
Photocatalytic hydrogen generation from water reduction using orchestrated photosensitizers†
Shu-Cheng
Yang
a,
Gang
Chang
b,
Guan-Jun
Yang
*c,
Yan-Jie
Wang
*d and
Baizeng
Fang
*d
aDepartment of Environmental Engineering, School of Energy and Power Engineering, Xi'an Jiaotong University, Xi'an, Shaanxi 710049, PR China
bDepartment of Chemistry, School of Science, Xi'an Jiaotong University, Xi'an, Shaanxi 710049, PR China
cState Key Laboratory for Mechanical Behavior of Materials, Xi'an Jiaotong University, Xi'an, Shaanxi 710049, PR China. E-mail: ygj@mail.xjtu.edu.cn; Tel: +86 2982663914
dDepartment of Chemical & Biological Engineering, University of British Columbia, 2360 East Mall, Vancouver, BC, Canada V6T 1Z3. E-mail: yjwang@chbe.ubc.ca; bfang@chbe.ubc.ca
First published on 16th October 2014
Abstract
Vinyl (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) is a functional group in chlorophyll molecules that are essential in natural photosynthesis, which is crucial for tuning the electronic properties and the interaction between chlorophyll and other biological cofactors. Approaches to apply a vinyl group in artificial photosynthesis have been reported to improve the electron transfer from an iridium complex photosensitizer (PS) to a colloidal platinum catalyst likely by chemical adsorption. However, versatile molecular design of Ir-PSs utilizing a vinyl group is still very important to further develop photocatalytically active compounds. Here we report two new Ir-PSs with a phenylvinyl (styrenyl) moiety in two different positions. Their hydrogen-generation performance was assessed in the presence of a triethylamine electron donor and a colloidal platinum catalyst. As expected, both the Ir-PSs show much higher turnover numbers than the non-styrenylic Ir-PS, and interestingly, the H2 generation longevity was clearly observed to be dependent on the styrenyl position.
CH2) is a functional group in chlorophyll molecules that are essential in natural photosynthesis, which is crucial for tuning the electronic properties and the interaction between chlorophyll and other biological cofactors. Approaches to apply a vinyl group in artificial photosynthesis have been reported to improve the electron transfer from an iridium complex photosensitizer (PS) to a colloidal platinum catalyst likely by chemical adsorption. However, versatile molecular design of Ir-PSs utilizing a vinyl group is still very important to further develop photocatalytically active compounds. Here we report two new Ir-PSs with a phenylvinyl (styrenyl) moiety in two different positions. Their hydrogen-generation performance was assessed in the presence of a triethylamine electron donor and a colloidal platinum catalyst. As expected, both the Ir-PSs show much higher turnover numbers than the non-styrenylic Ir-PS, and interestingly, the H2 generation longevity was clearly observed to be dependent on the styrenyl position.
1. Introduction
Energy and environment are two important topics at a global level. It is indispensable to seek green energy systems in order to solve the challenges of these two issues.1 Hydrogen is regarded as a clean energy resource2–6 which can be used in fuel cells while only producing water after consumption.7–10 So far, many efforts have been made to utilize solar energy and water to generate hydrogen.11–13 In nature, especially for some plants, green algae and cyanobacteria, this process occurs daily and it is driven by the cooperation of two large protein–cofactor complexes, i.e. photosystems I and II. The key biomolecule in photosystems I and II is chlorophyll which allows plants to absorb energy from light.14 Chlorophyll is a chlorin pigment with a magnesium ion at the centre. The chlorin ring embraces a long phytol chain and several peripheral functional groups, such as vinyl (–CHCH2) and formyl (–CHO).15 The functional groups are crucial for tuning the electronic and electrochemical properties and thereby the charge separation and transfer performance. This natural photochemical system is so perfect that it is truly a standard model for an artificial H2-generation system. In artificial photosystems, a key challenge is the development of more efficient photosensitizers (PSs) that are responsible for absorbing light and then transferring electrons to catalytic centers. The established effective PSs are known to include, for example, ruthenium,16 platinum,17 rhenium18 or heavy-atom (I, Se)-containing organic dyes.19 Recently, iridium complexes in particular have emerged as active PSs, especially those with the general formula [Ir(C^N)2(N^N)]+ (where C^N is a cyclometalating ligand and N^N is a bipyridyl ligand).20,21 In order to screen various Ir-PSs, a simple three-component photoreaction with the Ir-PS, the triethylamine (TEA) sacrificial electron donor, and the colloidal platinum catalyst was established.22,23 Following a reductive quenching pathway (as illustrated in Fig. 1A), the excited state (Ir-PS*) is first formed by light absorption and then reduced by using TEA to produce Ir-PS−. The latter can subsequently transfer an electron onto Pt to drive water reduction and hydrogen evolution. As the lowest unoccupied molecular orbitals of Ir-PSs are mainly located in the bipyridyl ligand,20 the electron transfer process from Ir-PS− to Pt is largely influenced by this ligand. Furthermore, this bipyridyl ligand was found to easily cleave from the Ir metal center, causing Ir-PS degradation, due to the fact that an electron populates the anti-bonding orbital in the Ir-PS− state.22 Therefore, it is important to modify the bipyridyl ligand in order to facilitate electron transfer to quickly quench the Ir-PS− state. For this purpose, Bernhard et al. reported a family of new Ir-PSs with vinyl (–CHCH2) on the bipyridyl ligand.24 During photocatalytic reactions, Ir-PSs are likely to be chemically adsorbed on the Pt surface, either kept intact or reduced. Although the exact Ir-PS species remains a challenge to be recognized due to the very low concentration in photocatalytic reactions, the strengthened bonding between Ir-PS and Pt was proven to benefit electron transfer, fast Ir-PS− quenching, and improved Ir-PS stability.24 As –CHCH2 happens to be a critical functional group in chlorophyll molecules, further molecular engineering of Ir-PSs using this group is of great interest. Here we describe the synthesis of two new Ir-PSs, 4-Styrenyl-Ir and 3-Styrenyl-Ir, with styrenyl (phenylvinyl) moieties in two different positions of the bipyridyl ligand, as displayed in their chemical structures shown in Fig. 1B. The replacement of vinyl by styrenyl is due to the much easier synthesis of the latter compounds. The as-synthesized styrenyl Ir-PSs show much improved turnover numbers (TONs) compared to their counterpart, which is the non-styrenylic Ir-PS (4-Me-Ir) in H2-generation reactions. Most importantly, the H2-generation stability is found to depend on the styrenyl position.
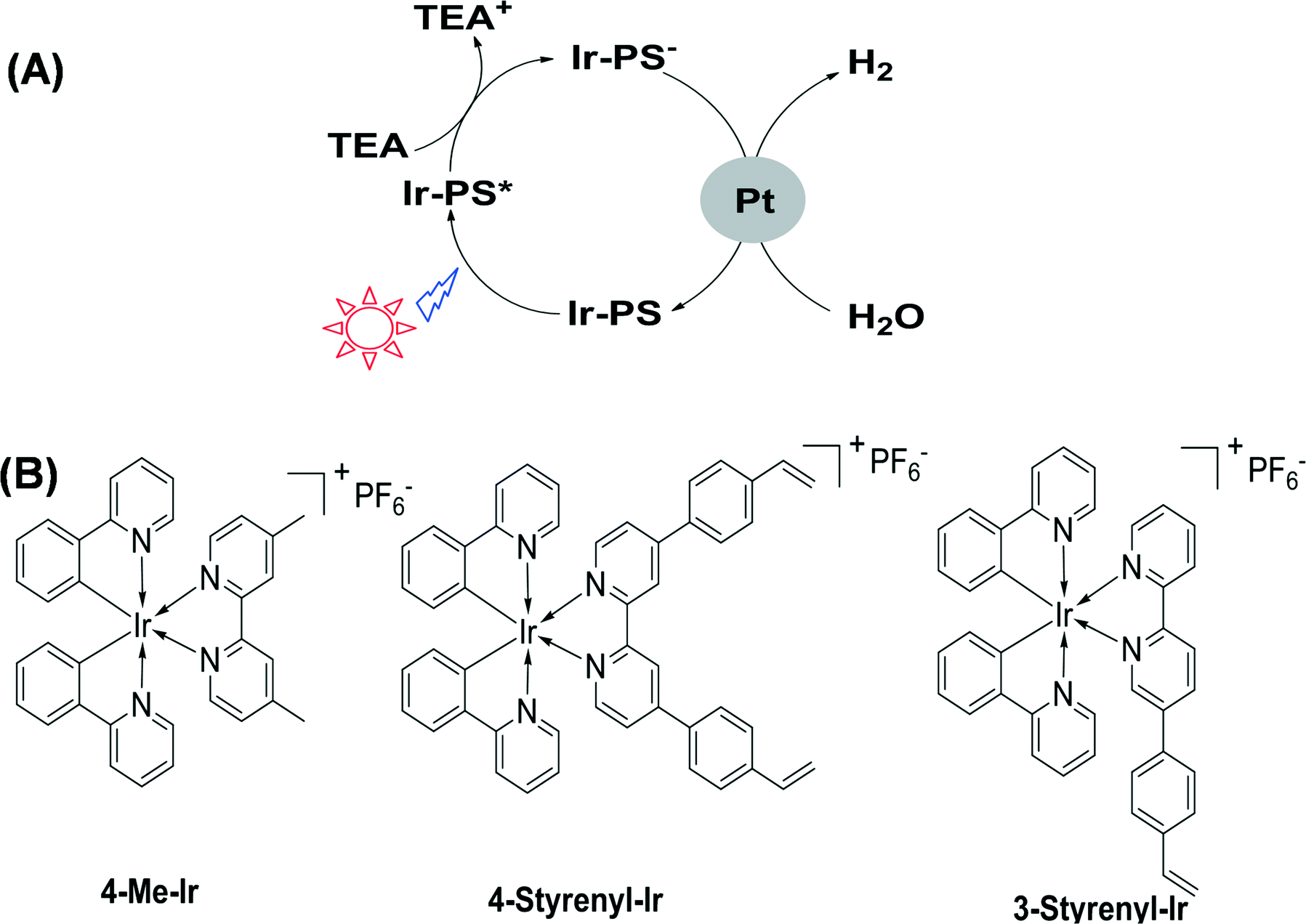 | ||
| Fig. 1 Schematic photocatalytic hydrogen-generation process (A) and chemical structures of Ir(III) complexes (B). | ||
2. Experimental section
2.1 Synthesis
The synthesis routes of the two styrenyl bipyridyl ligands (3 and 4) and the two Ir-PSs (4-Styrenyl-Ir and 3-Styrenyl-Ir) are illustrated in Fig. 2. The two styrenyl Ir-PSs were synthesized through a straightforward protocol,25,26 including the preparation of styrenyl bipyridyl ligands, the coordination with Ir and the final counterion exchange with KPF6. Specifically, the styrenyl ligands 3 and 5 were synthesised under Suzuki–Miyaura cross-coupling conditions27 using 4-vinylphenylboronic acid (2) with the respective bipyridyl bromide (1 or 4). They then reacted with a chloro-bridged Ir(III) dimer [Ir(ppy)2]Cl2 (ppy = phenylpyridine) in a mixed solvent of CH2Cl2/CH3OH at reflux, followed by ion exchange with KPF6 in CH3OH/H2O to afford 4-Styrenyl-Ir and 3-Styrenyl-Ir with good yields of 68% and 89%, respectively. The detailed synthesis of each compound can be seen in the ESI.†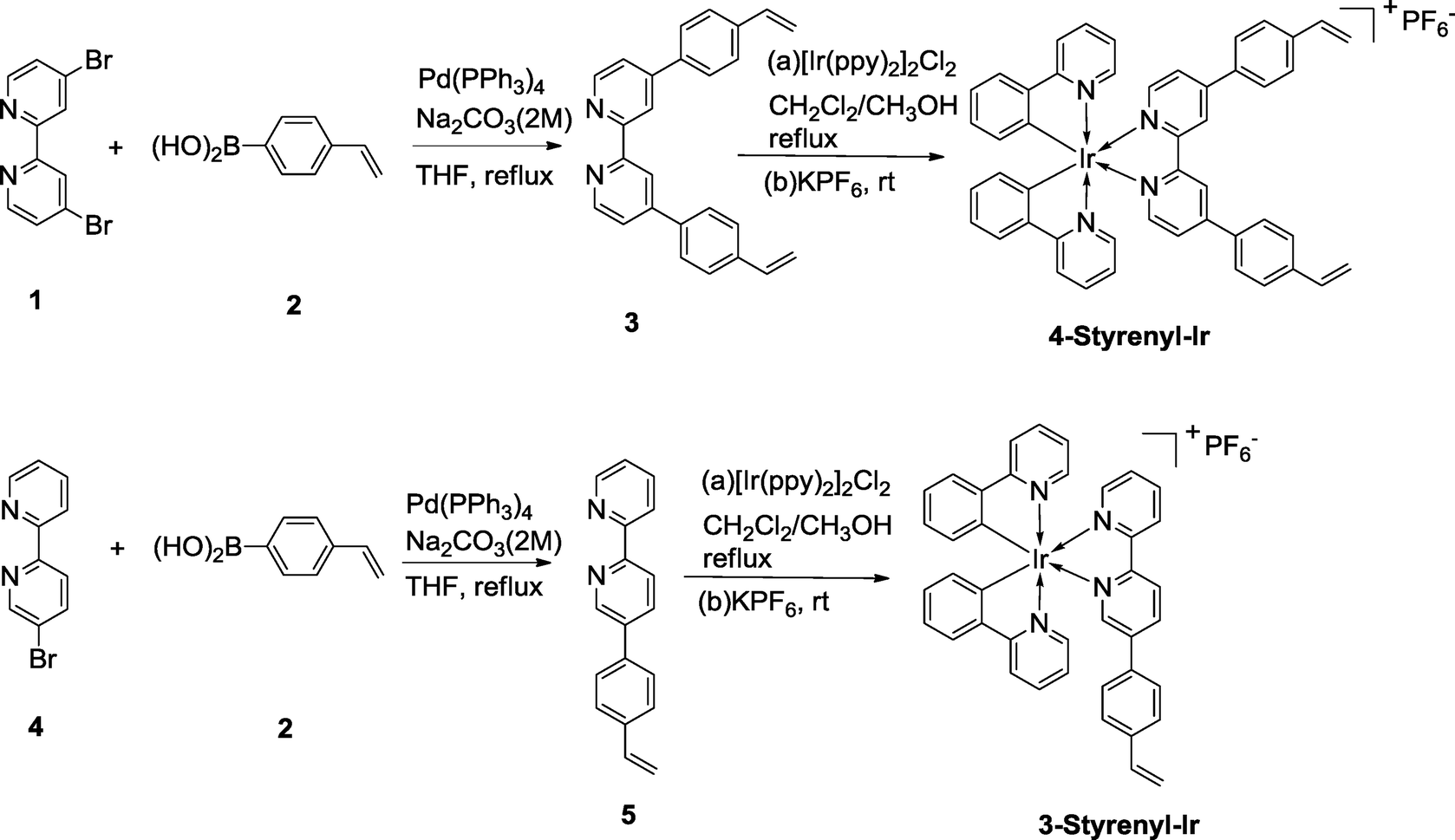 | ||
| Fig. 2 Illustration for the synthesis of the two Ir-PSs, 4-Styrenyl-Ir and 3-Styrenyl-Ir. | ||
2.2 Characterization
UV–vis absorption spectra were recorded using a Cary 500 UV–vis spectrophotometer.Steady-state photoluminescence (PL) was recorded using a SENS-9000 spectrophotometer.
Time-resolved PL was carried out using QM4 with time-correlated single-photon counting capability.
Cyclic voltammetry (CV) measurements were performed using a BASi Epsilon-EC workstation with anhydrous tetrahydrofuran (THF) solvent, glass carbon as the working electrode, platinum wire as the counter electrode, Ag/AgNO3 (0.1 M) as the reference electrode, an nBu4NBF6 (0.1 M) electrolyte and a ferrocene standard at a scan rate of 100 mV s−1.
2.3 Performance tests
The H2-generation efficiencies of the Ir-PSs were evaluated in a mixture of 0.50 μmol of PS, 0.30 μmol of K2PtCl4, 2.0 mL of TEA, 2 mL of H2O, and 6.0 mL of THF using a 150 W xenon lamp as the light source. The colloidal Pt catalyst was formed in situ from K2PtCl4 in the solution.21 The amount of evolved gas was measured with an automatic gas burette, and gases were analyzed by gas chromatography (Shimadzu).The incident photon-to-hydrogen conversion efficiency (IPCE) was determined by dividing the rate of evolved hydrogen gas (average in the first 30 min) with the flux of incident photons at a specific wavelength. A high-pressure mercury lamp (LLE-9, 50 W) was employed as the light source, and the light output was adjusted to 1.0 W for 365, 405, 436, 492 and 546 nm, respectively, with the assistance of light filters. The power output was determined by using a radiometer (FZ-A).
3. Results and discussion
The as-synthesized styrenyl Ir-PSs reveal very strong absorption in the UV region (Fig. 3A) and moderate absorption in the visible band (Fig. 3B), which correspond to the ligand-centered transitions and metal-to-ligand charge-transfer transitions (MLCT),28 respectively. More importantly, the styrenyl Ir-PSs show much higher absorption intensity at 300–400 nm than the non-styrenyl Ir-PS (4-Me-Ir) due to the extended π conjugation of the bipyridyl ligand.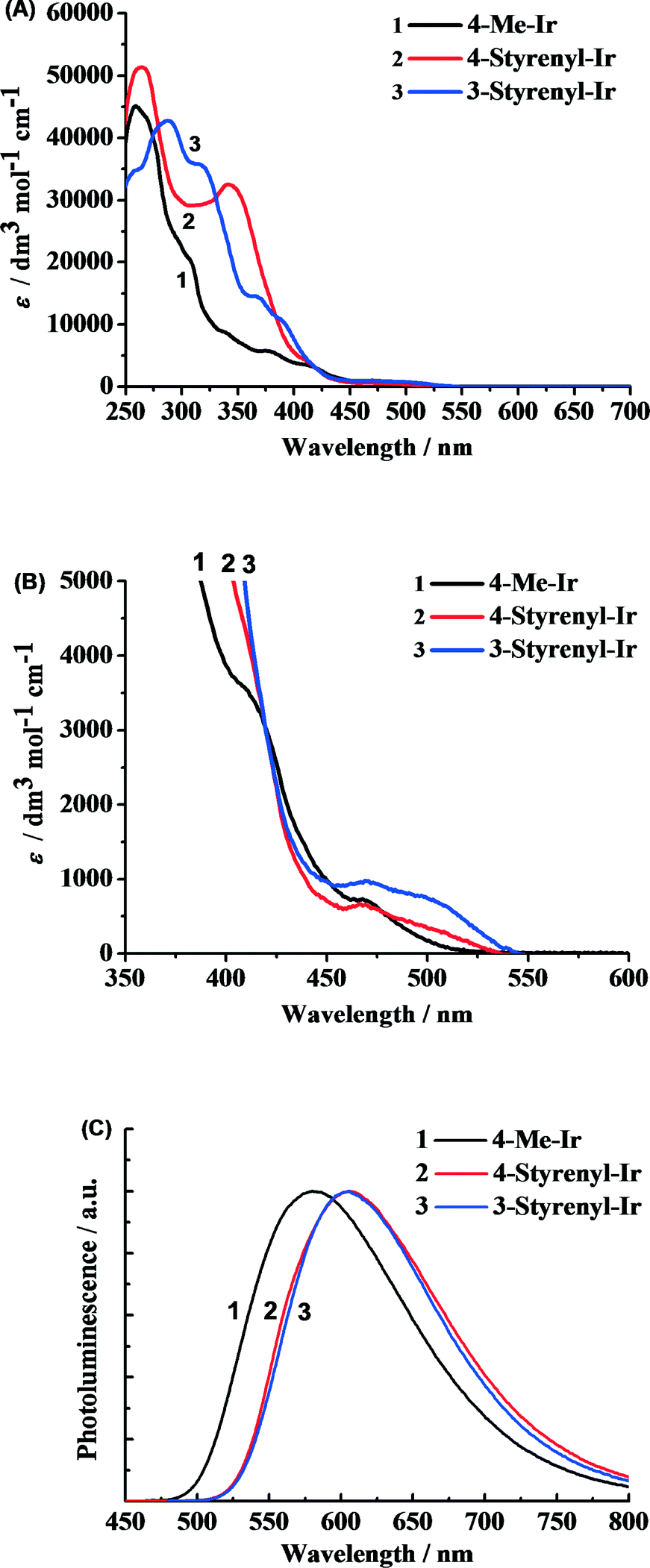 | ||
| Fig. 3 Absorption spectra of Ir(III) complexes in CH2Cl2 at room temperature (A) and the expansion at the low-energy transitions (B) and photoluminescence spectra of Ir-PSs in CH2Cl2 with excitation at 410 nm (C). | ||
The PL spectra measured in argon-saturated CH2Cl2 solution are shown in Fig. 3C with the emission peak values reported in Table 1. All of the Ir-PSs show broad structureless emission indicative of the dominant 3MLCT transitions [i.e. C^N π orbital and Ir(III) d orbital to the N^N π* orbital].29 The two styrenyl Ir-PSs yield red-shifted emission by 31 nm, which is clearly ascribed to the extended conjugation. However, PL quantum yields (φPL) and excited-state lifetimes of the two styrenyl Ir-PSs exhibit only subtle differences from the non-styrenyl Ir-PS, suggesting similar radiative and non-radiative decay rates of the excited states.
| Ir(III) complex | λ e /nm | E g(opt)b/eV | φ PL | τ /μs | E(PS+/PS)e/V | E(PS/PS−)f/V | E(PS*/PS−)g/V |
|---|---|---|---|---|---|---|---|
| a Emission peak in CH2Cl2. b Optical bandgap. c Excited at 350 nm in degassed THF using quinine sulfate as reference. d Excited-state lifetime. e Oxidation potential. f Reduction potential. g Excited-state oxidation potential, calculated by E(PS/PS−) + Eg(opt). | |||||||
| 4-Me-Ir | 574 | 2.45 | 0.28 | 0.53 | +1.58 | −1.13 | +1.31 |
| 4-Styrenyl-Ir | 605 | 2.32 | 0.34 | 0.37 | +1.64 | −0.91 | +1.41 |
| 3-Styrenyl-Ir | 605 | 2.34 | 0.21 | 0.47 | +1.68 | −0.92 | +1.42 |
From electrochemical CV measurements (the plots are shown in Fig. S1 in the ESI†), styrenyl Ir-PSs exhibit a slight but reproducible increase in oxidation potentials (i.e., E(PS+/PS)) and a decrease in reduction potentials (i.e., E(PS/PS−)), as shown in Table 1. The excited-state oxidation potential (i.e., E(PS*/PS−)) calculated by E(PS/PS−) + Eg(opt)30 also presents a value only 0.1 V higher than that of the non-styrenylic Ir-PS. In photocatalytic reactions with the TEA electron donor and the platinum catalyst, the excited-state oxidation potentials of both Ir-PSs (ca. +1.4 V) are sufficient to accept electrons from TEA (E(TEA+/TEA) = +0.93 V vs. NHE),31 and the reduction potentials (ca. −0.9 V) are also enough to drive water reduction on Pt (E(H+/H2) = −0.71 V vs. NHE at pH 12).
Therefore, based on the experimental results of the absorption, PL, and CV measurements, the two styrenyl Ir-PSs should be promising for photocatalytic hydrogen generation from water.
The different Ir-PSs were tested for hydrogen generation using a mixture containing 0.50 μmol of PS, 0.30 μmol of K2PtCl4, 2.0 mL of TEA, 2.0 mL of H2O, and 6.0 mL of THF and a 150 W xenon lamp as the light source. The colloidal Pt catalyst was formed in situ from K2PtCl4 in the solution.21 Under illumination, evolved H2 gas was collected using a gas burette and the gas volumes were measured. The collected gas was further analyzed by gas chromatography (GC) and the volumes were converted to moles using the ideal gas equation. Besides traces of solvent, only hydrogen was found. The turnover number (TON) of the Ir-PS was calculated with respect to single-electron transfer processes. The turnover frequency (TOF) was estimated by TON in the initial 30 min, as the kinetic plots show linear trends in this region, as shown in Fig. 4.
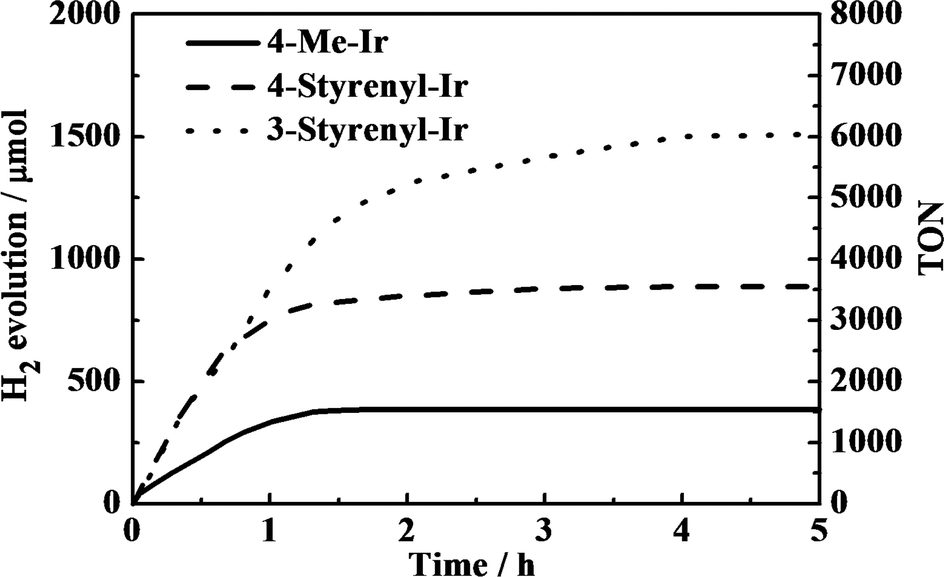 | ||
| Fig. 4 Representative kinetic traces of H2-generation photoreactions with Ir(III) complex photosensitizers. The H2-generation photoreactions were carried out using 0.5 μmol of PS, 0.30 μmol of K2PtCl4, 2.0 mL of TEA, 2.0 mL of H2O and 6.0 mL of THF. | ||
As shown in Fig. 4, an induction period is not clearly observed in the kinetic plots. This observation is in good agreement with the other investigations using Ir-PSs,24 which may imply the very fast formation of active platinum centers.21 Additionally, the absence of an induction period suggests that the styrenyl Ir-PSs themselves have been active before their reduced derivatives are formed on the Pt surface.
The kinetic traces clearly demonstrate that the two styrenyl Ir-PSs enable faster H2 evolution in the early stage than the non-styrenylic Ir-PS, giving double TOFs, as shown in Table 2. As a result of higher TOFs of the as-synthesized styrenyl Ir-PSs, their final TONs are much higher than those of the non-styrenyl Ir-PS. This can be attributed to the improved UV absorption, increased excited-state oxidation potentials, and enhanced adsorption of styrenyl Ir-PS molecules.24
| Ir(III) complex | V(H2)a/mL | V(H2)a/μmol | TONb | TOFc/min−1 |
|---|---|---|---|---|
| a The H2 generation photoreactions were carried out using 0.5 μmol of PS, 0.30 μmol of K2PtCl4, 2.0 mL of TEA, 2.0 mL of H2O, and 6.0 mL of THF. b n(H)/n(PS). c Calculated from TON in the first 30 min. | ||||
| 4-Me-Ir | 8.6 ± 1.3 | 716 ± 54 | 1432 ± 108 | 29 ± 3 |
| 4-Styrenyl-Ir | 21.2 ± 2.1 | 1777 ± 88 | 3553 ± 175 | 69 ± 5 |
| 3-Styrenyl-Ir | 36.0 ± 3.7 | 3017 ± 154 | 6033 ± 308 | 70 ± 3 |
To elucidate the effect of light absorption on hydrogen generation, the IPCE of the system was determined, in which an Hg vapor lamp whose wavelengths and power output can be modulated separately was used. The light output was adjusted to 0.5 W for 365, 405, 436, 492 and 546 nm, and the rate of photons emitted at these wavelengths was calculated. By comparing the photon flux with the rate of the produced hydrogen atoms, the efficiency of the system for each individual wavelength was calculated. The obtained IPCE spectra of the Ir-PSs are shown in Fig. 5. For all of the as-synthesized Ir-PSs, the IPCE levels off at ca. 20–23% in a wavelength range of 365–492 nm and then decreases dramatically to nearly zero at 546 nm. Interestingly, the IPCE values for the various Ir-PSs are very close at each individual wavelength. This suggests that the superior hydrogen-generation performance of the styrenyl Ir-PSs to the non-styrenyl Ir-PS is not a function of light absorption under the investigated conditions. However, the much higher Ir-PS concentration in the IPCE measurements can lead to a larger self-quenching rate of the excited Ir-PS (i.e., Ir-PS*) than that in the kinetics tests. Therefore, a direct link between the IPCE results shown in Fig. 5 and the kinetics shown in Fig. 4 is impossible.
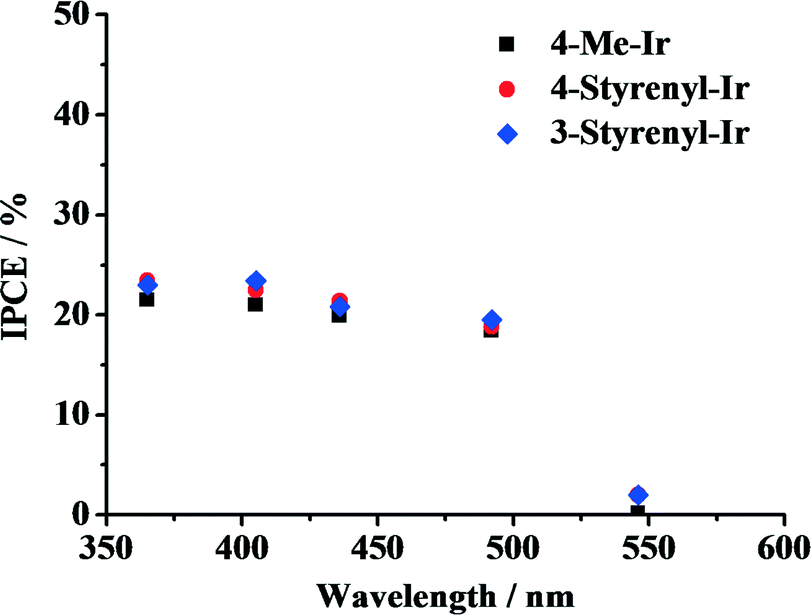 | ||
| Fig. 5 IPCE shown as a function of wavelength for the various Ir-PSs. The photoreactions were carried out using 20 μmol of PS, 0.30 μmol of K2PtCl4, 2.0 mL of TEA, 2.0 mL of H2O and 6.0 mL of THF. | ||
To examine the effect of increased excited-state oxidation potential E(PS*/PS−) on the hydrogen-generation activity, Stern–Volmer PL quenching measurements were performed for the as-synthesized Ir-PSs. Since E(PS*/PS−) determines the reduction of Ir-PS* by TEA, Stern–Volmer PL quenching measurements of the various Ir-PSs by TEA can give insights into the contribution from E(PS*/PS−) values. The PL quenching efficiencies by TEA follow the steady-state Stern–Volmer equation PL0/PL = 1 + KSV [TEA],32 where PL0 is the initial solution photoluminescence of the PS, PL is the solution photoluminescence of the PS at the quencher concentration [TEA], and KSV is the Stern–Volmer constant. The Stern–Volmer plots are shown in Fig. 6 and KSV values are calculated from the slopes. As can be seen, the KSV values (3.7–4.3) are also very close for these three Ir-PSs, implying that the increase of E(PS*/PS−) by ca. 0.1 V for the styrenyl Ir-PSs has no clear benefit on the reaction rate of PS* reduction. Therefore, it is conclusive that the increased TOF of hydrogen generation for the styrenyl Ir-PSs cannot be related to the excited-state oxidation potential.
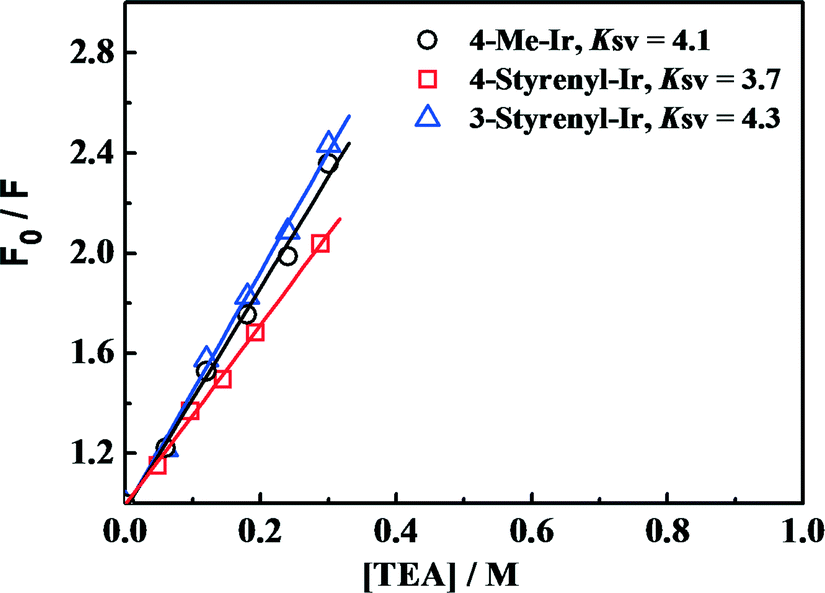 | ||
Fig. 6 Steady-state Stern–Volmer plots for the various Ir-PSs (1.0 μmol in 10 mL of THF/H2O at 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as a function of [TEA]. :1) as a function of [TEA]. | ||
Based on the above analysis, we therefore ascribe the increased hydrogen-generation rate for the styrenyl Ir-PSs to the enhanced adsorption of styrenyl Ir-PS molecules. It is assumed that the vinyl group in the styrenyl moiety is reduced on the Pt surface during photoreactions and thereby forms a self-assembled molecular layer by chemical adsorption, as illustrated in Fig. 7. The adsorption not only facilitates the assembly of Ir-PS molecules on the Pt catalytic surface but also promotes the electron transfer between them.
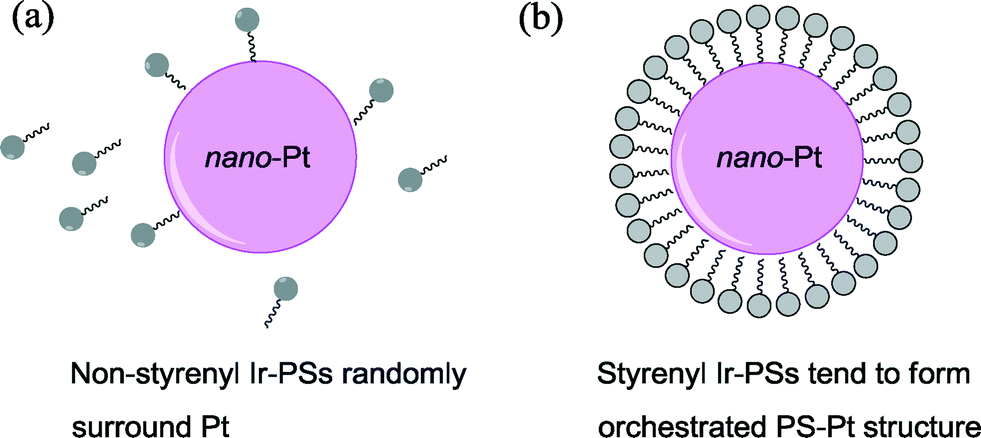 | ||
| Fig. 7 Schematic diagram of the interaction between the Pt catalyst and the non-styrenyl (a) or styrenyl (b) Ir-PSs. | ||
Most importantly, 3-Styrenyl-Ir shows higher stability in photoreactions than 4-Styrenyl-Ir, which enables the former Ir-PS to achieve a nearly doubled final TON (up to 6000). As indicated previously,22 the stability is mainly related to the quenching rate of Ir-PS−, in which the electron in the anti-bonding orbital is prone to cleave the bipyridyl ligand from the Ir metal center. The superior stability of 3-Styrenyl-Ir over 4-Styrenyl-Ir suggests that the quenching of Ir-PS− is a function of the styrenyl position in the Ir complex. We did not ascribe the stability difference to the number of styrene in the bipyridyl ligands as one or two vinyl groups on the Ir-PSs presented no difference in previous reports.24 It is likely that the monolayer structure of Ir-PS molecules on the Pt surface is determined by the position of styrene and therefore leads to different electron transfer performance. Significantly, the comparison of the two new styrenyl Ir-PSs has provided a good hint on future molecular engineering for innovative photocatalytic compounds.
4. Conclusions
In summary, in this study, two new Ir-PSs with styrenyl moiety pendants have been developed and explored as photosensitizers for photocatalytic hydrogen generation from water reduction. Photocatalytic hydrogen-generation reactions in the presence of a TEA electron donor and a colloidal Pt catalyst reveal that the inclusion of a styrenyl moiety significantly increases the TOF of Ir-PSs. Most importantly, higher photocatalytic stability was observed for the Ir-PS with a styrenyl moiety in the meta position of the bipyridyl ligand. These findings can be related to the status of Ir-PS molecules adsorbed chemically on the Pt surface. This warrants future inquiry into vinylic PSs. We will continue to study these materials and will elucidate structure–property relationships. The final high TONs of styrenyl Ir-PSs herein justify investment in these efforts.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 51308453), the Fundamental Research Funds for the Central Universities of China and the National Program for Support of Top-notch Young Professionals.References
- B. Fang, A. Bonakdarpour, K. Reilly, Y. Xing, F. Taghipour and D. Wilkinson, ACS Appl. Mater. Interfaces, 2014, 6, 15488–15498 CAS.
- Y. Xing, B. Fang, A. Bonakdarpour, S. Zhang and D. Wilkinson, Int. J. Hydrogen Energy, 2014, 39, 7859–7867 CrossRef CAS PubMed.
- Z. Wu, B. Fang, Z. Wang, C. Wang, Z. Liu, F. Liu, W. Wang, A. Alfantazi, D. Wang and D. Wilkinson, ACS Catal., 2013, 3, 2101–2107 CrossRef CAS.
- B. Fang, J. Kim, M. Kim and J. Yu, Acc. Chem. Res., 2013, 46, 1397–1406 CrossRef CAS PubMed.
- Z. Wu, B. Fang, A. Bonakdarpour, A. Sun, D. Wilkinson and D. Wang, Appl. Catal., B, 2012, 125, 59–66 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- B. Fang, M. Kim, J. Kim, M. Song, Y. Wang, H. Wang, D. Wilkinson and J. Yu, J. Mater. Chem., 2011, 21, 8066–8073 RSC.
- B. Fang, N. Chaudhari, M. Kim, J. Kim and J. Yu, J. Am. Chem. Soc., 2009, 131, 15330–15338 CrossRef CAS PubMed.
- B. Fang, J. Kim, M. Kim and J. Yu, Chem. Mater., 2009, 21, 789–796 CrossRef CAS.
- B. Fang, J. Kim and J. Yu, Electrochem. Commun., 2008, 10, 659–662 CrossRef CAS PubMed.
- W. Teoh, J. Scott and R. Amal, J. Phys. Chem. Lett., 2012, 3, 629–639 CrossRef CAS.
- P. Kamat, J. Phys. Chem. Lett., 2012, 3, 663–672 CrossRef CAS.
- S. Fukuzumi, Y. Yamada, T. Suenobu, K. Ohkubo and H. Kotani, Energy Environ. Sci., 2011, 4, 2754–2766 CAS.
- P. Jordan, P. Fromme, H. Witt, O. Klukas, W. Saenger and N. Krauβ, Nature, 2001, 411, 909–917 CrossRef CAS PubMed.
- J. Thornber, Annu. Rev. Plant Physiol., 1975, 26, 127–158 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- P. Du, J. Schneider, G. Luo, W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 4952–4962 CrossRef CAS PubMed.
- B. Probst, M. Guttentag, A. Rodenberg, P. Hamm and R. Alberto, Inorg. Chem., 2011, 50, 3404–3412 CrossRef CAS PubMed.
- T. McCormick, B. Calitree, A. Orchard, N. Kraut, F. Bright, M. Detty and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 15480–15483 CrossRef CAS PubMed.
- L. Tinker, N. McDaniel, P. Curtin, C. Smith, M. Ireland and S. Bernhard, Chem. – Eur. J., 2007, 13, 8726–8732 CrossRef CAS PubMed.
- P. Curtin, L. Tinker, C. Burgess, E. Cline and S. Bernhard, Inorg. Chem., 2009, 48, 10498–10506 CrossRef CAS PubMed.
- B. DiSalle and S. Bernhard, J. Am. Chem. Soc., 2011, 133, 11819–11821 CrossRef CAS PubMed.
- F. Gärtner, S. Denurra, S. Losse, A. Neubauer, A. Boddien, A. Gopinathan, A. Spannenberg, H. Junge, S. Lochbrunner, M. Blug, S. Hoch, J. Busse, S. Gladiali and M. Beller, Chem. – Eur. J., 2012, 18, 3220–3225 CrossRef PubMed.
- S. Metz and S. Bernhard, Chem. Commun., 2010, 46, 7551–7553 RSC.
- F. Gärtner, D. Cozzula, S. Losse, A. Boddien, G. Anikumar, H. Junge, T. Schulz, N. Marquet, A. Spannenberg, S. Gladiali and M. Beller, Chem. – Eur. J., 2011, 17, 6998–7006 CrossRef PubMed.
- M. Lowry, W. Hudson, R. Pascal and S. Bernhard, J. Am. Chem. Soc., 2004, 126, 14129–14135 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- S. Wu, J. Ling, S. Lai, M. Huang, C. Cheng and I. Chen, J. Phys. Chem. A, 2010, 114, 10339–10344 CrossRef CAS PubMed.
- M. Lepeltier, T. Lee, K. Lo, L. Toupet, H. Bozec and V. Guerchais, Eur. J. Inorg. Chem., 2005, 2005, 110–117 CrossRef.
- E. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed.
- N. McDaniel and S. Bernhard, Dalton Trans., 2010, 39, 10021–10030 RSC.
- D. Olley, E. Wren, G. Vamvounis, M. Fernee, X. Wang, P. Burn, P. Meredith and P. Shaw, Chem. Mater., 2011, 23, 789–794 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the synthesis for ligand 3, 4-Styrenyl-Ir, ligand 5 and 3-Styrenyl-Ir, CV plots for 4-Me-Ir, 4-Styrenyl-Ir and 3-Styrenyl-Ir. See DOI: 10.1039/c4cy01112k |
| This journal is © The Royal Society of Chemistry 2015 |