of ethanol on cobalt catalysts,7 but it was not clear whether an alloy was formed during reaction. In the PdZn system it has been shown that the formation of the intermetallic PdZnβ phase (Zn:Pd atomic ratio of 1) was critical for obtaining a high activity and selectivity for methanol steam reforming to produce H2.17 But the stability of the PdZn intermetallic has not been tested under aqueous phase conditions, hence the main objective of this study was to investigate the behavior of the intermetallic PdZn phase for H2 production during ethanol APR.
Two types of carbon supports were used, a commercial carbon black and carbon nanotubes prepared in house. The aim of this study was to: (I) investigate the effect of Zn on the catalytic performance of Pd and (II) the effect of metal particle size and support structure (carbon black and carbon nanotubes) on the performance of the PdZn bimetallic catalysts in the APR of ethanol. We were especially interested in the yield of H2 and the relative CH4/CO2 selectivity of these catalysts as well as the formation of other oxygenated byproducts. As we show in this paper, the addition of Zn enhanced the H2 yield per mole of ethanol reacted and led to CH4/CO2 ratios less than 1. Furthermore, large PdZn particles (>5 nm) provide higher TOFs and carbon nanotubes provide the highest reactivity. No CO is found during APR of ethanol, indicating these catalysts have high WGS reactivity.
2. Experimental
2.1. Catalyst preparation
PdZn bimetallic catalysts supported on carbon black (CB) with different Zn loadings were prepared by co-impregnation. First, the commercial carbon black (Vulcan XC 72R) was functionalized in 50% HNO3 at 90 °C for 14 h, followed by filtration and washing in water till a pH = 7 was reached in the filtrate. The carbon black was next dried at 120 °C for 12 h. The catalysts were prepared via incipient wetness impregnation in two steps. The Pd and Zn nitrates were used as catalyst precursors. The monometallic catalyst contained 5 wt.% Pd while the bimetallic catalyst contained the same loading of Pd but we also added 3.1 wt.% Zn and 5.0 wt.% Zn to study the role of Zn/Pd ratio (atomic ratio 1:1 and 1:1.6). Subsequently, the sample was kept at room temperature for 12 h in air, followed by drying at 120 °C for 12 h. Then the sample was pre-reduced at 500 °C for 2 h in a flow of 7% H2/N2. The bimetallic catalysts are denoted as PdZn1 and PdZn1.6 indicating the atomic ratio of Zn/Pd in the catalysts. The catalyst name also shows the support, CB – carbon black and CNT – carbon nanotubes. To study the role of crystallite size, we prepared 5 wt.% Pd/CB catalysts with different Pd particle sizes. The 5 wt.% Pd/CB catalysts with small Pd particle size (Pd/CB) was obtained by the impregnation of Pd nitrate on carbon black, followed by H2 reduction at 250 °C for 2 h in a flow of 7% H2/N2. The 5 wt.% Pd/CB catalyst with large Pd particle size (Pd/CB–H) was obtained by a reduction at 500 °C for 12 h in a flow of 7% H2/N2 after the impregnation.
To study the effect of particle size on the carbon nanotubes (CNTs) supported catalysts we prepared two bimetallic PdZn catalysts with Pd loadings of 1 and 5 wt.% and a Zn to Pd atomic ratio of 1. Carbon nanotubes were prepared by chemical vapor deposition as described in a previous study.18 The pore size of obtained CNTs is 5–15 nm and they were functionalized in 50% HNO3 at 90 °C for 8 h. The preparation procedure for these catalysts was similar to that for PdZn1/CB, described above. These catalysts were labeled as PdZn1/CNT-1 (1 wt.% Pd) and PdZn1/CNT (5 wt.% Pd), respectively.
2.2. Catalyst characterization
N2 physisorption experiments were performed using a Quantachrome Autosorb-1 instrument. Prior to the experiment, the sample was outgassed at 200 °C for 6 h. The surface area was obtained using the BET method using adsorption data over a relative pressure range from 0.05 to 0.30. The total pore volumes were calculated from the amount of N2 adsorbed at a relative pressure of 0.99. X-ray powder diffraction (XRD) patterns for the catalysts were recorded with a Scintag Pad V diffractometer using Cu Kα radiation and a Ni filter. For some of the catalysts we also used a Rigaku Smart Lab XRD. Scanning transmission electron microscopy (STEM) was carried out in a JEOL 2010F microscope. The powders were deposited on holey carbon support films after being dispersed in ethanol. An electron probe, diameter of 0.2 nm, was scanned over the specimen, and electrons scattered at high angles were collected to form the image in HAADF (High Angle Annular Dark Field) mode. The histogram of particle size distribution (Pd and PdZn) was obtained by counting 200–300 particles in the STEM images.We tried to use H2 chemisorption at 100 °C to count the number of surface Pd sites in these catalysts. The numbers were not consistent with the dispersion obtained from TEM images because of the weak chemisorption on the PdZn catalysts. CO chemisorption is not recommended for Pd catalysts because of variations in the ratio of bridge bonded and linearly adsorbed CO with particle size and Zn content. Since it is difficult to count sites in Pd and PdZn catalysts via chemisorption, we used CO oxidation reactivity to determine the number of Pd sites in these catalysts. The product analysis during CO oxidation was carried out on a Varian CP-4900 Micro-GC utilizing a TCD detector with 20 mg of sample being used for each experiment. The sample was reduced at 250 °C in situ for 2 h in 6% H2/He followed by a cool down in 6% H2/He to room temperature. The samples were then exposed to CO oxidation conditions which used 1.5 mL min−1 CO, 1 mL min−1 O2, and 75 mL min−1 He (~2% CO). The temperature was ramped to 300 °C with a ramp rate of 2 °C min−1 and sampling performed every 3 min. The runs were repeated to ensure reproducible behavior and the reactivity in the second run was used to determine the number of exposed surface Pd atoms as explained in more detail in the ESI.†
In previous work,19 it was shown that PdZn catalysts show higher specific reactivity than Pd caused by a weakening of the CO bond due to added Zn. But these catalysts show rapid deactivation due to the surface oxidation of Zn in the PdZn alloy. After reaching a steady state, the specific reactivity of Pd and PdZn was comparable.19 Our experimental procedure involving two CO oxidation runs to 300 °C ensured that the catalyst had reached its steady state behavior. Hence, we could use the previously reported CO oxidation rates and TOFs on Pd and PdZn to estimate the number of exposed Pd atoms in our supported catalysts (see ESI† for further detail).19 The estimates of numbers of surface atoms obtained from CO oxidation reactivity agreed with the average particle diameters obtained using TEM. Using these results, the turnover frequency (TOF s−1) for aqueous-phase reforming could then be calculated. Furthermore, it should be pointed out that the supports CB, CNT, ZnO/CB and ZnO/CNT are not active for CO oxidation at T < 300 °C.
2.3. Catalyst reactivity during aqueous-phase reforming of ethanol
Reaction studies for aqueous-phase reforming of ethanol were conducted over a temperature range of 200–260 °C and 65 bar pressure (controlled with a back pressure regulator) in an up-flow tubular reactor. Before reaction measurements, the catalyst (0.2 g) was reduced at 250 °C under flowing H2 (20 cm3 (STP) min−1) at atmospheric pressure for 4 h. Subsequently, a feed containing 10 wt.% ethanol in water (0.003 mL min−1) was pumped through the reactor. Before the back pressure regulator, we introduced a stream of Ar (10 cm3 (STP) min−1) to sweep the gas products to the GC. The effluent gas stream was composed of H2, CO2, CH4, C2H4, C2H6, CH3CHO, C2H5OH and was analyzed with an online GC (VARIAN CP-3800 GC) equipped with a FID and a TCD. A gas–liquid separation trap at room temperature (in line before the back pressure regulator) was used to collect the liquid. The liquid included CH3CH2OH, CH3CHO, CH3COOH and CH3COCH3 and the composition was quantified by the GC on the FID using a CP-PoraBOND Q capillary column. All reactions were carried out for 100 h and no deactivation was found over this period. The first data point was recorded after 24 h of reaction by which time steady state behavior was obtained, and the H2 yield was stable after that. The ethanol conversion (X), product selectivity (S) and H2 production were calculated based on the following equations: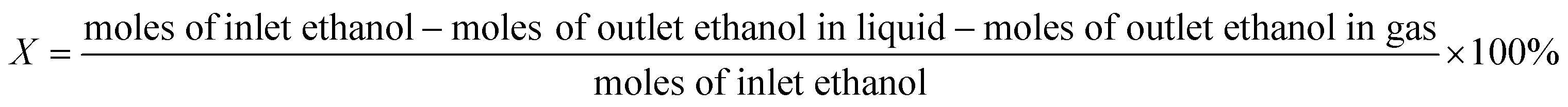 | (1) |
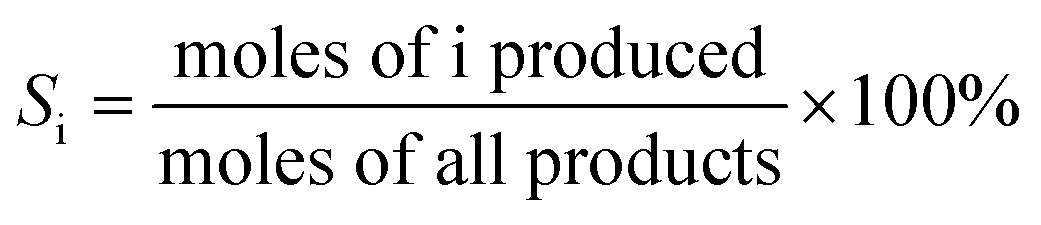 | (2) |
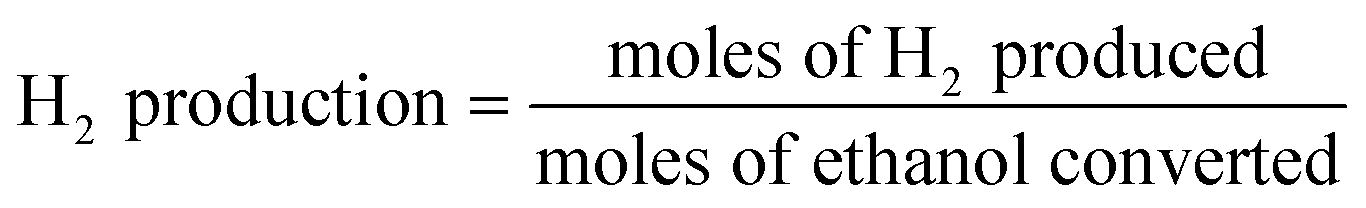 | (3) |
3. Results and discussion
3.1. Catalyst characterization
The N2 physisorption data and the metal dispersion and particle size for the as-prepared catalysts are shown in Table 1. For the PdZn/CB catalysts with different Zn loadings, the BET surface areas were in the range of 160–175 m2 g−1 and no significant change in the pore volumes of the catalysts was found by varying the Zn loading. The PdZn1/CNT shows lower surface area and pore volume in comparison with the bimetallic PdZn/CB catalysts due to the low surface area of the CNT support. As explained in the experimental section and the ESI,† the metal dispersion and particle size was derived by using CO oxidation reactivity.| Sample | Surface area (m2 g−1) | Total pore volume (cm3 g−1) | Dispersionb (%) | Particle size (nm) | ||
|---|---|---|---|---|---|---|
| CO oxidationb | XRD | TEM | ||||
| a All catalysts had a loading of 5 wt.% Pd, except the catalyst in the last row which contained 1 wt.% Pd. b The number of surface Pd sites was obtained from CO oxidation at 185 °C (see ESI) and the particle size was estimated using the formula: d = 100/D, where d is particle size in nm and D is dispersion. | ||||||
| Pd/CB | 175 | 0.72 | 37.6 | 2.7 | 4.1 | 2.5 |
| Pd/CB–H | 170 | 0.7 | 16.0 | 6.3 | 8.7 | 4.7 |
| PdZn1/CB | 161 | 0.78 | 41.0 | 2.4 | 3.8 | 2.9 |
| PdZn1.6/CB | 156 | 0.64 | 33.5 | 3.0 | 4.5 | 3.1 |
| PdZn1/CNT | 95.4 | 0.29 | 4.0 | 25 | 30 | 8.5 |
| PdZn1/CNT-1 | 98.1 | 0.32 | 28.6 | 3.5 | 4.6 | 2.1 |
The XRD patterns of monometallic Pd/CB before and after reaction are shown in Fig. 1. We also show the XRD pattern of the Pd/CB–H catalyst (pretreated at 500 °C for 12 h in a flow of H2/N2) to increase the crystallite sizes, hence it shows a sharp Pd diffraction peak (Fig. 1c). The broad peak seen in the Pd/CB catalyst at 25° corresponds to the (002) plane of the graphitic carbon. The peak at 40.2° comes from metallic Pd and it is very broad due to the small particle size (Fig. 1a). Fig. 1 also shows the XRD pattern after reaction for the Pd/CB indicating modest growth in particle size after reaction (Fig. 1b).
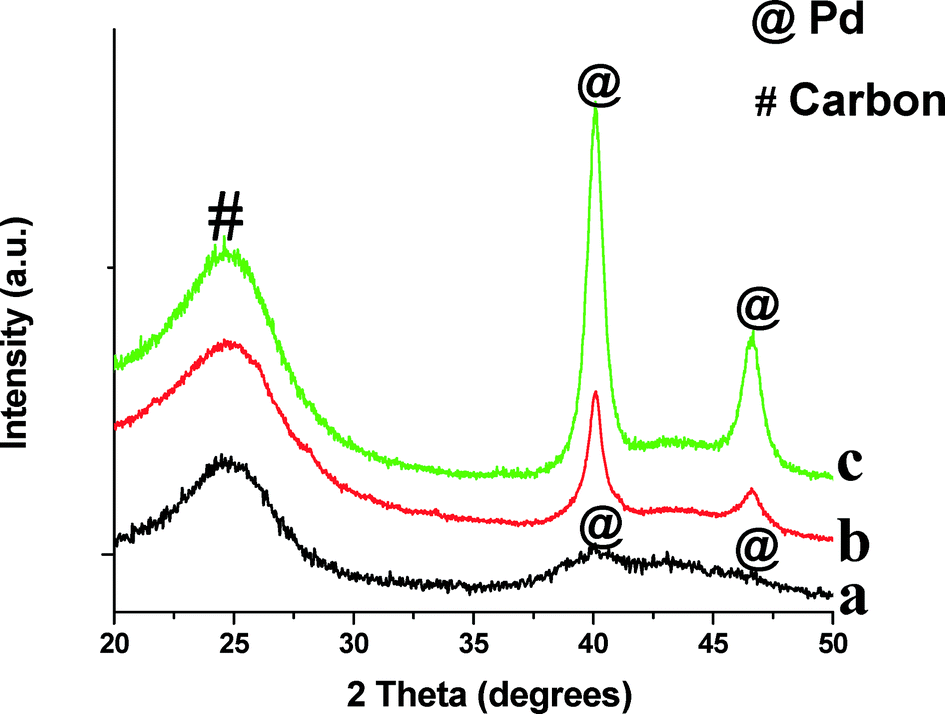 | ||
| Fig. 1 XRD patterns of monometallic Pd catalysts supported on carbon black (CB) before and after ethanol APR reaction: (a) Pd/CB; (b) Pd/CB after APR reaction; (c) Pd/CB–H aged in H2 at 500 °C. | ||
The XRD patterns of bimetallic PdZn catalysts are shown in Fig. 2. For PdZn1/CB (Fig. 2a and insert), the XRD pattern shows two diffraction peaks at 41° and 44° which correspond to the intermetallic L10 PdZnβ phase (Zn:Pd = 1, ICDD 006-0620).20 Increasing the Zn loading to 5 wt.% (Zn:Pd = 1.6) leads to the appearance of crystalline ZnO coexisting with the PdZnβ phase (Fig. 2b and insert). Due to the lower surface area of the CNT support, the bimetallic PdZn1/CNT catalyst with 5 wt.% Pd had larger particles, hence well-defined diffraction peaks of PdZnβ phase can be seen (Fig. 2e). The higher crystallinity of the support leads to the sharper graphite peaks in the CNT supported catalysts (Fig. 2d and e). Since the average particle size was significantly greater than the other catalysts, we also prepared a catalyst with 1 wt.% Pd loading to provide particle sizes comparable to the other supports. The XRD pattern of this catalyst PdZn1/CNT-1 shows broad diffraction peaks of the PdZnβ phase (Fig. 2d) comparable to those seen on the CB support. The low Pd loading enhances the relative intensity of the carbon peaks from the CNT support, therefore we have drawn vertical lines to show clearly the PdZn (111) and those from the CNT support (Fig. S4, ESI† provides a larger figure to show clearly the CNT peaks in relation to PdZn). The XRD patterns of the PdZn catalysts after reaction show very little change confirming the stability of this catalyst (Fig. 2c and f).
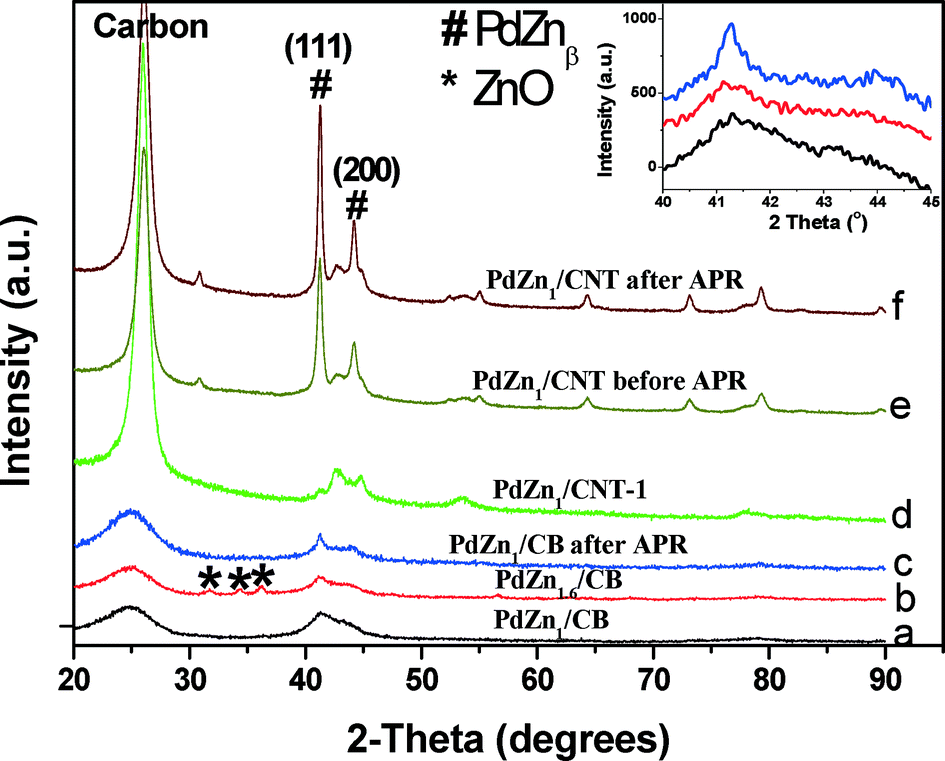 | ||
| Fig. 2 XRD patterns of bimetallic PdZn catalysts supported on different carbons before and after aqueous-phase reforming (APR) of ethanol: (a) PdZn1/CB; (b) PdZn1.6/CB; (c) PdZn1/CB after APR; (d) PdZn1/CNT-1 before APR; (e) PdZn1/CNT before APR; (f) PdZn1/CNT after APR, and the zoom of patterns a–c at 40–45° (insert). | ||
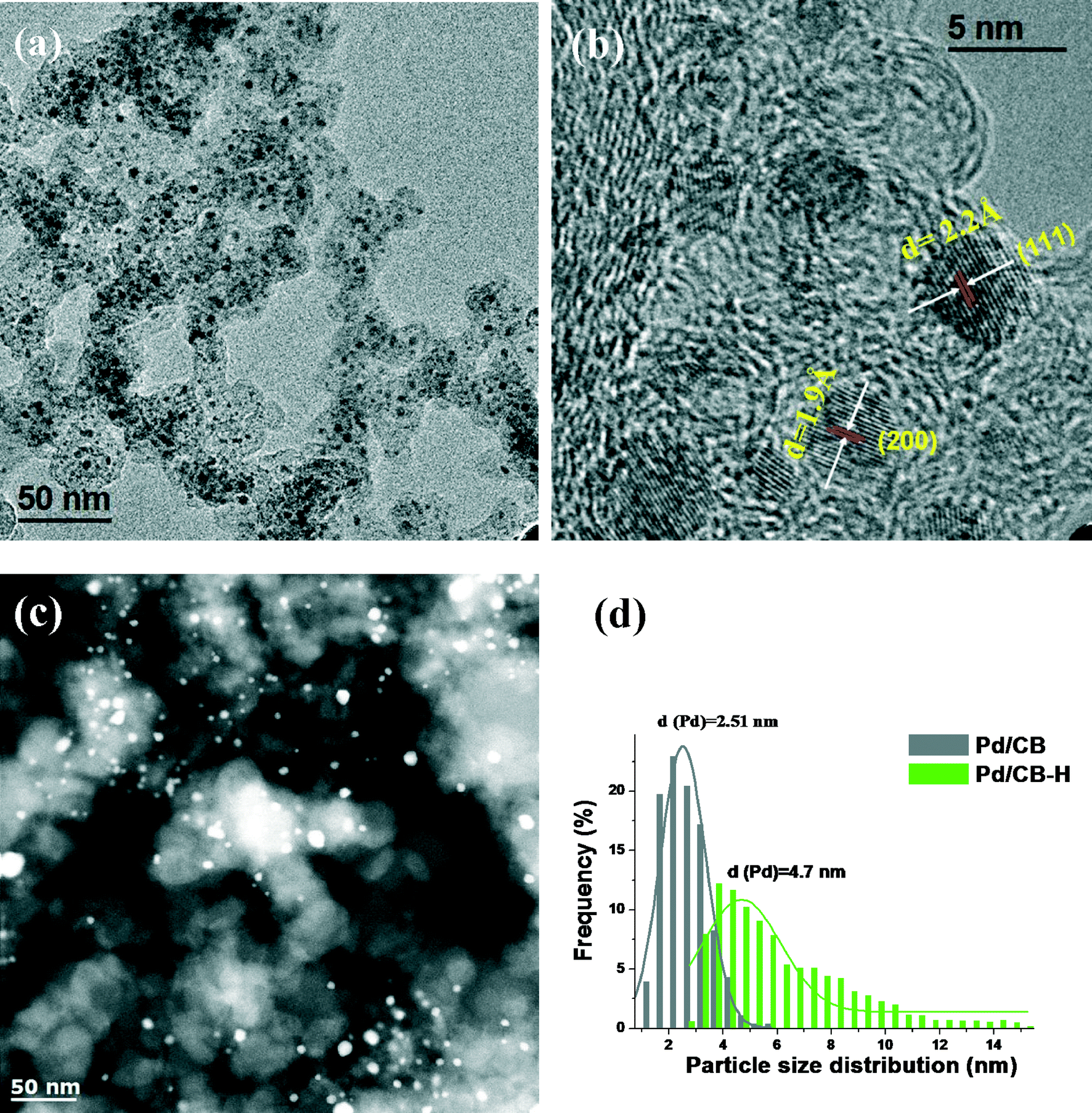 | ||
| Fig. 3 Representative TEM/STEM images of Pd/CB catalysts with different Pd particle sizes: (a) and (b) Pd/CB; (c) Pd/CB–H (pretreated at 500 °C for 12 h in a flow of 7% H2/N2 mixture); (d) particle size distribution diagrams. | ||
TEM images of the bimetallic PdZn/CB catalysts are shown in Fig. 4. The average particle size for PdZn1/CB is 2.88 nm (Fig. 4a and b, insert) and is consistent with the broad XRD peaks (Fig. 2a) and with the dispersion from CO oxidation which was 41%. The HRTEM image of this sample can be indexed to the (200) plane of the PdZnβ phase (Fig. 4b). Keeping the Pd loading at 5 wt.% but increasing the Zn loading to 5 wt.% (PdZn1.6/CB) caused only a small increase in particle size to 3.1 nm (Fig. 4c). The HRTEM image indexes to the (111) and (200) planes of PdZnβ in this catalyst (Fig. 4c, insert). The slight increase in particle size is consistent with a small drop in the dispersion (Table 1). The majority of the particles are smaller than 4 nm in these two catalysts prepared on the carbon black support.
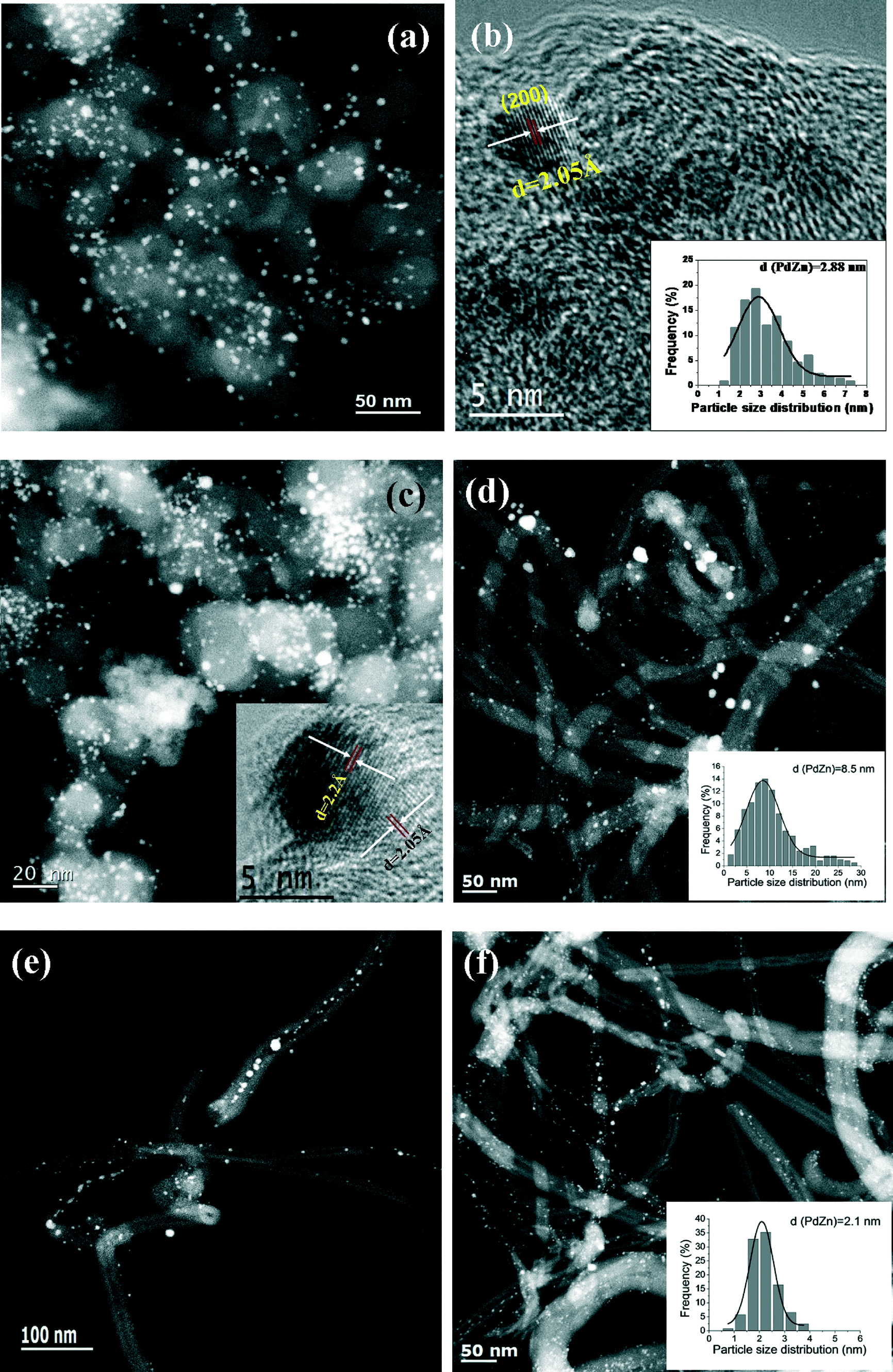 | ||
| Fig. 4 Representative TEM/STEM images of bimetallic PdZn catalysts supported on carbon black and carbon nanotubes: (a) and (b) PdZn1/CB; (c) PdZn1.6/CB; (d) PdZn1/CNT; (e) STEM image of PdZn1/CNT showing a significant fraction of the PdZnβ particles located inside the CNT pores; (f) STEM image and particle size distribution of PdZn1/CNT-1 with 1 wt.% Pd loading. | ||
We also prepared catalysts with similar weight loading with the CNT support. However, because of the lower surface area, the average PdZnβ particle size was much larger than that on the carbon black. For the PdZn1/CNT catalyst with 5 wt.% Pd loading, the STEM image shows that the PdZnβ particle sizes were in the range of 2–30 nm (Fig. 4d, average size = 8.5 nm), which is consistent with the XRD result showing sharp, well defined peaks of the intermetallic PdZnβ phase. We found that the PdZnβ particles were also located inside the CNT pores in this PdZn1/CNT catalyst (Fig. 4e and S5, ESI†). The estimated dispersion based on CO oxidation reactivity suggests a particle size of 25 nm which is much greater than the TEM estimate. We attribute the lower dispersion due to the presence of PdZn particles inside the pores, and the large size of the particles may lead to some pore blocking leading to poor accessibility during CO oxidation. Since the particle sizes are much larger for this CNT supported catalyst than the catalysts on the CB support, we also prepared a catalyst with 1 wt.% Pd loading. The STEM image of this PdZn1/CNT-1 catalyst shows highly-dispersed bimetallic PdZnβ particles (Zn:Pd = 1) with an average size of 2.1 nm (Fig. 4f, insert). The particle size distribution (Fig. 4f, insert) for this catalyst is similar to that of the carbon black supported catalyst shown in Fig. 4a and c and is also consistent with the dispersion shown in Table 1.
 | ||
| Fig. 5 Representative STEM images and particle size histograms of spent Pd and PdZn catalysts after ethanol aqueous-phase reforming: (a) PdZn1/CB; (b) PdZn1.6/CB; (c) PdZn1/CNT-1; (d) PdZn1/CNT. | ||
For the bimetallic particles the reaction caused very little change in the particle size distribution and only the slightest sharpening of the XRD peaks (see Fig. 2). This suggests that the intermetallic PdZnβ phase and the carbon supports, are hydrothermally stable. The spent PdZn1/CB bimetallic catalysts shows PdZnβ particles with an average size of 3.0 nm after APR reaction (Fig. 5a), which is also slightly larger than that on PdZn1/CB before reaction. We did not observe significant change for the particle size of the spent PdZn1.6/CB, PdZn1/CNT-1 and PdZn1/CNT (Fig. 5b–d), as compared to that of the catalysts before reaction. Moreover, the STEM-EDX results did not show significant change for the Pd/Zn ratios on the bimetallic catalysts after APR reaction, compared to the catalysts before APR reaction (see Fig. S7 in ESI†).
3.2. Aqueous-phase reforming of ethanol
| Sample | X (%) | TOF (min−1) | H2 production (mol mol−1 C2H5OH converted) | Product distribution (mol%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CH4 | CO2 | Alkanes | CH3CHO | CH3COOH | CH3OH | CH3COCH3 | ||||
| a Reaction conditions: 250 °C, 65 bar, ethanol/water = 10%, pump rate = 0.003 mL min−1, catalytic performance data were collected at steady state (after 24 h). | ||||||||||
| Pd/CB | 16.9 | 1.24 | 0.23 | 55.6 | 32.2 | 2.9 | 2.9 | 0.58 | 2.9 | 2.9 |
| Pd/CB–H | 15.5 | 2.67 | 0.37 | 54.8 | 35.1 | 1.9 | 3.2 | 0.8 | 1.0 | 3.5 |
| PdZn1/CB | 10.8 | 0.72 | 1.1 | 33.3 | 31.3 | 3.98 | 1.5 | 22.9 | 1.0 | 5.97 |
| PdZn1.6/CB | 8.8 | 0.72 | 1.7 | 40.6 | 35.5 | 1.02 | 6.6 | 15.2 | 0.5 | 0.51 |
| PdZn1/CNT | 29.5 | 23.3 | 1.9 | 23.8 | 34.6 | 4.32 | 0.22 | 36.8 | 0.11 | 0.22 |
| PdZn1/CNT-1 | 14.7 | 2.45 | 1.0 | 29.6 | 37 | 4.7 | 0.1 | 28.5 | 0 | 0.1 |
Effect of Zn addition to Pd. The Pd/CB catalyst showed a CH4 selectivity of 55.6% and CO2 selectivity of 32.2% with very low H2 yield (<0.4 mol per mole ethanol reacted). Addition of Zn and the formation of the PdZnβ phase led to decreased CH4 selectivity, increased CO2 selectivity and the H2 yield increased up to 1.9 moles per mole of ethanol consumed. The bimetallic PdZn/CB catalysts showed higher amounts of oxygenated products in the liquid phase, especially acetic acid. The results indicate that the PdZnβ phase is able to dehydrogenate ethanol while the monometallic Pd leads to the decomposition of ethanol. The high CH4 content is also a result of the hydrogenation of CO or CO2 to CH4 on Pd, which does not occur on the PdZn catalysts.
Effect of support. We studied two different morphologies of carbon support: the carbon black contains graphitized spherical particles while the carbon nanotubes have well defined cylindrical pores with pore sizes of 5–15 nm. The bimetallic PdZnβ particles supported on CNTs show higher conversion and higher specific reactivity than the particles on the CB support. We ascribe the higher specific reactivity for bimetallic PdZn/CNT catalysts to a confinement effect since significant fraction of the particles are located within the CNT pores (Fig. 4e and S5, ESI†).22 It is known that carbon black contains small amounts of S, however the small amounts of S in the CB did not influence the catalytic reactivity for the dehydration of n-butanol in previous work.14 Hence we conclude that the higher reactivity of CNT supports observed in this study may be related to the pore confinement effects. We also see a particle size effect as explained in the next section.
It is also noteworthy that the CH4/CO2 ratio for the PdZn/CNT is <1, which is lower than that for those on the CB supports (Table 2). An explanation is that the acetone produced can be reformed to produce CO2 during the ESR.4 This is consistent with the lower acetone selectivity for the PdZn/CNT catalyst, as seen in Table 2. Another explanation for the lower CH4/CO2 is due to the decarboxylation of surface intermediate acetaldehyde on the PdZn/CNT catalysts.15
Particle size effect. The average particle sizes of Pd and bimetallic PdZn catalysts obtained from CO oxidation, XRD and TEM are summarized in Table 1. It can be seen that a good agreement was found for the average particle sizes obtained from different techniques, which allowed us to discuss the particle size effect in APR reaction. Based on the data, the Pd/CB–H catalyst has larger Pd particles than Pd/CB. The ethanol APR results for these two catalysts are comparable indicating that the effect of particle size on the monometallic catalyst is not significant. But on the bimetallic catalysts, we see a significant particle size effect. The catalyst with the large PdZnβ particles exhibited a significantly higher specific reactivity than smaller PdZnβ. Our TEM results revealed that all the PdZnβ particles exhibit a spherical morphology with well-defined facets visible only on the larger particles. The larger particles are located in the pores of the carbon and could lead to pore blocking and errors in counting sites. However, since we find that the total reactivity per gram catalyst (and % conversion) is much higher on the catalyst with larger particles. Therefore the larger particles of PdZn must be more reactive. A similar particle size effect has been suggested previously for methanol steam reforming.23
Reaction pathways. The observed product distributions suggest two limiting behaviors, that of monometallic Pd which provides very low yield of H2 (~0.2 mole H2 per mole of ethanol reacted) and CH4/CO2 ~1.5 and bimetallic PdZn which leads to higher yields of H2 (~2 mole H2 per mole ethanol reacted) and CH4/CO2 ≤1. Some of the catalysts exhibit intermediate behavior (~1 mol H2 per mole ethanol reacted). Based on our observed product distributions, we propose the following series of steps during aqueous-phase reforming of ethanol over Pd and PdZn bimetallic catalysts (Scheme 1).
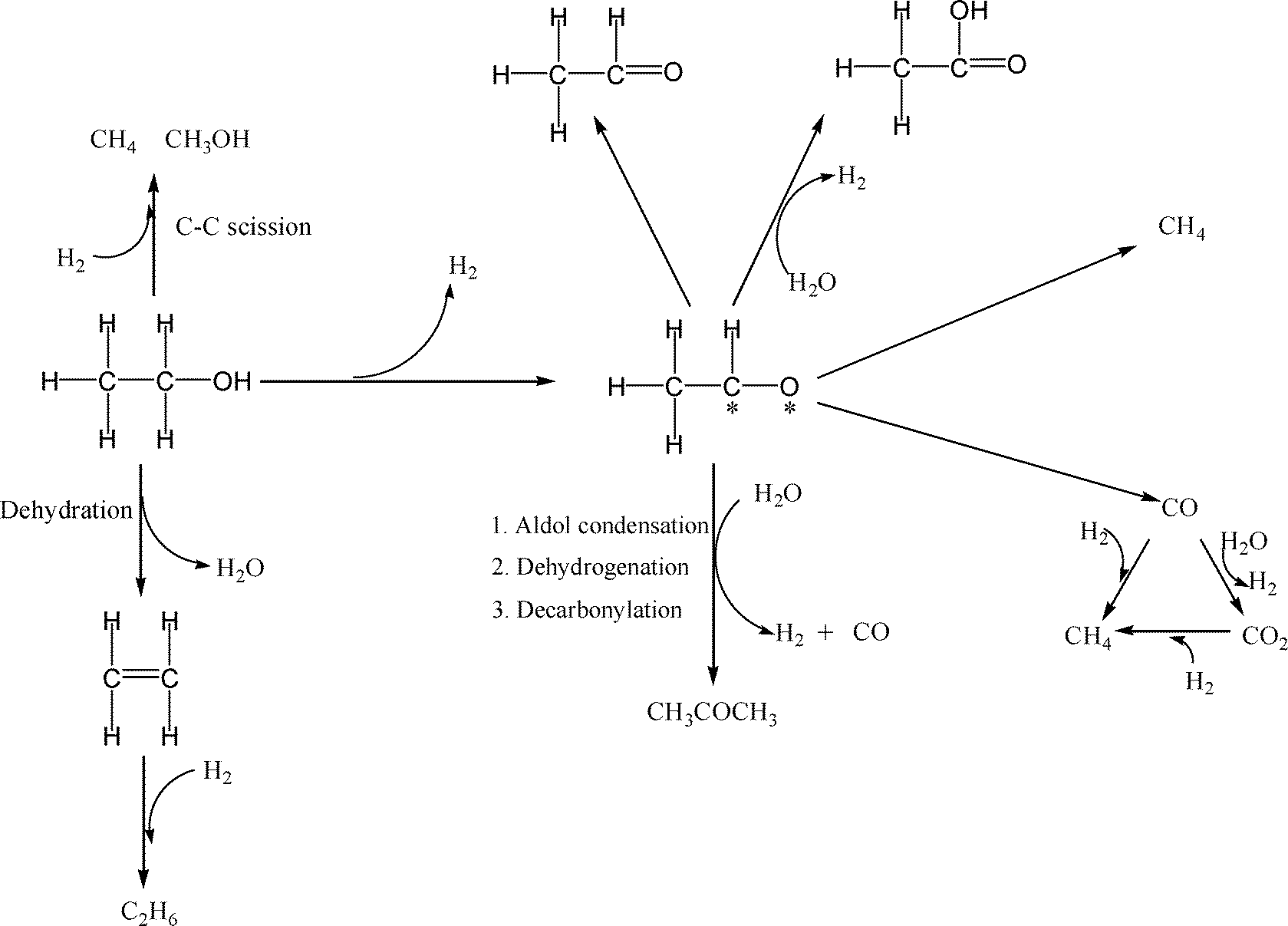 | ||
| Scheme 1 Proposed APR mechanism of ethanol on monometallic Pd and bimetallic PdZn catalysts. | ||
The bimetallic PdZn catalysts show the maximum observed yield of 2 moles of H2 per mole of ethanol converted. The overall reaction stoichiometry to get this yield of H2 is shown in eqn (1). Mechanistically, the first step is the dehydrogenation of ethanol to acetaldehyde to yield the first mole of H2. Decomposition of the acetaldehyde leads to formation of CO and CH4 yielding a 1:1 ratio of CH4 and CO. The CO reacts with water due to the high water gas shift activity of these catalysts, yielding CO2 and the second mole of H2. On PdZn catalysts we see significant amounts of oxygenated products in the liquid phase, hence we infer that the acetaldehyde can also dehydrogenate to acetic acid, leading to additional H2 formation. However, none of these steps would explain the observed CO2/CH4 ratio being greater than 1, which is only possible if a decarbonylation step is also involved.15 Hence, we indicate the path leading to acetone formation, which then allows us to account for all of the products observed during the APR of ethanol. Other pathways leading to the minor products such as ethane and methanol are also shown in Scheme 1. The overall C and H balance is consistent with this mechanistic picture (Fig. S8, ESI†).
In contrast to the bimetallic catalyst, the monometallic Pd/CB shows a significantly higher CH4/CO2 ratio and a very low yield of H2 per mole ethanol. This indicates that the H2 is being consumed to produce CH4 on the Pd catalyst, as revealed in eqn (2) and (3). The hydrogenation of CO2 to CH4 has been suggested to occur during ethanol aqueous-phase reforming on a Ru/C catalyst where the resulting CH4/CO2 ratio was 3:1.15 The observed CH4/CO2 ratio and the low yield of H2 on the monometallic Pd can be accounted by a combination of eqn (1) and (3).
| CH3CH2OH + H2O → CO2 + 2H2 + CH4 | (1) |
| Hydrogenation: CO + 3H2 → CH4 + H2O | (2) |
| CO2 + 4H2 → CH4 + 2H2O | (3) |
The intermediate behavior (between that of monometallic Pd and bimetallic PdZnβ) of some of the catalysts (yield of ~1 mol H2 per mole ethanol reacted) can result from altered selectivity due to depletion of the Zn from the near surface region of the PdZn nanoparticles, as seen during methanol steam reforming.24 We attribute the intermediate H2 yield to the inability to form the ordered intermetallic tetragonal L10 phase, but rather Zn alloyed randomly within fcc Pd (the α phase). When excess Zn was present as in the PdZn1.6/CB catalyst we see the expected behavior of the PdZnβ phase. Another catalyst that shows intermediate behavior is the lower weight loading PdZn/CNT-1 catalyst which has very small particles. We also saw previously that very small particles of PdZnβ were not selective to CO2 formation during methanol steam reforming which might be related to altered electronic properties.23 The ordered intermetallic tetragonal PdZn has very different local density of states near the Fermi level, resembling Cu rather than Pd. But the fcc PdZn alloy shows very different selectivity. In summary, large, crystalline particles of the PdZnβ phase yield the highest specific reactivity and selectivity for APR of ethanol.
4. Conclusions
The performance of monometallic Pd and bimetallic PdZn catalysts supported on both carbon black (CB) and carbon nanotubes (CNTs) was evaluated for H2 production during aqueous-phase reforming (APR) of ethanol at low temperature (250 °C). All of the Pd and bimetallic PdZn catalysts showed high water-gas shift activity since no CO was detected in the products. The CB and CNTs were found to be hydrothermally stable under the liquid phase hydrothermal conditions used for these experiments (250 °C and 65 bar). The PdZnβ particles were also stable, with minimal growth in crystallite size. The TOF for ethanol steam reforming and the product H2 was affected by the PdZnβ particle size, with the most effective catalysts being those where the nanoparticles were located in the pores of the carbon nanotubes. It is possible that pore-confinement within the CNT plays a role since a large fraction of the large PdZnβ particles were located within the pores of the nanotubes. Among the catalysts studied, we conclude that PdZnβ in carbon nanotubes provide an excellent catalyst for the aqueous phase reforming of ethanol, yielding CO-free H2 at low temperatures (250 °C) with a yield of 2 moles of H2 per mole of ethanol reacted. However, the selectivity towards H2 is considerably lower than the theoretical value of 6 H2 per mole ethanol converted. This is a limitation of the low temperature route which does not allow for complete oxidation of both carbon atoms in the ethanol molecule and results in the formation of oxygenated products.Acknowledgements
This work is supported by DOE grant DE-FG02-05ER15712 and the Center for Biorenewable Chemicals (CBiRC) supported by NSF under no. EEC-0813570. We thank Eric Peterson for help with XRD, Jay McCabe for CO oxidation reactivity and Dr. H. Pham for assistance with N2 physisorption measurements.References
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- D. R. Palo, R. A. Dagle and J. D. Holladay, Chem. Rev., 2007, 107, 3992–4021 CrossRef CAS PubMed.
- L. V. Mattos, G. Jacobs, B. H. Davis and F. B. Noronha, Chem. Rev., 2012, 112, 4094–4123 CrossRef CAS PubMed.
- J. Sun, D. Mei, A. M. Karim, A. K. Datye and Y. Wang, ChemCatChem, 2013, 5, 1299–1303 CrossRef CAS PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2005, 56, 171–186 CrossRef CAS PubMed.
- J. Llorca, N. S. Homs, J. Sales and P. R. R. de la Piscina, J. Catal., 2002, 209, 306–317 CrossRef CAS.
- G. W. Huber and J. A. Dumesic, Catal. Today, 2006, 111, 119–132 CrossRef CAS PubMed.
- H. N. Pham, A. E. Anderson, R. L. Johnson, K. Schmidt-Rohr and A. K. Datye, Angew. Chem., Int. Ed., 2012, 51, 13163–13167 CrossRef CAS PubMed.
- B. Roy, K. Artyushkova, H. N. Pham, L. Li, A. K. Datye and C. A. Leclerc, Int. J. Hydrogen Energy, 2012, 37, 18815–18826 CrossRef CAS PubMed.
- R. M. Ravenelle, J. R. Copeland, W.-G. Kim, J. C. Crittenden and C. Sievers, ACS Catal., 2011, 1, 552–561 CrossRef CAS.
- A. L. Jongerius, J. R. Copeland, G. S. Foo, J. P. Hofmann, P. C. A. Bruijnincx, C. Sievers and B. M. Weckhuysen, ACS Catal., 2013, 3, 464–473 CrossRef CAS.
- H. Xiong, H. N. Pham and A. K. Datye, J. Catal., 2013, 302, 93–100 CrossRef CAS PubMed.
- H. Xiong, M. Nolan, B. H. Shanks and A. K. Datye, Appl. Catal., A, 2014, 471, 165–174 CrossRef CAS PubMed.
- S. Rabe, M. Nachtegaal, T. Ulrich and F. Vogel, Angew. Chem., Int. Ed., 2010, 49, 6434–6437 CrossRef CAS PubMed.
- P. Ferrin, D. Simonetti, S. Kandoi, E. Kunkes, J. A. Dumesic, J. K. Nørskov and M. Mavrikakis, J. Am. Chem. Soc., 2009, 131, 5809–5815 CrossRef CAS PubMed.
- B. Halevi, E. J. Peterson, A. Roy, A. DeLariva, E. Jeroro, F. Gao, Y. Wang, J. M. Vohs, B. Kiefer, E. Kunkes, M. Hävecker, M. Behrens, R. Schlögl and A. K. Datye, J. Catal., 2012, 291, 44–54 CrossRef CAS PubMed.
- H. Xiong, M. A. M. Motchelaho, M. Moyo, L. L. Jewell and N. J. Coville, J. Catal., 2011, 278, 26–40 CrossRef CAS PubMed.
- R. S. Johnson, A. DeLaRiva, V. Ashbacher, B. Halevi, C. J. Villanueva, G. K. Smith, S. Lin, A. K. Datye and H. Guo, Phys. Chem. Chem. Phys., 2013, 15, 7768–7776 RSC.
- A. Karim, T. Conant and A. Datye, J. Catal., 2006, 243, 420–427 CrossRef CAS PubMed.
- J. Llorca, P. R. D. L. Piscina, J. Sales and N. Homs, Chem. Commun., 2001, 641–642 RSC.
- X. Pan and X. Bao, Acc. Chem. Res., 2011, 44, 553–562 CrossRef CAS PubMed.
- R. Dagle, Y.-H. Chin and Y. Wang, Top. Catal., 2007, 46, 358–362 CrossRef CAS.
- T. Conant, A. M. Karim, V. Lebarbier, Y. Wang, F. Girgsdies, R. Schlögl and A. Datye, J. Catal., 2008, 257, 64–70 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00914b |
| This journal is © The Royal Society of Chemistry 2015 |