
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Icosahedral metallacarborane/carborane species derived from 1,1′-bis(o-carborane)†‡
Gobika
Thiripuranathar
,
Wing Y.
Man
,
Cesar
Palmero
,
Antony P. Y.
Chan
,
Bernhard T.
Leube
,
David
Ellis
,
David
McKay
 ,
Stuart A.
Macgregor
,
Laure
Jourdan
,
Georgina M.
Rosair
and
Alan J.
Welch
*
,
Stuart A.
Macgregor
,
Laure
Jourdan
,
Georgina M.
Rosair
and
Alan J.
Welch
*
Institute of Chemical Sciences, Heriot-Watt University, Edinburgh, UK EH14 4AS. E-mail: a.j.welch@hw.ac.uk; Tel: +44 (0)131 451 3217
First published on 9th February 2015
Abstract
Examples of singly-metallated derivatives of 1,1′-bis(o-carborane) have been prepared and spectroscopically and structurally characterised. Metallation of [7-(1′-1′,2′-closo-C2B10H11)-7,8-nido-C2B9H10]2− with a {Ru(p-cymene)}2+ fragment affords both the unisomerised species [1-(1′-1′,2′-closo-C2B10H11)-3-(p-cymene)-3,1,2-closo-RuC2B9H10] (2) and the isomerised [8-(1′-1′,2′-closo-C2B10H11)-2-(p-cymene)-2,1,8-closo-RuC2B9H10] (3), and 2 is easily transformed into 3 with mild heating. Metallation with a preformed {CoCp}2+ fragment also affords a 3,1,2-MC2B9-1′,2′-C2B10 product [1-(1′-1′,2′-closo-C2B10H11)-3-Cp-3,1,2-closo-CoC2B9H10] (4), but if CoCl2/NaCp is used followed by oxidation the result is the 2,1,8-CoC2B9-1′,2′-C2B10 species [8-(1′-1′,2′-closo-C2B10H11)-2-Cp-2,1,8-closo-CoC2B9H10] (5). Compound 4 does not convert into 5 in refluxing toluene, but does do so if it is reduced and then reoxidised, perhaps highlighting the importance of the basicity of the metal fragment in the isomerisation of metallacarboranes. A computational study of 1,1′-bis(o-carborane) is in excellent agreement with a recently-determined precise crystallographic study and establishes that the {1′,2′-closo-C2B10H11} fragment is electron-withdrawing compared to H.
Introduction
1,1′-Bis(o-carborane), the trivial name for [1-(1′-1′,2′-closo-C2B10H11)-1,2-closo-C2B10H11] (Fig. 1), is the simplest bis(carborane) species, comprising two ortho-carborane units connected by a C–C bond.1 It was first synthesised by insertion of diacetylene into B10 frameworks2 but it is also produced from the CuCl2-mediated coupling reactions of mono- or di-lithiated salts of ortho-carborane,3 although yields by this route are somewhat compromised by the additional formation of C–B and B–B linked isomers. CuCl2-coupling was also used to make 1,1′-bis(m-carborane)3,4 and 1,1′-bis(p-carborane),4,5 the latter an important starting point for the construction of “carborods”, rigid-rod oligomers of para-carborane. Ref. 5(a) also notes that 1,1′-bis(p-carborane) can be prepared by CuCl-coupling, an idea subsequently used by Xie to afford an improved yield of 1,1′-bis(o-carborane).6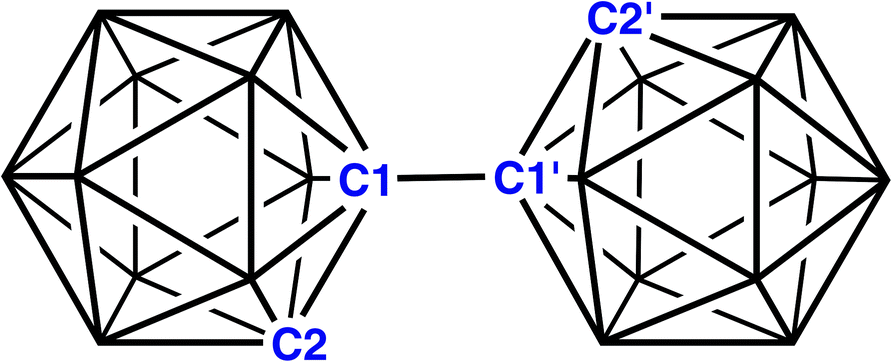 | ||
| Fig. 1 1,1′-bis(o-carborane). | ||
Although 1,1′-bis(o-carborane) has been known for many years, its chemistry remains underdeveloped. Double deprotonation forms a dianionic chelating ligand which has been used to complex a variety of transition-metal cations7 and also an {AsMe} fragment.8 Mono- and di-deboronation (single and double “decapitation”, respectively) of 1,1′-bis(o-carborane) has also been reported.9 In addition, 1,1′-bis(o-carborane) has been reduced with both 2e and 4e.10 In solution, [PPh3Me]+ and [(15-crown-5)3Na2]2+ salts of the 2e reduced species are identical, whilst in the solid state the anion of the [PPh3Me]+ salt has two partially-open 4-atom CBCB faces11 and the anion of the [(15-crown-5)3Na2]2+ salt has one 4-atom CBCB face which is partially-open and one 5-atom CBCBB face which is rather more open.10 Double protonation of the 4e reduced form and subsequent work-up caused the linking C atoms to adopt bridging positions on B–B edges above nido 11-vertex cages, in a similar manner to the protonation and work-up of [7,9-nido-C2B10H12]2− affording [μ9,10-CH2-7-nido-CB10H11]−.12
Prior to our recent research13,14 the only metallacarboranes derived from bis(carboranes) of which we are aware are two 2,1,8-MC2B9-1′,2′-C2B10 species15,16 and two bis(metallacarboranes),17 one of 3,1,2-MC2B9-3′,1′,2′-MC2B9 geometry and the other of 3,1,2-MC2B9-2′,1′,8′-MC2B9 geometry.18
Recently we explored the consequences of 4e reduction and metallation of bis(o-carborane). Reduction and metallation with {Ru(p-cymene)}2+ fragments (p-cymene = η-C10H14, 1-iPr,4-MeC6H4) led unexpectedly to a 13-vertex metallacarborane/12-vertex carborane species and cleavage of an aromatic C–C bond under ambient conditions.13 Reduction and metallation with {CoCp}2+ fragments (Cp = η-C5H5) afforded racemic and meso diastereoisomers of the 13-vertex metallacarborane/13-vertex metallacarborane species [1-(1′-4′-Cp-4′,1′,6′-closo-CoC2B10H11)-4-Cp-4,1,6-closo-CoC2B10H11].14
In this contribution we report the monodeboronation and subsequent metallation with {ML} fragments (L = η-bonded polyene) of 1,1′-bis(o-carborane) leading to 12-vertex metallacarborane/12-vertex carborane products with both non-isomerised [1-(1′-1′,2′-closo-C2B10H11)-3-L-3,1,2-closo-MC2B9H10] and isomerised [8-(1′-1′,2′-closo-C2B10H11)-2-L-2,1,8-closo-MC2B9H10] architectures. We describe detailed spectroscopic and structural studies of these products and investigate the isomerisation between them.
Results and discussion
Monodeboronation of 1,1′-bis(o-carborane) with one equivalent of KOH in refluxing EtOH, according to the procedure outlined by Hawthorne et al.,9 followed by cation metathesis, afforded the anion [7-(1′-1′,2′-closo-C2B10H11)-7,8-nido-C2B9H11]− ([1]−), as either the [HNMe3]+ or [BTMA]+ salt (BTMA = benzyltrimethylammonium) in good yields. The 1H NMR spectrum of [1]− shows, in addition to the resonances associated with the appropriate cation, two CHcage resonances of equal integral at δ ca. 4.4 and 2.0 ppm. The former is assigned to the {closo-C2B10} component and the latter to the {nido-C2B9} component with reference to the spectra of 1,1′-bis(o-carborane) and [7,8-nido-C2B9H12]−.19The 11B{1H} NMR spectrum of [1]− consists of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:5:2:3:2:1:1:1:1 pattern between δ –4 and –36 ppm. A 11B{1H}-11B{1H} COSY spectrum of [HNMe3][1] in (CD3)2CO was obtained in an attempt to assign these resonances to {closo-C2B10} or {nido-C2B9} components. By analogy with the spectra of [7,8-nido-C2B9H12]−19 and 1,2-closo-C2B10H12,20 it seems reasonable to suggest that the two highest frequency resonances are due to the {closo-C2B10} cage and the four lowest frequency resonances are due to the {nido-C2B9} cage, but beyond this it was not possible to deconvolute the entire spectrum of [1]− unambiguously.
:1:1:5:2:3:2:1:1:1:1 pattern between δ –4 and –36 ppm. A 11B{1H}-11B{1H} COSY spectrum of [HNMe3][1] in (CD3)2CO was obtained in an attempt to assign these resonances to {closo-C2B10} or {nido-C2B9} components. By analogy with the spectra of [7,8-nido-C2B9H12]−19 and 1,2-closo-C2B10H12,20 it seems reasonable to suggest that the two highest frequency resonances are due to the {closo-C2B10} cage and the four lowest frequency resonances are due to the {nido-C2B9} cage, but beyond this it was not possible to deconvolute the entire spectrum of [1]− unambiguously.
The salt [HNMe3][1] is a convenient starting point for the synthesis of MC2B9-C2B10 products by deprotonation then metallation, following the protocol established for the first metallacarborane by Hawthorne et al.21
Following deprotonation of [HNMe3][1] with n-BuLi in THF and addition of [RuCl2(p-cymene)]2, yellow [1-(1′-1′,2′-closo-C2B10H11)-3-(p-cymene)-3,1,2-closo-RuC2B9H10] (2) and colourless [8-(1′-1′,2′-closo-C2B10H11)-2-(p-cymene)-2,1,8-closo-RuC2B9H10] (3) were isolated in yields of 8 and 19%, respectively. Both compounds were initially characterised by elemental microanalysis and EI mass spectrometry, the latter clearly showing the molecular ion peaks as a characteristic envelope due to the two naturally-occurring boron isotopes.
In the 1H spectrum of a freshly-prepared CDCl3 solution of 2 are CHcage resonances at δ 4.03 and 3.91 but these are too close to each other to speculate which is due to the carborane and which is due to the ruthenacarborane. The 1H NMR spectrum of 2 also confirms overall molecular asymmetry with two integral-3 doublets (and not one integral-6 doublet) for the CH(CH3)2 protons of the p-cymene ligand. The 11B{1H} NMR spectrum of 2 consists of ten resonances between δ 2.8 and −17.2 with relative integrals 1:1:2:1:2:2:4:2:1:3 from high frequency to low frequency.
With time, solutions of 2 show clear evidence for a slow transformation of 2 into an isomer 3, a compound which was originally isolated along with 2 from the initial reaction. A THF solution of 2 heated to reflux for two hours reveals its complete conversion to 3, with 58% of the compound being recovered following work-up involving thin layer chromatography (TLC). In 3 there is a significantly greater separation of the CHcage resonances, which now appear at δ 3.64 and 2.63. Since only the ruthenacarborane part of 2 has changed in its isomerisation into 3 we tentatively assign the lower frequency resonance, δ 2.63, as arising from CHcage in the {RuC2B9} portion of 3. Once again the resonances due to the p-cymene ligand reveal the overall molecular structure to be asymmetric. In the 11B{1H} NMR spectrum of 3 are ten resonances between δ −1.0 and −20.4 with integrals in the relative ratios 2:2:1:2:1:6:2:1:1:1.
In addition to {Ru(arene)}22 a common transition-metal fragment in metallacarborane chemistry is {CoCp}. There are two different ways to introduce this fragment to afford a CpCoC2Bx metallacarborane, (i) reaction of the [C2Bx]2− dianion with CoCl2/NaCp (i.e. in situ generation of the {CoCp} fragment) followed by oxidation (CoII → CoIII)23 or (ii) reaction of the [C2Bx]2− dianion with [CpCo(CO)I2] (i.e. using a “preformed” {CoCp} fragment).24 In reaction with [C2B9H11]2− both approaches lead to exactly the same product, but we have found that this is not the case starting from [1]−.
Deprotonation of [HNMe3][1] followed by addition of [CpCo(CO)I2] affords, on work-up, the isomer [1-(1′-1′,2′-closo-C2B10H11)-3-Cp-3,1,2-closo-CoC2B9H10] (4) as an orange solid. Microanalysis and mass spectrometry confirm the molecular formula. In the 1H NMR spectrum are three singlets at δ 5.86 (5H, Cp), 4.24 (1H) and 4.03 (1H), the last two relatively broad and arising from the cage CH atoms. In the 11B NMR spectrum are nine resonances in a 1:1:5:1:2:5:2:1:1 pattern, lying between δ 6.5 and −15.9 ppm.
To our surprise, treatment of deprotonated [HNMe3][1] with CoCl2/NaCp followed by aerial oxidation yielded an isomer of 4, the 2,1,8-1′,2′ species [8-(1′-1′,2′-closo-C2B10H11)-2-Cp-2,1,8-closo-CoC2B9H10] (5). This yellow product has, as well as the expected singlet for the Cp protons, cage CH resonances at lower frequency than in 4, δ 3.59 and 2.73. In 4 the {CoC2B9} part of the molecule has a 3,1,2-CoC2 heteroatom pattern whilst in 5 it is 2,1,8-CoC2. In the corresponding reference compound [3-Cp-3,1,2-closo-CoC2B9H11] the cage CH atoms resonate at δ 4.08 (CDCl3) and in [2-Cp-2,1,8-closo-CoC2B9H11] they resonate at δ 2.73 and 2.47,25 on the basis of which we tentatively assign the signal at δ 2.73 in 5 to the {2,1,8-CoC2B9} fragment. The 11B NMR spectrum of 5 reveals eleven resonances between δ 1.7 and −17.7 in a 1:2:1:1:1:2:6:1:2:1:1 pattern of integrals. Note that in the synthesis of 4 a trace amount of 5 is also detected (see Experimental) and that in the synthesis of 5 a trace amount of 4 is observed.
Given that the 3,1,2-RuC2B9-1′,2′-C2B10 species 2 easily transforms to its isomer 2,1,8-RuC2B9-1′,2′-C2B103 on heating to reflux in THF we attempted to thermally isomerise the 3,1,2-CoC2B9-1′,2′-C2B10 species 4, expecting it to convert into 2,1,8-CoC2B9-1′,2′-C2B105. However, even in refluxing toluene for five hours there is no evidence that 4 converts into 5 by thermolysis. We later show from crystallographic studies that, at least as far as we can tell, compounds 2 and 4 suffer similar degrees of intramolecular steric crowding, implying that the different isomerisation characteristics of 2 and 4 cannot be explained by steric factors.
When [1]2− is treated with [CpCo(CO)I2] a {CoIIICp}2+ fragment is introduced to the dianion, affording the non-isomerised 4. However, when [1]2− is treated with CoCl2/NaCp the reacting fragment is {CoIICp}+. This generates the 19e monoanion [CpCoII(C2B9H10)(C2B10H11)]−, which is then oxidised to the 18e, isomerised, CoIII species 5. We therefore added one equivalent of electrons to 4 at room temperature and, after stirring for one hour, oxidised the product aerially. Only 5 was detected by 1H and 11B NMR spectroscopies. This strongly implies that in the bulk synthesis of 5, a [3,1,2-CoC2B9-1′,2′-C2B10]− species is formed first (as would be expected from the reaction between a {CoCp}+ cation and a [7,8-C2B9-1′,2′-C2B10]2− anion) and that this 19e anionic intermediate then isomerises before it is oxidised. Taken together with the facile isomerisation of the 3,1,2-RuC2B9-1′,2′-C2B10p-cymene species 2 these observations highlight that the basicity of the metal fragment, and not just its steric bulk, might be important in effecting a 3,1,2-MC2B9 to 2,1,8-MC2B9 isomerisation.
For compounds 2, 4 and 5 we attempted to identify which 11B resonances were due to which part of the molecule ({MC2B9} or {C2B10}) from 11B{1H}–11B{1H} COSY spectra but, as was the case with [1]−, it proved impossible to do this unambiguously. In Table 1 we list the weighted average 11B chemical shifts, <δ(11B)>, of the conjoined species 1,1′-bis(o-carborane), 2, 4 and 5 along with those of their “components”, [1,2-closo-C2B10H12],20 [3-(p-cymene)-3,1,2-closo-RuC2B9H11],26 [3-Cp-3,1,2-closo-CoC2B9H11]23 and [2-Cp-2,1,8-closo-CoC2B9H11].25 Although the spectra of 1,1′-bis(o-carborane)3,6 and all these “components” have been reported previously we have remeasured some of them here in CDCl3 for internal consistency. Note that we have not included compound 3 in this Table since its {MC2B9} component, [2-(p-cymene)-2,1,8-closo-RuC2B9H11], is not currently known.
| Compound | <δ(11B)> |
|---|---|
| a For 1,1′-bis(o-carborane), 1,2-closo-C2B10H12, 3-(p-cymene)-3,1,2-closo-RuC2B9H11, 3-Cp-3,1,2-closo-CoC2B9H11 and 2-Cp-2,1,8-closo-CoC2B9H11 individual 11B chemical shifts are given in Table S1 (ESI). | |
| 1,1′-bis(o-carborane) | –8.9 |
| [1-(1′-1′,2′-closo-C2B10H11)-3-(p-cymene)-3,1,2-closo-RuC2B9H10] (2) | –9.1 |
| [1-(1′-1′,2′-closo-C2B10H11)-3-Cp-3,1,2-closo-CoC2B9H10] (4) | –6.7 |
| [8-(1′-1′,2′-closo-C2B10H11)-2-Cp-2,1,8-closo-CoC2B9H10] (5) | –7.9 |
| 1,2-closo-C2B10H12 | –10.7 |
| 3-(p-cymene)-3,1,2-closo-RuC2B9H11 | –10.5 |
| 3-Cp-3,1,2-closo-CoC2B9H11 | –7.3 |
| 2-Cp-2,1,8-closo-CoC2B9H11 | –7.7 |
These data show that when 1,1′-bis(o-carborane) and the metallacarborane-carborane species 2, 4 and 5 are “constructed” from their constituent parts the <δ(11B)> value for the “product” lies to high frequency of the (weighted) average of that of the two “components”. For 5 the <δ(11B)> value is very close (and slightly to low frequency of) to that for the metallacarborane component, whilst for 1,1′-bis(o-carborane), 2 and 4 the <δ(11B)> value is actually to high frequency of that of both components.
A shift to higher frequency of the average 11B resonance implies, overall, that the B nuclei in these last two conjoined cages are deshielded, and therefore δ+, relative to those in the individual components. A comparative computational study of [1,2-closo-C2B10H12] and 1,1′-bis(o-carborane) supports this conclusion. By DFT calculation we find effectively no preference in 1,1′-bis(o-carborane) between conformations with C2–C1–C1′–C2′ torsion angles of 108° and 180° (Fig. 2). In terms of only the electronic energy the 108° conformation is favoured by 0.2 kcal mol−1, whereas if zero point energy is included the 180° conformation is preferred by 0.5 kcal mol−1. The barrier to free rotation about the C1–C1′ bond is only ca. 10 kcal mol−1, corresponding to a transition state at a C2–C1–C1′–C2′ torsion angle of 0°. Computational models are listed in the ESI.‡ It is very satisfying to note that our recent definitive crystallographic study of 1,1′-bis(o-carborane) found that the non-linking C atom is equally disordered between vertices 2 and 3 (and, by symmetry, 2′ and 3′).1 This means that in the crystal any one molecule of 1,1′-bis(o-carborane) is equally likely to have a C–C–C–C torsion angle of 180° (C2–C1–C1′–C2′) or 108° (C3–C1–C1′–C2′), in perfect agreement with the results of the DFT study. The computed C1–C1′ distance in the 180° conformation is 1.542 Å, and in the 108° conformation it is 1.540 Å. Experimentally C1–C1′ is 1.5339(11) Å.1
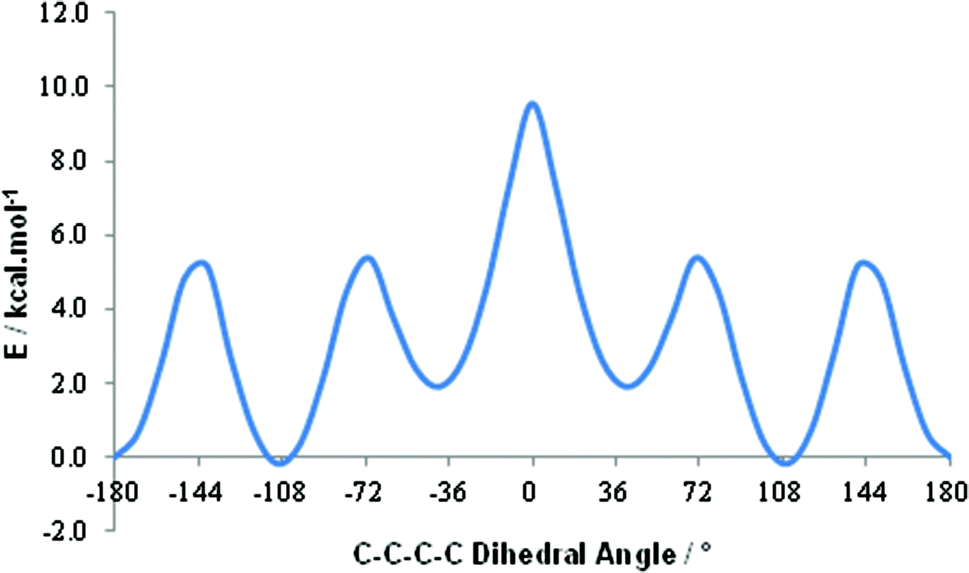 | ||
| Fig. 2 Plot of energy vs. dihedral angle for 1,1′-bis(o-carborane) from DFT calculation where the C2–C1–C1′–C2′ torsion angle was subjected to a relaxed scan from −180° to 0° and the resulting data points mirrored to illustrate full 360° rotation. | ||
In Table 2 we list the natural charges for atoms in [1,2-closo-C2B10H12] and 1,1′-bis(o-carborane), the latter in the 180° conformation. In [1,2-closo-C2B10H12] the C atoms carry a charge of −0.56 and the B atoms an average charge of −0.06. H bonded to C is +0.36 whilst the average charge of H bonded to B is +0.10. In 1,1′-bis(o-carborane) the negative charge on both C atoms decreases (C1, the substituted atom, −0.31; C2, −0.51) and the B atoms are also less negative (average charge −0.04). The remaining C-bonded H atom carries a charge of +0.32 and the average charge on H bound to B is +0.09. Thus substitution of one of the C-bound H atoms in [1,2-closo-C2B10H12] by a {1′,2′-closo-C2B10H11} unit causes all the polyhedral atoms (both C and B) in the original cage to become less negatively charged. At the same time there is an opposite, but smaller, change in the charges on the H atoms bonded to the polyhedral atoms, which become slightly less positively charged. The overall charge on the {C2B10H11} fragment changes from −0.36 in [1,2-closo-C2B10H12] to precisely zero in 1,1′-bis(o-carborane).27 In brief the {1′,2′-closo-C2B10H11} substituent is electron-withdrawing compared to H. This conclusion is consistent with our analysis of the <δ(11B)> values for 1,1′-bis(o-carborane) and for 2.
| Atom | Charge | |
|---|---|---|
| (a) 1,2-closo-C2B10H12 | ||
| C1, C2 | −0.56 |
|
| B3, B6 | +0.13 | |
| B4, B5, B7, B11 | −0.03 | |
| B8, B10 | −0.19 | |
| B9, B12 | −0.17 | |
| H1, H2 | +0.36 | |
| H3, H6 | +0.08 | |
| H4, H5, H7, H11 | +0.10 | |
| H8, H10 | +0.11 | |
| H9, H12 | +0.10 | |
| (b) 1,1′-bis(o-carborane) | ||
| C1 | −0.31 |
|
| C2 | −0.51 | |
| B3, B6 | +0.15 | |
| B4, B5 | −0.01 | |
| B7, B11 | −0.01 | |
| B8, B10 | −0.18 | |
| B9 | −0.16 | |
| B12 | −0.14 | |
| H2 | +0.32 | |
| H3, H6 | +0.08 | |
| H4, H5 | +0.08 | |
| H7, H11 | +0.09 | |
| H8, H10 | +0.10 | |
| H9 | +0.10 | |
| H12 | +0.09 | |
Salt [BTMA][1] and compounds 2–5 were also studied crystallographically. In [BTMA][1] (Fig. 3) the C2B10 cage is ordered but the C2B9 cage is disordered with positions 3 and 12 partially occupied by boron. The second C atom of the nido cage is ordered, however, and the C8–C7–C1′–C2′ torsion angle is 177.2(6)°. The linking C7–C1′ bond length is 1.514(9) Å.
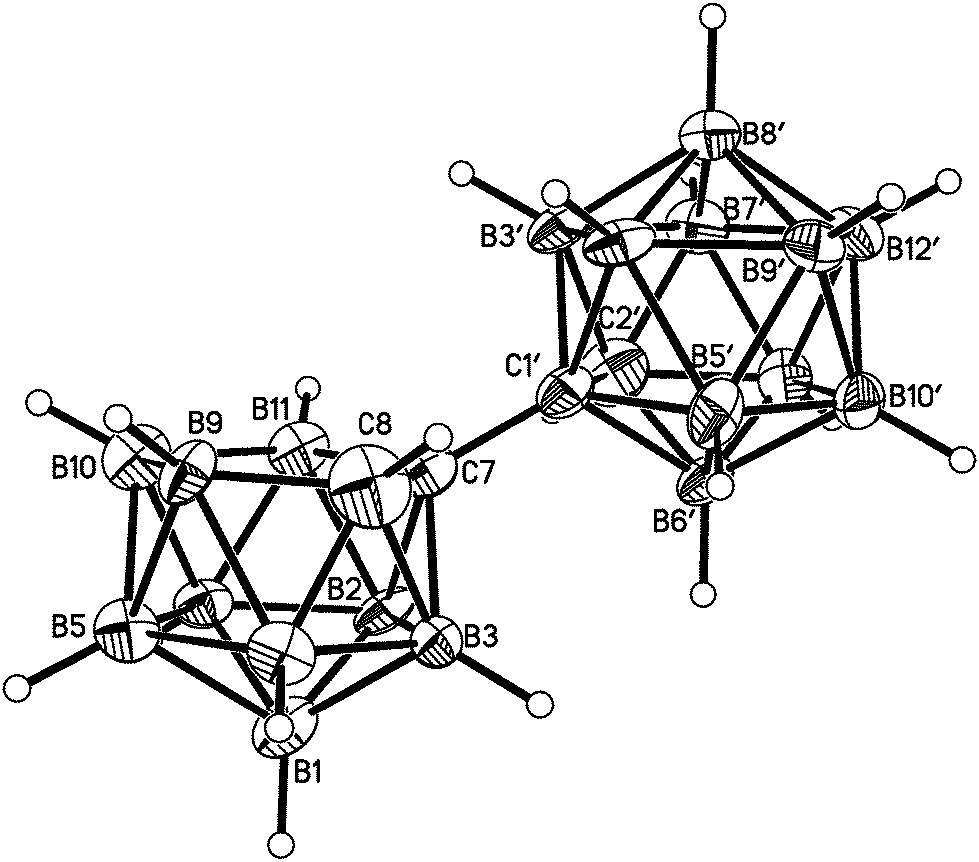 | ||
| Fig. 3 Perspective view of the anion in the salt [BTMA][1] and atom numbering scheme. In the unprimed cage there is partial disorder of B3, part of which appears as the 12th atom of an icosahedron (not shown for clarity); partial occupancies are B3 0.548(10) and B12 0.452(10). The H atom bridging on the open face of the unprimed cage was not located. Displacement ellipsoids are drawn at the 40% probability level except for H atoms. | ||
Perspective views of single molecules of the 3,1,2-RuC2B9-1′,2′-C2B10 species 2, its 2,1,8-RuC2B9-1′,2′-C2B10 analogue 3, and the equivalent cobalt species 4 and 5 are presented in Fig. 4–7, respectively. Since compounds 2, 4 and 5 are composed of {3,1,2-closo-RuC2B9} (compound 2), {3,1,2-closo-CoC2B9} (compound 4) and {2,1,8-closo-CoC2B9} (compound 5) icosahedra conjoined to {1,2-closo-C2B10} icosahedra, and all these individual components have previously been studied crystallographically, we have used the Structure Overlay tool in Mercury28 to calculate individual atom and overall fragment root-mean-square (rms) misfits between the components of 2, 4 and 5 and the corresponding literature molecules (there is currently no structural study of a 2-(arene)-2,1,8-closo-RuC2B9H11 species in the literature and so a similar exercise cannot be undertaken for compound 3). The results, summarised in Table 3, clearly show that for the {3,1,2-MC2B9} fragments the greatest misfit is at the metal vertex, ca. 0.08–0.09 Å, and that the misfit at C1 (the position of substitution) is also relatively large, ca. 0.06–0.08 Å. The overall misfit for {3,1,2-MC2B9} is typically 0.038–0.040 Å. In contrast the misfit for the {2,1,8-MC2B9} fragment is considerably less with an overall misfit of only 0.012 Å, the greatest individual misfit, 0.025 Å, occurring at C8 (the position of substitution) and no other atom having a misfit >0.018 Å. The {C2B10} fragments fit better with their reference molecule, the overall misfit here being 0.02–0.03 Å, and it is always C1′ or C2′ that has the largest individual misfit, typically 0.05–0.06 Å.
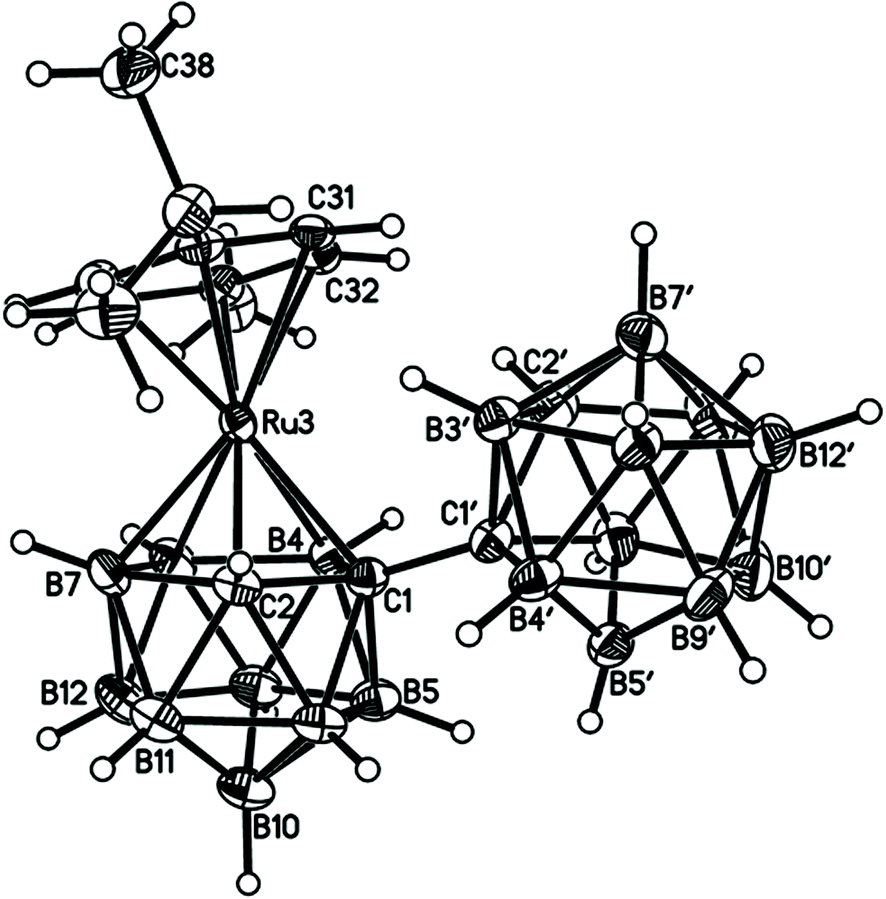 | ||
| Fig. 4 Perspective view of compound 2 and atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level except for H atoms. | ||
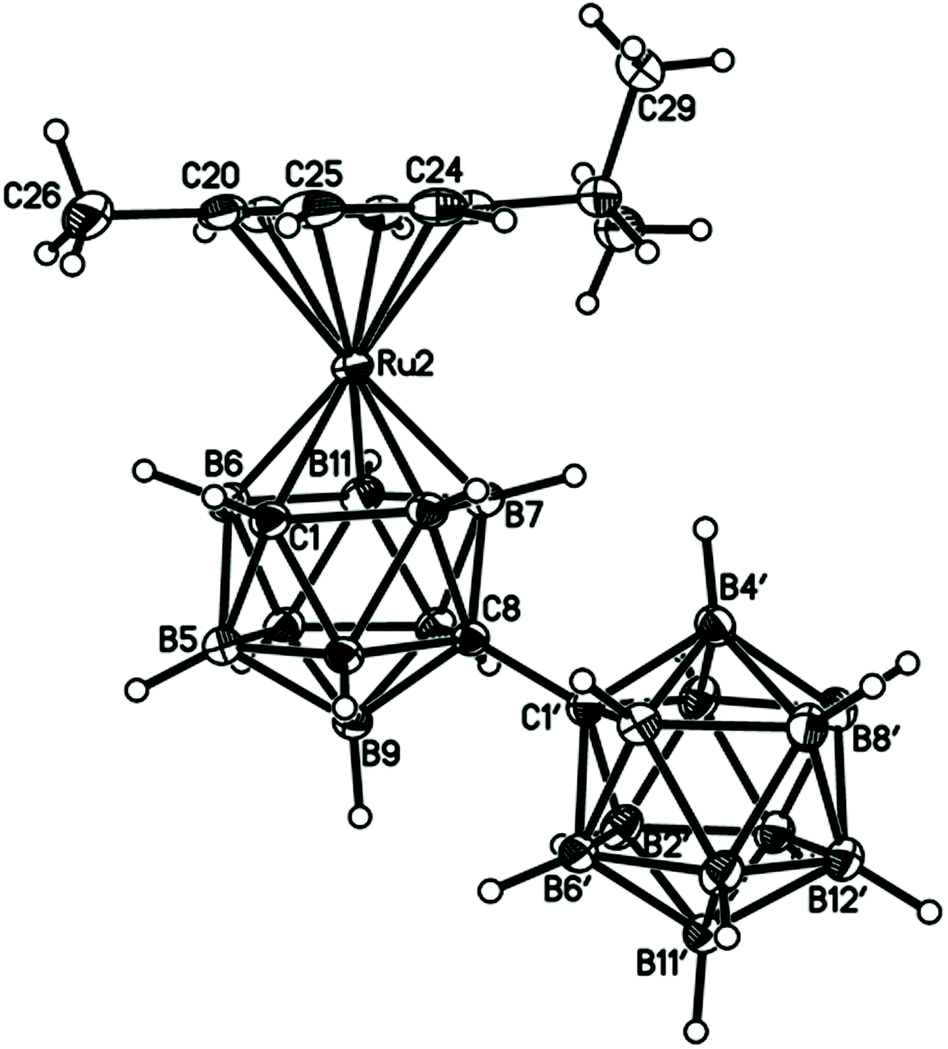 | ||
| Fig. 5 Perspective view of compound 3 and atom numbering scheme. Position 2′ is 0.446(19)B + 0.554(19)C, with complementary occupations at position 3′. Displacement ellipsoids as for Fig. 4. | ||
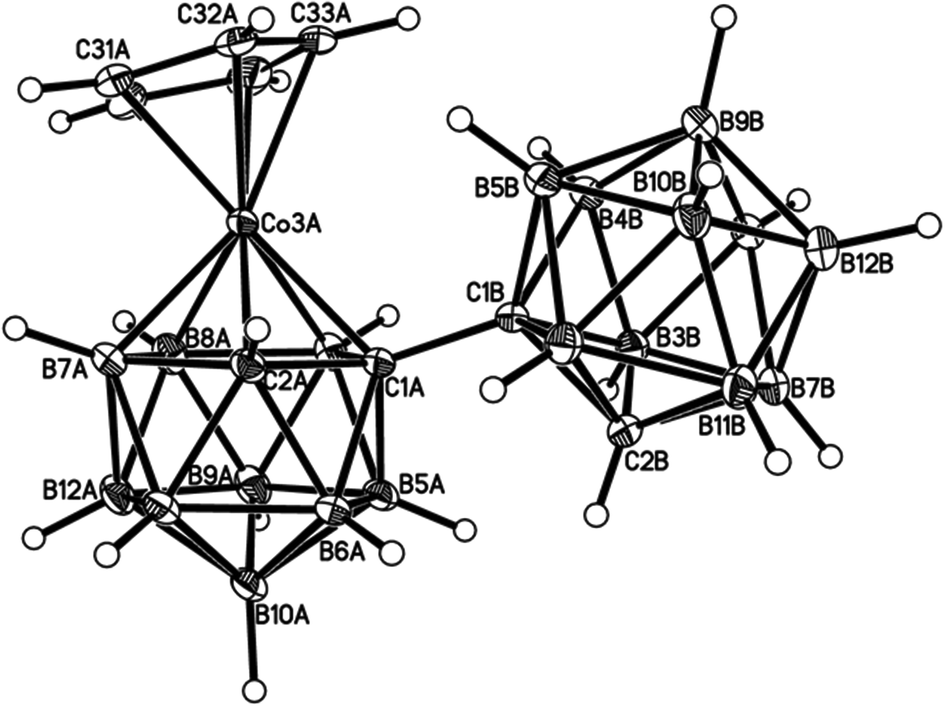 | ||
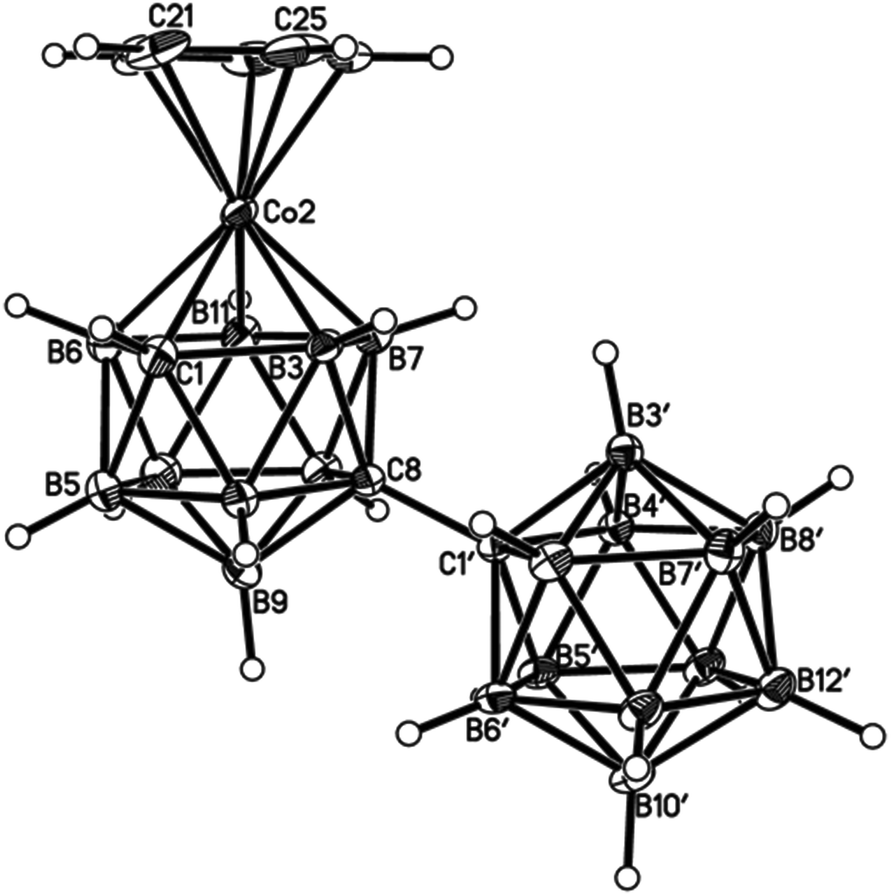
| Fig. 6 Perspective view of one of two crystallographically-independent molecules (molecule AB) of compound 4 and atom numbering scheme. Displacement ellipsoids as for Fig. 4. | ||
 | ||
| Fig. 7 Perspective view of compound 5 and atom numbering scheme. Displacement ellipsoids as for Fig. 4. | ||
| Compound 2a | Compound 4 (C–D)b | Compound 4 (C–D)b | Compound 5c | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| {RuC2B9} | Dev. | {C2B10} | Dev. | {CoC2B9} | Dev. | {C2B10} | Dev. | {CoC2B9} | Dev. | {C2B10} | Dev. | {CoC2B9} | Dev. | {C2B10} | Dev. |
| a The reference {RuC2B9} compound is 3-(p-cymene)-3,1,2-closo-RuC2B9H11 (ref. 26). CCDC refcode ODOGAQ. b The reference {CoC2B9} compound is 3-Cp-3,1,2-closo-CoC2B9H11 (ref. 45). CCDC refcode DUBDIN. c The reference {CoC2B9} compound is 2-Cp-2,1,8-closo-CoC2B9H11 (ref. 25). CCDC refcode NOWMIX00. a–c In all cases the reference {C2B10} compound is 1,2-closo-C2B10H12 (ref. 46). CCDC refcode TOKGIJ. We arbitrarily used the molecule containing C13 and C14 (CCDC numbering) matching C13 with C1′. | |||||||||||||||
| C1 | 0.066 | C1′ | 0.054 | C1 | 0.074 | C1′ | 0.059 | C1 | 0.076 | C1′ | 0.054 | C1 | 0.004 | C1′ | 0.032 |
| C2 | 0.036 | C2′ | 0.056 | C2 | 0.039 | C2′ | 0.020 | C2 | 0.037 | C2′ | 0.017 | Co2 | 0.012 | C2′ | 0.056 |
| Ru3 | 0.083 | B3′ | 0.039 | Co3 | 0.081 | B3′ | 0.009 | Co3 | 0.091 | B3′ | 0.009 | B3 | 0.006 | B3′ | 0.013 |
| B4 | 0.033 | B4′ | 0.012 | B4 | 0.024 | B4′ | 0.016 | B4 | 0.025 | B4′ | 0.016 | B4 | 0.006 | B4′ | 0.014 |
| B5 | 0.025 | B5′ | 0.047 | B5 | 0.028 | B5′ | 0.013 | B5 | 0.020 | B5′ | 0.014 | B5 | 0.008 | B5′ | 0.024 |
| B6 | 0.038 | B6′ | 0.017 | B6 | 0.025 | B6′ | 0.008 | B6 | 0.037 | B6′ | 0.008 | B6 | 0.010 | B6′ | 0.044 |
| B7 | 0.008 | B7′ | 0.008 | B7 | 0.016 | B7′ | 0.022 | B7 | 0.012 | B7′ | 0.015 | B7 | 0.018 | B7′ | 0.011 |
| B8 | 0.016 | B8′ | 0.009 | B8 | 0.010 | B8′ | 0.005 | B8 | 0.006 | B8′ | 0.010 | C8 | 0.025 | B8′ | 0.007 |
| B9 | 0.009 | B9′ | 0.009 | B9 | 0.008 | B9′ | 0.010 | B9 | 0.011 | B9′ | 0.006 | B9 | 0.000 | B9′ | 0.010 |
| B10 | 0.025 | B10′ | 0.005 | B10 | 0.013 | B10′ | 0.009 | B10 | 0.019 | B10′ | 0.009 | B10 | 0.005 | B10′ | 0.006 |
| B11 | 0.013 | B11′ | 0.017 | B11 | 0.011 | B11′ | 0.011 | B11 | 0.009 | B11′ | 0.017 | B11 | 0.016 | B11′ | 0.009 |
| B12 | 0.006 | B12′ | 0.013 | B12 | 0.014 | B12′ | 0.017 | B12 | 0.015 | B12′ | 0.016 | B12 | 0.005 | B12′ | 0.013 |
| Overall | 0.038 | Overall | 0.030 | Overall | 0.037 | Overall | 0.021 | Overall | 0.039 | Overall | 0.020 | Overall | 0.012 | Overall | 0.025 |
It is clear from Fig. 4 and 6 that a consistent feature of the 3,1,2-MC2B9-1′,2′-C2B10 structures is a pronounced bend-back of the arene or Cp ligand in a direction away from the C2B10 substituent on C1. This structural feature is undoubtedly the result of intramolecular steric crowding, which also likely contributes to the relatively large misfit values of the metal atoms in 2 and 4. The ligand bend-back is conveniently quantified by θ, the dihedral angle between the plane of the ligand C atoms (arene or Cp) and the plane defined by B5B6B11B12B9 (the lower pentagonal belt usually taken as the reference plane in 3,1,2-MC2B9 icosahedra).29 For 2θ is 16.08(9)° whilst for 4θ is 15.83(8)° (molecule A–B) and 16.34(8)° (molecule C–D; in 4 there are two crystallographically-independent molecules A–B and C–D where the first letter refers to the CoC2B9 cage and second letter to the C2B10 cage). C1–C1′ distances in 2 and 4 are 1.545(3), 1.549(2) (A–B) and 1.550(2) Å (C–D), respectively. All these are significantly longer that the C1–C1′ distance in 1,1′-bis(o-carborane), 1.5339(11) Å,1 again a reflection of the steric crowding in 2 and 4.
In the 2,1,8-MC2B9-1′,2′-C2B10 compounds 3 and 5 significant intramolecular steric crowding is removed since the C2B10 substituent to the MC2B9 cage is now at position 8 and so not adjacent to the metal atom. Consequently the arene or Cp ring plane lies effectively parallel to the lower pentagonal belt, now the C8B4B5B10B12 plane [θ is only 0.27(5)° in 3 and 2.19(7)° in 5], and the C8–C1′ distances are 1.5294(17) and 1.5329(16)Å, respectively, slightly shorter than or identical to the intercage C–C distance in 1,1′-bis(o-carborane).1
The gross similarities between the structures of 2 and 4 (similar ligand bend-back angles, similar C–C1′ distances) imply that, to a first approximation, they are equally sterically crowded. However, whilst 2 is relatively easily isomerised to 3 by gentle heating, even prolonged heating to reflux of 4 in toluene does not convert it into 5; rather 4 has to be reduced to the anion [4]− which then isomerises (presumably to [5]−) at room temperature, affording 5 on aerial oxidation. This reduction-induced isomerisation of metallacarboranes has precedent in the literature.30 Thus, as already has been noted, it appears that the basicity of the metal fragment, and not simply the steric crowding it affords, is important in determining the ease of 3,1,2-MC2B9 to 2,1,8-MC2B9 isomerisation in these species. Given that it is generally accepted that cobaltacarboranes are more susceptible to isomerisation than ruthenacarboranes, at least for 13-vertex species,31 this is an interesting observation and one that we will address more fully in future contributions.32
Conclusions
Examples of 12-vertex metallacarborane/carborane compounds, MC2B9-C2B10, derived from single deboronation and then metallation of 1,1′-bis(o-carborane), have been prepared and characterised. Both non-isomerised 3,1,2-MC2B9-1′,2′-C2B10 and isomerised 2,1,8-MC2B9-1′,2′-C2B10 isomers have been isolated. For M = {Ru(p-cymene)} the isomerisation of the former to the latter is effected by gentle heating. In contrast, the non-isomerised form with M = {CoCp} does not isomerise in refluxing toluene but readily isomerises as the result of 1e reduction followed by reoxidation.Experimental
Synthesis
Experiments were performed under dry, oxygen free N2, using standard Schlenk techniques, although subsequent manipulations were sometimes performed in the open laboratory. All solvents were freshly distilled under nitrogen from the appropriate drying agents immediately before use (CH2Cl2 [DCM], CaH2: THF and 40–60 petroleum ether; sodium wire) or were stored over 4 Å molecular sieves and were degassed (3 × freeze–pump–thaw cycles) before use. Preparative TLC employed 20 × 20 cm Kieselgel F254 glass plates. NMR spectra at 400.1 MHz (1H) or 128.4 MHz (11B) were recorded on a Bruker DPX-400 spectrometer from CDCl3 or (CD3)2CO solutions at room temperature. Electron impact mass spectrometry (EIMS) was carried out using a Finnigan (Thermo) LCQ Classic ion trap mass spectrometer at the University of Edinburgh. Elemental analyses were conducted using an Exeter CE-440 elemental analyser at Heriot-Watt University. The starting materials 1,1′-bis(o-carborane),6 [Ru(p-cymene)Cl2]2,33 [Ru(η-C6H6)Cl2]234 and CpCo(CO)I235 were prepared by literature methods or slight variations thereof. All other reagents were supplied commercially.
[HNMe3][1]: Yield 0.37 g, 64%. C7H32B19N requires C 25.0, H 9.60, N 4.17. Found for [HNMe3]1: C 24.7, H 9.71, N 4.04%. 11B{1H} NMR [(CD3)2CO], δ −3.9 (1B), −6.0 (1B), −8.8 (1B), −10.4 (5B), −11.2 (sh., 2B), −13.5 (3B), −16.8 (2B), −19.0 (1B), −22.7 (1B), −33.9 (1B), −35.3 (1B). 1H NMR [(CD3)2CO], δ 4.36 (s, 1H, CHcage), 3.22 (s, 9H, N(CH3)3), 1.99 (s, 1H, CHcage).
[BTMA][1]: Yield 0.55 g, 74%. C14H38B19N requires C 39.5, H 8.99, N 3.29. Found for [BTMA]1: C 41.5, H 9.15, N 3.25%. 11B{1H} NMR [(CD3)2CO], δ −4.2 (1B), −6.2 (1B), −9.0 (1B), −10.6 (5B), −11.5 (sh., 2B), −13.8 (3B), −17.0 (2B), −19.2 (1B), −22.8 (1B), −33.2 (1B), −35.5 (1B). 1H NMR [(CD3)2CO], δ 7.75–7.45 (m, 5H, C6H5), 4.75 (s, 2H, CH2), 4.35 (s, 1H, CHcage), 3.35 (s, 9H, N(CH3)3), 1.95 (s, 1H, CHcage).
:70 afforded a yellow band (Rf = 0.47) subsequently identified as [1-(1′-1′,2′-closo-C2B10H11)-3-(p-cymene)-3,1,2-closo-RuC2B9H10] (2) (0.024 g, 8%) and a colourless band (Rf = 0.51) identified as [8-(1′-1′,2′-closo-C2B10H11)-2-(p-cymene)-2,1,8-closo-RuC2B9H10] (3) (0.057 g, 19%).
2: C14H35B19Ru requires C 33.0, H 6.92. Found for 2: C 32.5, H 7.17%. 11B{1H} NMR [CDCl3], δ 2.8 (1B), 0.5 (1B), −2.8 (2B), −3.9 (sh., 1B), −7.2 (2B), −8.8 (2B), −10.7 (4B), −12.6 (2B), −14.4 (1B), −17.2 (3B). 1H NMR [CDCl3], δ 6.11–5.96 (m, 4H, C6H4), 4.03 (s, 1H, CHcage), 3.91 (s, 1H, CHcage), 3.04 (app. septet, 1H, CH(CH3)2), 2.49 (s, 3H, CH3), 1.37 (d, 3H, CH(CH3)2), 1.35 (d, 3H, CH(CH3)2). EIMS: envelope centred on m/z 510 (M+).
3: C14H35B19Ru requires C 33.0, H 6.92. Found for 3: C 33.0, H 6.82%. 11B{1H} NMR [CDCl3], δ −1.0 (2B), −2.8 (2B), −4.1 (1B), −4.9 (2B), −8.0 (1B), −10.1 (6B), −13.4 (2B), −16.2 (1B), −19.2 (1B), −20.4 (1B). 1H NMR [CDCl3], δ 5.94–5.84 (m, 4H, C6H4), 3.64 (s, 1H, CHcage), 2.81 (app. septet, 1H, CH(CH3)2), 2.63 (s, 1H, CHcage), 2.31 (s, 3H, CH3), 1.30 (d, 3H, CH(CH3)2), 1.28 (d, 3H, CH(CH3)2). EIMS: envelope centred on m/z 510 (M+).
:70, to afford a colourless band at Rf = 0.51 identified as 3 (0.014 g, 58%) by 1H and 11B NMR spectroscopies.
:70, Rf = 0.28) purification by column chromatography using the same eluent gave, on removal of solvent, an orange powder (0.038 g, 13%), subsequently identified as [1-(1′-1′,2′-closo-C2B10H11)-3-Cp-3,1,2-closo-CoC2B9H10] (4). C9H26B19Co requires C 27.1, H 6.57. Found for 4: C 26.5, H 6.67%. 11B{1H} NMR [CDCl3], δ 6.5 (1B), 2.5 (1B), −2.6 (5B), −4.4 (1B), −8.0 (2B), −9.7 (5B), −12.3 (2B), −14.2 (1B), −15.9 (1B). 1H NMR [CDCl3], δ 5.86 (s, 5H, C5H5), 4.24 (s, 1H, CHcage), 4.03 (s, 1H, CHcage). EIMS: envelope centred on m/z 399 (M+).
*A trace amount of a yellow spot (Rf = 0.34) identified as [8-(1′-1′,2′-closo-C2B10H11)-2-Cp-2,1,8-closo-CoC2B9H10] (5) was also observed and its identity confirmed via1H NMR spectroscopy.
:50, Rf = 0.69) purification by column chromatography using the same eluent gave, on removal of solvent, a yellow powder (0.117 g, 49%), subsequently identified as [8-(1′-1′,2′-closo-C2B10H11)-2-Cp-2,1,8-closo-CoC2B9H10] (5). C9H26B19Co requires C 27.1, H 6.57. Found for 6: C 27.1, H 6.75%. 11B{1H} NMR [CDCl3], δ 1.7 (1B), 0.0 (2B), −0.9 (1B), −2.5 (1B), −3.8 (1B), −6.1 (2B), −9.9 (6B), −11.8 (1B), −13.2 (2B), −16.8 (1B), −17.7 (1B). 1H NMR [CDCl3], δ 5.50 (s, 5H, C5H5), 3.59 (s, 1H, CHcage), 2.73 (s, 1H, CHcage). EIMS: envelope centred on m/z 399 (M+).
**A trace amount of an orange spot (Rf = 0.60) identified as 4 was also observed and its identity confirmed via1H NMR spectroscopy.
:70, led to the recovery of 4 (0.020 g, 53%).
Crystallography
Diffraction-quality crystals of salt [BTMA][1] and compounds 2, 3, 4 and 5 were afforded by slow diffusion of a CH2Cl2 solution of the appropriate species and 40–60 petroleum ether at −30 °C. Intensity data for all except 4 were collected on a Bruker X8 APEXII diffractometer using Mo-Kα X-radiation, with crystals mounted in inert oil on a cryoloop and cooled to 100 K by an Oxford Cryosystems Cryostream. Compound 4 afforded crystals too small for our in-house system and consequently data were collected at the National Crystallographic Service at the University of Southampton at 100 K on a Rigaku AFC12 diffractometer operating with Mo-Kα X-radiation. Indexing, data collection and absorption correction were performed using the APEXII suite of programs.36 Structures were solved by direct methods (SHELXS37 or OLEX238) and refined by full-matrix least-squares (SHELXL).37Cage C atoms not involved in the intercage link were identified by a combination of (i) the examination of refined (as B) isotropic thermal parameters, (ii) the lengths of cage connectivities, (iii) the Vertex-Centroid Distance Method39 and (iv) the Boron-H Distance Method,40 with all four methods affording excellent mutual agreement.
The anion in [BTMA][1] is partially disordered. The C2B10 cage is fully ordered but the C2B9 cage has one B atom disordered between two sites, B3 and B12, with SOFs 0.548(10) and 0.452(10) respectively. Atoms B3 and B12 were refined with an isotropic thermal parameter fixed at 0.03 Å2. There is also partial disorder in 3 between atoms C2′ and B3′ (C2B10 cage), successfully modelled with vertex 2 being 0.446(19)C + 0.554(19)B, with complementary SOFs at vertex 3.
In [BTMA][1] it was not possible to locate the (disordered) bridging H atom associated with the open face of the nido cage and final refinement with constrained BH and CcageH atoms (B–H = Ccage–H = 1.12 Å) afforded better agreement than that with these H atoms allowed to refine. In the BTMA cation the H atoms were constrained to Cphenyl–H = 0.95 Å, Csecondary–H = 0.99 Å, Cmethyl–H = 0.98 Å. For all other structures BH and CcageH atoms were allowed to refine positionally whilst other H atoms were constrained to idealised geometries; Caromatic–H = 1.00 Å, CCp–H = 1.00 Å, Ctertiary–H = 1.00 Å, Cmethyl–H = 0.98 Å. All H displacement parameters, Uiso, were constrained to be 1.2 × Ueq (bound B or C) except Me H atoms [Uiso(H) = 1.5 × Ueq C(Me)]. Table 4 contains further experimental details.
| [BTMA][1] | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Formula | C14H38B19N | C14H35B19Ru | C14H35B19Ru | C9H26B19Co | C9H26B19Co |
| M | 425.84 | 509.88 | 509.88 | 398.62 | 398.62 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/c | P21/n | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| a/Å | 18.851(9) | 11.5653(7) | 10.9051(9) | 6.7993(5) | 12.6472(6)(4) |
| b/Å | 10.072(4) | 14.1222(9) | 16.9528(14) | 14.4533(10) | 6.6422(3) |
| c/Å | 13.477(6) | 15.1116(10) | 13.8437(11) | 20.3575(14) | 23.8175(10) |
| α (°) | 90 | 90 | 90 | 89.609(3) | 90 |
| β (°) | 97.068(13) | 91.611(4) | 105.039(4) | 85.554(3) | 95.642(2) |
| γ (°) | 90 | 90 | 90 | 89.158(3) | 90 |
| U/Å3 | 2540(2) | 2467.2(3) | 2471.7(4) | 1994.3(2) | 1991.10(16) |
| Z, Z′ | 4, 1 | 4, 1 | 4, 1 | 4, 2 | 4, 1 |
| F(000)/e | 896 | 1032 | 1032 | 808 | 808 |
| D calc/Mg m−3 | 1.114 | 1.373 | 1.370 | 1.328 | 1.330 |
| μ(Mo-Kα)/mm−1 | 0.052 | 0.640 | 0.639 | 0.853 | 0.855 |
| θ max (°) | 20.84 | 27.47 | 33.53 | 27.48 | 29.57 |
| Data measured | 14879 |
34686 |
52456 |
26471 |
37451 |
| Unique data, n | 2651 | 5604 | 9614 | 9102 | 5581 |
| R int | 0.2172 | 0.0431 | 0.0388 | 0.0378 | 0.0368 |
| R, wR2 (obs. data) | 0.0867, 0.1912 | 0.0309, 0.0689 | 0.0288, 0.0639 | 0.0334, 0.0821 | 0.0325, 0.0732 |
| S | 1.005 | 1.029 | 1.031 | 1.070 | 1.086 |
| Variables | 308 | 373 | 374 | 649 | 325 |
| E max, Emin/e Å−3 | 0.27, −0.26 | 0.68, −0.71 | 0.96, −1.37 | 0.66, −0.34 | 0.36, −0.24 |
Calculations
All geometries were optimised without constraints using Gaussian 03, Revision D.0141 employing the BP86 functional42 and 6-31G** basis sets for B, C and H atoms.43 Analytical frequency calculations were used to confirm geometries as minima or transition states. The transition state was further characterised through IRC calculations.44Acknowledgements
We thank ORSAS (GS) and the EPSRC (DE and DMcK supported by project EP/E02971X/1, WYM supported by project EP/I031545/1) for funding. We also thank the UK National Crystallography Service (University of Southampton) for collecting intensity data on compound 4, and Dr Dmitry Perekalin (INEOS-RAS, Moscow, Russia) for useful discussion. CP is an Erasmus exchange student from the Universidad de Zaragoza, Spain, BTL is an Erasmus exchange student from the Philipps-Universität Marburg, Germany, and LJ is a Stagier exchange student from the IUT de Rouen, France.References
- W. Y. Man, G. M. Rosair and A. J. Welch, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, 462 CAS.
- (a) J. A. Dupont and M. F. Hawthorne, J. Am. Chem. Soc., 1964, 86, 1643 CrossRef CAS; (b) T. E. Paxon, K. P. Callahan and M. F. Hawthorne, Inorg. Chem., 1973, 12, 708 CrossRef.
- X. Yang, W. Jiang, C. B. Knobler, M. D. Mortimer and M. F. Hawthorne, Inorg. Chim. Acta, 1995, 240, 371 CrossRef CAS.
- L. I. Zakharkin and A. I. Kovredov, Izv. Akad. Nauk SSSR, Ser. Khim., 1973, 1428 CAS.
- (a) X. Yang, W. Jiang, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 1992, 114, 9719 CrossRef CAS; (b) J. Müller, K. Baše, T. F. Magnera and J. Michl, J. Am. Chem. Soc., 1992, 114, 9721 CrossRef.
- S. Ren and Z. Xie, Organometallics, 2008, 27, 5167 CrossRef CAS.
- (a) D. A. Owen and M. F. Hawthorne, J. Am. Chem. Soc., 1970, 92, 3194 CrossRef CAS; (b) D. A. Owen and M. F. Hawthorne, J. Am. Chem. Soc., 1971, 93, 873 Search PubMed; (c) D. E. Harwell, J. McMillan, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1997, 36, 5951 CrossRef CAS PubMed.
- A. I. Yanovsky, N. G. Furmanova, Y. T. Struchkov, N. F. Shemyakin and L. I. Zakharkin, Izv. Akad. Nauk SSSR, Ser. Khim., 1979, 1523 Search PubMed.
- M. F. Hawthorne, D. A. Owen and J. W. Wiggins, Inorg. Chem., 1971, 10, 1304 CrossRef CAS.
- T. D. Getman, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1992, 31, 101 CrossRef CAS.
- T. D. Getman, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 1990, 112, 4594 CrossRef.
- (a) G. B. Dunks, R. J. Wiersema and M. F. Hawthorne, J. Chem. Soc., Chem. Commun., 1972, 899 RSC; (b) G. B. Dunks, R. J. Wiersema and M. F. Hawthorne, J. Am. Chem. Soc., 1973, 95, 3174 CrossRef CAS.
- D. Ellis, D. McKay, S. A. Macgregor, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2010, 49, 4943 CrossRef CAS PubMed.
- D. Ellis, G. M. Rosair and A. J. Welch, Chem. Commun., 2010, 46, 7394 RSC.
- J. A. Doi, E. A. Mizusawa, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1984, 23, 1482 CrossRef CAS.
- J. A. Long, T. B. Marder, P. E. Behnken and M. F. Hawthorne, J. Am. Chem. Soc., 1984, 106, 2979 CrossRef CAS.
- P. E. Behnken, T. B. Marder, R. T. Baker, C. B. Knobler, M. R. Thompson and M. F. Hawthorne, J. Am. Chem. Soc., 1985, 107, 932 CrossRef CAS.
- A B8–B9′ linked species, [8-(9′-3′-Cp-3′,1′,2′-closo-CoC2B9H10)-3-Cp-3,1,2-closo-CoC2B9H10] has been reported but was prepared from the reaction between [3-Cp-3,1,2-closo-CoC2B9H11] and S8/AlCl3, not from a bis(carborane); J. G. Planas, C. Viñas, F. Teixidor, M. E. Light and M. B. Hursthouse, J. Organomet. Chem., 2006, 691, 3472 CrossRef CAS.
- J. Buchanan, E. J. M. Hamilton, D. Reed and A. J. Welch, J. Chem. Soc., Dalton Trans., 1990, 677 RSC.
- M. A. Fox, J. A. K. Howard, A. K. Hughes, J. M. Malget and D. S. Yufit, J. Chem. Soc., Dalton Trans., 2001, 2263 RSC.
- M. F. Hawthorne, D. C. Young and P. A. Wegner, J. Am. Chem. Soc., 1965, 87, 1818 CrossRef CAS.
- M. P. Garcia, M. Green, F. G. A. Stone, R. G. Somerville, A. J. Welch, C. E. Briant, D. N. Cox and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 1985, 2343 RSC.
- C. J. Jones and M. F. Hawthorne, Inorg. Chem., 1973, 12, 606 Search PubMed.
- A. Burke, R. McIntosh, D. Ellis, G. M. Rosair and A. J. Welch, Collect. Czech. Chem. Commun., 2002, 67, 991 CrossRef CAS.
- W. Y. Man, S. Zlatogorsky, H. Tricas, D. Ellis, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2014, 53, 12222 CrossRef CAS PubMed.
- M. E. Lopez, M. J. Edie, D. Ellis, A. Horneber, S. A. Macgregor, G. M. Rosair and A. J. Welch, Chem. Commun., 2007, 2243 RSC.
- Since 1,1′-bis(o-carborane) is neutral overall and symmetric about the C1–C1′ mid-point, the two halves individually must be neutral.
- F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef . A free download of Mercury is available from the Cambridge Crystallographic Data Centre, http://www.ccdc.cam.ac.uk/.
- D. M. P. Mingos, M. I. Forsyth and A. J. Welch, J. Chem. Soc., Dalton Trans., 1978, 1363 RSC.
- e.g. (a) T. E. Paxson, M. K. Kaloustian, G. M. Tom, R. J. Wiersema and M. F. Hawthorne, J. Am. Chem. Soc., 1972, 94, 4882 CrossRef CAS; (b) T. P. Hanusa and L. J. Todd, Polyhedron, 1985, 4, 2063 CrossRef CAS.
- e.g. (a) D. Ellis, M. E. Lopez, R. McIntosh, G. M. Rosair, A. J. Welch and R. Quenardelle, Chem. Commun., 2005, 1348 RSC; (b) S. Zlatogorsky, D. Ellis, G. M. Rosair and A. J. Welch, Chem. Commun., 2007, 2178 RSC.
- D. Mandal, G. Thiripuranathar, W. Y. Man, G. M. Rosair and A. J. Welch, work in progress.
- M. A. Bennett, T.-N. Huang, T. W. Matheson and A. K. Smith, Inorg. Synth., 1982, 21, 74 CrossRef CAS.
- M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 233 RSC.
- S. A. Frith and J. L. Spencer, Inorg. Synth., 1990, 28, 273 CrossRef CAS.
- Bruker AXS APEX2, version 2009-5, Bruker AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- A. McAnaw, G. Scott, L. Elrick, G. M. Rosair and A. J. Welch, Dalton Trans., 2013, 42, 645 RSC.
- A. McAnaw, M. E. Lopez, D. Ellis, G. M. Rosair and A. J. Welch, Dalton Trans., 2014, 43, 5095 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian Inc., Wallingford, CT, USA, 2004.
- (a) H. L. Schmider and A. D. Becke, J. Chem. Phys., 1998, 108, 9624 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822 CrossRef.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS; (b) P. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- (a) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS; (b) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS.
- D. E. Smith and A. J. Welch, Organometallics, 1986, 5, 760 CrossRef CAS.
- M. G. Davidson, T. G. Hibbert, J. A. K. Howard, A. Mackinnon and K. Wade, Chem. Commun., 1996, 2285 RSC.
Footnotes |
| † In memory of Professor Kenneth Wade. |
| ‡ Electronic supplementary information (ESI) available: Table S1; 11B NMR chemical shifts for key reference compounds. Tables S2–S5; computational models. CCDC 1042151–1042155 (salt [BTMA][1] and compounds 2–5, respectively). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00081e |
| This journal is © The Royal Society of Chemistry 2015 |