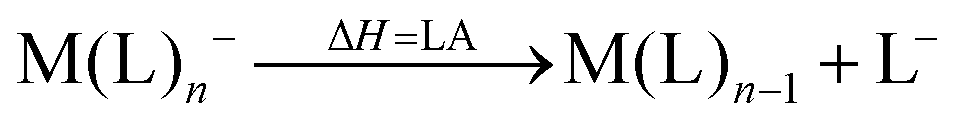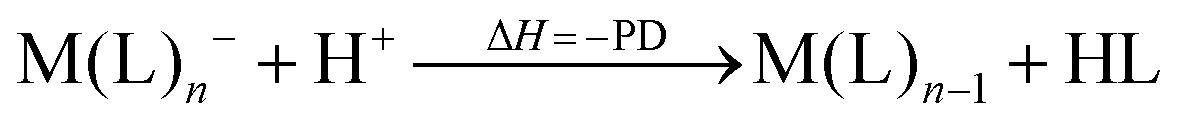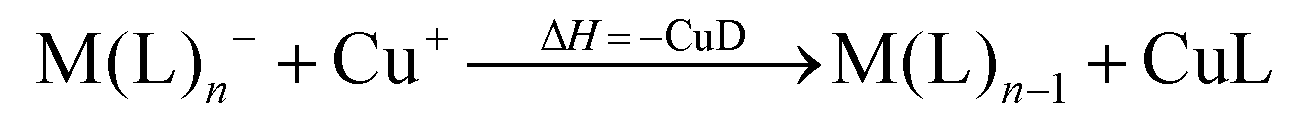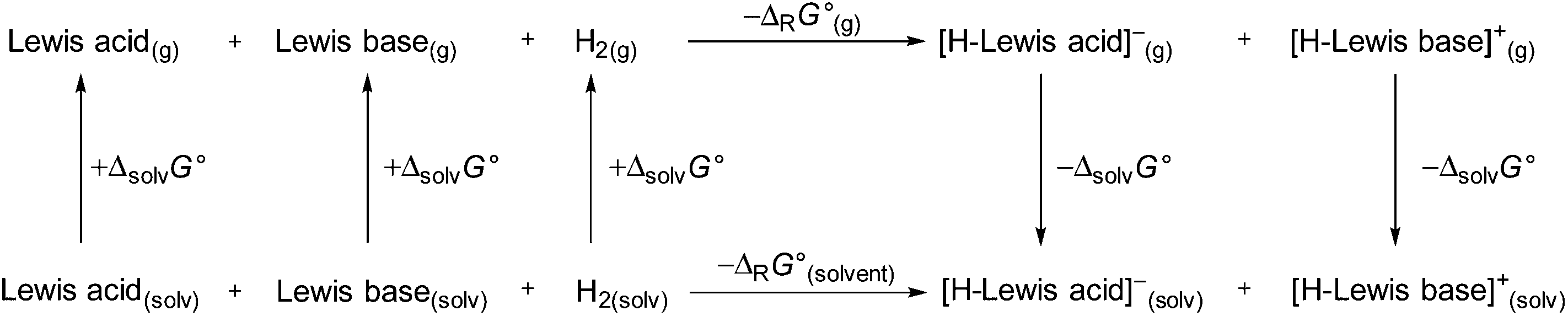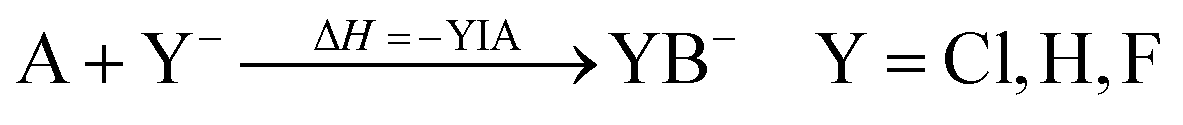DOI:
10.1039/C4DT02822H
(Paper)
Dalton Trans., 2015,
44, 7489-7499
From unsuccessful H2-activation with FLPs containing B(Ohfip)3 to a systematic evaluation of the Lewis acidity of 33 Lewis acids based on fluoride, chloride, hydride and methyl ion affinities†
Received
16th September 2014
, Accepted 12th March 2015
First published on 13th March 2015
Abstract
The possibility of obtaining frustrated Lewis pairs (FLPs) suitable for H2-activation based on the Lewis acid B(Ohfip)31 (Ohfip = OC(H)(CF3)2) was investigated. In this context, the crystal structure of 1 as well as the crystal structure of the very weak adduct 1·NCMe was determined. When reacting solutions of 1 with H2 (1 bar) and selected phosphanes, amines, pyridines and N-heterocyclic carbenes, dihydrogen activation was never observed. Without H2, adduct formation with 1 was observed to be an equilibrium process, regardless of the Lewis base adduct. Thus, the thermodynamics of H2 activation of 1 in comparison with the well-known B(C6F5)3 was analyzed using DFT calculations in the gas phase and different solvents (CH2Cl2, ortho-difluorobenzene and acetonitrile). These investigations indicated that FLP chemistry based on 1 is considerably less favored than that with B(C6F5)3. This is in agreement with control NMR experiments indicating hydride transfer from [H–B(Ohfip)3]− upon reaction with B(C6F5)3, giving [H–B(C6F5)3]− and B(Ohfip)3 in toluene and also MeCN. Induced by these unsuccessful reactions, the Lewis acidity towards HSAB hard and soft ions was investigated for gaining a deeper insight. A unified reference system based on the trimethylsilyl compounds Me3Si–Y (Y = F, Cl, H, Me) and their respective ions Me3Si+/Y− calculated at the G3 level was chosen as the anchor point. The individual ion affinities were then assessed based on subsequent isodesmic reactions calculated at a much less expensive level (RI-)BP86/SV(P). This method was validated by systematic calculations of smaller reference systems at the frozen core CCSD(T) level with correlation effects extrapolated to a full quadruple-ζ basis. Overall, 33 common and frequently used Lewis acids were ranked with respect to their FIA, CIA, HIA and MIA (fluoride/chloride/hydride/methyl ion affinity).
Introduction
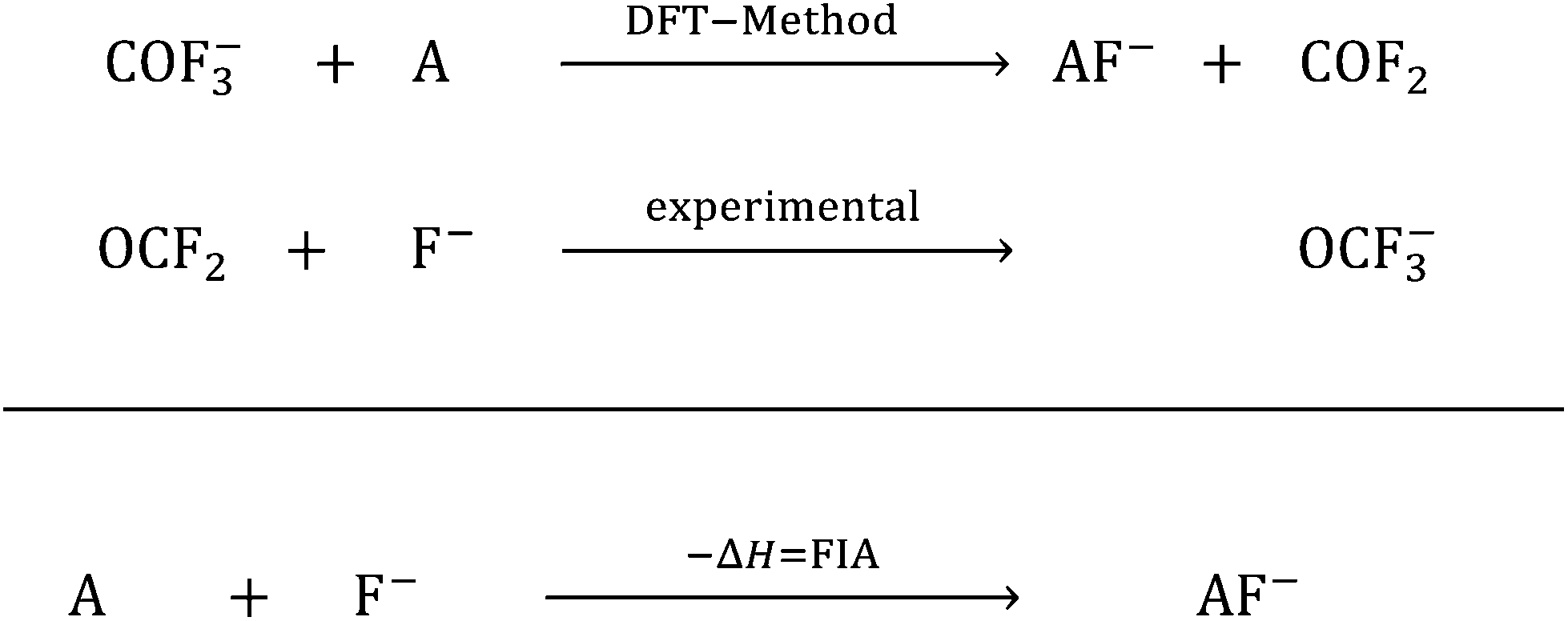
In the past few years, Lewis acids, especially their strongest representatives, have been of great interest and have found applications in catalysis, ionization, rearrangement reactions and bond heterolysis reactions.1–6 Naturally, tabulating the strengths of Lewis acids is very important and useful for estimating the potency of a given Lewis acid. But in contrast to the strength of Brønsted acids, which are typically measured experimentally and ranked within one homogeneous medium on the basis of the well-known pH and pKa scales that can be set as absolute by using the correct reference state and anchor points, the strength of Lewis acids depends on the formation of a Lewis acid–base pair.7–9 In this respect Brønsted acidity is a special case of Lewis acidity, in which only one type of Lewis acid (the proton) interacts with a large variety of Lewis bases free of choice. Thus, it is only possible to determine the absolute strength of a Lewis acid with respect to a well-defined Lewis base. Towards this aim, in 1984 the fluoride ion affinity (FIA) was introduced by Bartlett et al. to classify the strength of the Lewis acid A by the enthalpy that is released by binding a fluoride ion.10 This concept was continued by many others.11–20 To avoid the problems that appear in the calculation of a “naked fluoride ion”, this approach was improved by using the experimental FIA of OCF2 of 209 kJ mol−1 as an anchor point in a (pseudo-)isodesmic reaction (eqn (1)).21| | 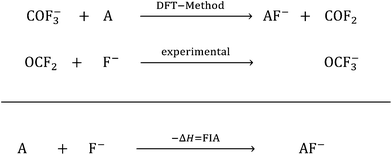 | (1) |
Using this method the strengths of many Lewis acids were classified.22 However, the fluoride ion is a hard base in the HSAB sense,23 so the FIA may be deceptive for HSAB soft Lewis acids. Thus, it is advisable to compare methods with different approaches to find a convenient Lewis acid. Therefore, analogous to the FIA, the affinities to ions of varying hardness (i.e., Cl−, H− and CH3− = Me−) were used to rate the strengths of Lewis acids.17,24,25 Unfortunately, there is no consistent reference system which would allow comparing trends between the different methods and lead to a broadly applicable Lewis acid scale.
However, not only Lewis acids are in the focus of current interest, but also weakly coordinating anions (WCAs) have become an indispensable tool in chemistry and have found versatile applications. Boron26–29 or aluminum30,31 based WCAs were used for lithium ion batteries. Other WCAs stabilized reactive cations, e.g. [CX3]+ (X = Cl, Br, I),14,32,33 [PnXm]+ (X = Br, I),34 [N5]+,35 [P9]+,36 [AuXe4]2+,37 [H(Et2O)2]+,38 [Zn2]2+,39 benzidine radical cations,40 [Cu(S12)(S8)]+,42 [tBu3Si-Ga-SitBu3]+,43 triarylsilylium or germylium ions,44 protonated benzene,45 and the 2-norbornyl cation.46 Weakly bound complexes like those of ethene with copper,48 silver49 or gold are other examples.50 Ion-like silylium ions coordinated to the carborates41 or the recent Me3Si-F-Al(OC((CF3)3)3 present strong Lewis acids.47 Further WCA applications are catalytic C–F activation,51 ionic liquids52–58 and electrochemistry.59–67 Clearly the stability of a typical WCA [M(L)n]− (M = Lewis acidic central atom of valency n − 1; L = univalent residue) is related to the strength of the underlying Lewis acid M(L)n−1. In earlier work, we judged the relative stabilities of WCAs based on calculations for a representative set of WCAs and their parent Lewis acids.18 They were assessed by the ligand affinity (LA), the decomposition in the presence of a hard (proton decomposition, PD) and a soft electrophile (copper decomposition, CuD), the position of the HOMO, the HOMO–LUMO gap as well as the FIA of the Lewis acid parent to the WCA (eqn (2)–(4)).
| | 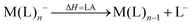 | (2) |
| | 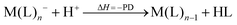 | (3) |
| | 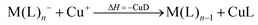 | (4) |
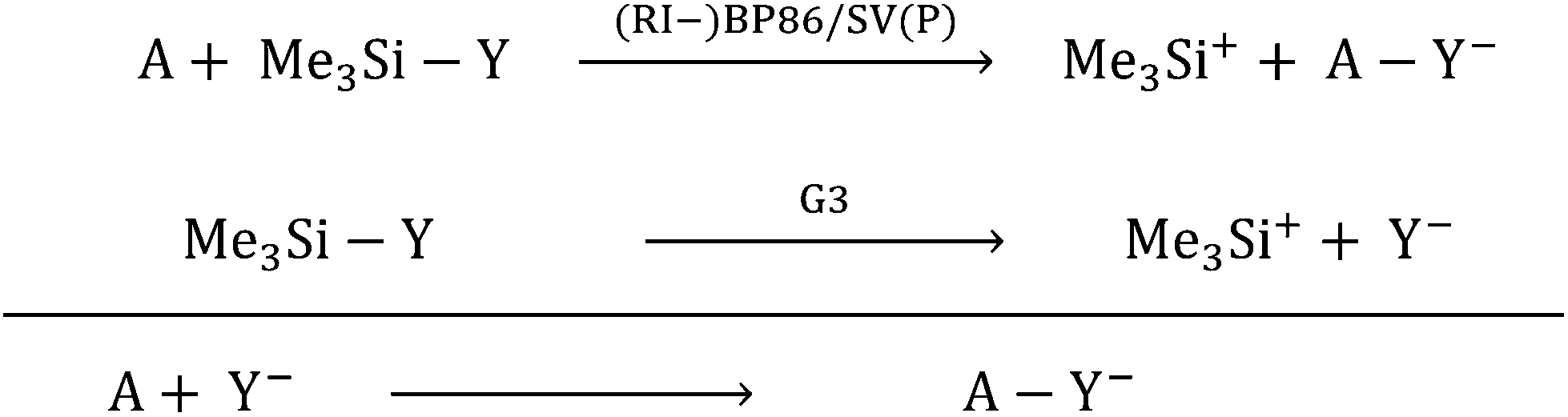
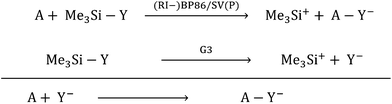
With the current contribution, we augmented the known scale18 for the most common Lewis acids (A) with respect to their CIA (chloride ion affinity), HIA (hydride ion affinity) and MIA (methyl ion affinity) in addition to the FIA. For all XIAs (X = F, C, H, Me), we chose a unified reference system. It is based on the trimethylsilyl compounds Me3SiY (Y = F, Cl, H, Me) and their respective ions calculated at the G3 level as the anchor point. Thus, the relative values of the Lewis acidity towards different bases (Y−) are comparable based on unified reference reactions that were obtained at a highly correlated level, while the residual calculations of the in part very large molecules were assessed based on subsequent isodesmic reactions calculated at a much less expensive level ((RI-)BP86/SV(P)), eqn (5)).
| |  | (5) |
To further validate the data, we performed systematic calculations of the smaller reference systems at the frozen core CCSD(T) level with correlation effects extrapolated to a full quadruple-ζ basis.68–71 The error bar of this methodology was reported to be below 1 kJ mol−1,69 and thus serves as a validation of the simpler isodesmic procedure according to eqn (5) that, for size reasons, had to be applied for larger Lewis acids. Furthermore, we expanded the known WCA stability scale with the PD and CuD of a series of hitherto not explored WCAs [M(L)n]−. All calculations were done in the gas phase.
Results and discussion
The background of these investigations has been experiments to activate hydrogen with the Lewis acid tris(2H-hexafluoroisopropoxy)borane (B(Ohfip)31), which has found applications in electrochemistry.72 We investigated the possibility of obtaining frustrated Lewis pair (FLP) chemistry73 based on this Lewis acid and reacted it with H2 and selected phosphanes, amines and N-heterocyclic carbenes (NHCs), but we have never observed a reaction. Even upon the addition of very strong Lewis bases, adduct formation was partly only occurring in equilibrium. By contrast, the respective anion [B(Ohfip)4]− is known57,74 as a rather stable WCA, and the FIA of the B(Ohfip)3 acid, calculated according to the procedure in eqn (5), is with 384 kJ mol−1 rather large. Moreover, during the course of the synthesis of [B(Ohfip)4]− from Na[BH4] and HO-hfip, the [H–B(Ohfip)3]− anion is a stable intermediate that needs many hours of reflux to further completely react with HO-hfip to give the symmetric borate. For both reasons, the high FIA of the Lewis acid and the known stability of the [H–B(Ohfip)3]− anion, we did not expect these unsuccessful reactions and therefore started to perform calculations to investigate and compare the Lewis acidity towards HSAB-different ions for gaining a deeper insight. In the following we first describe our experiments before turning to the general calculations on a wide range of Lewis acids.
Synthesis and characterization
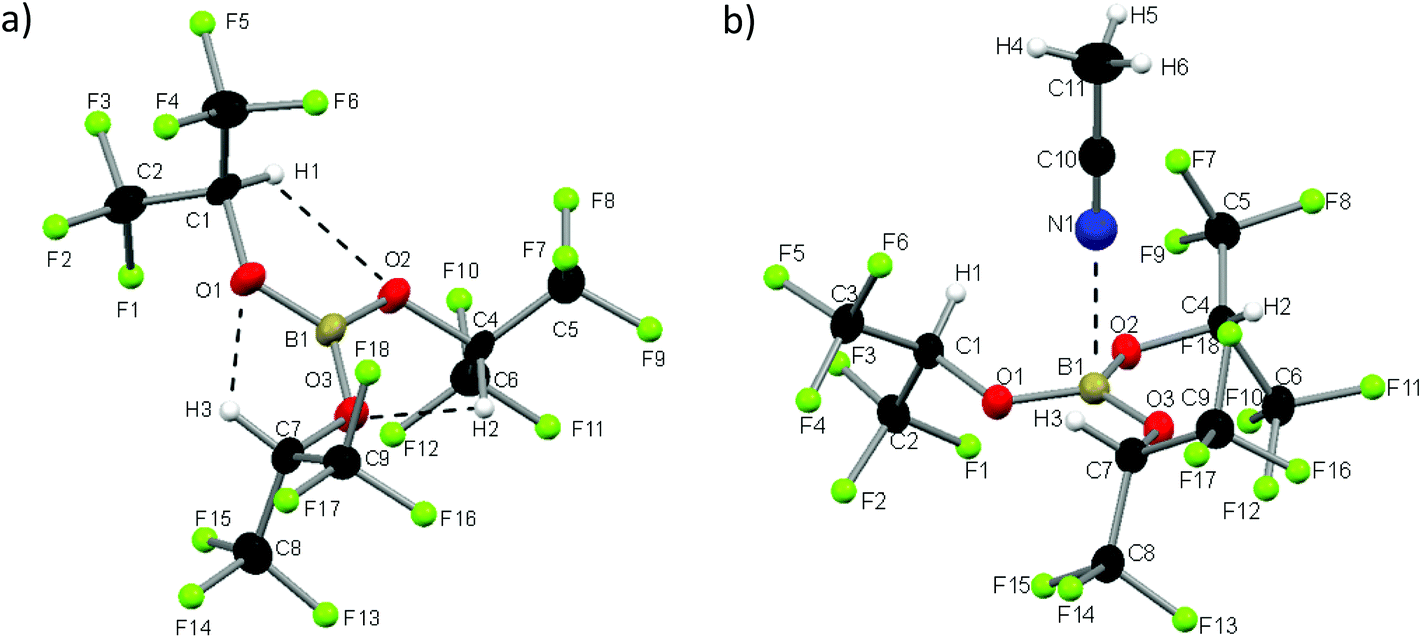
We synthesized 1 according to the literature and obtained white plate-like crystals melting at 32° C.72 The Lewis acid 1 crystallizes in the monoclinic space group P21/c and exists, in contrast to the dimeric heavier homologue Al(Ohfip)3, as a monomer.75 The sum of the three O–B–O angles adds up to 360°, so apparently the orbital interactions of the π-system between the boron and oxygen atoms are maximized and the B–O distances of 136 pm are shortened by 11 pm compared to the distances in the THF adduct of Na[B(Ohfip)4].57 The C–O distances are with 141 pm nearly unchanged compared to those of HO-hfip.76 Interestingly, the central B(OCH)3 unit resides in a plane. This might be attributed to (i) the steric demand of the hfip residues and (ii) the formation of three weak intramolecular (C–)H⋯O hydrogen bonds (dHO = 198 pm). For each ligand, one CF3-group is above and the other is below this central plane. From a multitude of crystallization experiments with a large variety of neutral Lewis bases, we obtained one very weak adduct by dissolving 1 in acetonitrile (1·NCMe) and cooling the solution slowly down to −40 °C. The compound 1·NCMe crystallizes in the trigonal space group R3c. To the best of our knowledge we report the longest distance between boron and a nitrogen atom of an acetonitrile molecule (248 pm, Fig. 1).
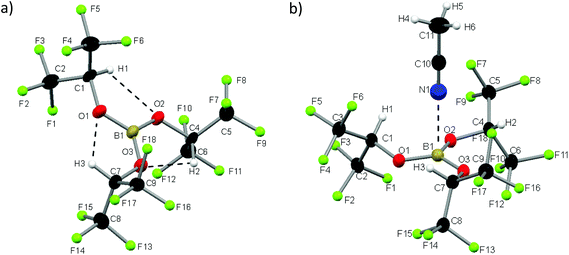 |
| | Fig. 1 Molecular structures of 1 and 1·NCMe; thermal ellipsoids are shown at the 50% probability level. Fluorine atoms are drawn as spheres of arbitrary radius for clarity. Selected distances [pm] and angles [°]: (1): B1–O1 = 136.0(4), B1–O2 = 135.8(4), B1–O3 = 135.9(4), O1–C1 = 141.6(3), O2–C2 = 141.6(3), O3–C3 = 141.1(3), O1–H1 = 198.1, O2–H2 = 198.0, O3–H3 = 197.8, O1–B1–O2 = 120.5(3), O2–B1–O3 = 119.7(3), O1–B1–O3 = 119.8(3), B1–O1–C1 = 122.3(2), B1–O2–C4 = 121.7(2), B1–O3–C7 = 122.3(2); (1·NCMe): B1–N1 = 248.4, B1–O1 = 136.97(9), O1–C1 = 140.46(16), O1–H1 = 197.0, N1–H1 = 299.4, O1–B1–O2 = 119.57(3), B1–O1–C1 = 121.31(9). | |
We were rather astonished to see this weak interaction, as the boron nitrogen distances in related complexes like MeCN·BCl3, MeCN·B(C6F5)3 or the acetonitrile adduct of the perfluoroaryldiborane C6F4-1,2-(B(C6F5)2)2 are 156, 162 and 161 pm.77–79 The boron atoms of these complexes are distorted tetrahedrally coordinated. The hitherto longest boron nitrogen distance was measured in the mixed crystal MeCN·(B(CH2Ph)3)0.92(Ga(CH2Ph)3)0.08 and amounts to 178 pm.80 The O–B–O angles in 1·NCMe amount on average to 119.6° and add up to 358.7°, which signals an almost ideal trigonal planar geometry. The averaged B–O bond length is 137 pm and hence only very slightly widened compared to 1 (136 pm). The acetonitrile molecule of 1·NCMe lies on a pseudo C3 axis.
FLP chemistry
We continued our investigation by testing the ability of 1 to activate hydrogen with different FLP compounds. To analyze the thermodynamics of the hydrogen activation of 1 in comparison with the well-known B(C6F5)3 the standard Gibbs energies were calculated in the gas phase and different solvents (COSMO solvation model) by using a Born–Fajans–Haber cycle (Fig. 2, Table 1).
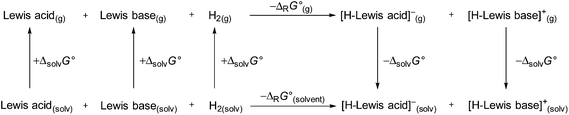 |
| | Fig. 2 Born–Haber–Fajans cycle to access the ΔrG°(g) and ΔrG°(solvent) of 1 and B(C6F5)3 in various solvents. | |
Table 1 ΔrG°(g) and ΔrG°(solvent) of the H2-activation of selected Lewis acid/base-pairs in various solvents (BP86/SV(P)) and calculated according to the cycle in Fig. 2; NHC = 1,3-dimethyl-4,5-diphenyl imidazole-2-ylidene, IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene
|
Vs.
1
|
ΔrG°(g) |
ΔrG°(CH2Cl2) |
ΔrG°(o-difluoro-benzene) |
ΔrG°(MeCN) |
|
Lewis base
|
| PMePh2 |
474 |
341 |
332 |
324 |
| PtBu3 |
464 |
191 |
174 |
155 |
| NEt3 |
420 |
132 |
114 |
94 |
| 2,6-Lutidine |
475 |
148 |
127 |
113 |
| NHPh2 |
480 |
162 |
141 |
120 |
| NHC |
313 |
167 |
158 |
163 |
| IDipp |
310 |
53 |
37 |
3 |
|
Vs. B(C6F5)3 |
ΔrG°(g) |
ΔrG°(CH2Cl2) |
ΔrG°(o-difluoro-benzene) |
ΔrG°(MeCN) |
|
Lewis base
|
| PMePh2 |
305 |
196 |
188 |
181 |
| PtBu3 |
292 |
54 |
39 |
22 |
| NEt3 |
247 |
−5 |
−21 |
−39 |
| 2,6-Lutidine |
308 |
25 |
6 |
−14 |
| NHPh2 |
384 |
273 |
266 |
259 |
| NHC |
140 |
30 |
23 |
15 |
| IDipp |
137 |
−84 |
−98 |
−114 |
In agreement with our futile experimental efforts, Table 1 shows that FLP chemistry based on 1 is considerably less favored than that with B(C6F5)3. Furthermore, these investigations suggested that polar solvents promote H2 activation and therefore acetonitrile was inter alia selected as a solvent. However, we have to point out that even the very weak adduct formation in 1·MeCN might shut down FLP chemistry. To cope with this concern, we have done several test reactions also in chlorinated solvents like CH2Cl2 and did not observe any desired chemistry with H2 but noted that the liquid B(Ohfip)3 forms two immiscible phases in CH2Cl2. Thus, the investigations were discarded and we concentrated on MeCN, despite its problems. To obtain an experimental confirmation of the relative HIA of the two boron Lewis acids B(Ohfip)3 and B(C6F5)3, we did react K+[HB(Ohfip)3]− with B(C6F5)3 and expected to get neutral B(Ohfip)3 and K+[HB(C6F5)3]−. This isodesmic reaction was calculated by (RI–)BP86/SV(P) to be exothermic (–135 kJ mol−1) and exergonic (–149 kJ mol−1) in the gas phase, in agreement with our expectation from Table 1. Our NMR scale experiments in MeCN and deuterated toluene confirmed the results of the calculations. After the addition of K+[HB(Ohfip)3]− to a solution of B(C6F5)3 in toluene-d8 immediately a white solid precipitated (mainly K+[HB(C6F5)3]−). The broad singlet at 17.7 ppm in the 11B NMR spectra of the solution revealed that B(Ohfip)3 had formed, but only a very weak signal of scarcely soluble K+[HB(C6F5)3]− (at around −25 ppm, 1JH,B ≈ 84 Hz) and no remaining B(C6F5)3 or [HB(Ohfip)3]− was observed. After removing most of the solution with a syringe, deuterated MeCN was added to examine the now soluble precipitate. The 11B NMR spectrum shows mainly the doublet of [HB(C6F5)3]− at the typical values δ11B = −25.4 (1JH,B 93 Hz). Since it was not possible to get rid of all the solution, still a small signal of B(Ohfip)3 was present in the spectra. It seems as if a complete conversion takes place in toluene; however, due to the presence of both B(Ohfip)3 and [HB(C6F5)3]− in acetonitrile, small amounts of [HB(Ohfip)3]− are being formed again. This was backed by the results of the same reaction in acetonitrile instead of toluene, in which the reaction reaches equilibrium, but largely lies on the side of [HB(C6F5)3]−. This seems reasonable, since B(C6F5)3 should be less reactive in acetonitrile due to strong adduct formation, while B(Ohfip)3 is only marginally stabilized by MeCN.
Experiments directed towards FLP chemistry
We started experimental investigations with several common Lewis bases. All following reactions were carried out under an argon atmosphere in NMR tubes closed by a J. Young valve according to the following procedure: compound 1 and the different phosphanes, amines and NHCs were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry in d3-acetonitrile and characterized by NMR spectroscopy. Subsequently the reaction mixtures were evacuated, exposed to hydrogen pressure of one bar, and were then again NMR spectroscopically analyzed. We started our attempts by using phosphanes, since e.g. B(C6F5)3 (i) forms an adduct with PPh3,81 (ii) in combination with (C6F5)Ph2P it leads to a FLP system capable of reversibly activating hydrogen82 and (iii) a mixture with PtBu3 is able to activate terminal alkynes.83 Here PPh3, PtBu3 and PMePh2 did not form classical adducts with 1, and there is no sign of a hydrogen activation (e.g. doublet 1JPH in the 31P NMR, doublet 1JBH splitting in the 11B NMR spectrum: expected for [HB(Ohfip)3]− 127 Hz at δ = 7.7 ppm). To eliminate the possibility that the reaction occurs very slowly, in a second approach, 1 was stirred with PPh3 over twenty days and exposed to H2 at 1 bar, but without any noticeable reaction. Of all the tested bases collected in Table 1, 1 only forms an adduct visible in the NMR with NEt3. However, this adduct only exists in equilibrium. Also testing the ability to activate hydrogen failed in all instances. No adduct formation and no hydrogen cleavage were detected by the combination of 1 and NHPh2 or 2,6-lutidin, which are able to activate hydrogen in combination with B(C6F5)3.84 Since it appeared that the reactivity of 1 is not sufficient to activate H2 with typical Lewis bases like phosphanes and amines, we used the strongly basic NHCs 1,3-dimethyl-4,5-diphenyl-imidazol-2-ylidene and 1,3-di(2,6-diisopropylphenyl)imidazol-2-ylidene. They form classical adducts with 1, which were observed in the 11B NMR spectra at 0.8/1.3 ppm, but no detectable hydrogen activation occurred.
:1 stoichiometry in d3-acetonitrile and characterized by NMR spectroscopy. Subsequently the reaction mixtures were evacuated, exposed to hydrogen pressure of one bar, and were then again NMR spectroscopically analyzed. We started our attempts by using phosphanes, since e.g. B(C6F5)3 (i) forms an adduct with PPh3,81 (ii) in combination with (C6F5)Ph2P it leads to a FLP system capable of reversibly activating hydrogen82 and (iii) a mixture with PtBu3 is able to activate terminal alkynes.83 Here PPh3, PtBu3 and PMePh2 did not form classical adducts with 1, and there is no sign of a hydrogen activation (e.g. doublet 1JPH in the 31P NMR, doublet 1JBH splitting in the 11B NMR spectrum: expected for [HB(Ohfip)3]− 127 Hz at δ = 7.7 ppm). To eliminate the possibility that the reaction occurs very slowly, in a second approach, 1 was stirred with PPh3 over twenty days and exposed to H2 at 1 bar, but without any noticeable reaction. Of all the tested bases collected in Table 1, 1 only forms an adduct visible in the NMR with NEt3. However, this adduct only exists in equilibrium. Also testing the ability to activate hydrogen failed in all instances. No adduct formation and no hydrogen cleavage were detected by the combination of 1 and NHPh2 or 2,6-lutidin, which are able to activate hydrogen in combination with B(C6F5)3.84 Since it appeared that the reactivity of 1 is not sufficient to activate H2 with typical Lewis bases like phosphanes and amines, we used the strongly basic NHCs 1,3-dimethyl-4,5-diphenyl-imidazol-2-ylidene and 1,3-di(2,6-diisopropylphenyl)imidazol-2-ylidene. They form classical adducts with 1, which were observed in the 11B NMR spectra at 0.8/1.3 ppm, but no detectable hydrogen activation occurred.
To investigate whether the Li+ salt of the [HB(Ohfip)3]− anion and a protonated Lewis base like [HNEt3]+Cl− are compatible, we dissolved both salts in acetonitrile, and tested whether they are compatible in solution or if they release elemental hydrogen. However, even after stirring for one week, no gas formation occurred and the NMR signals remained unchanged. Thus, no regeneration of the original Lewis acid/base pairs occurred. The formation of LiCl was never observed in these reactions and it appears that either the salts dissolve as tight ion pairs with no chance of ion exchange (unlikely in polar MeCN) or that the small amounts of LiCl remain metastable as [LiCl(donor)x]y aggregates in solution. Overall, we have to note that H2 activation might still be possible with this system, e.g. with bulkier NHCs as well as HD gas to look for scrambling and H2 formation in the 1H NMR spectrum, but at least not under our tested reaction conditions.
Quantum chemical investigations
Ion affinities of Lewis acids A.
Earlier and the following calculations to determine ion affinities were based on isodesmic reactions (eqn (1) and (5)) with the BP86 functional85–87 and the SV(P) basis,88 which represent good agreement between costs and accuracy. This method allows fast access to large molecules, which cannot be calculated at correlated levels. Basis reactions of the respective ion affinity calculations were the ion affinities of Me3Si+ against Y− giving Me3SiY (Y = F, Cl, H, Me), which were calculated at the reliable G3 level (Table 2).
Table 2 Calculated reference reaction values (G3 level) as anchor points to determine FIA, CIA, HIA and MIA values
| Reference systems |
ΔrH° [kJ mol−1] |
| Me3Si–F → Me3Si+ + F− |
+958 |
| Me3Si–Cl → Me3Si+ + Cl− |
+759 |
| Me3Si–H → Me3Si+ + H− |
+959 |
| Me3Si–Me → Me3Si+ + Me− |
+1000 |
Validation study.
During our investigation to improve these scales by unifying the reference system and adding up the CIA, HIA and MIA values, we noticed a larger discrepancy in our earlier published FIA values for BI3, AlCl3 and AlI3.22 To validate and confirm the BP86/SV(P) values, we carried out RI-MP2 structure optimizations with TURBOMOLE89,90 and def2-QZVPP basis sets91 with the corresponding RI-C auxiliary bases for all atoms.92 Based on these structures, gas phase reaction energies were calculated according to eqn (6) with single point calculations at the CCSD(T)(FC)/double-ζ level plus an MP2 extrapolation of the correlation energy from double-ζ to quadruple-ζ basis sets with Gaussian 09 (ESI, S-Table 1†).93| | 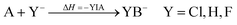 | (6) |
This approach was published earlier by Klopper et al.68,69 and has successfully been used by our group to study protonation equilibria.7–9
The CIA and FIA values in Table 3 agree well with the values calculated by BP86/SV(P); however, some of the values may be off by up to 17 kJ mol−1. The BP86/SV(P) HIA values of the boron halides, B(CN)3 and B(OH)3 nicely suit, since the B–H bond is less polar. Nevertheless, if the central atom is not a second row element, the bond becomes more polar or hydridic, so the HIA values may be approximately 20 kJ mol−1 too high. In order to get better values we also calculated the HIA values with BP86/SVP with a polarization function at the hydrogen, but those values were even inferior to those calculated with BP86/SV(P). Also orienting BP86/TZVP calculations were inferior to the simple BP86/SV(P) method. Thus, for simplicity and to be applicable for a larger set of compounds, we used the BP86/SV(P) method. Since the discrepancy in the HIA values from the absolute values is always in one direction, the relative HIA values at the simpler BP86/SV(P) level are still suitable for discussion.
Table 3 Overview of the CIA, HIA and FIA values [in kJ mol−1] of chosen representative Lewis acids calculated via CCSD(T)(FC)/double-ζ level plus MP2 extrapolation to quadruple-ζ. The values in parentheses give the discrepancy to the affinity values calculated viaeqn (5) at the BP86/SV(P) level
| Lewis acid |
CIA |
HIA |
FIA |
|
Does not form a complex.
[SbF5H]− is not stable and would decompose to SbF4− + HF; this value is given in brackets.
|
| BF3 |
151 (5) |
297 (−2) |
346 (4) |
| BCl3 |
195 (12) |
395 (5) |
384 (8) |
| BBr3 |
219 (7) |
440 (2) |
425 (−16) |
| AlF3 |
308 (1) |
388 (−36) |
482 (11) |
| AlCl3 |
320 (3) |
428 (−22) |
502 (4) |
| AlBr3 |
324 (−2) |
425 (−39) |
505 (−5) |
| GaF3 |
319 (14) |
444 (−18) |
447 (13) |
| GaCl3 |
299 (5) |
446 (−19) |
429 (−5) |
| GaBr3 |
295 (1) |
445 (−24) |
426 (−13) |
| PF5 |
165 (−14) |
400 (−17) |
380 (−17) |
| PCl5 |
179 (2) |
468 (−14) |
393 (1) |
| AsF5 |
237 (−14) |
461 (−24) |
434 (4) |
| SbF5 |
333 (−8) |
530 [517]b (−32/−13) |
495 (2) |
| B(CN)3 |
363 (12) |
583 (−3) |
540 (−10) |
| B(OH)3 |
|
163 (−7) |
208 (4) |
Ion affinity scale.
With this cadre of verified data, we calculated the FIA, CIA, HIA and MIA for a large set of 33 Lewis acids A through a set of isodesmic reactions as given in eqn (5) and with respect to the ion affinities of Me3Si–Y (Y = F, Cl, H, Me) at the G3 level (Table 5).
The HIA and MIA values of main group III halides (Table 4) follow similar trends and rise for the heavier halogen atoms. If we take a look at the affinity values of AlF3 and GaF3 there is not such a large difference compared to AlF3 and BF3, since aluminum and gallium have nearly the same size and aluminum is just a little bit more electropositive than gallium. However, they differ in their affinity values towards soft or hard Lewis bases. AlF3 favors the hard fluoride ion, while GaF3 prefers softer Lewis bases like Me− or H− anions, which confirms that aluminum trifluoride is harder than gallium trifluoride. Nevertheless, if the halide ligands become heavier, gallium turns out scaled by aluminum, which indicates that the GaX3 Lewis acids are more stable and less acidic. This could mean that the overlap of the π orbitals is more pronounced and hence the π back bonding is stronger. To summarize, in this row, the AlX3 compounds are the strongest Lewis acids with respect to fluoride and chloride, but towards the hydride ion, GaX3 acids are stronger and with respect to the methanide ion similar to the aluminum acids. The CIA and FIA values of PF5 and PCl5 are quite low and nearly identical, which may be a result of the electrostatic repulsion of the six rather hard halides in the octahedral complexes. For the strongest classical Lewis acid SbF5, two HIA and MIA values are given, since [SbF5H]− and [SbF5Me]− are not stable and would decompose.
Table 4 Overview of the calculated CIA, HIA, FIA and MIA values of representative Lewis acids. [SbF5H]− and [SbF5Me]− are less stable and would decompose to SbF4− + HF and SbF4− + MeF, respectively; this value is given in brackets. TMS-F-Al(OC(CF3)3)3 can be attacked, depending on the nucleophile, at the Si or the Al atom and decomposes either to TMS-Y + F-Al(OC(CF3)3)3 or TMS-F + Y-Al(OC(CF3)3)3. The values for the nucleophilic attack at the Al atom are given in parentheses
| Lewis acid |
LUMO [eV] |
CIA [kJ mol−1] |
HIA [kJ mol−1] |
FIA [kJ mol−1] |
MIA [kJ mol−1] |
Lewis acid |
LUMO [eV] |
CIA [kJ mol−1] |
HIA [kJ mol−1] |
FIA [kJ mol−1] |
MIA [kJ mol−1] |
|
The anion containing a B–Y–B bridge is thermodynamically favored.
The most stable isomer was used.
The experimental crystal structure was used as the start geometry.94 Unstable compounds and the values of unstable compounds are given in italics.
|
|
M–X
|
|
|
|
|
|
B(Ohfip)3 |
−0.42 |
141 |
348 |
384 |
387 |
| BF3 |
−0.29 |
146 |
299 |
342 |
355 |
Al(OC(CF3)3)3 |
−1.51 |
352 |
490 |
543 |
530 |
| BCl3 |
−2.38 |
183 |
391 |
405 |
436 |
B(OTeF5)3 |
−6.78 |
325 |
556 |
552 |
602 |
| BBr3 |
−2.88 |
213 |
438 |
441 |
477 |
As(OTeF5)5 |
−7.30 |
403 |
710 |
559 |
753 |
| BI3 |
−3.41 |
261 |
505 |
493 |
540 |
Sb(OTeF5)5 |
−7.62 |
465 |
746 |
625 |
809 |
| AlF3 |
−2.24 |
306 |
423 |
471 |
464 |
M–C
|
|
|
|
|
|
| AlCl3 |
−2.18 |
318 |
450 |
498 |
490 |
B(CN)3 |
−5.97 |
351 |
587 |
551 |
610 |
| AlBr3 |
−2.48 |
326 |
464 |
510 |
502 |
B(CF3)3 |
−4.77 |
358 |
583 |
556 |
614 |
| AlI3 |
−3.01 |
347 |
497 |
535 |
535 |
B(C6F5)3 |
−3.93 |
236 |
484 |
452 |
483 |
| GaF3 |
−3.41 |
306 |
462 |
434 |
491 |
Al(C6F5)3 |
−3.07 |
348 |
483 |
536 |
518 |
| GaCl3 |
−3.13 |
294 |
464 |
434 |
493 |
Ga(C6F5)3 |
−3.28 |
307 |
479 |
453 |
502 |
| GaBr3 |
−3.34 |
295 |
470 |
438 |
498 |
B(C12F9)3 |
−3.95 |
190 |
452 |
431 |
415 |
| GaI3 |
−3.74 |
310 |
495 |
457 |
523 |
B(C6H3(CF3)2)3 |
−4.04 |
281 |
486 |
482 |
504 |
| PF5 |
−1.53 |
179 |
417 |
398 |
456 |
B(C10F7)3 |
−3.91 |
265 |
519 |
483 |
519 |
| PCl5 |
−4.73 |
178 |
483 |
392 |
507 |
F4C6(1,2-(B(C6F5)2)2 |
−4.11 |
309 |
536a |
523 |
493a |
| AsF5 |
−4.22 |
251 |
485 |
430 |
527 |
B2(C6F5)2(C6F4)2 |
−4.68 |
271 |
514 |
477 |
524 |
| SbF5 |
−5.83 |
341 |
562 [531] |
493 |
607 [544] |
PF2(C2F5)3 |
−2.42 |
157b |
428b |
388c |
426b |
|
M–O
|
|
|
|
|
|
SiMe2CH2CB11Cl11 |
−3.03 |
397 |
590 |
597 |
627 |
| B(OH)3 |
−0.13 |
|
170 |
204 |
220 |
TMS-F-Al(OC(CF3)3)3 |
−1.21 |
259 (267) |
459 (407) |
458 |
459 (407) |
Putting boron acids in context.
As expected, B(OH)3 is a quite weak Lewis acid and since [B(OH)3Cl]− is experimentally unknown, we disregarded this anion from our calculations. Compound 1 reveals relatively low ion affinity values. Compared to B(C6F5)3 the FIA value of 1 is just 68 kJ mol−1 lower, but the HIA value is 136 kJ mol−1 lower. This huge difference in the HIA value may explain our experimentally observed results. Similarly, the HIA of B(C6F5)3 is nearly identical with that one of the versus fluoride considerably stronger Lewis acid Al(C6F5)3. B(C12F9)3 and B(C6H3(CF3)2)3 show the same effect, which implies that boron Lewis acids stabilize the hydride ion better than other Lewis acids A with more electropositive central atoms like AI. These results are in agreement with HSAB-arguments: 1 and Al(C6F5)3 are considerably harder than the softer boranes with B–C bonds and thus the affinities of the softer boranes are maximal with respect to the softer hydride ions, whereas those of the harder acids are maximal for hard bases like fluoride. The low CIA and FIA, MIA values of B(C12F9)3 result from the sterically demanding perfluorinated biphenyl ligands, which hinder the access to the Lewis acid center. Since F4C6(1,2-(B(C6F5)2)2 has two nearby Lewis acid atoms, small ions like fluoride and hydride prefer a bridged geometry, but the difference between the bridged and non-bridged geometry is smaller than 1 kJ mol−1.
Group 14 acids.
Main group IV is represented by the silylium zwitterion SiMe2CH2CB11Cl11,95 and the Janus-headed Lewis acid TMS-F-Al(OC(CF3)3)3.47 The ion affinity of the silylium zwitterion is boosted by around 140 kJ mol−1 compared to the silicon atom in neutral TMS-F-Al(OC(CF3)3)3, which itself features IAs only slightly inferior to B(C6F5)3.
Lewis superacids.
Since SbF5 is viewed as the strongest conventional Lewis acid, the FIA value of monomeric SbF5 is used to determine Lewis superacids: “Molecular Lewis acids, which are stronger than monomeric SbF5 in the gas phase are Lewis superacids.”96 This approach is simple, and since strong Lewis acids and the fluoride anion are typically hard, it gives a nice overview of Lewis acids by their relative strengths. The FIA values of monomeric AlCl3, AlBr3, AlI3, Al(OC(CF3)3)3, B(OTeF5)5, As(OTeF5)5, Sb(OTeF5)3, B(CN)3, B(CF3)3, Al(C6F5)3, F4C6(1,2-(B(C6F5)2)2 and SiMe2CH2CB11Cl11 excel the FIA value of SbF5 and may be classified as Lewis superacids. However, P-based acids like PF2(C2F5)3 possess, compared to PF5, slightly lower CIA, FIA and MIA values, due to the steric demand of the C2F5 groups in the acid–base complexes. The HIA of PF2(C2F5)3 is a bit higher, since H− is small and the steric effect of the C2F5 groups is overcompensated by its electron withdrawing effect.
Relationship between ion affinity values and the LUMO level.
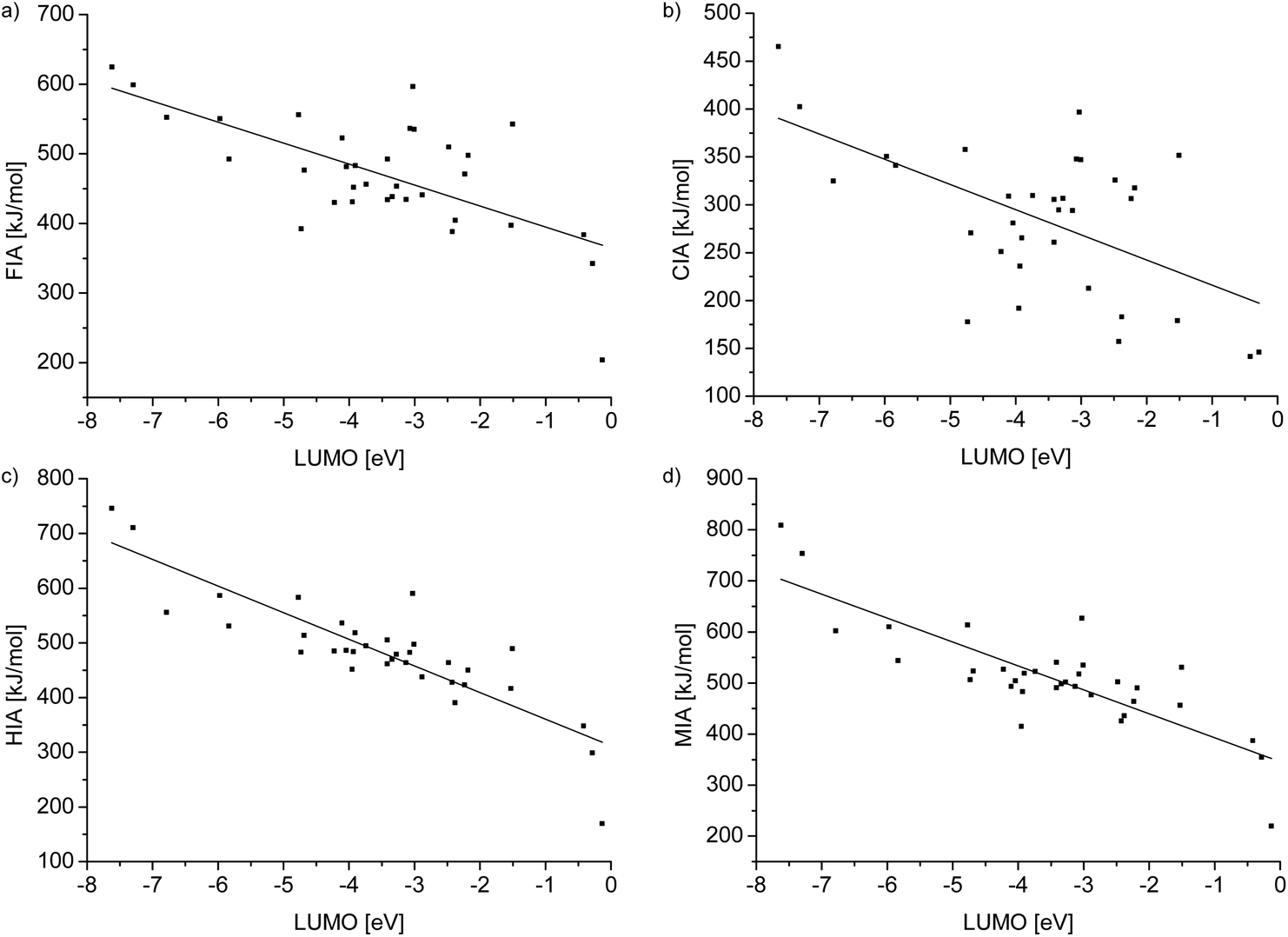
To gain a deeper insight into the relationship between the ion affinity values and the LUMO levels of the Lewis acids in Table 4, we plotted the values and added a regression line in each case (see Fig. 3).
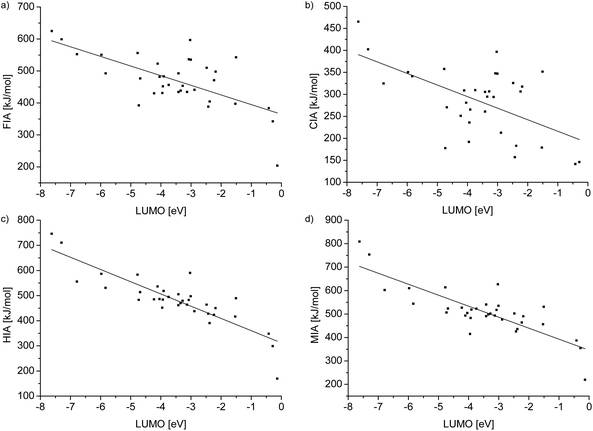 |
| | Fig. 3 Plots showing the regression lines of the relationships between the ion affinity values and the LUMO level of the Lewis acids collected in Table 4. Linear regression of ion affinity values against the LUMO level for (a): y = −30.14(6.05)x + 364.61(24.86), R2 = 0.41, (b): y = −26.31(6.71)x + 189.68(26.85), R2 = 0.31, (c): y = −48.68(5.06)x + 311.86(19.98), R2 = 0.73 and (d): y = −46.90(5.75)x + 345.77(22.68), R2 = 0.67. | |
By trend, the gradient shows that a low LUMO level is accompanied by a high ion affinity value. However, a low LUMO level is not automatically connected with a high ion affinity value. Furthermore, R2 and the value of the gradient of the FIA and CIA values are, compared to the corresponding values of the HIA and MIA values, significantly smaller. These facts are consistent with Pearson's HSAB concept, since interactions of the soft hydride and methyl ions with Lewis bases are more orbital based and consequently strongly influenced by the LUMO levels. Therefore, the HIA and MIA values are in a distinctive relationship with the LUMO values. Especially the HIA, with significantly less steric influence on the result, can be well predicted by the LUMO level of a given Lewis acid and vice versa. On the other hand, the interaction of the hard fluoride and chloride ions has a high ionic contribution and thus it is less connected to the LUMO energies. At first sight, the FIA would be expected to have an even lower R2 value than the CIA, because the fluoride ion is harder than the chloride ion. However, the chemistry of fluoride is sometimes exceptional: since the fluoride atom is smaller than the chloride, it is possible that the overlapping of the involved orbitals is improved and steric effects are less developed. Therefore, it obtains a more covalent character and fits the trend slightly better than chloride.
Stability of WCAs based on FIA, PD, CuD, and HOMO levels and the HOMO–LUMO gap.
If the above mentioned Lewis acids are expanded by an L− ligand, the related WCAs are obtained. We determined the stability towards decomposition, oxidation and reduction for most of the WCAs that relate to the Lewis acids in Table 4. The higher the FIA of the acid, the more stable is the WCA towards ligand abstraction. To rate the stability of a WCA towards attack of a hard (H+) and a soft electrophile (Cu+) the isodesmic decomposition reactions (eqn (2) and (3)) were calculated to obtain the proton decomposition (PD) and the copper decomposition (CuD). Herein we show, instead of the previously used ΔrU values, the ΔrG° values of the PD and CuD; hence they are closer to laboratory conditions.
The entropy S of the H+ and Cu+ cations was calculated using the Sackur–Tetrode equation.97–99 Since a gaseous anion and a gaseous cation react to give two neutral species, the PD and CuD are both exothermic. The less negative the PD and CuD values are, the more stable is the WCA against electrophilic attack.18 The lower the HOMO energy, the more resistant is an anion towards oxidation and hence the electron is harder to remove. The HOMO–LUMO gap is related to its resistance towards reduction. The larger the gap, the more stable is the WCA towards gaining an electron. The data in Table 5 cannot be taken as absolute, but since the same calculation methods were used, relative trends will definitely be correct.
Table 5 Calculated properties of WCAs: FIA of the parent Lewis acid as shown in Table 4
| Anion with M–X bonds |
FIA [kJ mol−1] |
PD [kJ mol−1] |
CuD [kJ mol−1] |
HOMO [eV] |
Gap [eV] |
| [BF4]− |
342 |
−1212 |
−540 |
−1.799 |
10.820 |
| [BCl4]− |
405 |
−1237 |
−629 |
−1.708 |
7.975 |
| [BBr4]− |
441 |
−1225 |
−631 |
−1.787 |
5.820 |
| [BI4]− |
493 |
−1190 |
−613 |
−2.127 |
3.620 |
| [AlF4]− |
471 |
−1083 |
−411 |
−2.510 |
8.016 |
| [AlCl4]− |
498 |
−1102 |
−493 |
−2.546 |
7.054 |
| [AlBr4]− |
510 |
−1105 |
−511 |
−2.505 |
5.685 |
| [AlI4]− |
535 |
−1095 |
−519 |
−2.658 |
3.973 |
| [GaF4]− |
434 |
−1118 |
−446 |
−2.677 |
7.048 |
| [GaCl4]− |
434 |
−1125 |
−516 |
−2.604 |
5.501 |
| [GaBr4]− |
438 |
−1127 |
−533 |
−2.532 |
4.369 |
| [GaI4]− |
457 |
−1112 |
−536 |
−2.673 |
3.051 |
| [PF6]− |
398 |
−1163 |
−491 |
−2.673 |
8.801 |
| [PCl6]− |
392 |
−1242 |
−633 |
−2.246 |
1.929 |
| [AsF6]− |
430 |
−1129 |
−457 |
−3.150 |
6.282 |
| [SbF6]− |
493 |
−1065 |
−393 |
−3.911 |
5.134 |
| |
|
With M–O bonds
|
| [B(Ohfip)4]− |
384 |
−1212 |
−526 |
−3.377 |
7.056 |
| [Al(OC(CF3)3)4]− |
543 |
−1077 |
−413 |
−4.096 |
6.737 |
| [B(OTeF5)4]− |
552 |
−1098 |
−496 |
−5.547 |
2.126 |
| [As(OTeF5)6]− |
559 |
−1053 |
−452 |
−6.129 |
1.983 |
| [Sb(OTeF5)6]− |
625 |
−999 |
−398 |
−6.460 |
2.181 |
| |
|
With M–C bonds
|
| [B(CN)4]− |
551 |
−1092 |
−438 |
−4.182 |
6.818 |
| [B(CF3)4]− |
556 |
−1143 |
−411 |
−3.527 |
9.069 |
| [B(C6F5)4]− |
452 |
−1263 |
−567 |
−3.120 |
4.214 |
| [Al(C6F5)4]− |
536 |
−1224 |
−528 |
−3.304 |
4.251 |
| [Ga(C6F5)4]− |
453 |
−1246 |
−550 |
−3.308 |
4.332 |
| [B(C12F9)4]− |
431 |
−1231 |
−534 |
−3.517 |
3.339 |
| [B(C6H3(CF3)2)4]− |
482 |
−1250 |
−527 |
−3.798 |
3.930 |
| [B(C10F7)4]− |
483 |
−1236 |
−540 |
−3.098 |
2.795 |
| [F4C6(1,2-(B(C6F5)2)2)(C6F5)]− |
523 |
−1325 |
−629 |
−3.207 |
1.969 |
| [B2(C6F5)3(C6F4)2]− |
477 |
−1259 |
−563 |
−3.284 |
2.548 |
Among the [MX4]− anions (M = B, Al, Ga; X = F, Cl, Br, I) the [MF4]− and [MCl4]− anions are exceptionally stable towards reduction (see gap), and the [MF4]− and [MI4]− anions have an increased resistance against an attack of a soft or a hard nucleophile (see PD and CuD). This confirms that especially fluorine is a suitable ligand at the central atom to enhance the properties of a WCA. Usually, the HOMO level rises with increasing weight of the central atoms and ligands. The alkoxyaluminate [Al(OC(CF3)3)4]− possesses distinguished thermodynamic stability values paired with a simple straightforward synthesis. Compared to the teflate based anions it offers similar values (see FIA, PD, CuD, and HOMO levels), but it is significantly more reduction resistant. These values underline its current role in chemistry to stabilize highly reactive cations.14,33,38,39,42,43,48–50,100,101 In addition, Table 5 includes a series of WCAs with (mainly fluorinated) organic ligands. Of those, [B(CN)4]− and [B(CF3)4]− offer very good WCA properties, although [B(CF3)4]− allows for additional decomposition pathways.102 Typically, the fluorination of ligands increases the stability values of borate based anions.18 Apart from that, the stability values of all the WCAs including M–C bonds are quite similar.
Conclusions
Attempts to use the conveniently available B(Ohfip)3 as a Lewis acid component in FLP chemistry systems failed in our hands for a wide range of neutral Lewis bases and with all the used reaction conditions. However, it may well be possible using the right bases (e.g. bulky NHCs), solvents other than MeCN and using HD to study the exchange reactions. To investigate this unexpected result, we introduced and validated a Lewis acid scale based on a consistent reference system (Me3Si–Y/Me3Si+/Y−; Y = Cl, H, F, Me). Validation was performed at the highly reliable ccsd(t)/DZ→QZ level for a subset of 15 smaller Lewis acids MXn. These values with an error bar below 1 kJ mol−1 are currently the best available benchmark calculations on Lewis acidity for these systems. With the consistent reference system and a set of isodesmic reactions, we calculated the CIA, HIA, FIA and MIA values of 33 common and frequently used, partly rather large Lewis acids (Table 4). With this approach, comparable ion affinity values were obtained for four different Lewis bases Y− of differing HSAB hardness. For any given Lewis acid, the consistent ion affinity scale may be extended by performing only five low level calculations and using the herein established reference system. In addition, we evaluated the stability of WCAs that are based on the herein investigated Lewis acids, by calculating their LUMO energies, HOMO–LUMO gap, proton decomposition (PD) and the copper decomposition reaction (CuD). Overall, the reference data collected in this work will be of great help to rationalize experimental findings in all areas of chemistry exploiting Lewis acidity towards hard or soft bases or the selection of a suitable WCA counterion for a given process.
Experimental data
Techniques and instruments
All reactions were carried out under an inert atmosphere by using standard vacuum and Schlenk techniques or a glovebox with an argon atmosphere (H2O and O2 <1 ppm). Special J. Young NMR tubes sealed with Teflon valves were used to exclude air and moisture. All solvents were dried over CaH2 or P4O10 and distilled afterwards. NMR data were recorded from solutions in d8-toluene or d3-acetonitrile at room temperature on a BRUKER AVANCE II+ 400 MHz WB spectrometer. 1H and 13C chemical shifts are given with respect to TMS, 19F NMR spectra to fluorotrichloromethane, 11B NMR spectra to the boron-trifluoride-diethyl-ether-complex, 31P NMR spectra to a 85% phosphoric(V) acid solution and 7Li NMR spectra to 9.7 M LiCl in D2O. Data collections for X-ray structure determinations were performed on a Rigaku Spider image plate system or a BRUKER APEX II Quazar CCD diffractometer at 100 and 110 K, respectively, with MoKα radiation. The single crystals were mounted in perfluoroether oil on a MiTeGen Micromount™. B(Ohfip)3 has CCSD deposition number 1004582 and the MeCN-adduct 1004583.
Acknowledgements
This work was supported by Albert-Ludwigs-Universität Freiburg, the ERC in the Project UniChem and by the DFG in the Normalverfahren. We would like to thank Fadime Bitgül for the measurement of the NMR spectra, Boumahdi Benkmil B.Sc. for his support regarding single X-ray crystallography, and Peter Reiser for performing some preliminary calculations during his advanced lab practical with Nils Trapp.
References
- S. J. Geier, P. A. Chase and D. W. Stephan, Chem. Commun., 2010, 46, 4884–4886 RSC
 .
.
- T. Krahl and E. Kemnitz, J. Fluorine Chem., 2006, 127, 663–678 CrossRef CAS PubMed .
- S. N. Kessler and H. A. Wegner, Org. Lett., 2010, 12, 4062–4065 CrossRef CAS PubMed .
- M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2006, 45, 1683–1684 CrossRef CAS PubMed .
- A. W. Schmidt and H. J. Knolker, Synlett, 2010, 2207–2239 CAS .
- D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535–1539 CAS .
- D. Himmel, S. K. Goll, I. Leito and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 6885–6888 CrossRef CAS PubMed .
- D. Himmel, S. K. Goll, I. Leito and I. Krossing, Chem. – Eur. J., 2011, 17, 5808–5826 CrossRef CAS PubMed .
- D. Himmel, S. K. Goll, I. Leito and I. Krossing, Chem. – Eur. J., 2012, 18, 9333–9340 CrossRef CAS PubMed .
- T. E. Mallouk, G. L. Rosenthal, G. Muller, R. Brusasco and N. Bartlett, Inorg. Chem., 1984, 23, 3167–3173 CrossRef CAS .
- H. D. B. Jenkins, H. K. Roobottom and J. Passmore, Inorg. Chem., 2003, 42, 2886–2893 CrossRef CAS PubMed .
- K. O. Christe and H. D. B. Jenkins, J. Am. Chem. Soc., 2003, 125, 9457–9461 CrossRef CAS PubMed .
- T. S. Cameron, R. J. Deeth, I. Dionne, H. B. Du, H. D. B. Jenkins, I. Krossing, J. Passmore and H. K. Roobottom, Inorg. Chem., 2000, 39, 5614–5631 CrossRef CAS .
- I. Krossing, A. Bihlmeier, I. Raabe and N. Trapp, Angew. Chem., Int. Ed., 2003, 42, 1531–1534 CrossRef CAS PubMed .
- H. D. B. Jenkins, I. Krossing, J. Passmore and I. Raabe, J. Fluorine Chem., 2004, 125, 1585–1592 CrossRef CAS PubMed .
- L. A. Muck, A. Y. Timoshkin and G. Frenking, Inorg. Chem., 2012, 51, 640–646 CrossRef PubMed .
- A. Y. Timoshkin and G. Frenking, Organometallics, 2008, 27, 371–380 CrossRef CAS .
- I. Krossing and I. Raabe, Chem. – Eur. J., 2004, 10, 5017–5030 CrossRef CAS PubMed .
- A. Kraft, J. Beck and I. Krossing, Chem. – Eur. J., 2011, 17, 12975–12980 CrossRef CAS .
- A. Kraft, N. Trapp, D. Himmel, H. Bohrer, P. Schluter, H. Scherer and I. Krossing, Chem. – Eur. J., 2011, 18, 9371–9380 CrossRef PubMed .
- K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and J. A. Boatz, J. Fluorine Chem., 2000, 101, 151–153 CrossRef CAS .
- L. O. Muller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quinones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659–7663 CrossRef PubMed .
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS .
- M. T. Mock, R. G. Potter, D. M. Camaioni, J. Li, W. G. Dougherty, W. S. Kassel, B. Twamley and D. L. DuBois, J. Am. Chem. Soc., 2009, 131, 14454–14465 CrossRef CAS PubMed .
- E. R. Clark, A. Del Grosso and M. J. Ingleson, Chem. – Eur. J., 2013, 19, 2462–2466 CrossRef CAS PubMed .
- J. Barthel, M. Wuhr, R. Buestrich and H. J. Gores, J. Electrochem. Soc., 1995, 142, 2527–2531 CrossRef CAS PubMed .
- J. Barthel, R. Buestrich, H. J. Gores, M. Schmidt and M. Wuhr, J. Electrochem. Soc., 1997, 144, 3866–3870 CrossRef CAS PubMed .
- J. Barthel, R. Buestrich, E. Carl and H. J. Gores, J. Electrochem. Soc., 1996, 143, 3565–3571 CrossRef CAS PubMed .
- J. Barthel, R. Buestrich, E. Carl and H. J. Gores, J. Electrochem. Soc., 1996, 143, 3572–3575 CrossRef CAS PubMed .
- S. M. Ivanova, B. G. Nolan, Y. Kobayashi, S. M. Miller, O. P. Anderson and S. H. Strauss, Chem. – Eur. J., 2001, 7, 503–510 CrossRef CAS .
- S. Tsujioka, B. G. Nolan, H. Takase, B. P. Fauber and S. H. Strauss, J. Electrochem. Soc., 2004, 151, A1418–A1423 CrossRef CAS PubMed .
- H. P. A. Mercier, M. D. Moran, G. J. Schrobilgen, C. Steinberg and R. J. Suontamo, J. Am. Chem. Soc., 2004, 126, 5533–5548 CrossRef CAS PubMed .
- A. J. Lehner, N. Trapp, H. Scherer and I. Krossing, Dalton Trans., 2010, 40, 1448–1452 RSC .
- M. Gonsior, I. Krossing, L. Muller, I. Raabe, M. Jansen and L. van Wullen, Chem. – Eur. J., 2002, 8, 4475–4492 CrossRef CAS .
- K. O. Christe, W. W. Wilson, J. A. Sheehy and J. A. Boatz, Angew. Chem., Int. Ed., 2001, 40, 2947–2947 CrossRef .
- T. Kochner, T. A. Engesser, H. Scherer, D. A. Plattner, A. Steffani and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6529–6531 CrossRef PubMed .
- S. Seidel and K. Seppelt, Science, 2000, 290, 117–118 CrossRef CAS .
- I. Krossing and A. Reisinger, Eur. J. Inorg. Chem., 2005, 1979–1989 CrossRef CAS .
- S. Schulz, D. Schuchmann, I. Krossing, D. Himmel, D. Blaser and R. Boese, Angew. Chem., Int. Ed., 2009, 48, 5748–5751 CrossRef CAS PubMed .
- X. Y. Chen, B. B. Ma, X. Y. Wang, S. X. Yao, L. C. Ni, Z. Y. Zhou, Y. Z. Li, W. Huang, J. Ma, J. L. Zuo and X. P. Wang, Chem. – Eur. J., 2012, 18, 11828–11836 CrossRef CAS PubMed .
- C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef CAS PubMed .
- G. Santiso-Quinones, R. Brückner, C. Knapp, I. Dionne, J. Passmore and I. Krossing, Angew. Chem., Int. Ed., 2009, 48, 1133–1137 CrossRef CAS PubMed .
- A. Budanow, T. Sinke, J. Tillmann, M. Bolte, M. Wagner and H. W. Lerner, Organometallics, 2012, 31, 7298–7301 CrossRef CAS .
- A. Schäfer, M. Reißmann, S. Jung, A. Schäfer, W. Saak, E. Brendler and T. Müller, Organometallics, 2013, 32, 4713–4722 CrossRef .
- F. Scholz, D. Himmel, L. Eisele, W. Unkrig and I. Krossing, Angew. Chem., Int. Ed., 2014, 53, 1689–1692 CrossRef CAS PubMed .
- F. Scholz, D. Himmel, F. W. Heinemann, P. V. Schleyer, K. Meyer and I. Krossing, Science, 2013, 341, 62–64 CrossRef CAS PubMed .
- M. Rohde, L. O. Müller, D. Himmel, H. Scherer and I. Krossing, Chem. – Eur. J., 2014, 20, 1218–1222 CrossRef CAS PubMed .
- G. Santiso-Quinones, A. Reisinger, J. Slattery and I. Krossing, Chem. Commun., 2007, 5046–5048 RSC .
- I. Krossing and A. Reisinger, Angew. Chem., Int. Ed., 2003, 42, 5725–5728 CrossRef CAS PubMed .
- J. Schaefer, D. Himmel and I. Krossing, Eur. J. Inorg. Chem., 2013, 2712–2717 CrossRef CAS .
- G. Meier and T. Braun, Angew. Chem., Int. Ed., 2009, 48, 1546–1548 CrossRef CAS PubMed .
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS .
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed .
- A. Bosmann, G. Francio, E. Janssen, M. Solinas, W. Leitner and P. Wasserscheid, Angew. Chem., Int. Ed., 2001, 40, 2697–2699 CrossRef .
- T. Timofte, S. Pitula and A. V. Mudring, Inorg. Chem., 2007, 46, 10938–10940 CrossRef CAS PubMed .
- I. Raabe, K. Wagner, K. Guttsche, M. K. Wang, M. Gratzel, G. Santiso-Quinones and I. Krossing, Chem. – Eur. J., 2009, 15, 1966–1976 CrossRef CAS PubMed .
- S. Bulut, P. Klose and I. Krossing, Dalton Trans., 2011, 40, 8114–8124 RSC .
- F. Scholz, D. Himmel, H. Scherer and I. Krossing, Chem. – Eur. J., 2013, 19, 109–116 CrossRef CAS PubMed .
- M. G. Hill, W. M. Lamanna and K. R. Mann, Inorg. Chem., 1991, 30, 4687–4690 CrossRef CAS .
- P. G. Gassman and P. A. Deck, Organometallics, 1994, 13, 1934–1939 CrossRef CAS .
- P. G. Gassman, J. R. Sowa, M. G. Hill and K. R. Mann, Organometallics, 1995, 14, 4879–4885 CrossRef CAS .
- L. Pospisil, B. T. King and J. Michl, Electrochim. Acta, 1998, 44, 103–108 CrossRef CAS .
- R. J. LeSuer and W. E. Geiger, Angew. Chem., Int. Ed., 2000, 39, 248–250 CrossRef CAS .
- N. Camire, U. T. Mueller-Westerhoff and W. E. Geiger, J. Organomet. Chem., 2001, 637, 823–826 CrossRef .
- F. Barriere, N. Camire, W. E. Geiger, U. T. Mueller-Westerhoff and R. Sanders, J. Am. Chem. Soc., 2002, 124, 7262–7263 CrossRef CAS PubMed .
- N. Camire, A. Nafady and W. E. Geiger, J. Am. Chem. Soc., 2002, 124, 7260–7261 CrossRef CAS PubMed .
- W. E. Geiger and F. Barriere, Acc. Chem. Res., 2010, 43, 1030–1039 CrossRef CAS PubMed .
- W. Klopper and H. P. Luthi, Mol. Phys., 1999, 96, 559–570 CrossRef CAS .
- A. D. Boese, J. M. L. Martin and W. Klopper, J. Phys. Chem. A, 2007, 111, 11122–11133 CrossRef CAS PubMed .
- P. Jurecka and P. Hobza, Chem. Phys. Lett., 2002, 365, 89–94 CrossRef CAS .
- P. Jurecka and P. Hobza, J. Am. Chem. Soc., 2003, 125, 15608–15613 CrossRef CAS PubMed .
- H. S. Lee, X. Q. Yang, C. L. Xiang, J. McBreen and L. S. Choi, J. Electrochem. Soc., 1998, 145, 2813–2818 CrossRef CAS PubMed .
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed .
-
L. Alvarez, Li[B(OC(H)(CF3)2)4] – Synthesis, Optimization and Characterization of its Electrolytes, University of Freiburg, Freiburg, 2013 Search PubMed.
- M. Kuprat, R. Kuzora, M. Lehmann, A. Schulz, A. Villinger and R. Wustrack, J. Organomet. Chem., 2010, 695, 1006–1011 CrossRef CAS PubMed .
- A. Berkessel, J. A. Adrio, D. Huettenhain and J. M. Neudorfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS PubMed .
- B. Swanson, D. F. Shriver and J. A. Ibers, Inorg. Chem., 1969, 8, 2182–2189 CrossRef CAS .
- H. Jacobsen, H. Berke, S. Doring, G. Kehr, G. Erker, R. Frohlich and O. Meyer, Organometallics, 1999, 18, 1724–1735 CrossRef CAS .
- P. A. Chase, L. D. Henderson, W. E. Piers, M. Parvez, W. Clegg and M. R. J. Elsegood, Organometallics, 2006, 25, 349–357 CrossRef CAS .
- B. Neumuller and F. Gahlmann, Z. Anorg. Allg. Chem., 1992, 612, 123–129 CrossRef .
- G. C. Welch, T. Holtrichter-Roessmann and D. W. Stephan, Inorg. Chem., 2008, 47, 1904–1906 CrossRef CAS PubMed .
- L. Greb, P. Ona-Burgos, B. Schirmer, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2012, 51, 10164–10168 CrossRef CAS PubMed .
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396–8397 CrossRef CAS PubMed .
- S. J. Geier and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 3476–3477 CrossRef CAS PubMed .
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS .
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef .
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 34, 7406–7406 CrossRef .
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef PubMed .
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS .
- O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS PubMed .
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC .
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC .
-
G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford, CT, 2004 .
- G. Laus, A. Schwarzler, P. Schuster, G. Bentivoglio, M. Hummel, K. Wurst, V. Kahlenberg, T. Lorting, J. Schutz, P. Peringer, G. Bonn, G. Nauer and H. Schottenberger, Z. Naturforsch., B: Chem. Sci., 2007, 62, 295–308 CrossRef CAS .
- R. Ramirez-Contreras, N. Bhuvanesh, J. Zhou and O. V. Ozerov, Angew. Chem., Int. Ed., 2013, 52, 10313–10315 CrossRef CAS PubMed .
- L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quinones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659–7663 CrossRef PubMed .
- H. Tetrode, Ann. Phys., 1912, 38, 434–442 CrossRef CAS .
- O. Sackur, Ann. Phys., 1911, 36, 958–980 CrossRef .
- O. Sackur, Ann. Phys., 1913, 40, 67–86 CrossRef CAS .
- T. Köchner, T. A. Engesser, H. Scherer, D. A. Plattner, A. Steffani and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6529–6531 CrossRef PubMed .
- I. Raabe, D. Himmel, S. Mueller, N. Trapp, M. Kaupp and I. Krossing, Dalton Trans., 2008, 946–956 RSC .
- M. Finze, E. Bernhardt, M. Zahres and H. Willner, Inorg. Chem., 2004, 43, 490–505 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data are included in the supporting information, as well as detailed energies, vibrational frequencies, xyz-coordinates, solvation energy calculations, HOMO–LUMO-gaps and other data on the computations (>700 pages). CCDC 1004582 and 1004583. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02822h |
| ‡ Dr Nils Trapp is now working with ETH Zürich. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence