DOI:
10.1039/C4DT02884H
(Paper)
Dalton Trans., 2015,
44, 4994-5003
Terbium-based time-gated Förster resonance energy transfer imaging for evaluating protein–protein interactions on cell membranes†
Received
23rd September 2014
, Accepted 14th January 2015
First published on 14th January 2015
Abstract
Fluorescence imaging of cells and subcellular compartments is an essential tool to investigate biological processes and to evaluate the development and progression of diseases. In particular, protein–protein interactions can be monitored by Förster resonance energy transfer (FRET) between two proximal fluorophores that are attached to specific recognition biomolecules such as antibodies. We investigated the membrane expression of E- and N-cadherins in three different cell lines used as model systems to study epithelial to mesenchymal transition (EMT) and a possible detection of circulating tumour cells (CTCs). EMT is a key process in cancer metastasis, during which epithelial markers (such as E-cadherin) are down-regulated in the primary tumour whereas mesenchymal markers (such as N-cadherin) are up-regulated, leading to enhanced cell motility, intravasation, and appearance of CTCs. Various FRET donor–acceptor pairs and protein recognition strategies were utilized, in which Lumi4-Tb terbium complexes (Tb) and different organic dyes were conjugated to several distinct E- and N-cadherin-specific antibodies. Pulsed excitation of Tb at low repetition rates (100 Hz) and time-gated (TG) imaging of both the Tb-donor and the dye-acceptor photoluminescence (PL) allowed efficient detection of the EMT markers as well as FRET in the case of sufficient donor–acceptor proximity. Efficient FRET was observed only between two E-cadherin-specific antibodies and further experiments indicated that these antibodies recognized the same E-cadherin molecule, suggesting a limited accessibility of cadherins when they are clustered at adherens junctions. The investigated Tb-to-dye FRET systems provided reduced photobleaching compared to the AlexaFluor 488-568 donor–acceptor pair. Our results demonstrate the applicability and advantages of Tb-based TG FRET for efficient and stable imaging of antibody–antibody interactions on different cell lines. They also reveal the limitations of interpreting colocalization on cell membranes in the case of lacking FRET signals.
Introduction
Circulating tumour cells (CTCs), i.e. cells in the bloodstream originating from a solid tumour, are currently actively studied as a potential source of information on the tumour, its genetic alterations, and its response to treatment.1 One major difficulty is to identify CTCs since they are present at a very low abundance in comparison with white blood cells (typically 10−5–10−6). For tumours of epithelial origin, which constitute the major types of cancer, it is assumed that cells cannot efficiently egress from the tumour unless they undergo an epithelial to mesenchymal transition (EMT), which reduces the strength of cell–cell interactions and endow them with migratory capacities (Fig. 1A).2 One of the hallmarks of EMT is the replacement of E-cadherin (epithelial marker) by N-cadherin (mesenchymal marker) at the surface of cells. Cadherins are transmembrane proteins that play a crucial role in cell–cell interactions, mostly through the organization of adherens junctions made of dynamic patches of these molecules.3 Within a patch, molecules are organized through cis interactions between cadherins (i.e. between molecules in the same membrane) and trans interactions (i.e. between molecules on apposed membranes).4 While classical cadherins mediate homophilic interactions, heterophilic interactions between E- and N-cadherin have been observed in vitro and in some cases in vivo.5 However, it is unclear how E- and N-cadherins are distributed in the cell membrane when they are coexpressed. By itself, the expression of N-cadherin cannot be used to detect CTCs since hematopoietic cells also express the protein. On the other hand, relying solely on E-cadherin expression would bias the study in favour of purely epithelial cells, which are unlikely to be the most aggressive ones. The only FDA (U.S. Food and Drug Administration)-approved test for CTC detection (Cell Search from Johnson & Johnson)6 relies on the expression of epithelial markers and detects fewer cells than an assay that enriches in CTC on the basis of their larger size compared to most hematopoietic cells (ISET from Rarecells).7 While during development EMT seems to be a toggle between two states, cancer cells behave in a less clear cut manner and can simultaneously express epithelial and mesenchymal markers.8–10 Indeed, we have observed that some tumour cell lines express both E- and N-cadherins, in agreement with recent publications.11 Being able to detect cells coexpressing E- and N-cadherins would allow to monitor the presence of cells that are likely to be more relevant to the metastatic process than purely epithelial cells.
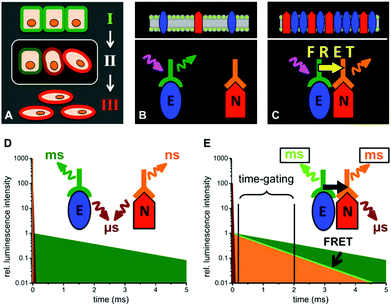 |
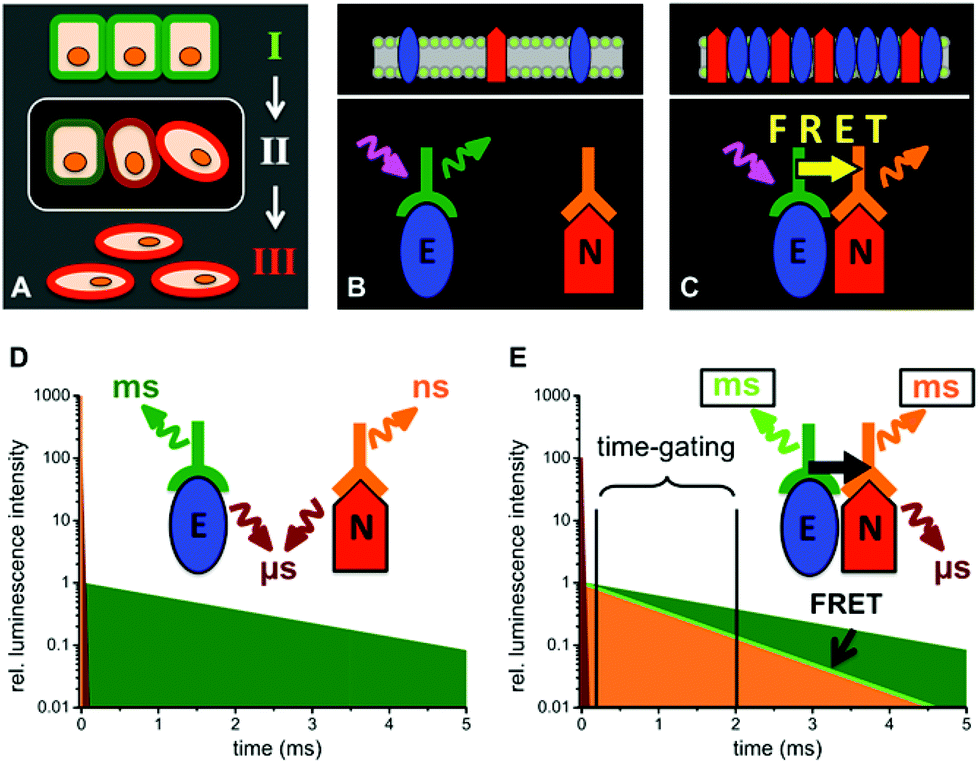
| | Fig. 1 Schematic presentations of EMT (A, cells in epithelial (I), intermediate (II) and mesenchymal (III) state) and the distinction between E-cadherin and N-cadherin coexpression (B and D) and E/N-cadherin clustering (C and E) by TG FRET using Tb-donor antibody (green) excitation (magenta arrow) and dye-acceptor antibody (orange) emission (orange arrow). (D) When E- and N-cadherins are not in a close distance (>20 nm) UV-excitation of the immunostained cells leads to long-lifetime Tb PL (green), a strong nano- to microsecond autofluorescence (brown) and a weak short-lived (nanoseconds) acceptor PL. (E) Clustering of E- and N-cadherins brings the Tb-donor and dye-acceptor in close proximity, which causes Tb-to-dye FRET. As the FRET efficiency ηFRET depends on the PL decay times of the pure Tb-donor (τD) and the one of the Tb-dye donor–acceptor pair (τDA) by ηFRET = 1 − (τDA/τD),23 FRET leads to millisecond Tb and dye emission (light green and orange, respectively) that is shorter than the pure Tb PL decay time. TG detection of the FRET quenched Tb-donor PL and FRET-sensitized dye-acceptor PL intensities several microseconds after the excitation pulse leads to specific PL signals for E/N clustering. | |
While the detection of E- and N-cadherin expression with antibodies coupled to two distinct fluorophores should in principle allow the identification of cells with an intermediate epithelial/mesenchymal phenotype, such an approach cannot provide any information about the E/N-cadherin distances and their possible interactions in clusters. Alternatively, one can combine immunolabelling with Förster resonance energy transfer (FRET) to reveal with a high specificity the presence of the two molecules in close proximity. FRET is a non-radiative energy transfer from an excited donor to a proximal (ca. 1 to 20 nm) ground-state acceptor and requires spectral overlap of donor emission and acceptor absorption.12–15 Thus FRET could produce an ideal signature of E/N-cadherin clustering if the two molecules were close enough to each other in the cell membrane. Many donor–acceptor pairs using organic dyes, metal complexes, nanoparticles, fluorescent proteins, and other fluorophores are available for FRET experiments.16–19 In a typical cellular imaging setup for the analysis of protein–protein interactions the donor and acceptor fluorophores are conjugated to two different biological recognition molecules (antibodies in most cases),20,21 which are specific for the two interacting proteins. Once the antibodies bind to their protein targets, donor and acceptor can interact by FRET, which can be detected by quenching of the donor photoluminescence (PL) and/or sensitization of the acceptor PL.22,23 One major drawback of conventional dyes and fluorescent proteins is their susceptibility to photobleaching,24–27 which makes long excitation and emission cycles almost impossible and causes difficulties in FRET analysis due to donor and/or acceptor bleaching. Because changes in donor and acceptor PL intensities and/or lifetimes are used for FRET analysis, alterations due to photobleaching can strongly interfere with the analysis of FRET signals. Luminescent nanoparticles, such as semiconductor quantum dots (QDs), can overcome photobleaching problems.27,28 However, their relatively large sizes compared to biomolecules may perturb biological function and although QDs allow for relatively large Förster distances (R0, donor–acceptor distance at which the FRET efficiency is 50%)29 and high FRET efficiencies in case of direct attachment of dyes to water-soluble uncoated QDs,30 the often applied thick polymer or lipid-based surface coatings result in increased donor–acceptor distances.31–33
An alternative approach consists in using time-gated (TG) or time-resolved (TR) imaging with lanthanides, taking advantage of their long PL lifetimes (in the μs to ms range). Such imaging techniques involve pulsed excitation at low repetition rates (Hz to kHz range), which leads to fewer excitation-emission cycles per unit time than for fluorophores with short (ns range) PL lifetimes, such as organic dyes, and therefore to a reduced photobleaching. Moreover, PL detection can be performed several microseconds after the excitation pulse when autofluorescence of other components (e.g. endogenous fluorophores in biological samples, in particular in tissues)34,35 has already occurred and therefore significantly reduce background signals. TG and TR microscopy approaches with long-lived fluorophores were already developed more than 20 years ago36,37 but they have never become standard imaging tools. Technological advances and improved lanthanide-based fluorophores have led to a recent revival of time-gated imaging, which includes applications on standard wide-field microscopes equipped with pulsed UV excitation sources and time-gated cameras,38–42 time-gated scanning luminescence using optical choppers and PMT detection,43 time-gated orthogonal scanning automated microscopy (OSAM),44 which was also applied for multiplexed imaging using upconversion nanocrystals,45 and pinhole shifting lifetime imaging microscopy (PSLIM) and temporal sampling lifetime imaging microscopy (TSLIM) for the use on conventional confocal laser scanning microscopes in both TG and TR imaging.46 Apart from detection of only the lanthanide PL, FRET from lanthanide donors to different dye acceptors has also been demonstrated in several spectroscopy and imaging studies.46–54
Here, we present an extensive TG-FRET imaging investigation of different model cell lines, which express E-cadherin, N-cadherin, or both, using immunostaining with luminescent Tb complexes (Tb) as FRET donors and various organic dyes as FRET acceptors to demonstrate the usability of TG-FRET imaging for the detection of protein–protein interactions at the cell membranes. Moreover, we also show the limitations of FRET imaging to interpret these interactions when no FRET signal is detected.
Results and discussion
E- and N-cadherin expression in the different cell lines
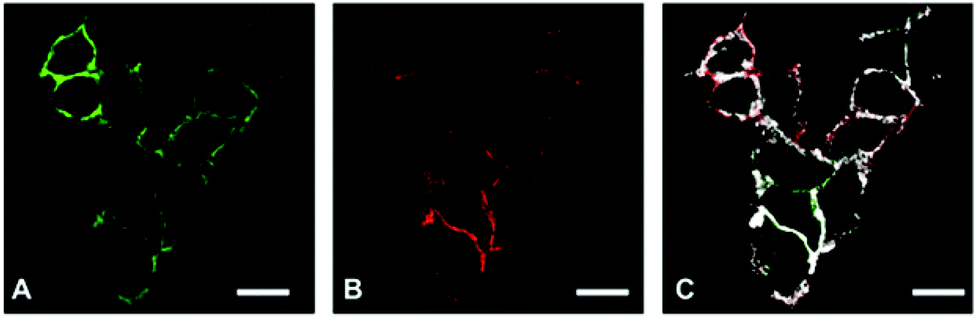
The main goal of our biological study was to distinguish between a simple coexpression of E- and N-cadherins (Fig. 1 B and D) and cadherin clustering at the FRET-imaging scale of ca. 1 to 10 nm (Fig. 1 C and E). For this purpose we investigated three tumour-derived cell lines: MCF-7 (from a breast carcinoma) expressing only E-cadherin, A549 (from a lung carcinoma) expressing E- and N-cadherin, and M4-T (from a melanoma) expressing only N-cadherin. MCF-7 was selected to evaluate the performance of our TG-FRET imaging system. We expected to be able to detect FRET between two E-cadherins for the MCF-7 cells because it was reported that E-cadherins at adherens junctions are packed in a quasi-crystalline structure with a mean distance between molecules of ca. 7 to 8 nm.55,56 Flow cytometry experiments (Fig. S1 in the ESI†) confirmed the expression of E-cadherin but not N-cadherin at the cell membrane of MCF-7 cells. A549 cells were selected due to their ability of expressing both E- and N-cadherin as confirmed by confocal microscopy (Fig. 2) and flow cytometry (Fig. S2 in the ESI†). The confocal microscopy images clearly show coexpression of both cadherins on the cell membranes but as the spatial resolution is diffraction-limited they do not allow a determination of the E/N-cadherin distances. Because most dye acceptors were used to label N-cadherin-specific antibodies we selected the purely N-cadherin expressing M4-T cells to compare Tb-to-dye FRET to dye-to-dye FRET for an evaluation of background fluorescence suppression and photobleaching.
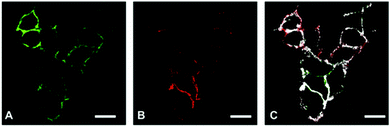 |
| | Fig. 2 Confocal images of A549 cells, which coexpress E-cadherin (A, FITC dye) and N-cadherin (B, AlexaFluor 594 dye). The overlay image (C) shows colocalization (indicated by the white pixels) with Mander's overlap coefficients of M1 = 0.95 (fraction of AF594 pixels overlapping FITC pixels) and M2 = 0.48 (fraction of FITC pixels overlapping AF594 pixels) but does not contain any information about the E/N-cadherin distances (E/N-cadherin clusters). Scale bars correspond to 20 μm. | |
Antibody-fluorophore conjugates and FRET properties
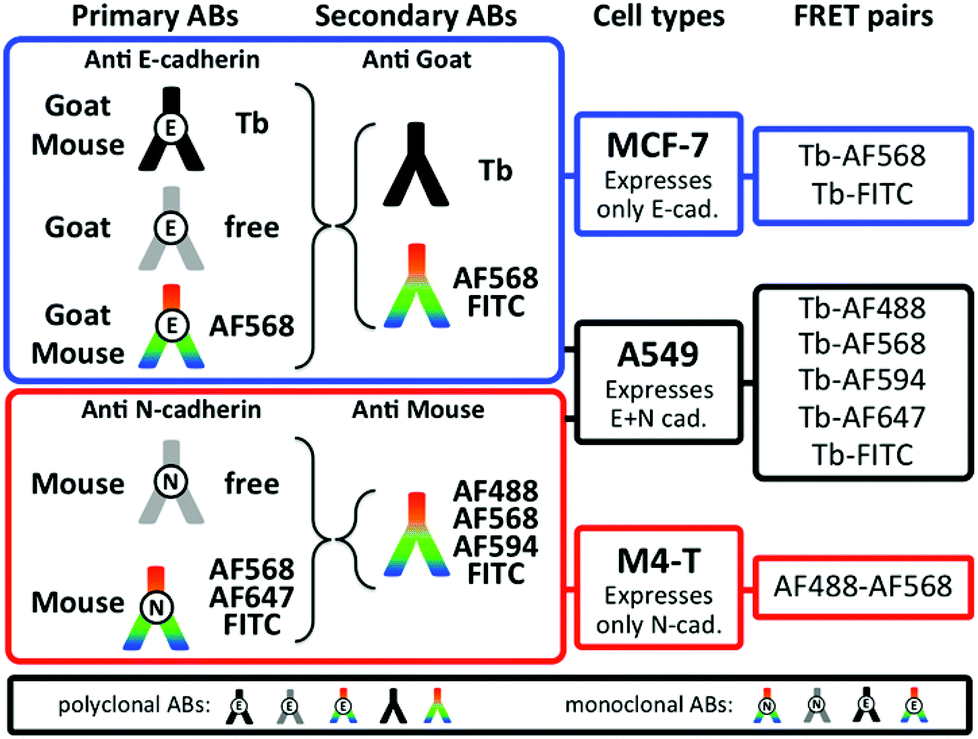
FRET imaging studies using immunostaining with donor and acceptor antibodies require many control experiments to allow determination of the origin of the different donor and acceptor PL signals as well as decreased PL signals, which can possibly occur due to insufficient staining or too large donor–acceptor distances (no FRET) caused by the protein expression levels and/or the FRET antibodies. We tested many different antibodies for efficient E- and N-cadherin targeting and selected those with the best selectivity to be combined with the Tb-complex Lumi4®-Tb57 as FRET donor and different dyes as FRET acceptors. Some of the antibodies were readily available as dye-conjugates whereas others were conjugated in-house (cf. Experimental section). It should be noted that despite its strong and well-known susceptibility to photobleaching fluorescein isothiocyanate (FITC) is still a frequently applied dye for fluorescence immunostaining because many antibody-FITC conjugates are commercially available. Fig. 3 shows the different combinations of primary and secondary antibodies with Tb and various dyes for specific recognition of E- or N-cadherin and the resulting FRET pairs. E-cadherin was selected to be stained with Tb donor antibodies for studying both E-to-E and E-to-N cadherin FRET experiments between Tb and dye antibodies. Accordingly, one type of goat polyclonal and one type of mouse monoclonal primary antibodies were conjugated with Tb and AlexaFluor 568 (AF568) and secondary antibodies were labelled with Tb (anti-goat), Alexa Fluor dyes AF488, AF568, AF594, or FITC (anti-goat and anti-mouse). For N-cadherin (acceptor protein) a mouse monoclonal primary antibody was conjugated with AF568, AF647, or FITC and the same dye-conjugated anti-mouse antibodies as for E-cadherin staining were used as secondary antibodies. The various combinations led to two different Tb-dye FRET pairs for TG imaging of E-cadherin expression on MCF-7 cells, five different Tb-dye FRET pairs for TG imaging of E- and N-cadherin expression on A549 cells, and one dye–dye FRET pair for steady-state (SS) imaging of N-cadherin expression on M4-T cells.
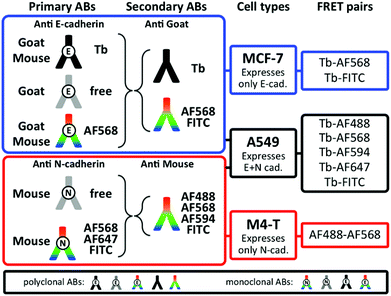 |
| | Fig. 3 Overview of available Lumi4-Tb and dye antibody conjugates, cell lines, and FRET pairs used for PL imaging experiments in our study. The black box on the bottom shows the different antibodies that were available as polyclonals and/or monoclonals. | |
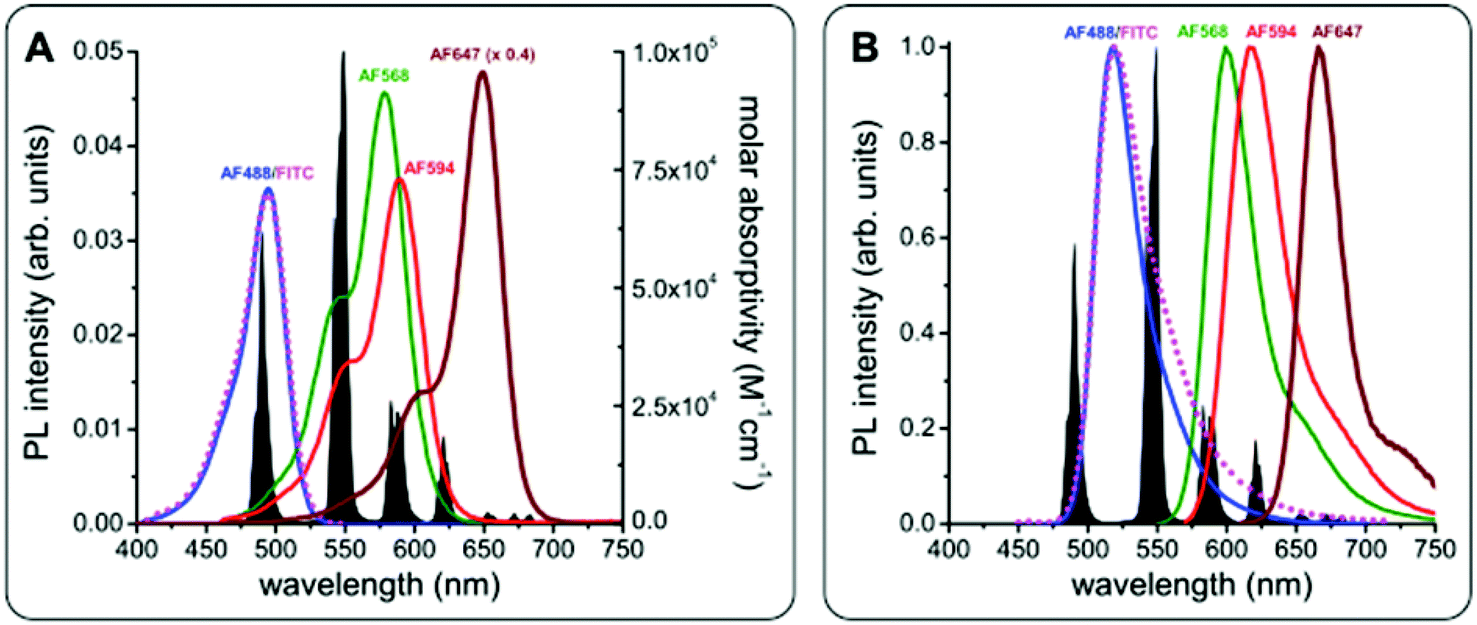
The chemical and photophysical properties of Lumi4-Tb and its advantages as Tb FRET donors have been discussed in detail elsewhere.54,57,58 Briefly, they consist of unpolarized emission and extremely long excited-state lifetimes of 2.6 ms (Fig. S1 in the ESI†), which justify the orientation factor approximation of κ2 = 2/3, and multiple and well-separated PL emission bands, which allow FRET to several different dyes for spectral multiplexing. The absorbance and emission spectra of Tb and the different dyes are shown in Fig. 4. Using the spectral overlap of Tb donor emission and the dye acceptor absorbance, the Förster distances were calculated by eqn (1),
| | | R0 = 0.02108(κ2ΦDn−4J)1/6 nm | (1) |
where
κ2 = 2/3 is the orientation factor between the Tb emission and dye absorption transition dipole moments,
ΦD = 0.75 is the PL quantum yield of the central Tb-ion,
n = 1.33 is the refractive index of the aqueous buffer, and
J (in M
−1 cm
−1 nm
4) is the spectral overlap integral as defined by
eqn (2),
where
ĪD(
λ) (with ∫
ĪD(
λ) = 1) is the intensity normalized emission of Tb and
εA(
λ) is the molar absorptivity (or extinction coefficient) of the dye. The
R0 values of the different Tb-dye FRET pairs are in the 5 to 6 nm range (
Fig. 4), which should provide a maximum detectable distance of approximately 10 to 12 nm (2 ×
R0).
23
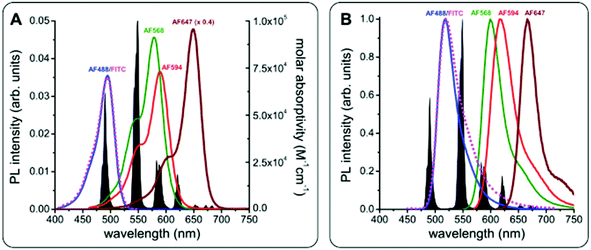 |
| | Fig. 4 PL emission spectrum of Tb (black in A and B) and absorbance (A) and PL emission (B) spectra of FITC (magenta), AF488 (blue), AF568 (green), AF594 (red), and AF647 (brown, molar absorptivity spectrum in A multiplied by 0.4 for better visibility of all spectra). Förster distances of the different FRET pairs were calculated using eqn (1) and (2): R0(Tb-FITC) = 4.9 nm, R0(Tb-AF488) = 4.9 nm, R0(Tb-AF568) = 6.1 nm, R0(Tb-AF594) = 5.9 nm, R0(Tb-AF647) = 5.9 nm, and R0(AF488-AF568) = 6.2 nm. | |
TG imaging for evaluating TG-FRET and E/E-cadherin clustering on MCF-7 cell membranes
To demonstrate the efficiency of TG Tb-to-dye FRET for imaging protein–protein interactions, we first performed a series of experiments on MCF-7 cells. Fixed cells were incubated for 3 h with primary antibodies against E-cadherin and for an additional 2 h with secondary antibodies. The cells were then washed and mounted on microscopy slides for imaging. As shown in Fig. 5A, TG imaging of MCF-7 cells targeted with both polyclonal Tb- and dye-labelled primary antibodies led to efficient FRET (positive TG dye signals upon Tb excitation). Similar experiments were performed with secondary Tb and dye antibodies, which also led to bright FRET signals (Fig. 5B and Fig. S4 in the ESI†) and very good evidence for efficient Tb-to-dye FRET.
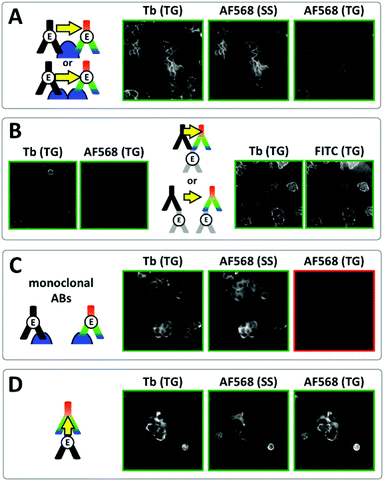 |
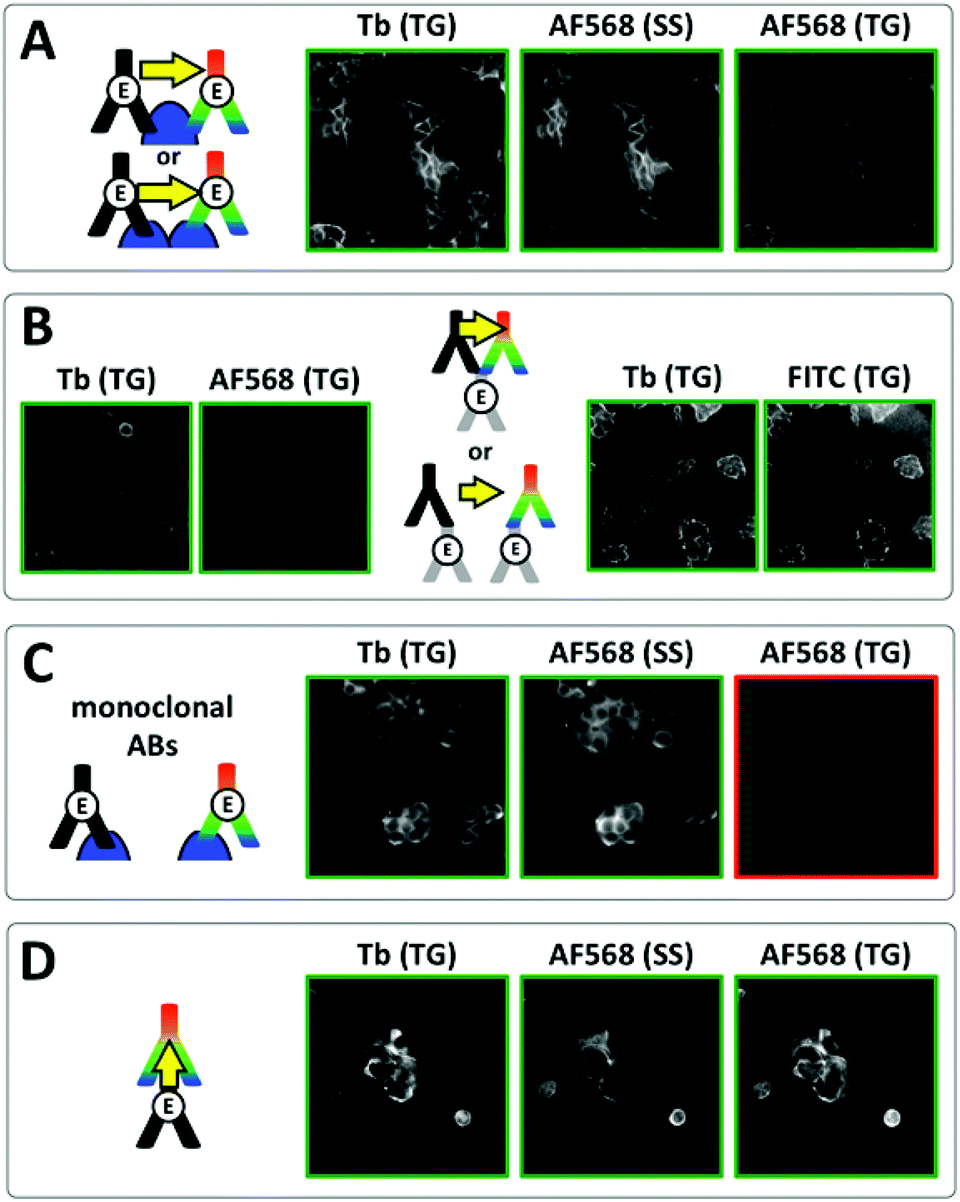
| | Fig. 5 Investigation of E/E-cadherin clustering in MCF-7 cells by Tb-to-dye FRET. For better clarity, positive signal images have green frames whereas negative signal images have red frames. (A) Time-gated (TG, 0.01–2.51 ms after excitation pulse) and steady-state (SS, excited at 520 ± 14 nm) images in the Tb detection channel (Tb, 542 ± 10 nm) and the AF568 detection channel (AF568, 607 ± 5 nm) using polyclonal primary Tb and AF568 antibodies resulted in positive TG Tb, SS AF568 and TG AF568 (FRET) PL signals, which could be caused by antibody-protein recognition on the same or different E-cadherins (top or bottom scheme, respectively). (B) Similar results as in A were found when using unlabelled polyclonal antibodies against E-cadherin and Tb and AF568 (left) or FITC (right) secondary antibodies, which offers again two possible binding scenarios (top and bottom scheme, respectively). (C) Using monoclonal Tb and AF568 primary antibodies led to efficient costaining but no FRET signal (TG AF568) due to too large distances (>ca. 12 nm) between Tb and AF568 antibodies (scheme). (D) For a verification of efficient TG Tb-to-dye FRET Tb primary antibodies and AF568 secondary antibodies (against the Tb primaries) were used for immunostaining. Efficient costaining as well as FRET due to antibody–antibody recognition (scheme) are clearly visible in the TG Tb, SS AF568, and TG AF568 PL images, respectively. Control experiments using only primary Tb antibodies showed that the TG AF568 signal is not caused by spectral crosstalk from the Tb PL (Fig. S7 in the ESI†). | |
As shown in the schemes of Fig. 5A and B, such positive FRET signals may arise from binding of the two antibodies to either the same protein (E-cadherin or primary antibody) or to two different ones. To reveal which binding scenario was responsible for the FRET signals we performed a series of control experiments. Time-resolved spectroscopy using unlabelled primary E-cadherin antibodies and secondary Tb and dye antibodies in solution (without any cells) showed that several secondary antibodies could bind to the same primary antibody, which was evidenced by FRET-sensitization of the dye acceptor secondary antibodies by the Tb donor secondary antibodies (Fig. S5 and S6 in the ESI†). We further prepared Tb- and dye-labelled monoclonal antibodies against E-cadherin. In contrast to the polyclonal antibodies, which can bind to different epitopes of E-cadherin and therefore enable multiple-antibody binding to the same E-cadherin, monoclonal antibodies are specific against the same epitope, which strongly reduces the possibility of multiple-antibody binding to the same E-cadherin. Despite very bright Tb and dye antibody staining on the same cells, no FRET was detected (Fig. 5C). Taking into account the expected distances of ca. 7 to 8 nm between clustered E-cadherins at adherens junctions (vide supra), this result was somewhat unexpected. A possible explanation for the lack of FRET is the unavailability of two different clustered E-cadherins for efficient antibody recognition. Such steric hindrance does not exist for unclustered E-cadherins or E-cadherins located on cell surfaces outside of cell junctions, where the density of molecules is much lower. Therefore multiple polyclonal antibody binding to the same E-cadherin or primary antibody (and FRET) becomes possible (top schemes in Fig. 5A and B), whereas the monoclonal antibodies can bind only to single E-cadherins, which are separated by distances beyond the FRET range (scheme in Fig. 5C).
Despite the limitations of our FRET analysis to quantify the distances between E-cadherins in fixed MCF-7 cells, the successful TG-FRET imaging experiments for FRET donor and acceptor antibody binding to the same protein (E-cadherin or primary antibody) clearly demonstrate the feasibility of efficient protein proximity detection using TG Tb-to-dye FRET microscopy. This was further confirmed by efficient FRET on the MCF-7 cells between Tb-labelled primary antibodies and dye-labelled secondary antibodies (Fig. 5D).
TG imaging for evaluating E/N-cadherin clustering on A549 cell membranes
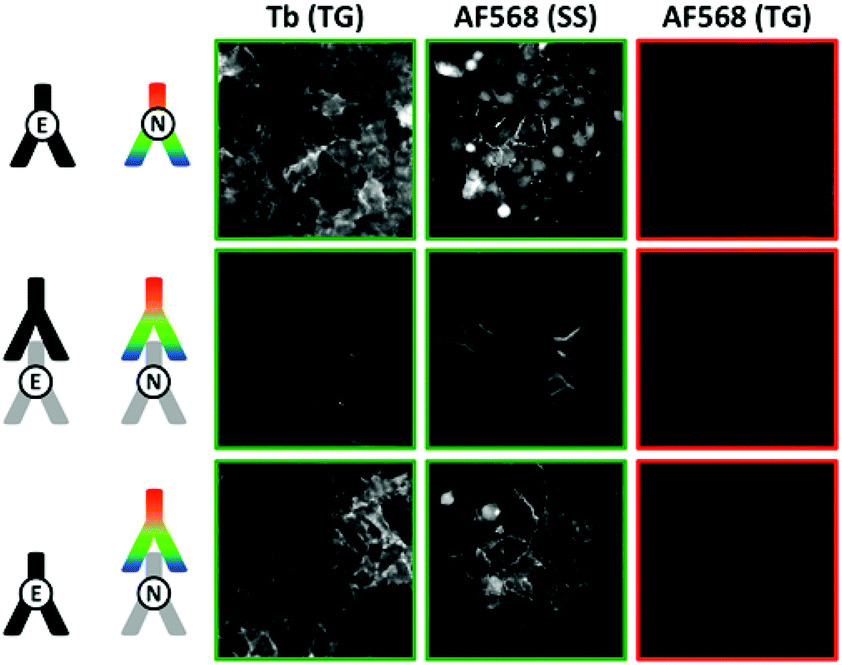
For the different TG-FRET imaging experiments to study E- and N-cadherin co-expression, the immunolabelling was performed similarly to the one of MCF-7 cells. Fig. 6 (and Fig. S8 to S10 in the ESI†) shows TG and SS images of various antibody combinations for FRET immunostaining. Independent of the applied combination of Tb- and dye-labelled antibodies, A549 cells showed positive PL signals for both E-cadherin (TG Tb signals) and N-cadherin (SS dye signals) immunostaining, with a clear membrane localization and some intracellular signal. Despite this double protein expression, FRET from Tb to dyes (TG dye signals) could not be observed in any of the various antibody combinations. Taking into account the results from the MCF-7 cell imaging, the absence of FRET was certainly caused by too long distances between the Tb and dye antibodies. From the biological point of view the lack of FRET signals prevents any quantification of colocalization and/or clustering. Nevertheless, different qualitative interpretations are possible. First, E- and N-cadherins may not be coexpressed in clusters, which would result in E–N cadherin distances beyond the maximum detectable FRET distance of ca. 10 to 12 nm. Second, clustered E- and N-cadherins may be in such a close proximity that efficient binding of the antibodies in E/N clusters is not possible (or at least very inefficient). Third, the antibodies may be able to bind to the cadherins but the antibody-protein recognition sites place the two antibodies at a distance beyond the detectable FRET range.
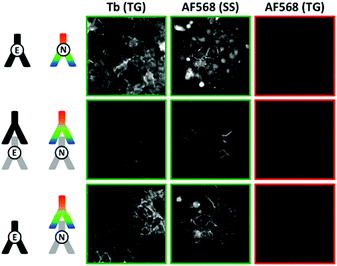 |
| | Fig. 6 TG (0.01–2.51 ms after excitation pulse) and SS (excited at 520 ± 14 nm) images in the Tb detection channel (Tb, 542 ± 10 nm) and the AF568 detection channel (AF568, 607 ± 5 nm) of different FRET-pair antibody combinations to detect a possible E/N-cadherin clustering. Costaining of Tb-antibodies (against E-cadherin) and AF568-antibodies (against N-cadherin) is clearly visible in the TG Tb and SS AF568 PL images. However, no FRET signal (TG AF568 signal) was detected, which shows that Tb-donor and dye-acceptor are not in close (<ca. 12 nm) proximity. For better clarity, positive signal images have green frames whereas negative signal images have red frames. Control experiments using FITC, AF647, AF488, and AF594 antibodies against N-cadherin as acceptors and different primary/secondary antibody combinations led to the same results (Fig. S8 to S10 in the ESI†). | |
Comparison with steady-state dye-to-dye FRET imaging using N-cadherin expression on M4-T cell membranes
Apart from short-lived autofluorescence background of the cells as one possible disadvantage of SS imaging, many dyes are prone to photobleaching, which limits their application for imaging cellular processes on longer time scales. During initial TG-FRET imaging experiments, in which we recorded first Tb-TG, dye-SS, and then dye-TG images, we discovered a significant decrease of the dye PL intensity during the acquisition of the SS images. We therefore adapted our acquisition series and always took the TG-images (of both Tb and dyes) before the SS images, which significantly improved the TG-dye image acquisition. It should be noted that different dyes provide different photostabilities and that the conditions of the cell medium may be optimized by deoxygenation or the addition of different chemicals. However, the selection of matching FRET pairs, the varying availability of selective antibodies conjugated with different dyes, and a possible sensitivity of the cells to chemical treatment (in view towards live cell or in vivo imaging) usually limit the possibilities to achieve a fully optimized system. We therefore decided to perform a comparison of Tb-to-dye FRET imaging and dye-to-dye FRET imaging under similar conditions, which implied to use the same acceptor dye AF568 and identical staining procedures. To also match the Förster distance to the Tb-to-dye system we selected the AF488-AF568 donor–acceptor pair with a Förster distance of approximately 6.2 nm.19
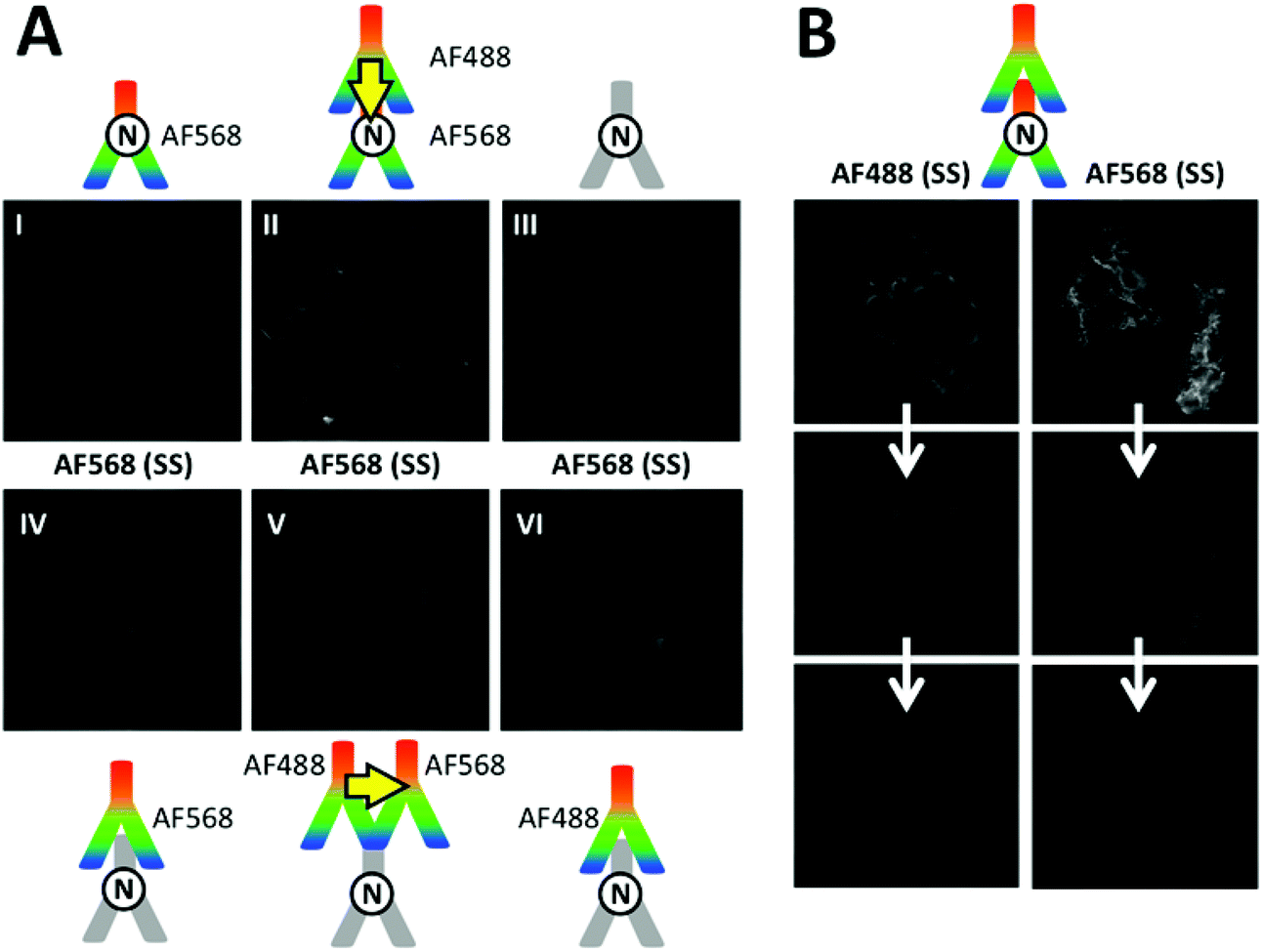
The fluorescence images of M4-T cells in Fig. 7A show SS AF568 PL signals upon AF488 excitation at 438 ± 12 nm. This wavelength range is not optimal for AF488 excitation but it was selected to avoid any significant direct AF568 excitation (Fig. 4A), which is an important requirement for SS FRET experiments to distinguish between direct and FRET-sensitized acceptor PL. Although the most efficient FRET scenario of direct primary–secondary antibody binding (cf. Fig. 5D) provided the brightest PL signal (Fig. 7A II), unconjugated primaries (Fig. 7A III), AF568 primaries (Fig. 7A I), AF568 secondaries (Fig. 7A IV), AF488 secondaries (Fig. 7A VI), and both FRET secondaries (Fig. 7A V, with lower PL intensity than the non-FRET images IV and VI) also provided more or less strong PL signals in the AF568 detection channel upon AF488 excitation. Such unwanted PL background signals impede the analysis of FRET because they cannot be distinguished from the FRET signals. In addition to background PL, photobleaching is very problematic for both measurements over long time periods and the quantitative analysis of FRET. Fig. 7B shows a series of images of photobleaching of the AF488- and AF568-labelled antibodies against N-cadherin in M4-T cells. Both AF488 (upon AF488 excitation) and AF568 (upon AF568 excitation) show strongly decreased PL intensities within only three image acquisition cycles. Similar bleaching was found in M4-T cells that were stained with only one of the antibodies (AF488 or AF568, Fig. S11 in the ESI†). The necessity to image donor and acceptor PL on the same cells in a serial manner (unless image splitters for the simultaneous detection of two colours are used) further complicates a precise evaluation of dye-to-dye FRET because the acceptor PL images contain a convolution of donor photobleaching and FRET sensitization by the photobleached donor (Fig. S12 in the ESI†). It should be noted that the use of a different cell line (M4-T) and the detection of only N-cadherins does not allow a direct comparison with the TG Tb-to-dye FRET images taken on A549 and MCF-7 cells using E- and N-cadherin immunostaining. Nevertheless, the similar experimental conditions demonstrate the problematic PL background and photobleaching issues in dye-to-dye FRET imaging and underline the efficiency of TG Tb-to-dye FRET imaging with high signal-to-background ratios, stable PL intensities, and high photostability over longer measurement times compared to standard SS imaging with organic dyes.
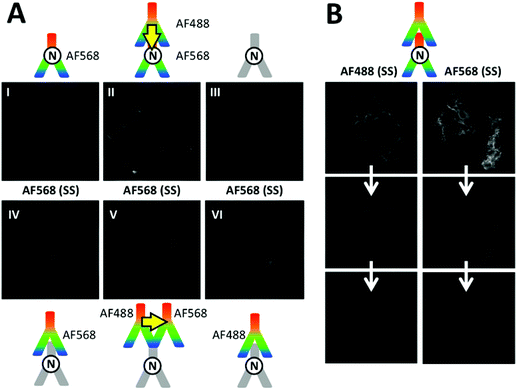 |
| | Fig. 7 (A) SS images of different combinations of AF488 donor antibodies with AF568 acceptor antibodies on M4-T cells were recorded in the AF568 PL emission channel upon excitation of AF488 (438 ± 12 nm, no significant direct excitation of AF568). Although the two possible AF488–AF568 FRET combinations (II and V) led to positive PL signals, the dye–dye FRET pair could not provide clear evidence for FRET because direct excitation of AF568 (I and IV), spectral crosstalk of AF488 PL in the AF568 detection channel (VI), and autofluorescence of immunostained cells without any dyes (III) also led to positive PL signals. Control experiments using specific excitation of only AF488 antibodies and AF568 antibodies confirmed that both dye-labelled antibodies were bound to the cell membranes (Fig. S12 in the ESI†). (B) Strong photobleaching (three serial image acquisitions from top to bottom) of both AF488 (excitation via 438 ± 12 nm and detection via 522 ± 6 nm transmission filters, 100 ms acquisition per image) and AF568 (excitation via 542 ± 10 nm and detection via 607 ± 5 nm transmission filters, 350 ms acquisition per image) with both dye-labelled antibodies against N-cadherin or with only one type of each dye-labelled antibody (Fig. S11 in the ESI†). | |
Conclusions
In this study we have shown that TG Tb-to-dye FRET microscopy can be an efficient imaging tool for the analysis of protein–protein interactions. In particular, the strong reduction of short-lived PL background from sample autofluorescence and direct acceptor excitation and of donor spectral crosstalk in combination with low excitation repetition rates and reduced photobleaching provide significant advantages of Tb-to-dye FRET analysis compared to conventional dye-to-dye systems. Concerning A549 cells used as a model of membrane E- and N-cadherin co-expression for a possible distinction of CTCs, we could not detect FRET using immunostaining with Tb and dye antibody conjugates. There are two main interpretations of these results: (i) although E-and N-cadherins colocalize at distances that cannot be accessed with diffraction-limited optical resolution (Fig. 2), they are in fact separated by distances larger than our FRET range (max. 12 nm), (ii) the dense packing achieved in adherens junctions does not allow antibody binding to adjacent molecules. This second possibility is supported by the absence of FRET between two E-cadherin molecules in MCF-7 cells although in culture these cells have extensive cell–cell contacts through adherens junctions. In spite of the numerous studies on cadherins there is only one conclusive report of FRET between two cadherins,59 which is based on the expression of engineered N-cadherins that contain fluorescent proteins inserted in the second extracellular domain next to the dimerization domain. Notably, this approach was designed to circumvent the issue of accessibility by using genetically modified cells expressing the fluorescent reporters. Moreover, when the fluorescent protein was inserted in a domain proximal to the membrane the FRET efficiency was greatly reduced, which illustrated that the packed cadherins are in an extended conformation in agreement with the large distance between the two membranes of the apposed cells at adherens junction (15 to 30 nm).4 Although our TG-FRET study could unfortunately not provide conclusive results concerning the clustering of E- and/or N-cadherins on the membranes of different cell lines, efficient FRET between Tb and dye antibodies bound to the same target (E-cadherin or primary antibody) demonstrated the advantages of TG Tb-to-dye FRET in comparison to dye-to-dye FRET using the well-known AF488–AF568 FRET-pair for cellular imaging. We believe that these results will be even more relevant for FRET imaging in tissues, which suffer from significantly higher autofluorescence background, and that TG Tb-to-dye FRET can serve to efficiently image protein–protein interactions via immunolabelling with antibodies that are able to target complementary epitopes at distances below ca. 12 nm.
Experimental
Cell culture and immunofluorescence labelling for confocal microscopy
Cells were cultured on glass coverslips at the bottom of 12-well plates for 48 h in Dulbecco's Modified Eagle Medium (DMEM-Glutamax, Life Technologies) with 10% FBS and antibiotics (Penicillin–Streptomycin, Life Technologies) at 37 °C and 5% CO2. Then cells were fixed with 3% formaldehyde. Formaldehyde was removed and the wells were rinsed with 1 M glycine, followed by two washes with blocking solution (10 mM HEPES with 2% fetal bovine serum, FBS) and a final incubation with the blocking solution for 30 min at 37 °C. After removal of the blocking solution, 60 μL of either 1/100 (anti-E-cadherin) or 1/200 (anti-N-cadherin) dilution of the primary antibodies (see references below) in HEPES were added. The coverslips were then rinsed three times with HEPES and exposed for 1 h at 37 °C to 60 μL of the fluorophore-labelled secondary antibodies at 1/100 dilution in HEPES. Slides were mounted with Fluoromount-G (ref. 0100-01, Southern Biotech, Alabama, USA).
Cell culture and immunofluorescence labelling for time-gated imaging
Cells were grown on coverslips for 48 h in DMEM (Sigma-Aldrich) supplemented with 10% FBS, 2 mM L-glutamine (Life Technologies) and antibiotics (1× Anti Antibiotic-Antimycotic, Gibco) at 37 °C with 5% CO2. The cells were fixed in 4% formaldehyde in PBS and rinsed with a 1 M glycine solution. The cells were blocked for 30 min with 2% FBS in PBS. The samples were then incubated with antibody solutions at 37 °C (primaries 3 h, secondaries 2 h) for the appropriate experiment (see antibodies below), rinsed with PBS, and mounted on microscopy slides using Fluoro-Gel (Electron Microscopy Sciences). Primary antibodies: Anti-E cadherin goat polyclonal (ref. AF648, R&D Systems, Minneapolis, USA), anti-N Cadherin [8C11] antibody (ref. ab19348, Abcam, Cambridge, UK), pan Cadherin Antibody (3F4) (ref. H00000999-M01, Novus Biologicals). Secondary antibodies: donkey anti-goat IgG-FITC (ref. sc-2024, Santa Cruz Biotechnology Inc., Dallas, USA), Alexa Fluor® 594 Donkey Anti-Mouse IgG(H + L) (ref. A-21203, Life Technology, USA), Alexa Fluor 488 Donkey Anti-Mouse IgG (H + L) (ref. A-21202 Life Technology, USA), Donkey Anti-Mouse IgG H&L (FITC) preadsorbed (ref. ab7057 Abcam).
Antibody conjugation
Primary and secondary antibodies, (AF648 from R&D Systems, H00000999-M01 from Novus Biologicals, ab19348, ab7120 and ab7056 from Abcam) were labelled with amine-reactive dyes (Alexa Fluor 568 NHS ester and Alexa Fluor 647 NHS ester from Life Technologies and NHS-Fluorescein from Thermo Fisher Scientific) or TbL4 (Lumi4®-Tb-NHS, Lumiphore), both in concentration excess to the antibody solutions, in 100 mM carbonate buffer at pH 9.0. The mixtures were incubated while rotating at 25 rpm (Intelli-Mixer, ELMI) for 5 h at room temperature. The samples were purified using 30 kDa filter centrifugal devices (Amicon Ultra 0.5 mL filters) and stored in 100 mM Tris-HCl buffer, pH 7.2.
Confocal laser-scanning immunofluorescence microscopy
Cells were imaged using a TCS SP2 inverted microscope (Leica Mikrosysteme, Wetzlar, Germany) using a 63×/1.32 NA immersion oil objective. LCS MicroLab (Leica) and ImageJ (National Institute of Health, USA) softwares were used for acquisition and image processing respectively. FITC dye was excited with the wavelength 488 nm line of an argon laser; FITC selected detection range was 520–540 nm. AF594 dye was excited with the wavelength 543 nm line of a helium–neon laser, and the selected detection range was 610–650 nm. The exposure time was 5 μs per pixel for all scans.
Steady-state (SS) and time-gated (TG) widefield immunofluorescence microscopy
Cells were imaged using an inverted microscope (Olympus IX71). For steady-state fluorescence images the samples were excited using a mercury lamp (X-Cite 120Q, Lumen Dynamics) and images acquired with a scientific CMOS camera (PCO). For time-gated images the samples were excited from on top by a pulsed laser at 100 Hz (Spectra-Physics), triggering an ICCD camera (PI-MAX3, Princeton Instruments). The settings for acquisition were kept at 10 μs delay, 2.5 ms gatewidth, 400–800 gates per exposure and gain 100. Image processing was done using ImageJ (National Institute of Health, USA, http://imagej.nih.gov/ij/).
Acknowledgements
We thank Lumiphore, Inc. for the gift of Lumi4®-Tb-NHS reagent and Christian Auclair and Gérald Peyroche for fruitful discussions. This work was supported by the French « Investment for the Future » program managed by Agence Nationale de la Recherche, grant ANR-10-Nano-05.
References
- T. A. Yap, D. Lorente, A. Omlin, D. Olmos and J. S. de Bono, Clin. Cancer Res., 2014, 20, 2553 CrossRef CAS PubMed.
- J. P. Thiery, H. Acloque, R. Y. J. Huang and M. A. Nieto, Cell, 2009, 139, 871 CrossRef CAS PubMed.
- W. Meng and M. Takeichi, Cold Spring Harbor Perspect. Biol., 2009, 1 Search PubMed.
- J. Brasch, O. J. Harrison, B. Honig and L. Shapiro, Trends Cell Biol., 2012, 22, 299 CrossRef CAS PubMed.
- P. Katsamba, K. Carroll, G. Ahlsen, F. Bahna, J. Vendome, S. Posy, M. Rajebhosale, S. Price, T. M. Jessell, A. Ben-Shaul, L. Shapiro and B. H. Honig, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11594 CrossRef CAS PubMed.
- M. Cristofanilli, G. T. Budd, M. J. Ellis, A. Stopeck, J. Matera, M. C. Miller, J. M. Reuben, G. V. Doyle, W. J. Allard, L. W. M. M. Terstappen and D. F. Hayes, New Engl. J. Med., 2004, 351, 781 CrossRef CAS PubMed.
- F. Farace, C. Massard, N. Vimond, F. Drusch, N. Jacques, F. Billiot, A. Laplanche, A. Chauchereau, L. Lacroix, D. Planchard, S. Le Moulec, F. Andre, K. Fizazi, J. C. Soria and P. Vielh, Br. J. Cancer, 2011, 105, 847 CrossRef CAS PubMed.
- M. Yu, A. Bardia, B. S. Wittner, S. L. Stott, M. E. Smas, D. T. Ting, S. J. Isakoff, J. C. Ciciliano, M. N. Wells, A. M. Shah, K. F. Concannon, M. C. Donaldson, L. V. Sequist, E. Brachtel, D. Sgroi, J. Baselga, S. Ramaswamy, M. Toner, D. A. Haber and S. Maheswaran, Science, 2013, 339, 580 CrossRef CAS PubMed.
- W. L. Tam and R. A. Weinberg, Nat. Med., 2013, 19, 1438 CrossRef CAS PubMed.
- A. Lecharpentier, P. Vielh, P. Perez-Moreno, D. Planchard, J. C. Soria and F. Farace, Br. J. Cancer, 2011, 105, 1338 CrossRef CAS PubMed.
- T. Celia-Terrassa, O. Meca-Cortes, F. Mateo, A. Martinez de Paz, N. Rubio, A. Arnal-Estape, B. J. Ell, R. Bermudo, A. Diaz, M. Guerra-Rebollo, J. J. Lozano, C. Estaras, C. Ulloa, D. Alvarez-Simon, J. Mila, R. Vilella, R. Paciucci, M. Martinez-Balbas, A. Garcia de Herreros, R. R. Gomis, Y. Kang, J. Blanco, P. L. Fernandez and T. M. Thomson, J. Clin. Invest., 2012, 122, 1849 CAS.
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd edn, Springer, New York, 2006 Search PubMed.
-
I. L. Medintz and N. Hildebrandt, FRET-Förster Resonance Energy Transfer. From Theory to Applications, Wiley-VCH, Weinheim, 2013 Search PubMed.
-
B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence: Principles and Applications, Wiley-VCH, Weinheim, 2nd edn, 2012 Search PubMed.
-
N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern molecular photochemistry of organic molecules, University Science Books, 2010 Search PubMed.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562 CrossRef CAS PubMed.
- B. Hötzer, I. L. Medintz and N. Hildebrandt, Small, 2012, 8, 2297 CrossRef PubMed.
-
K. E. Sapsford, B. Wildt, A. Mariani, A. B. Yeatts and I. L. Medintz, in FRET-Förster Resonance Energy Transfer. From Theory to Applications, ed. I. L. Medintz and N. Hildebrandt, Wiley-VCH, Weinheim, 2013 Search PubMed.
-
A. G. Byrne, M. M. Byrne, G. Coker, K. Boeneman-Gemmill, C. Spillman, I. L. Medintz, S. L. Sloan and B. W. van der Meer, in FRET-Förster Resonance Energy Transfer. From Theory to Applications, ed. I. L. Medintz and N. Hildebrandt, Wiley-VCH, Weinheim, 2013 Search PubMed.
-
G. T. Hermanson, Bioconjugate Techniques, Academic Press, London, 3rd edn, 2013 Search PubMed.
-
R. P. Haugland, The Molecular Probes® Handbook - A Guide to Fluorescent Probes and Labeling Technologies, Life Technologies Corporation, USA, 11th edn, 2010 Search PubMed.
-
A. Periasamy and R. N. Day, Molecular imaging: FRET microscopy and spectroscopy, Oxford University Press, Inc., New York, 2005 Search PubMed.
-
N. Hildebrandt, in FRET-Förster Resonance Energy Transfer. From Theory to Applications, ed. I. L. Medintz and N. Hildebrandt, Wiley-VCH, Weinheim, 2013 Search PubMed.
- L. Y. Lee, S. L. Ong, J. Y. Hu, W. J. Ng, Y. Feng, X. Tan and S. W. Wong, Appl. Environ. Microbiol., 2004, 70, 5732 CrossRef CAS PubMed.
- J. Mahmoudian, R. Hadavi, M. Jeddi-Tehrani, A. R. Mahmoudi, A. A. Bayat, E. Shaban, M. Vafakhah, M. Darzi, M. Tarahomi and R. Ghods, Cell J., 2011, 13, 169 CAS.
- J. E. Berlier, A. Rothe, G. Buller, J. Bradford, D. R. Gray, B. J. Filanoski, W. G. Telford, S. Yue, J. Liu, C.-Y. Cheung, W. Chang, J. D. Hirsch, J. M. Beechem, R. P. Haugland and R. P. Haugland, J. Histochem. Cytochem., 2003, 51, 1699 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763 CrossRef CAS PubMed.
-
W. R. Algar, H. Kim and U. J. Krull, in FRET-Förster Resonance Energy Transfer. From Theory to Applications, ed. I. L. Medintz and N. Hildebrandt, Wiley-VCH, Weinheim, 2013 Search PubMed.
- W. R. Algar, H. Kim, I. L. Medintz and N. Hildebrandt, Coord. Chem. Rev., 2014, 263–264, 65 CrossRef CAS PubMed.
- A. Yahia-Ammar, A. M. Nonat, A. Boos, J.-L. Rehspringer, Z. Asfari and L. J. Charbonnière, Dalton Trans., 2014, 43, 15583 RSC.
- K. D. Wegner, F. Morgner, E. Oh, R. Goswami, K. Susumu, M. H. Stewart, I. L. Medintz and N. Hildebrandt, Chem. Mater., 2014, 26, 4299 CrossRef CAS.
- K. D. Wegner, P. T. Lanh, T. Jennings, E. Oh, V. Jain, S. M. Fairclough, J. M. Smith, E. Giovanelli, N. Lequeux, T. Pons and N. Hildebrandt, ACS Appl. Mater. Interfaces, 2013, 5, 2881 CAS.
- K. D. Wegner, Z. Jin, S. Lindén, T. L. Jennings and N. Hildebrandt, ACS Nano, 2013, 7, 7411 CrossRef CAS PubMed.
- N. Billinton and A. W. Knight, Anal. Biochem., 2001, 291, 175 CrossRef CAS PubMed.
- M. S. Viegas, T. C. Martins, F. Seco and A. do Carmo, Eur. J. Histochem., 2007, 51, 59 CAS.
- H. J. Tanke, J. Microsc., 1989, 155, 405 CrossRef PubMed.
- G. Marriott, R. M. Clegg, D. J. Arndt-Jovin and T. M. Jovin, Biophys. J., 1991, 60, 1374 CrossRef CAS.
- L. J. Charbonnière, N. Hildebrandt, R. F. Ziessel and H.-G. Löhmannsröben, J. Am. Chem. Soc., 2006, 128, 12800 CrossRef PubMed.
- K. Hanaoka, K. Kikuchi, S. Kobayashi and T. Nagano, J. Am. Chem. Soc., 2007, 129, 13502 CrossRef CAS PubMed.
- M. Rajendran, E. Yapici and L. W. Miller, Inorg. Chem., 2014, 53, 1839 CrossRef CAS PubMed.
- H. E. Rajapakse, N. Gahlaut, S. Mohandessi, D. Yu, J. R. Turner and L. W. Miller, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13582 CrossRef CAS PubMed.
- R. Pal and A. Beeby, Methods Appl. Fluoresc. Spectrosc., 2014, 2, 037001 CrossRef.
- Y. Lu, D. Jin, R. C. Leif, W. Deng, J. A. Piper, J. Yuan, Y. Duan and Y. Huo, Cytometry, Part A, 2011, 79A, 349 CrossRef PubMed.
- Y. Lu, P. Xi, J. A. Piper, Y. Huo and D. Jin, Sci. Rep., 2012, 2, 837 Search PubMed.
- Y. Lu, J. Zhao, R. Zhang, Y. Liu, D. Liu, E. M. Goldys, X. Yang, P. Xi, A. Sunna, J. Lu, Y. Shi, R. C. Leif, Y. Huo, J. Shen, J. A. Piper, J. P. Robinson and D. Jin, Nat. Photonics, 2014, 8, 32 CrossRef CAS.
- A. Grichine, A. Haefele, S. Pascal, A. Duperray, R. Michel, C. Andraudd and O. Maury, Chem. Sci., 2014, 5, 3475 RSC.
- K. R. Kupcho, D. K. Stafslien, T. DeRosier, T. M. Hallis, M. S. Ozers and K. W. Vogel, J. Am. Chem. Soc., 2007, 129, 13372 CrossRef CAS PubMed.
- T. Kokko, T. Liljenbäck, M. T. Peltola, L. Kokko and T. Soukka, Anal. Chem., 2008, 80, 9763 CrossRef CAS PubMed.
- T. Kokko, L. Kokko and T. Soukka, J. Fluoresc., 2009, 19, 159 CrossRef CAS PubMed.
- S. H. Kim, J. R. Gunther and J. A. Katzenellenbogen, J. Am. Chem. Soc., 2010, 132, 4685 CrossRef CAS PubMed.
- T. Hilal, V. Puetter, C. Otto, K. Parczyk and B. Bader, J. Biomol. Screening, 2010, 15, 268 CrossRef CAS PubMed.
- H. E. Rajapakse and L. W. Miller, Methods Enzymol., 2012, 505, 329 CAS.
- D. Geißler, S. Stufler, H.-G. Löhmannsröben and N. Hildebrandt, J. Am. Chem. Soc., 2013, 135, 1102 CrossRef PubMed.
- D. Geißler, S. Lindén, K. Liermann, K. D. Wegner, L. J. Charbonnière and N. Hildebrandt, Inorg. Chem., 2014, 53, 1824 CrossRef PubMed.
- O. J. Harrison, X. Jin, S. Hong, F. Bahna, G. Ahlsen, J. Brasch, Y. Wu, J. Vendome, K. Felsovalyi, C. M. Hampton, R. B. Troyanovsky, A. Ben-Shaul, J. Frank, S. M. Troyanovsky, L. Shapiro and B. Honig, Structure, 2011, 19, 244 CrossRef CAS PubMed.
- B.-A. Truong Quang, M. Mani, O. Markova, T. Lecuit and P.-F. Lenne, Curr. Biol., 2013, 23, 2197 CrossRef CAS PubMed.
- J. Xu, T. M. Corneillie, E. G. Moore, G.-L. Law, N. G. Butlin and K. N. Raymond, J. Am. Chem. Soc., 2011, 133, 19900 CrossRef CAS PubMed.
- N. Hildebrandt, K. D. Wegner and W. R. Algar, Coord. Chem. Rev., 2014, 273–274, 125 CrossRef CAS PubMed.
- S. A. Kim, C.-Y. Tai, L.-P. Mok, E. A. Mosser and E. M. Schuman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9857 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Results of cadherin expression from flow cytometry (Fig. S1 and S2); PL decay curve and determination of PL decay time of Tb antibodies (Fig. S3); additional TG microscopy images of control experiments for E-cadherin expression on MCF-7 cells (Fig. S4); PL decay curves of solution-phase assays demonstrating Tb-antibody and dye-antibody binding to the same primary antibody (Fig. S5 and S6); additional TG and SS microscopy images showing that there is no crosstalk of Tb PL in the FRET channel (AF568) in the case of the E-cadherin cluster investigations using FRET and MCF-7 cells (Fig. S7); additional TG and SS microscopy images of control experiments for E/N-cadherin co-expression on A549 cells (Fig. S8 to S10); additional TG and SS microscopy images showing photobleaching, problems in serial donor–acceptor image acquisition, and successful co-labelling of N-cadherin with Tb and dye antibodies on M4-T cells (Fig. S11 and S12). See DOI: 10.1039/c4dt02884h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Niko
Hildebrandt
*a
b and
Niko
Hildebrandt
*a