
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular N-coordination in ketiminoboranes†‡
Catherine E.
Bacon
a,
Liz
Mansour
b,
John J.
Hayward
b and
Jeremy M.
Rawson
*b
aDepartment of Chemistry, The University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
bDepartment of Chemistry & Biochemistry, The University of Windsor, 401 Sunset Avenue, Windsor, Ontario, Canada N9B 3P4. E-mail: jmrawson@uwindsor.ca
First published on 16th February 2015
Abstract
Treatment of the imine PhC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NSiMe3)py with Et2BOMe or BF3·Et2O afforded bicyclic ketiminoboranes 4a and 4bvia intramolecular N-coordination. The basicity of the imine N is evidenced by their reactivity towards Brønsted and Lewis acids and the structures of 4a·HCl and 4b·BF3 are reported as well as the dipyridyl imine derivative 4c·HCl.
NSiMe3)py with Et2BOMe or BF3·Et2O afforded bicyclic ketiminoboranes 4a and 4bvia intramolecular N-coordination. The basicity of the imine N is evidenced by their reactivity towards Brønsted and Lewis acids and the structures of 4a·HCl and 4b·BF3 are reported as well as the dipyridyl imine derivative 4c·HCl.
The use of intramolecular coordination of a pyridyl group has been exploited in recent years as a route to novel main group heterocycles. For example Dyer and co-workers investigated the intramolecular N-coordination of 2-pyridyl-N-phosphino-imines (1) and found that an equilibrium existed between open and closed forms 1a and 1b.1 The 2-coordinate nitrogen in 1b is found to be sufficiently basic to form the adduct 1c with Lewis acidic AlCl3,1 whilst oxidation with [(iPr2N)2P][OTf] led to an unusual π-conjugated coupled products (1d and 1e) (Scheme 1) via an oxidative radical coupling process.2
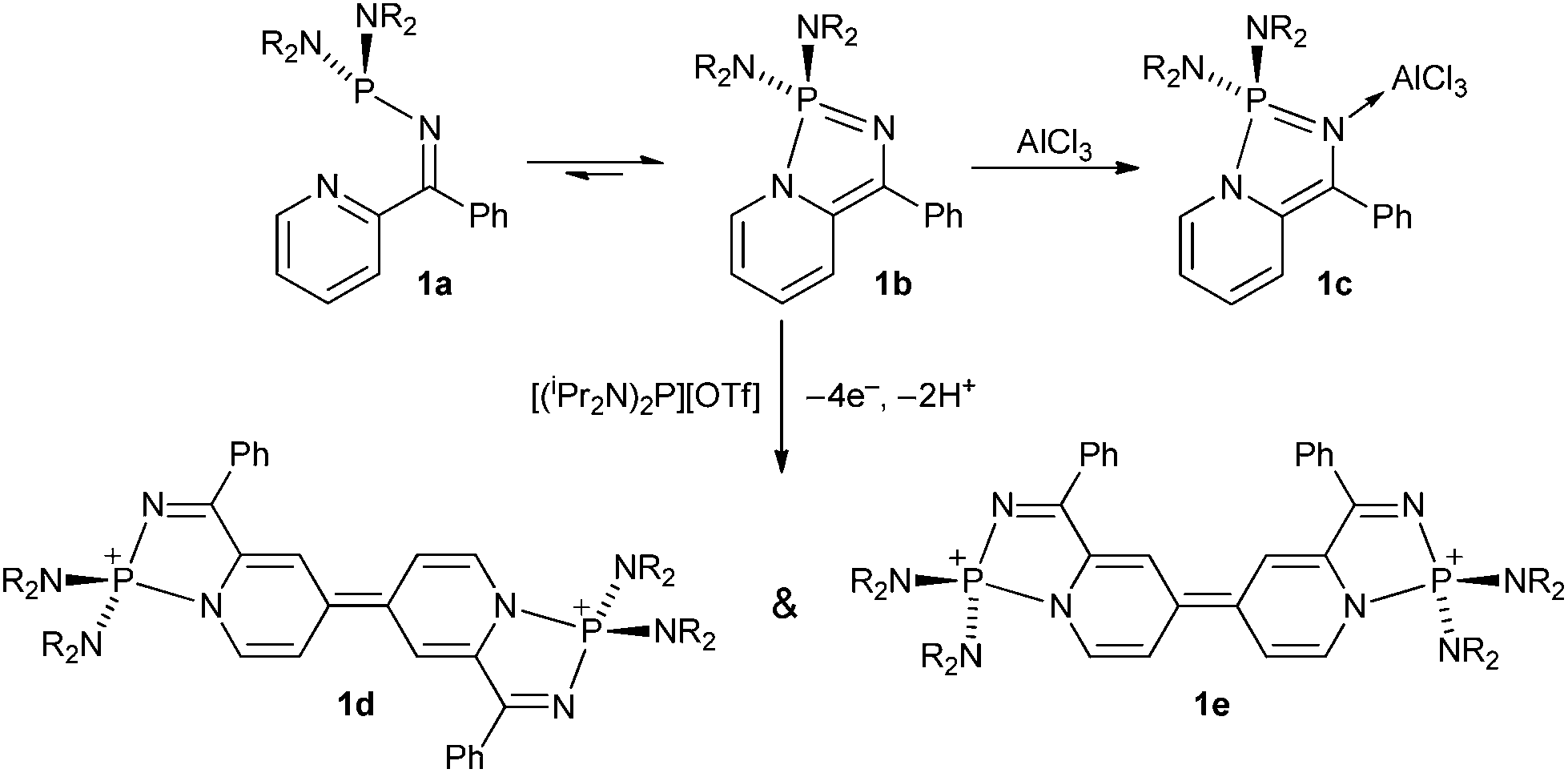 | ||
| Scheme 1 | ||
Studies on the chemistry of related group 16 compounds revealed similar behaviour between ring-open and ring-closed products (Scheme 2). For example, when X = Ar (E = S) the open-form 2a is favoured with a short intramolecular S⋯N contact whereas when X = Cl (E = S, Se) then the ring-closed form 2b was favoured.3 Work by Brusso and co-workers has revealed that at elevated temperatures ring-opening of these N-bridgehead thiadiazoles can occur.4 Similar intramolecular N-coordination has been implemented to generate hypervalent SiIV (3).5
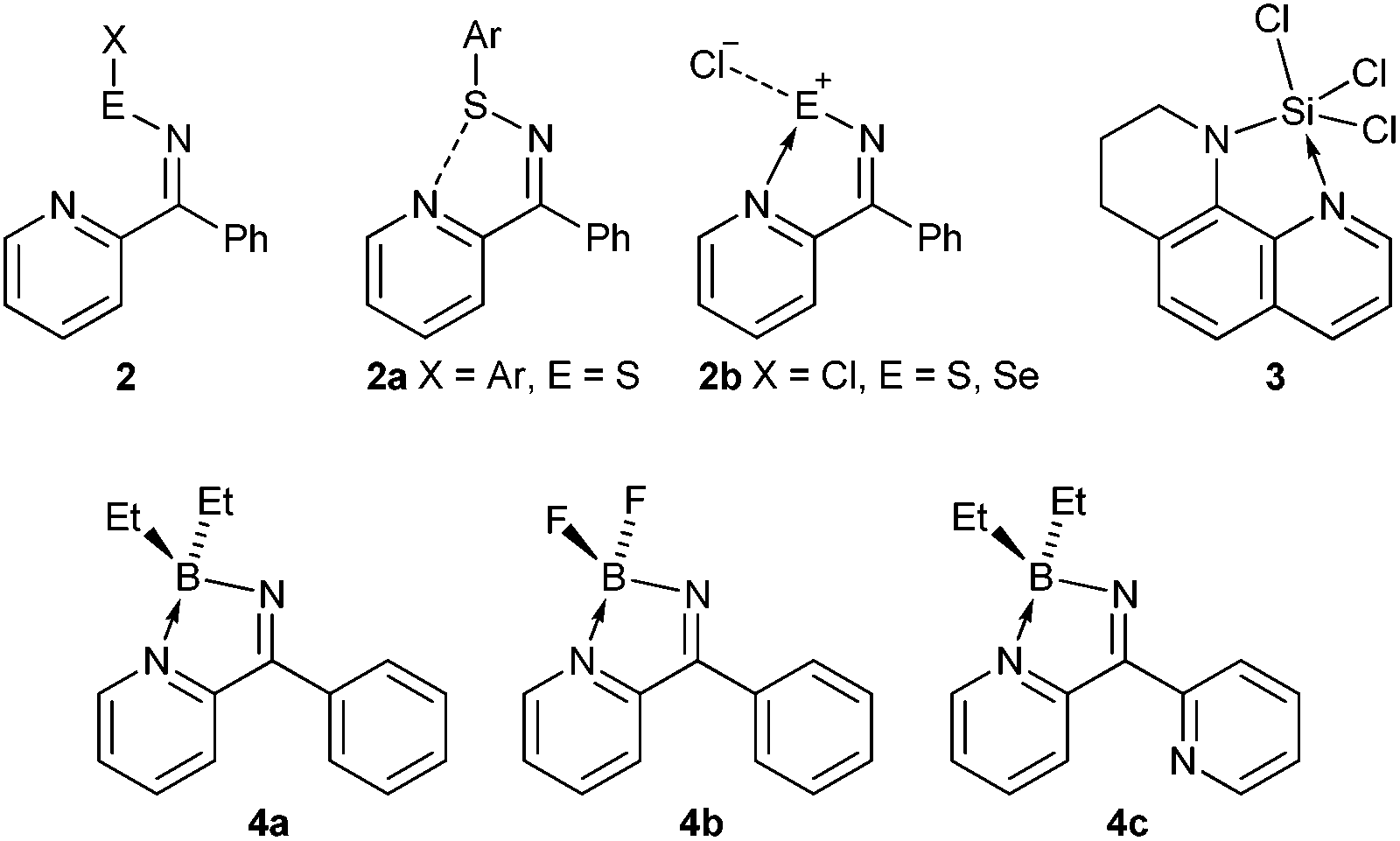 | ||
| Scheme 2 | ||
In these compounds the group 14/15/16 heteroatoms are all formally electron precise centres and intramolecular N-coordination makes them hypervalent affording some degree of lability between open and closed forms. Conversely group 13 elements are Lewis acidic and ring closure is expected to be strongly favoured. Ketiminoboranes, R2CN-BR2 were reported by Hawthorne, Wade and Lappert in the 1960's and are variously monomeric or dimeric depending upon substituents, with the monomeric ketimines R2CNBR2 formally isoelectronic with allene.6 In the current manuscript we describe the synthesis of ketiminoboranes in which the R group is capable of intramolecular coordination forming novel C/N/B heterocycles (4a–c). These heterocycles are similar to N,N′-boron chelate complexes, particularly derivatives of BODIPY, which have attracted considerable attention for their fluorescent properties,7 as dyes in photodynamic therapy,8 as well as photo-induced electron and energy transfer9 and as optical switches10inter alia. In the current paper we describe the generation of 4a–4c and find that the 2-coordinate imine nitrogen is strongly basic, permitting us to isolate and structurally characterise 4a·HCl, and 4b·BF3 and 4c·HCl.§
Compounds 4a–4c were prepared using a similar condensation reaction to that employed by Wade6e to prepare Ph2CNBPh2i.e. via the condensation of the imine Ar2CNSiMe3 with either Et2BOMe or BF3 OEt2. Crystals of 4a·HCl and 4c·HCl appeared over 3 days and were isolated by filtration (27–37% unoptimised isolated yield). The HCl presumably arises from adventitious hydrolysis of Me3SiCl. Crystals of 4b·BF3 were initially recovered in low yield from the reaction of PhC(NSiMe3)py with BF3·Et2O in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio but substantially improved yields (61%) were achieved using a 1:2 ratio. This suggests that the low solubility of the adduct favours crystallisation of the 1:2 product.
:1 ratio but substantially improved yields (61%) were achieved using a 1:2 ratio. This suggests that the low solubility of the adduct favours crystallisation of the 1:2 product.
The 1H NMR spectrum of 4a·HCl clearly reveals a broad singlet at 16.3 ppm consistent with N-protonation whilst the 11B NMR spectrum revealed a singlet at +8 ppm consistent with a tetrahedral B centre and a molecular ion peak at m/z = 251 with an isotope distribution pattern consistent with 4a·H+. The structure of 4a·HCl was determined by X-ray diffraction (Fig. 1) and found to crystallise as a THF solvate. The B–C bonds are unexceptional but the B–N bond lengths are slightly different (within 3 esd's) with the B1–N1 bond (1.561(5) Å) somewhat shorter than the formally dative pyridyl B–N bond (1.595(5) Å). Both are consistent with B–N single bond character (1.57–1.60 Å).11 The C10–N1 bond at 1.285(4) is short, consistent with significant imine character. At 3.058(3) Å the N1⋯Cl1 distance is consistent with a conventional N–H⋯Cl hydrogen-bonded contact.12
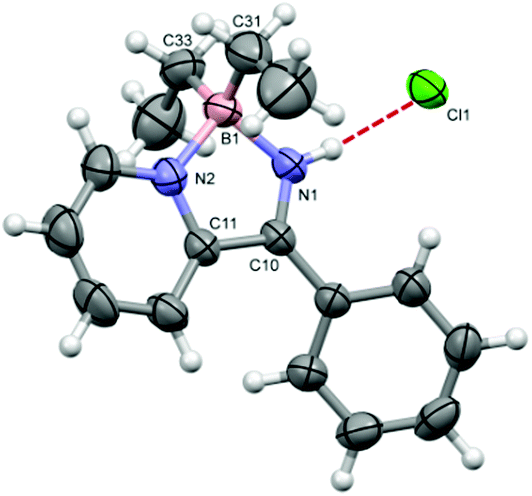 | ||
| Fig. 1 Molecular structure of 4a·HCl (THF solvent omitted for clarity) with thermal ellipsoids drawn at the 50% probability level. Selected bond lengths: B1–C31 1.598(6), B1–C33 1.598(5), B1–N1 1.561(5), B1–N2 1.595(5), N1–C10 1.285(4), N2–C11 1.355(4), C10–C11 1.476(4) Å. | ||
The 11B and 19F spectra of 4b·BF3 revealed four19F and four11B NMR resonances, the intensities of which varied depending upon solvent. In the 11B NMR in MeCN two triplet resonances are observed in the 4–8 ppm range corresponding to two chemically distinct BF2 environments, comparable with other 4-coordinate BN2F2 centres.10,13 In addition, a quartet at 0 ppm and a singlet at −1 ppm are observed (see ESI†). The quartet we tentatively assign to the N-coordinated BF3 in 4b·BF3 and the singlet as BF3 MeCN, based on chemical shift. These observations suggest a dynamic equilibrium (eqn (1)) in which the coordinated BF3 is labile in the presence of coordinating solvents. In the 19F NMR three resonances exhibit 11B hyperfine coupling (see ESI†) and the HMQC 2D NMR spectrum (Fig. 2) along with coupling constants confirms the assignments of the corresponding BF2 and BF3 groups. In the 19F NMR spectrum in MeCN the BF2 fluorine atoms in both 4b and 4b·BF3 appear around −159 ppm, reflecting very similar chemical environments whereas the BF3 resonances appear at −152 ppm (BF3·MeCN) and −141 ppm (4b·BF3). The resonance at −152 ppm appears as two signals in an approximate 4:1 ratio separated by 0.3 ppm and reflects the 11B and 10B isotopomers (∼80:20 natural abundance). In non-coordinating solvents such as benzene just two 11B resonances are detected suggesting displacement of BF3 in non-coordinating solvents is unfavourable and the structure of 4b·BF3 appears fully retained in solution.
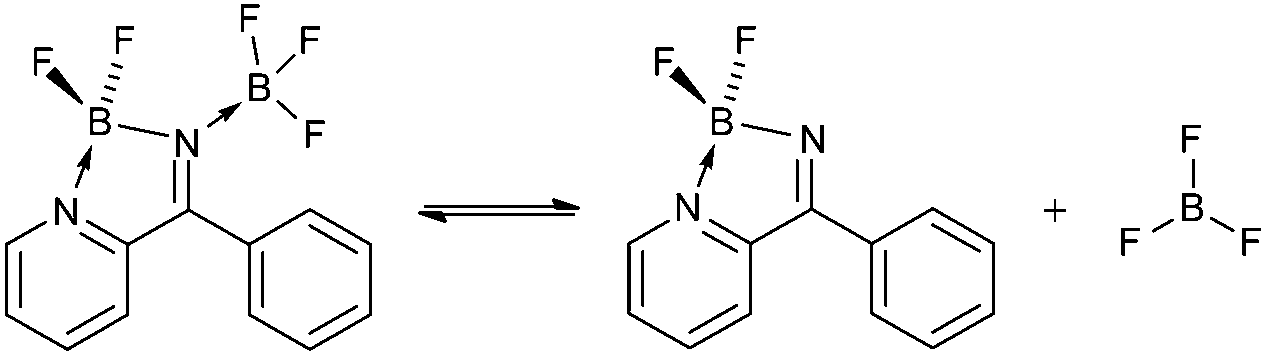 | (1) |
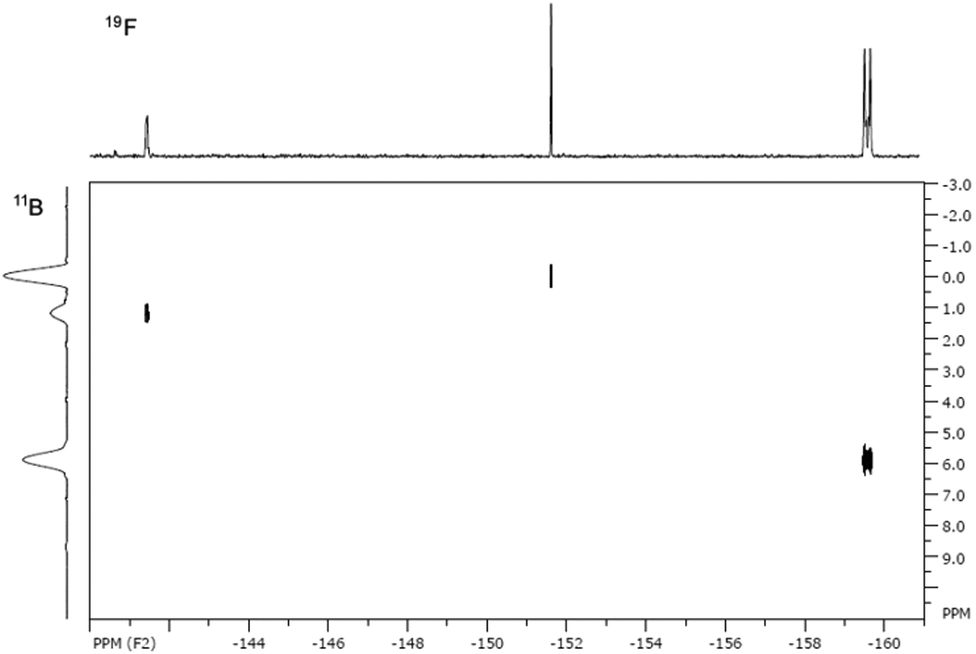 | ||
| Fig. 2 11B–19F HMQC NMR spectra of 4b·BF3 in MeCN. | ||
Crystals of 4b·BF3 were grown from the mother liquor on standing for 24–48 h. Single crystal structure determination revealed one molecule per asymmetric unit (Fig. 3). The heterocyclic C2N2B ring exhibits a similar geometry to the ethyl derivative with a longer B–N bond to the pyridyl nitrogen (1.600(2) Å) than to the imine nitrogen (1.574(2) Å) and a short imine-like CN bond (1.286(2) Å). These distances fall at the extremes of those reported previously for other C2N2B heterocycles with a pyridyl nitrogen atom coordinated to a BF2 group in which the dative bonds fall in the range 1.60–1.63 Å and the covalent B–N bonds fall in the range 1.50–1.57 Å.9,12 The exo B–N dative bond length to the BF3 group, at 1.600(2) Å, is identical to the dative pyridyl-N–B bond.
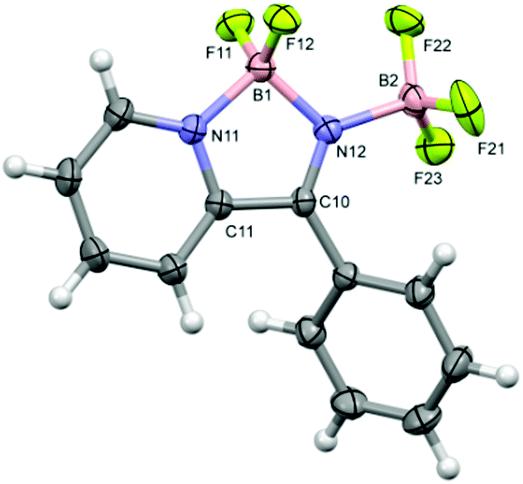 | ||
| Fig. 3 Molecular structure of 4b·BF3 with ellipsoids drawn at the 50% probability level. Selected bond lengths: B1–F11 1.366(2), B1–F12 1.364(2), B1–N11 1.600(2), B1–N12 1.574(2), N11–C11 1.349(2), C10–N12 1.286(2), C10–C11 1.490(2), N12–B2 1.600(2) B2–F21 1.377(2), B2–F22 1.377(2), B2–F23 1.366(2) Å. | ||
Theoretical calculations (DFT B3LYP/6-311G*) on the reaction of 4b with BF3 indicate adduct formation in the gas phase is favoured by 75 kJ mol−1 (see ESI†). Additional calculations along the B⋯N bond forming pathway reveal no significant activation energy barrier to formation of 4b·BF3. However stabilisation of the ‘free’ BF3 in coordinating solvents through adduct formation such as MeCN·BF3 or THF·BF3 is expected to destabilise 4b·BF3 with respect to loss of BF3. An NBO analysis revealed a bonding pattern best represented by the figure shown for 4b·BF3 (eqn (1)) (see ESI†). Notably the reaction of py2CO with Li[N(SiMe3)2]/Me3SiCl, followed by 1 equivalent of Et2BOMe afforded the pyridyl analogue, 4c·HCl in which the diazaborole nitrogen is protonated rather than the pyridyl nitrogen atom, reflecting the strongly basic nature of the diazaborole nitrogen atom (pKb = 5.6, calculated using DFT methods), cf. pyridine (pKb = 8.8).14 Synthetic details and crystallographic data for 4c·HCl are available as ESI.†
The current studies reflect the diversity of heterocyclic ring systems accessible by intramolecular N-coordination. Unlike the later p-block elements in which intramolecular coordination generates a hypervalent multi-centre bonding interaction, the electron poor boron centre adopts a 4-coordinate electron-precise centre upon intramolecular coordination. The resultant heterocycle offers a strongly basic nitrogen atom which affords similar acid–base chemistry to the N-pyridyl phosphine-imines.
Acknowledgements
We would like to thank EPSRC (C.E.B.), NSERC and the Canada Research Chairs program (J.J.H.) and the University of Windsor (L.M.) for financial support. In addition we are indebted to Dr M. Revington for assistance with the 2D multinuclear NMR studies.Notes and references
- D. A. Smith, A. S. Batsanov, K. Miqueu, J.-M. Sotiropoulos, D. C. Apperley, J. A. K. Howard and P. Dyer, Angew. Chem., Int. Ed., 2008, 47, 8674 CrossRef CAS PubMed.
- D. A. Smith, A. S. Batsanov, M. A. Fox, A. Beeby, D. C. Apperley, J. A. K. Howard and P. Dyer, Angew. Chem., Int. Ed., 2009, 48, 9109 CrossRef CAS PubMed.
- C. E. Bacon, D. J. Eisler, R. L. Melen and J. M. Rawson, Chem. Commun., 2008, 4924 RSC.
- A. A. Leitch, I. Korobkov, A. Assoud and J. L. Brusso, Chem. Commun., 2014, 50, 4934 RSC.
- See for example: (a) G. Klebe, J. W. Bats and K. Hensen, J. Chem. Soc., Dalton Trans., 1985, 1 RSC; (b) G. Klebe, J. W. Bats and H. Fuess, J. Am. Chem. Soc., 1984, 106, 5203 CrossRef.
- (a) M. F. Hawthorne, Tetrahedron, 1962, 17, 117 CrossRef CAS; (b) J. E. Lloyd and K. Wade, J. Chem. Soc., 1964, 1649 RSC; (c) I. Pattison and K. Wade, J. Chem. Soc. A, 1967, 1098 RSC; (d) V. A. Dorokhov and M. F. Lappert, Chem. Commun., 1968, 250 RSC; (e) J. R. Jennings, I. Pattison, C. Summerford, K. Wade and B. K. Wyatt, Chem. Commun., 1968, 250 RSC; (f) C. Summerford and K. Wade, J. Chem. Soc. A, 1969, 1487 RSC; (g) C. Summerford and K. Wade, J. Chem. Soc. A, 1970, 2010 RSC.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130 RSC; G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS PubMed.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77 RSC.
- (a) Y. Nagata and Y. Chujo, Macromol., 2008, 41, 3488 CrossRef CAS; (b) C. C. Cheng, W.-S. Yu, P.-T. Chou, S.-M. Peng, G.-H. Lee, P.-C. Wu, Y.-H. Song and Y. Chi, Chem. Commun., 2003, 2628 RSC.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2012, 134, 15221 CrossRef CAS PubMed.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry 3rd Edition, Pearson Ed, Harlow, England, 2008 Search PubMed.
- A search of the CSD (2013) afforded intermolecular hydrogen-bonded N⋯Cl distances in the range 2.939–3.298 Å (mean 3.12 Å) between three-coordinate N (R2NH) and Cl− ions.
- (a) K. Hensen, G. Klebe, J. W. Bats and H. Fuess, Fresenius’ J. Anal. Chem., 1982, 312, 24 CrossRef CAS; (b) G. Klebe, K. Hensen and H. Fuess, Chem. Ber., 1983, 116, 3125 CrossRef CAS; (c) J. J. Klappa, S. A. Geers, S. J. Schmidtke, L. A. MacManus-Spencer and K. McNeill, Dalton Trans., 2004, 883 RSC; (d) M. Mao, S. Xiao, J. Li, Y. Zou, R. Zhang, J. Pan, F. Dan, K. Zou and T. Yi, Tetrahedron, 2012, 68, 5037 CrossRef CAS PubMed; (e) M. Mao, R. Zhang, S. Xiao and J. Zou, J. Coord. Chem., 2011, 64, 3303 CrossRef CAS; (f) Q. Cao, S. Xiao, M. Mao, X. Chen, S. Wang, L. Li and K. Zou, J. Organomet. Chem., 2012, 717, 147 CrossRef CAS PubMed.
- N. Tro, T. D. Frigden and L. E. Shaw, Chemistry: A Molecular Approach, (Canadian Edition), Pearson Ed., 2013 Search PubMed.
Footnotes |
| † In memory of Ken Wade: Teacher, oft-times mentor, colleague and friend. His contributions in the field of structure and bonding in main group chemistry will continue into the future, but his guidance, encouragement and support for so many of the young academics he came in contact with will be sorely missed. |
| ‡ Electronic supplementary information (ESI) available: Full experimental details, details of the computational and crystallographic studies and crystallographic data in cif format. CCDC 1043411–1043413. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00196j |
| § Crystal data for 4a·HCl·THF monoclinic space group P21/c, M = 358.70. T = 240(2) K, a = 9.2150(2), b = 13.4773(3), c = 17.1221(4) Å, b = 97.697(2)°, V = 2107.29(8) Å3, Z = 4, Dc = 1.131 g cm−3, μ(Mo-Kα) = 0.191 mm−1. 21422 reflections measured (3.76 ≤ 2θ ≤ 29.98°) of which 6052 unique (Rint = 0.055). Final R1 (I > 2σ(I)) = 0.093, wR2 (all data) = 0.179 for 214 parameters. Max/min electron density +0.50/−0.48 e− Å−3. Crystal data for 4b·BF3 orthorhombic space group Pbca, M = 297.83. T = 173(2) K, a = 9.934(3), b = 12.290(4), c = 21.001(7) Å, V = 2564.0(14) Å3, Z = 8, Dc = 1.543 g cm−3, μ(Mo-Kα) = 0.142 mm−1. 25 Crystal data for 4c·HCl·THF monoclinic space group P21, M = 359.71. T = 180(2) K, a = 9.38350(10), b = 12.8596(3), c = 16.6196(3) Å, β = 99.1807(12)°, V = 1979.77(6) Å3, Z = 4, Dc = 1.207 g cm−3, μ(Mo-Kα) = 0.204 mm−1. 25 |
| This journal is © The Royal Society of Chemistry 2015 |