DOI:
10.1039/C5DT01022E
(Paper)
Dalton Trans., 2015,
44, 9756-9765
Synthesis of chelating diamido Sn(IV) compounds from oxidation of Sn(II) and directly from Sn(IV) precursors†
Received
13th March 2015
, Accepted 15th April 2015
First published on 22nd April 2015
Abstract
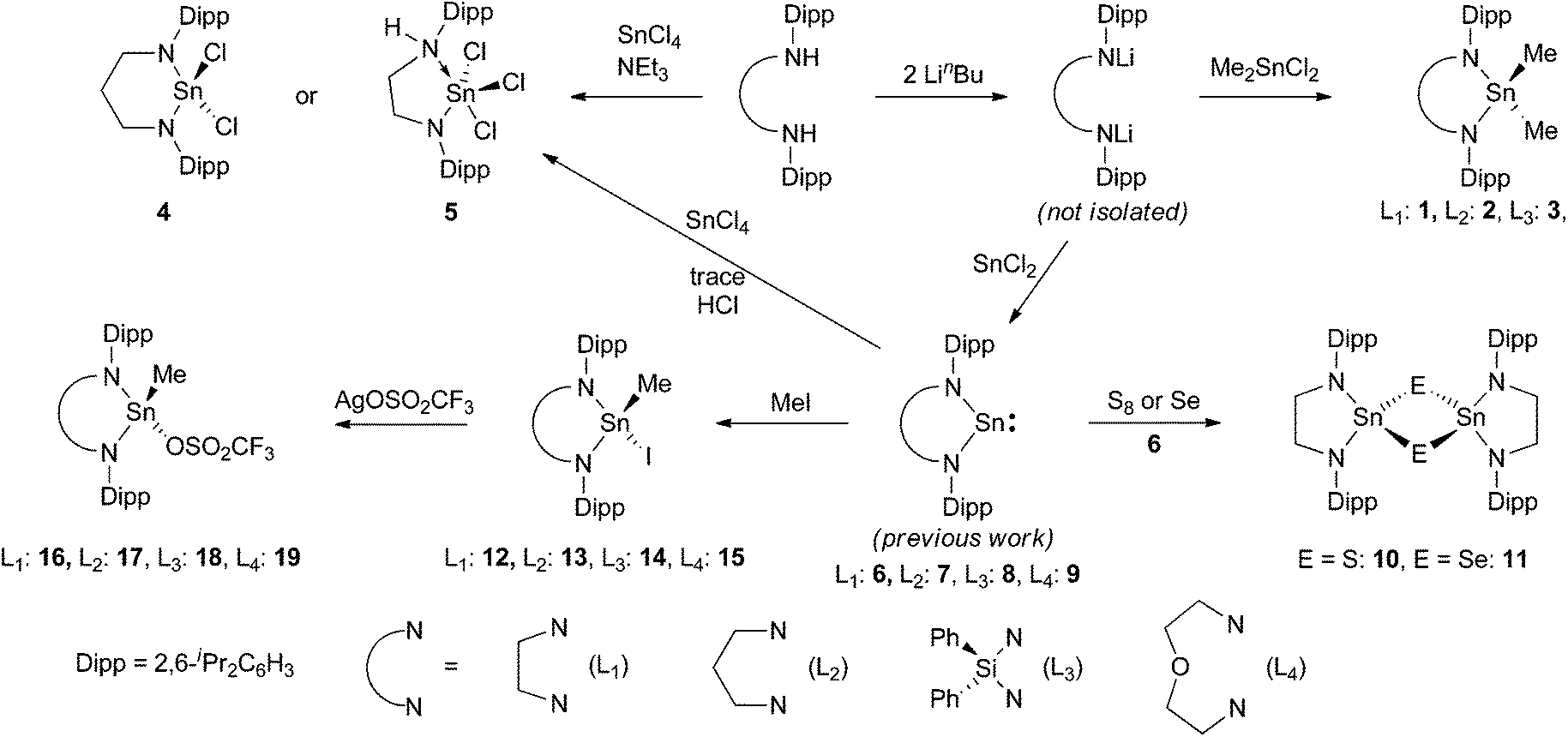
Three dimethyltindiamides containing chelating diamide ligands were synthesised from the reaction of the dilithiated diamine and Me2SnCl2; [SnMe2(L1)] 1 (L1 = κ2-N(Dipp)C2H4N(Dipp)), [SnMe2(L2)] 2 (L2 = κ2-N(Dipp)C3H6N(Dipp)) and [SnMe2(L3)] 3 (L3 = κ2-N(Dipp)SiPh2N(Dipp)), Dipp = 2,6-iPr2C6H3. Reaction of (L2)H2 with SnCl4 and NEt3 led to the formation of the diamidotin dichloride [SnCl2(L2)] 4 whereas reaction of (L1)H2 with SnCl4 and NEt3, or [Sn(L1)] with SnCl4, led to the exclusive formation of the amidotin trichloride [SnCl3{κ2-DippN(H)C2H4N(Dipp)}] 5. Reactions of [Sn(L1)] with sulfur and selenium formed [{Sn(L1)(μ-E)}2] (E = S 10 and Se 11). MeI reacted with N-heterocyclic stannylenes to generate the Sn(IV) addition products [Sn(Me)I(L1)] 12, [Sn(Me)I(L2)] 13, [Sn(Me)I(L3)] 14 and [Sn(Me)I(L4)] 15 (L4 = κ3-N(Dipp)C2H4OC2H4N(Dipp)), and subsequent reaction with AgOTf (OTf = OSO2CF3) generated the corresponding Sn(IV) triflates [Sn(Me)OTf(L1)] 16, [Sn(Me)OTf(L2)] 17 and [Sn(Me)OTf(L4)] 19 with [Sn(Me)OTf(L3)] 18 formed only as a mixture with unidentified by-products. All of the compounds were characterised by single crystal X-ray diffraction.
Introduction
The synthesis and reactivity of heteroleptic amidotin-halides are much less studied than the corresponding amidotinalkyls despite their obvious potential for further chemical transformations both via salt metathesis reactions and through protonolysis of the tin–amide bond. Previously Lappert and co-workers demonstrated the synthesis of the chloride-bridged dimeric Sn(II) species [Sn{N(SiMe3)2}(μ-Cl)]2 and [Sn{N(CMe2CH2)2CH2}(μ-Cl)]2 from the redistribution of acyclic amido-stannylenes with SnCl2 and these have proved to be useful starting materials.1,2 Additionally, Wrackmeyer and co-workers studied the oxidation of the stannylenes [Sn{N(SiMe3)2}2] and [Sn(κ2-N(tBu)Si2Me4N(tBu)] with SnCl4 by multinuclear NMR spectroscopy and found the reactions proceed with ease yielding tin(IV) dichloride species.3 In a more direct synthesis, diamine ligand precursors can be reacted with SnCl4 and NEt3 to give the corresponding diamidotindichloride species in good yield.3 Molecular structures were determined by X-ray crystallography for the five-membered ring dichlorotin species [SnCl2(κ2-N(tBu)Si2Me4N(tBu)] and also the ferrocenophane [SnCl2(κ2-N(SiMe3)C5H4FeC5H4N(SiMe3)].4 Two additional molecular structures of non-chelating diamidodichlorotin species are also known; [SnCl2{N(SiPhMe2)2}2] and [SnCl2{N(SiMe2NH)2SiMe2}2],5 however, for such potentially useful starting materials, relatively little is known about their chemistry. Oxidation reactions of tin(II) compounds are now an extensively studied area.6 Lappert and co-workers explored the oxidative addition chemistry of a number of haloalkanes with both alkyl- and amido-stannylenes and found them to be free radical in nature with the first step consisting of the transfer of an electron to the substrate, followed by abstraction of the halide to leave the tin and alkyl radicals to propagate the reaction.2 Substrates that add to give the corresponding Sn(IV) compounds include alkyl iodides, bromides and chlorides with the rate decreasing in the order I > Br > Cl, and for chloromethanes with CCl4 > HCCl3 > H2CCl2.7 Veith and co-workers observed oxidative addition of MeI and 1,2-dibromopentane to [Sn{κ2-N(tBu)SiMe2N(tBu)}], whereas other 1,2-dihaloalkanes were dehalogenated to give the corresponding unsaturated hydrocarbons.8 A cyclic alkyl stannylene was also found to react with MeI, along with the non-chelated ditin species formed as a by-product in the synthesis of the cyclic stannylene.9 Reactions of a related cyclic alkyl stannylene with a combination of an aryliodide and an alkene demonstrated different reactivity as addition to the double bond and allylic CH bond activation were both observed with the ratio sensitive to the steric bulk on the aryliodide and alkene.10
Oxidation with elemental halogens has also been investigated. For example, Br2 undergoes oxidative addition with [Sn{CH(SiMe3)2}2] to give the tin(IV) dibromide species,11 whilst oxidation of four-coordinate salen and bis(dimethylaminonaphthyl) stabilised stannylenes has also been observed with Br2 and I2 forming the tin(IV) dihalide species.12,13 Oxidation with elemental chalcogens allows the synthesis of tin-chalcogenides which have been shown to have remarkably diverse bonding,14 including the stabilisation of Sn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E (E = chalcogen) multiple bonding (for example, E = S,15 Se16 and Te17) when the substituents on the tin atom are large enough18 or the tin centres are higher-coordinate,19–22 and tin-chalcogenides are also of current interest as they belong to the group 14–16 semiconductors which have a low band gap.19,23 Bridging Sn–E–Sn units are observed when the steric bulk of the aryl groups were reduced,24 and reactions of elemental chalcogens with the divalent Sn(II) amides [Sn{N(SiMe3)2}2] and [Sn{κ2-N(tBu)SiMe2N(tBu)}] were shown to give Sn2E2 species containing four-membered rings.2,25–27
E (E = chalcogen) multiple bonding (for example, E = S,15 Se16 and Te17) when the substituents on the tin atom are large enough18 or the tin centres are higher-coordinate,19–22 and tin-chalcogenides are also of current interest as they belong to the group 14–16 semiconductors which have a low band gap.19,23 Bridging Sn–E–Sn units are observed when the steric bulk of the aryl groups were reduced,24 and reactions of elemental chalcogens with the divalent Sn(II) amides [Sn{N(SiMe3)2}2] and [Sn{κ2-N(tBu)SiMe2N(tBu)}] were shown to give Sn2E2 species containing four-membered rings.2,25–27
We have previously described the synthesis of saturated N-heterocyclic stannylenes,28 and in collaboration with the group of Hahn, N-heterocyclic plumbylenes.29 Saturated N-heterocyclic stannylenes were found to be much more thermally stable than the unsaturated N-heterocyclic stannylenes reported by Gudat and co-workers,30,31 and this has allowed the coordination chemistry of these species to be revealed.32 In this publication we report on tin(IV) chemistry of the same chelating diamide ligands formed by two routes; oxidation of the corresponding tin(II) species and synthesis directly from tin(IV) precursors generating new tin(IV) alkyl, halide, chalcogenide and triflate complexes.
Results and discussion
Dimethyldiamidotin compounds
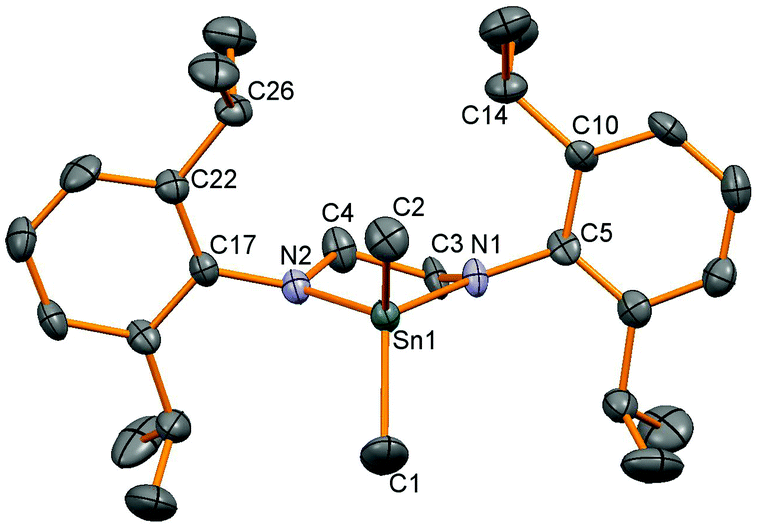
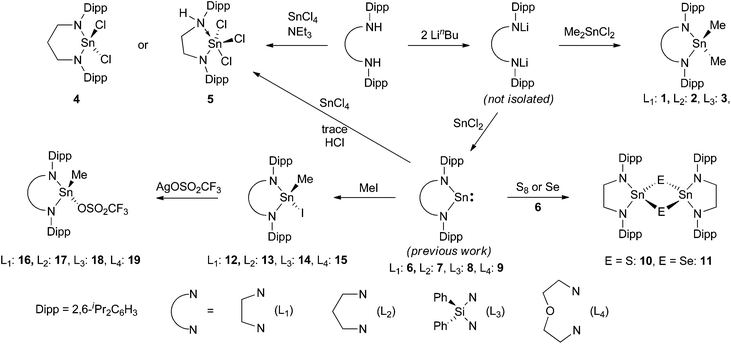
Three dimethyltindiamides containing bidentate diamido ligands were synthesised by reaction of the dilithiated diamines, generated from two equivalents of LinBu and the diamine as previously described,28 with Me2SnCl2 (Scheme 1). Reactions were performed in toluene for the more soluble lithium salts (Li2L1 and Li2L2), whilst diethyl ether was used for Li2L3. Crystallisation of the resulting tin(IV) products gave poor yields (12–35%) due to the very high solubility of all three compounds in hydrocarbon solvents despite the reactions proceeding with few by-products, but were enough for characterisation purposes. The diisopropylphenyl (Dipp) substituent in these ligands was a useful diagnostic tool in assessing the symmetry at the tin centre and potential rotation around the N–aryl bond. For compounds 1 and 2, 1H NMR spectroscopy revealed two sets of doublets for the isopropyl groups, along with a methine septet – an authentic septet for 2 due to equivalent 3J coupling to the inequivalent Me groups, but an apparent septet for 1 due to a 0.1 Hz difference in the analogous coupling – indicating that N–aryl bond rotation is slower than the timescale of the experiment at room temperature. When heated to 70 °C, compound 1 showed coalescence of the two doublets into a single doublet indicating onset of the high temperature regime averaging the two Me environments. 1H NMR spectroscopy of compound 3, on the other hand, only showed one doublet for the Dipp methyl groups indicating averaging of the two environments at room temperature. The effect of constraining the tin atom in a 4-, 5-, or 6-membered ring was probed by 119Sn NMR spectroscopy and showed a shift of the 119Sn resonance to higher frequency with decreasing N–Sn–N angle, 2: −3, 1: 33, 3: 98 ppm. X-ray crystallographic experiments were performed on single crystals of these three compounds, and a representative molecular structure is shown in Fig. 1 with selected data in Table 1 (see ESI† for figures of 2 and 3).
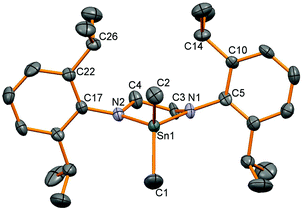 |
| | Fig. 1 Molecular structure of 1. Hydrogen atoms have been removed for clarity and thermal ellipsoids are set at 50% probability. | |
 |
| | Scheme 1 Synthesis of tin compounds. | |
Table 1 Selected bond distances (Å) and bond angles (°) for compounds 1–3
|
1
|
Sn(1)–N(1) |
2.000(4) |
N(1)–Sn(1)–N(2) |
84.2(2) |
| Sn(1)–N(2) |
2.028(4) |
C(1)–Sn(1)–C(2) |
114.3(2) |
| Sn(1)–C(1) |
2.127(6) |
|
|
| Sn(1)–C(2) |
2.107(5) |
|
|
|
2
|
Sn(1)–N(1) |
2.033(3) |
N(1)–Sn(1)–N(2) |
96.0(1) |
| Sn(1)–N(2) |
2.023(3) |
C(28)–Sn(1)–C(29) |
114.7(2) |
| Sn(1)–C(28) |
2.139(4) |
|
|
| Sn(1)–C(29) |
2.120(5) |
|
|
|
3
|
Sn(1)–N(1) |
2.056(3) |
N(1)–Sn(1)–N(2) |
74.9(1) |
| Sn(1)–N(2) |
2.061(3) |
C(37)–Sn(1)–C(38) |
112.5(2) |
| Sn(1)–C(37) |
2.122(4) |
|
|
| Sn(1)–C(38) |
2.133(4) |
|
|
All three structures show tetrahedral tin centres bound to two methyl groups and two nitrogen atoms from the diamide ligand. The Dipp groups are situated perpendicular to the N–Sn–N plane in order to minimise steric clashes and show the anticipated four methyl groups pointing towards the tin substituents and four towards the ligand backbone. The Sn–C bond lengths are all similar and unremarkable. The nitrogen atoms are all planar, and the Sn–N bond lengths are very similar to those observed in the corresponding Sn(II) compounds 6, 7 and 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 possibly reflecting a balance of decreasing bond lengths anticipated for Sn(IV) versus Sn(II) on covalent radii grounds, and the increase in bond length associated with higher coordination numbers. As anticipated, the largest effect was seen in the N–Sn–N angle which decreases with ring size; 96.0(1)° for 2, 84.2(2)° for 1 and 74.9(1)° for 3, a pattern which is almost identical to those seen in the corresponding tin(II) compounds.28
28 possibly reflecting a balance of decreasing bond lengths anticipated for Sn(IV) versus Sn(II) on covalent radii grounds, and the increase in bond length associated with higher coordination numbers. As anticipated, the largest effect was seen in the N–Sn–N angle which decreases with ring size; 96.0(1)° for 2, 84.2(2)° for 1 and 74.9(1)° for 3, a pattern which is almost identical to those seen in the corresponding tin(II) compounds.28
Synthesis and characterisation of amidotin(IV) chlorides
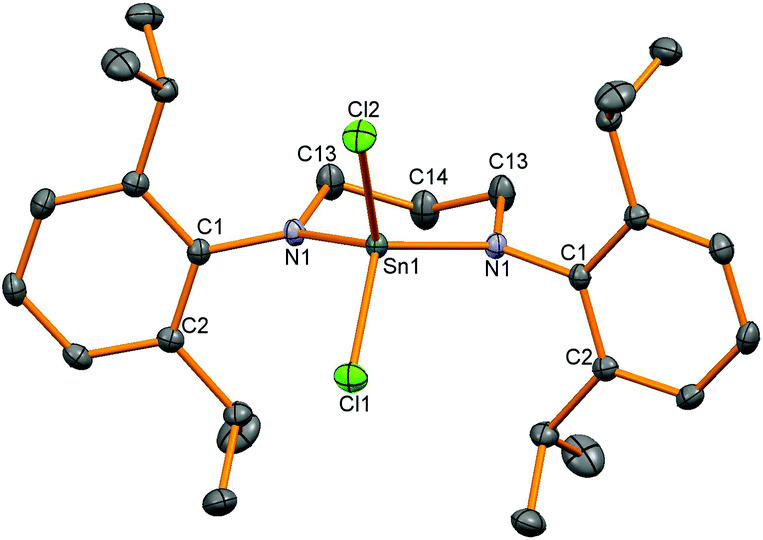
Synthetically, chloride substituents are very useful for salt metathesis reactions so access to these species was targeted via two methods; oxidation of a Sn(II) compound and reactions from Sn(IV) chloride. Following a literature precedent to generate amidotin chlorides,3 SnCl4 was reacted with diamine (L2)H2 but a stoichiometric ratio of 4 equivalents of NEt3 and 2.6 equivalents of SnCl4 were required to drive the reaction towards completion (as shown by 1H NMR spectroscopy). The product diamidotin dichloride 4 was obtained after recrystallization from n-hexane in 28% yield which was necessary to remove traces of unreacted diamine. 1H NMR spectroscopy showed the same pattern of two doublets and a methine septet as anticipated from slow rotation of the N–aryl bond and this did not coalesce upon heating to 65 °C. Using selective NOE experiments, the doublet at 1.30 ppm was assigned as the Dipp Me groups pointing towards the CH2 groups in the ligand backbone and 119Sn NMR spectroscopy showed a signal at lower frequency (−179 ppm) than 2. X-ray crystallographic experiments on a single crystal of 4 revealed a molecular structure in accordance with the solution NMR spectroscopic data with a tetrahedral tin centre bonded to two planar nitrogen atoms and two chloride substituents (Fig. 2). The Sn–N bond distances (Table 2) are slightly shorter than for the dimethyl analogue 2 and the two Sn–Cl bonds are of different length (Sn1–Cl1: 2.306 (1), Sn1–Cl2: 2.340(1) Å).
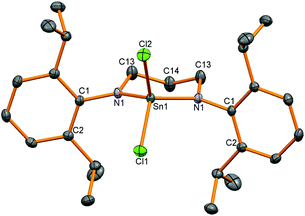 |
| | Fig. 2 Molecular structure of 4. Hydrogen atoms have been removed for clarity and thermal ellipsoids are set at 50% probability. | |
Table 2 Selected bond distances (Å) and bond angles (°) for compounds 4 and 5monoclinic
|
4
|
Sn(1)–N(1) |
1.981(1) |
N(1)–Sn(1)–N(1A) |
100.0(1) |
| Sn(1)–N(1A) |
1.981(1) |
Cl(1)–Sn(1)–Cl(2) |
100.6(1) |
| Sn(1)–Cl(1) |
2.306(1) |
|
|
| Sn(1)–Cl(2) |
2.340(1) |
|
|
|
5
|
Sn(1)–N(1) |
2.013(2) |
N(1)–Sn(1)–N(2) |
78.2(1) |
| Sn(1)–N(2) |
2.340(3) |
∑ (angles at N2) |
347.1 |
| Sn(1)–Cl(1) |
2.3665(9) |
|
|
| Sn(1)–Cl(2) |
2.3529(9) |
|
|
| Sn(1)–Cl(3) |
2.3206(10) |
|
|
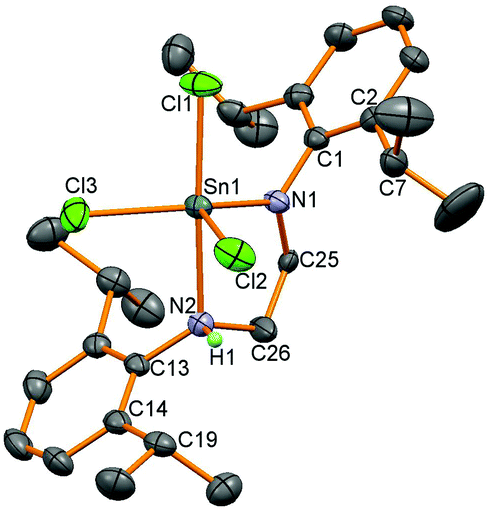
Two reactions were attempted to generate the analogous diamidotin dichloride using the L1 ligand backbone, but yielded instead 5 with three chloride substituents and the ligand functioning as a mixed amide/amine donor. Thus stannylene 6 reacted with SnCl4 (and presumably trace HCl in this reagent) to afford 5 in 25% yield; compound 5 was also synthesised from the reaction of (L1)H2, SnCl4 and NEt3 in 60% yield. In the stannylene oxidation reaction, one amido bond has been protonated whereas in the (L1)H2 reaction the second equivalent of HCl has not been removed by the NEt3 base. Asymmetry in the ligand was apparent from 1H NMR spectroscopy which revealed two different CH2 environments for the ligand backbone, a set of two doublets with a corresponding apparent septet for one Dipp group and a broad signal for the methyl groups on the other Dipp substituent. An N–H resonance is apparent at 5.46 ppm which integrates as one proton and couples to a CH2 group. 119Sn NMR spectroscopy showed a resonance at low frequency (−303 ppm) indicative of a higher coordination number at tin compared to 4. Single crystals of the product were grown from both reactions and crystallisation from different solvents showed the same molecular structure (Fig. 3) but in different polymorphs; triclinic from toluene and monoclinc from n-hexane. Data for the monoclinic polymorph are discussed and given in Table 2, data for both polymorphs are in the ESI.†
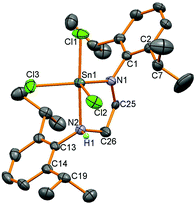 |
| | Fig. 3 Molecular structure of 5. All hydrogen atoms except the N–H have been removed for clarity and thermal ellipsoids are set at 50% probability. | |
The molecular structures of 5 all showed 5-coordinate tin centres in trigonal-bipyramidal geometries with the amido group situated in the equatorial plane and the amine substituent axial as identified by the longer tin–amine bond lengths (2.340(3) Å) and also by location by X-ray crystallography of the H atom attached to the nitrogen. The bond to the axial chloride substituent (2.3665(9) Å) is longer than the other two (2.3529(9) and 2.3206(10) Å) as observed in the other polymorph. These observations indicate that the difference in the length of the carbon backbone in L1 and L2 has led to different reactivity with the abstraction of the final equivalent of HCl by NEt3 hindered when L1 is used.
Stannylene oxidation with elemental chalcogens
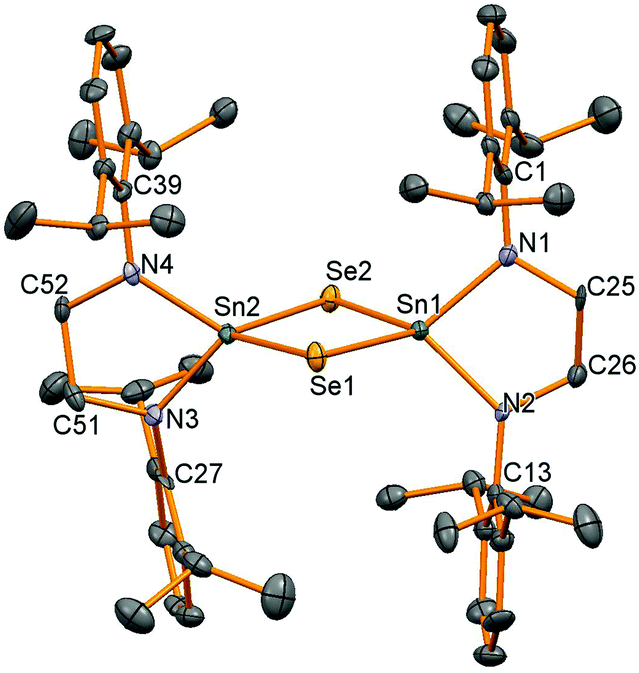
Oxidation of stannylene 6 with elemental sulfur or selenium was successfully carried out in thf solution, although oxidation using tellurium with or without additional PPh3 to form a more soluble tellurium transfer reagent was not successful. Good conversion to the chalcogenide products was achieved but traces of moisture also produced the diamine pro-ligand and upon recrystallisation to produce pure product yields of 20% were achieved for compounds 10 and 11, another indication of the high residual solubility of these compounds in hydrocarbon solvents even at low temperatures. 1H NMR spectroscopy showed very similar resonances for both compounds, but 119Sn NMR spectroscopy differentiated the two compounds with resonances at −78 ppm for 10 (2J(119)Sn–(117)Sn = 696 Hz) and −320 ppm for 11 with 77Se satellites (7.6% abundant, I = 1/2), 1JSn–Se = 1229 Hz, similar to that seen for (μ-Se)2[Sn{N(SiMe3)2}2]2 (−383 ppm, 1129 Hz).25
Single crystal X-ray diffraction experiments verified the bridged structure anticipated when the supporting ligand framework is not sterically encumbered enough to stabilise a SnE double bond. Both compounds are isostructural with a Sn2E2 diamond joining the two tetrahedral tin centres (Fig. 4, Table 3). The only major difference between the structures is the longer Sn–Se bond lengths (av. 2.528 Å in 11) compared to Sn–S (av. 2.407 Å in 10). The Sn–N, Sn–S and Sn–Se bonds are very similar to those seen before in the bridging species [(μ-E)Sn{N(SiMe3)2}2]2 (E = S: Sn–N 2.06 Å, Sn–S 2.416 Å; E = Se: Sn–N 2.049 Å, Sn–Se 2.541 Å, all averages)25 and [(μ-Se)Sn(NtBu)2SiMe2]2 (Sn–N 2.023(8) Å, Sn–Se 2.536(1) Å).27
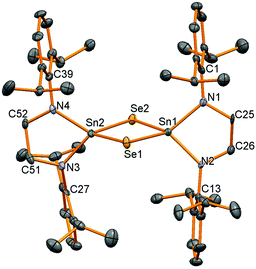 |
| | Fig. 4 Molecular structure of 11. All hydrogen atoms have been removed for clarity and thermal ellipsoids are set at 50% probability. | |
Table 3 Averaged bond distances (Å) and bond angles (°) for compounds 10 and 11 (two independent molecules in the unit cell)
|
10
|
Sn–N |
2.003 |
N–Sn–N |
85.8 |
| Sn–S |
2.407 |
S–Sn–S |
95.3 |
|
11
|
Sn–N |
2.002 |
N–Sn–N |
85.4 |
| Sn–Se |
2.528 |
Se–Sn–Se |
97.5 |
Oxidation of N-heterocyclic stannylenes with MeI
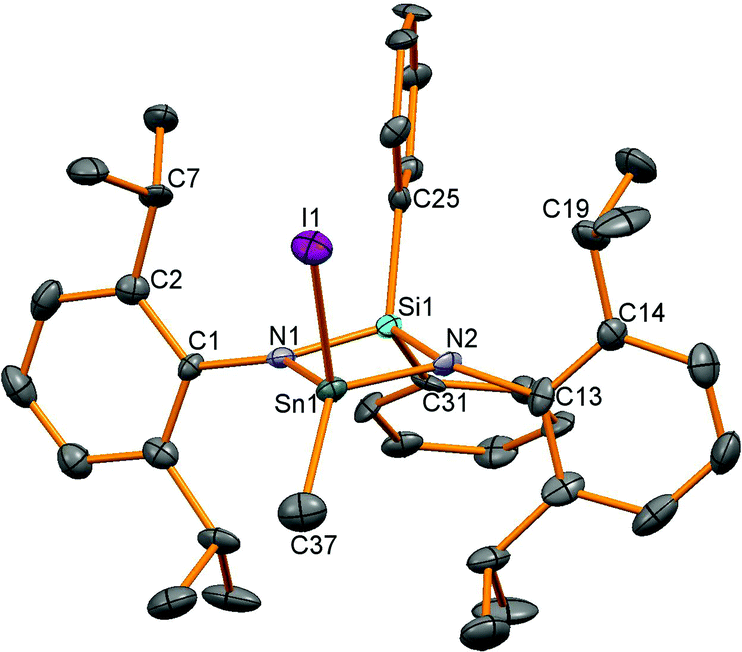
Oxidation of stannylenes with excess MeI in Et2O proceeded from isolated or in situ synthesised starting materials in a one pot reaction in reasonable yields of around 60% except for product 14 (37%). A satisfactory combustion analysis could not be obtained for 13 despite repeated attempts, and this was also the case for 10 and 16. This variable success could be due to the high moisture sensitivity of the tin–amide bond present in all the compounds leading to poor analyses. Oxidation of the Sn(II) compounds led to colourless or very-pale yellow tin(IV) compounds and 119Sn NMR spectroscopy showed a shift to lower frequency for resonances for all the compounds compared to the stannylenes (given in brackets, ppm): 12: −151 (6: 366), 13: −185 (7: 291), 14: −17 (8: 499), 15: −220 (9: 172). For compounds 12, 13 and 15, 1H NMR spectroscopy revealed four doublet and two septet resonances for the isopropyl groups indicative of reduced symmetry compared to the starting materials. For compound 14, only two doublet and one septet resonances were observed as fast N–Ar bond rotation on the NMR timescale (as seen in the dimethyl analogue 3) averages the inequivalent iPr groups (leading to one methine resonance), but with two different tin substituents, this process no longer averages the inequivalent Me positions. Single crystal X-ray diffraction experiments were performed for all of these compounds and revealed four coordinate tetrahedral centres for 12–14 (Fig. 5 shows 14 as a representative example), whereas 15, with the additional donor atom in the ligand backbone, is five coordinate with an additional contact from the oxygen in the ligand backbone (Fig. ESI 15†). The data for 14 were of poorer quality than the other structures due to difficulties in achieving good diffraction from the thin needle-like crystals.
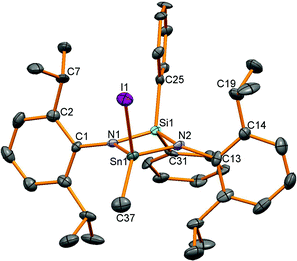 |
| | Fig. 5 Molecular structure of 14. All hydrogen atoms have been removed for clarity and thermal ellipsoids are set at 50% probability. | |
Bond lengths in these compounds are unremarkable (Table 4) and the N–Sn–N bond angles decrease from the wide bite-angle ligand L4 (15: 125.17(17)°) and then with decreasing chelate ring size; 13: 98.1, 12: 84.89(8), 14: 76.3(3)°.
Table 4 Selected bond distances (Å) and bond angles (°) for compounds 12–19
|
Average of two independent molecules.
Disordered Me and I groups (0.85:0.15). Only data for major component is given.
|
|
12
|
Sn–N |
2.005(2), 2.009(2) |
N–Sn–N |
84.89(8) |
| Sn–C |
2.134(2) |
C–Sn–I |
106.02(8) |
| Sn–I |
2.7184(2) |
∑ (angles at Sn, not I) |
330.8 |
|
16
|
Sn–N |
1.991(3), 1.973(3) |
N–Sn–N |
87.20(11) |
| Sn–C |
2.092(4) |
C–Sn–O |
103.36(13) |
| Sn–O |
2.133(2) |
∑ (angles at Sn, not OTf) |
347.4 |
|
13
|
Sn–N |
2.003 |
N–Sn–N |
98.1 |
| Sn–C |
2.110 |
C–Sn–I |
107.1 |
| Sn–I |
2.736 |
∑ (angles at Sn, not I) |
331.6 |
|
17
|
Sn–N |
1.986 |
N–Sn–N |
102.3 |
| Sn–C |
2.094 |
C–Sn–O |
102.1 |
| Sn–O |
2.128 |
∑ (angles at Sn, not OTf) |
348.4 |
|
14
|
Sn–N |
2.031(8), 2.032(9) |
N–Sn–N |
76.3(3) |
| Sn–C |
2.151(12) |
C–Sn–I |
105.2(3) |
| Sn–I |
2.7056(11) |
∑ (angles at Sn, not I) |
329.9 |
|
18
|
Sn–N |
2.018(2), 2.011(2) |
N–Sn–N |
77.56(8) |
| Sn–C |
2.099(3) |
C–Sn–O |
101.33(10) |
| Sn–O |
2.125(2) |
∑ (angles at Sn, not OTf) |
346.7 |
|
15
|
Sn–N |
2.046(4), 2.039(4) |
N–Sn–N |
125.17(17) |
| Sn–O |
2.518(4) |
C–Sn–I |
109.2(4) |
| Sn–C |
2.249(19) |
|
|
| Sn–I |
2.7041(8) |
|
|
|
19
|
Sn–N |
2.012(2), 2.025(2) |
N–Sn–N |
114.9(1) |
| Sn–O |
2.325(2) |
C–Sn–OTf |
100.20(11) |
| Sn–C |
2.113(3) |
|
|
| Sn–OTf |
2.148(2) |
|
|
Formation of tin triflates
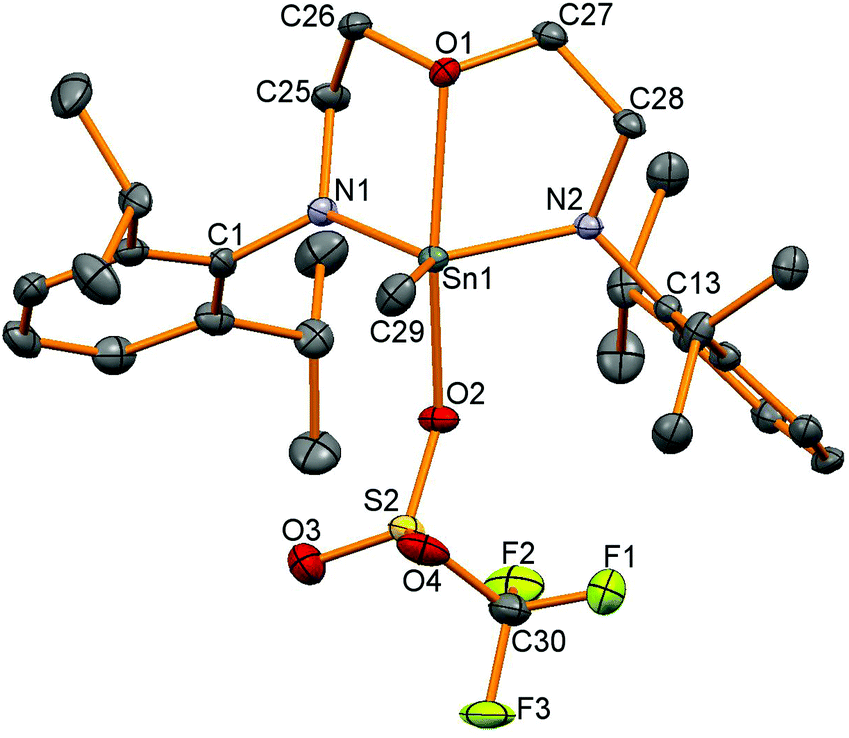
We were interested in using these tin iodides as precursors for generating cationic compounds with more weakly coordinating anions. Silicon compounds with the triflate anion (trifluoromethanesulfonate, OTf, [OSO2CF3]−) are potent Lewis acids and have gained widespread use in many organic transformations.33 Tin triflates34 are much less studied than silicon triflates but have been used as Lewis-acid catalysts in a handful of organic transformations.35 There are a number of structurally characterised tin triflates,12,36–43 and examples of four-coordinate tin triflates include [Sn{CH(SiMe3)2}3OTf] synthesised by protonation of the corresponding tin hydride using HOTf44 and another from protonation of a strained [2]ferrocenophane with a CH2Sn(tBu)2 bridge forming a four-coordinate tin triflate.45 Reactions of AgOTf with 12–15 were carried out generating the corresponding tin triflates 16, 17 and 19 but 1H NMR spectroscopy revealed that reactions to generate 18 also generated other unidentified by-products which could not be separated. Problems in generating tin triflates containing Si substituents have been noted before.46119Sn NMR spectroscopy showed resonances at −155 for 16, −95 for 17 and −152 ppm for 19 and 19F NMR spectroscopy showed very similar resonances for the triflate anion (16: −78, 17: −77, 19: −77 ppm). Single crystals were grown for all the products and selected data are given in Table 4. The tin–triflate bond lengths are similar for all the compounds (2.125(2)–2.148(2) Å) and shorter than in the related compound [Sn(Me)OTf{κ2-N(tBu)SiMe2N(tBu)}2]n (2.327 and 2.412 Å) which was found to be five-coordinate with bridging triflate anions.41 There are only very marginal differences in the Sn–N and Sn–C bond lengths compared to the methyliodide compounds for 16–18, however, compound 19 shows a much shorter oxygen contact with the backbone of ligand (19: Sn1–O1 2.325(2) Å, 15: Sn1–O1 2.518(4) Å). The geometry around the 4-coordinate tin atoms had shown the greatest change between the starting material and the product as the sum of the angles around the tin centre, excluding the iodide or triflate group, had increased from the iodide compounds (ca. 330° as expected for tetrahedral coordination) to ca. 347° indicative of a geometry intermediate between tetrahedral and trigonal planar with an apical OTf group (360° for trigonal planar atoms). The tin centre in 19 is five-coordinate (Fig. 6) and has a τ value47 of 0.64 (cf. 0.65 in 15).
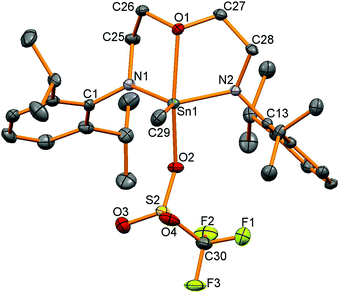 |
| | Fig. 6 Molecular structure of 19. All hydrogen atoms have been removed for clarity and thermal ellipsoids are set at 50% probability. | |
Experimental
General synthetic details
All air- and moisture-sensitive materials were weighed out, isolated, and stored in an argon-filled Saffron Beta glove box. Standard Schlenk-line techniques were used with filtrations carried out using Celite and a porosity 3 sinter. The solvents used were distilled HPLC grade and further dried and degassed using a commercially available solvent purification system (Anhydrous Engineering). Deuterated benzene was dried over potassium then vacuum transferred and kept over molecular sieves in the glove box. Elemental compositions (C, H and N) were determined by sealing samples in air-tight aluminium boats in a glove-box and were recorded on a Carlo Erba EA1108 CHN elemental analyser or by Mr Stephen Boyer (London Metropolitan University). Despite several attempts, satisfactory analyses could not be obtained for 3 out of the 14 compounds. This is most likely due to the high sensitivity of the tin–amide bond to trace water. Solution NMR spectroscopy samples were prepared using dry and degassed deuterated solvent in air-tight NMR tubes sealed with a Youngs tap. 119Sn and 29Si NMR spectra were run on Jeol Eclipse and Lambda 300 MHz spectrometers and referenced to external samples of SnMe4 and SiMe4 respectively. 1H and 13C NMR spectra were run on Jeol Eclipse 300, Lambda 300 and Delta 270 MHz spectrometers and were referenced to the internal solvent peaks. Septet is abbreviated sep. Mass spectrometry samples were run on a VG Autospec spectrometer. The N-heterocyclic stannylenes and diamines were synthesised as reported previously.28
General method for the synthesis of dimethyl tin compounds
The appropriate diamine (1.08 mmol) was dissolved in toluene (20 cm3), cooled to −78 °C and nBuLi (1.6 M in n-hexane, 1.4 cm3, 2.15 mmol) was added dropwise. The yellow solution was allowed to warm to room temperature and then left to stir overnight. Me2SnCl2 (0.237 g, 1.08 mmol) was added under positive pressure of nitrogen at 0 °C and the reaction was stirred overnight at room temperature, filtered and then recrystallised directly from a minimum amount of toluene.
Analytical data for [SnMe2(L1)] (1).
Using diamine (L1)H2, crystallised from toluene at −25 °C, yield: 67 mg, 0.13 mmol, 12%. 1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 7.10 (m, 6H, Ar-H), 3.81 (app. sep, 3JHH = 6.8 and 6.9 Hz, 4H, Dipp-CH), 3.41 (s with unresolved 119Sn and 117Sn satellites 3JHSn ≈ 29 Hz, CH2NSn), 1.27 (d, 3JHH = 6.8 Hz, 12H, Dipp-CH3), 1.21 (d, 3JHH = 6.9 Hz, 12H, Dipp-CH3), 0.19 (s with 119Sn and 117Sn satellites, 2JH(119)Sn = 62.0 Hz 2JH(117)Sn = 59.0 Hz, Sn-CH3). 13C NMR (75.4 MHz, 25 °C, C6D6): δ (ppm) 148.6 (Ar-Cipso), 146.8 (Ar-Cortho), 125.1 (Ar-Cpara), 123.7 (Ar-Cmeta), 57.2 (CH2N), 27.9 (Dipp-CH), 25.7 (Dipp-CH3), 24.5 (Dipp-CH3), −2.7 (Sn-CH3). 119Sn NMR (111.9 MHz, 25 °C, C6D6): δ (ppm) 33. Elemental analysis calculated for C28H44N2Sn (%): C 63.77, H 8.41 N 5.31. Found (%): C 64.35, H 8.56 N 5.25.
Analytical data for [SnMe2(L2)] (2).
Using diamine (L2)H2, crystallised from toluene after storage at −25 °C, yield: 0.256 g, 0.473 mmol, 35%. 1H NMR (300 MHz, C6D6): δ (ppm) 7.16–7.00 (m, 6H, Ar-H), 3.90 (sep, 3JHH = 6.9 Hz, 4H, Dipp-CH), 3.41–3.73 (m, 4H, NCH2), 1.97 (m, 2H, NCH2CH2CH2N), 1.37 (d, 3JHH = 6.9 Hz, 12H, Dipp-CH3), 1.29 (d, 3JHH = 6.9 Hz, 12H, Dipp-CH3), 0.16 (s with 119Sn and 117Sn satellites, 2JH(119)Sn = 61.4 Hz 2JH(117)Sn = 58.8 Hz, 6H, Sn-CH3). 13C NMR (75.4 MHz, 25 °C, C6D6): δ (ppm) 148.9 (Ar-Cipso), 147.0 (Ar-Cortho), 125.5 (Ar-Cpara), 123.8 (Ar-Cmeta), 56.7 (CH2N), 33.8 (NCH2CH2CH2N), 27.7 (Dipp-CH), 26.1 (Dipp-CH3), 24.7 (Dipp-CH3), −2.5 (Sn-CH3). 119Sn NMR (111.9 MHz, 25 °C, C6D6): δ (ppm) −3. Elemental analysis calculated for C29H46N2Sn (%): C 64.34 H 8.56 N 5.17. Found (%): C 64.08 H 7.88 N 4.97. Mass spectrometry (E.I.): m/z 543 (M+ + H), 527 (M+ − CH3 with correct isotopic distribution).
Analytical data for [SnMe2(L3)] (3).
Using diamine (L3)H2 (1.195 g, 2.23 mmol), Et2O (40 cm3) as the reaction solvent and n-hexane (30 cm3) as the extraction solvent gave a colourless solid (0.926 g, 61%) which was recrystallised from n-hexane in 15% yield. 1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 7.46–7.43 (m, 4H, Ar-H), 7.17 (m, 2H, Ar-H), 7.07–7.01 (m, 10H, Ar-H), 5.44 (sep, 3JHH = 6.8 Hz, 4H, Dipp-CH), 1.06 (d, 3JHH = 6.8 Hz, 24H, Dipp-CH3), 0.65 (s with 119Sn and 117Sn satellites 2JH(119)Sn = 58.9 Hz and 2JH(117)Sn = 56.3 Hz, 6 H, Sn-CH3). 13C NMR (75.4 MHz, 25 °C, C6D6): δ (ppm) 147.2, 142.4, 139.8, 135.5, 129.4, 127.4, 124.1, 124.0 (collection of Ar-C), 28.3 (Dipp-CH), 25.2 (Dipp-CH3), 3.9 (Sn-CH3). 29Si NMR (59.6 MHz, 25 °C, C6D6): δ (ppm) −22. 119Sn NMR (111.9 MHz, 25 °C, C6D6): δ (ppm) 98. Elemental analysis calculated for C38H50N2SiSn (%): C 66.96 H 7.39 N 4.11. Found (%): C 66.76 H 7.26 N 4.13.
Analytical data for [SnCl2(L2)] (4).
To a −78 °C solution of (L2)H2 (0.613 g, 1.55 mmol) and triethylamine (0.86 cm3, 6.2 mmol, 4 equiv.) in toluene (15 cm3) was added 1 M SnCl4 in n-hexane (4.0 cm3, 4.0 mmol, 2.6 equiv.) and the mixture was stirred for 3 days after which all of the volatiles were removed under vacuum. The residue was extracted into n-hexane (30 cm3) and filtered giving a clear, pale yellow solution which was concentrated under reduced pressure and pale yellow crystals formed after storage at −25 °C (0.255 g, 0.438 mmol, 28% yield). 1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 7.20–7.12 (m, 6H, Ar-H), 3.87 (sep, 3JHH = 7.0 Hz, 4H, Dipp-CH), 3.33 (t, 3JHH = 5.1 Hz, unresolved 119Sn and 117Sn satellites 3JHSn ≈ 35 Hz 4H, NCH2), 1.87 (quintet, 3JHH = 5.1 Hz, 2H, NCH2CH2CH2N), 1.43 (d, 3JHH = 7.0 Hz, 12H Dipp-CH3), 1.33 (d, 3JHH = 7.0 Hz, 12H, Dipp-CH3). 13C NMR (75.5 MHz, 25 °C, C6D6): δ (ppm) 149.0 (Ar-Cortho), 143.2 (Ar-Cipso), 127.8 (Ar-Cpara), 124.9 (Ar-Cmeta), 59.0 (NCH2 with unresolved 119Sn and 117Sn satellites 3JCSn ≈ 12 Hz), 34.0 (NCH2CH2CH2N), 28.8 (Dipp-CH), 26.4 (Dipp-CH3), 25.1 (Dipp-CH3). 119Sn NMR (111.9 MHz, 25 °C, C6D6): δ (ppm) −179. Elemental analysis calculated for C27H40Cl2N2Sn (%): C 55.70 H 6.92 N 4.81. Found (%): C 56.07 H 7.06 N 4.76.
[SnCl3{κ2-DippN(H)CH2CH2N(Dipp)}] (5).
Method A: To a −78 °C solution of {CH2N(Dipp)}2Sn (6) (0.390 g, 0.78 mmol) dissolved in n-hexane/toluene (25 cm3/10 cm3) was added a solution of SnCl4 in n-hexane (1 M, 0.78 cm3, 0.78 mol, 1 equiv.). The solution immediately turned cloudy and after stirring overnight all of the volatiles were removed under reduced pressure. The product was redissolved in n-hexane (30 cm3), filtered and concentrated under reduced pressure yielding pale yellow crystals after storage at 5 °C overnight (0.119 g, 0.20 mmol, 25% yield).
[SnCl3{κ2-DippN(H)CH2CH2N(Dipp)}] (5).
Method B: To a −78 °C solution of (L1)H2 (1.01 g, 2.66 mmol) and triethylamine (0.74 mL, 2 equiv., 5.31 mmol) in toluene (60 cm3) was added 1M SnCl4 in n-hexane (5.4 cm3, 5.4 mmol, 2 equiv.) and the mixture was stirred at room temperature for 2 days. Filtration gave a clear yellow solution which was concentrated under reduced pressure and the product crystallised at −25 °C (0.961 g, 1.59 mmol, 60%).
Analytical data for 5.
1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 7.27–7.16 (m, 3H, Ar-H), 7.06–6.99 (m, 3H, Ar-H), 5.46 (t, 3JHH = 7.2 Hz, 1H, NH), 3.66 (app. sep, 3JHH = 6.8 and 7.0 Hz, 2H, Dipp-CH), 3.21–3.13 (m, 4H, Dipp-CH and CH2), 2.97–2.91 (m, 2H, CH2), 1.53 (d, 3JHH = 7.0 Hz, 6H, Dipp-CH3), 1.35 (d, 3JHH = 6.8 Hz, 6H, Dipp-CH3), 1.26 (br. s, 12H, Dipp-CH3 attached to protonated N atom). 13C NMR (75.5 MHz, 25 °C, C6D6): δ (ppm) 148.7 (Ar-C), 142.0 (Ar-C), 141.6 (Ar-C), 138.8 (Ar-C), 127.7 (Ar-CH), 127.5 (Ar-CH), 125.3 (Ar-CH), 124.4 (Ar-CH), 50.6 (CH2), 49.6 (CH2), 29.4 (iPr-C), 28.7 (iPr-C), 25.6 (iPr-C), 24.8 (iPr-C), 24.5 (iPr-C). 119Sn NMR (111.9 MHz, 25 °C, C6D6): δ (ppm) −303 (s). Elemental analysis calculated for C26H39Cl3N2Sn (%): C 51.65 H 6.50 N 4.63. Found (%): C 51.73 H 6.59 N 4.79.
General method for oxidation with S8 and Se
Excess chalcogen (S8: 0.985 g, 3.8 mmol, Se: 238 mg, 3.0 mmol) was added to a −78 °C solution of 6 (1.91 g, 3.8 mmol and 238 mg, 0.48 mmol respectively) in thf and was then stirred at room temperature overnight. The solvent was removed under vacuum and the product was extracted into n-hexane (30 cm3) and filtered. The solution was concentrated under reduced pressure forming a crystalline solid after storage at −20 °C overnight.
Analytical data for [{Sn(L1)(μ-S)}2] (10).
Yield: 0.412 g, 0.39 mmol, 20%. 1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 7.18–7.06 (m, 6H, Ar-H), 3.67 (sep, 3JHH = 6.8 Hz, 4H, Dipp-CH), 3.37 (s with unresolved 119Sn and 117Sn satellites 3JHSn ≈ 57 Hz, 4H, CH2), 1.24 (d, 3JHH = 6.8 Hz, 12 H, Dipp-CH3), 1.17 (d, 3JHH = 6.8 Hz, 12H, Dipp-CH3). 13C NMR (75.5 MHz, 25 °C, C6D6): δ (ppm) 148.0 (Ar-Cortho), 143.1 (Ar-Cipso), 126.2 (Ar-Cpara), 124.3 (Ar-Cmeta), 55.4 (CH2), 28.4 (Dipp-CH), 25.4 (Dipp-CH3), 24.7 (Dipp-CH3). 119Sn NMR (112 MHz, 25 °C, C6D6): δ (ppm) −78 with 117Sn satellites 2JSnSn = 696 Hz.
Analytical data for [{Sn(L1)(μ-Se)}2] (11).
Yield: 56 mg, 0.049 mmol, 20%. 1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 7.17–7.07 (m, 6H, Ar-H), 3.68 (sep, 3JHH = 6.8 Hz, 4H, Dipp-CH), 3.34 (s with unresolved 119Sn and 117Sn satellites 3JHSn ≈ 58 Hz, 4H, CH2), 1.24 (d, 3JHH = 6.8 Hz, 12H, Dipp-CH3), 1.21 (d, 3JHH = 6.8 Hz, 12H, Dipp-CH3). 13C NMR (75.5 MHz, 25 °C, C6D6): δ (ppm) 148.0 (Ar-Cortho), 143.3 (Ar-Cipso), 126.2 (Ar-Cpara), 123.8 (Ar-Cmeta), 55.7 (CH2), 28.5 (Dipp-CH), 25.5 (Dipp-CH3), 24.9 (Dipp-CH3). 119Sn NMR (112 MHz, 25 °C, C6D6): δ (ppm) −320 with 77Se satellites 1JSnSe = 1229 Hz. Elemental analysis calculated for C52H76N4Se2Sn2 (%): C 54.19 H 6.65 N 4.86. Found (%): C 54.17 H 6.87 N 4.93.
Reactions with MeI
[Sn(Me)I(L1)] (12).
Method A: 6 (0.175 g, 0.35 mmol) was dissolved in Et2O (10 cm3) and MeI (0.057 cm3, 0.91 mmol) was added. The solution was stirred for 2.5 h, filtered and n-hexane (6 cm3) was added before concentration of the solution under reduced pressure to ca. 3 cm3. Crystalline material was obtained after placement at 4 °C overnight (0.10 g, 0.16 mmol, 46%).
[Sn(Me)I(L1)] (12). Method B, one pot reaction from the diamine: To a 0 °C solution of (L1)H2 (1.272 g, 3.34 mmol) dissolved in Et2O (40 cm3) was added nBuLi (1.6 M in n-hexane, 4.3 cm3, 6.68 mmol) which was allowed to stir at room temperature overnight. This was then added to a −78 °C suspension of SnCl2 (0.634 g, 3.34 mmol) in Et2O (20 cm3) which was then allowed to warm to room temperature and stirred for 3 h. The solution was filtered followed by addition of MeI (0.54 cm3, 8.69 mmol) and the reaction stirred overnight at room temperature. The solution was evaporated to dryness and the residue was extracted with n-hexane (40 cm3), filtered and the clear, pale yellow solution was reduced in volume to ca. 10 cm3. Upon cooling, pale yellow crystals formed (1.366 g, 2.14 mmol, 64% yield).
Analytical data for 12.
1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 7.20–7.17 (m, 6H, Ar-H), 4.15 (sep, 3JHH = 7.0 Hz, 2H Dipp-CH), 3.60 (m, 4H, overlapping Dipp-CH and NC(H)H-C(H)HN), 3.32 (m, 2H, NC(H)H-C(H)HN), 1.51 (d, 3JHH = 7.0, 6H, Dipp-CH3), 1.32 (d, 3JHH = 6.8, 6H, Dipp-CH3), 1.27 (d, 3JHH = 6.8, 6H, Dipp-CH3), 1.14 (d, 3JHH = 7.0, 6H, Dipp-CH3), 0.59 (s with 119Sn and 117Sn satellites 2JH(117)Sn = 77.5 Hz 2JH(119)Sn = 81.0 Hz, 3 H, Sn-CH3). 13C NMR (75.4 MHz, 25 °C, C6D6): δ (ppm) 149.6 (Ar-C), 147.4 (Ar-C), 144.6 (Ar-C), 126.1 (Ar-C), 124.7 (Ar-C), 123.5 (Ar-C), 56.7 (CH2NSn), 28.3 28.1 26.0 25.8 25.7 23.9 (iPr-C), 6.7 (Sn-CH3). 119Sn NMR (111.9 MHz, 25 °C, C6D6): δ (ppm) −151. Elemental analysis calculated for C27H41IN2Sn (%): C 50.73 H 6.47 N 4.38. Found (%): C 51.40 H 6.95 N 4.47.
[Sn(Me)I(L2)] (13).
Method A: 7 (0.827 g, 1.62 mmol) and MeI (0.26 cm3, 4.21 mmol) were reacted together. Very pale yellow crystals were isolated after crystallisation at 4 °C overnight (0.53 g, 0.81 mmol, 50%).
[Sn(Me)I(L2)] (13).
Method B: The diamine (L2)H2 (1.07 g, 2.7 mmol) was dilithiated and reacted with SnCl2 (0.512 g, 2.7 mmol) according to method B. MeI (0.6 cm3, 10 mmol) was added and pale yellow crystals were isolated after crystallization at −20 °C (1.08 g, 1.65 mmol, 61% yield).
Analytical data for 13.
1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 7.17–7.05 (m, 6 H, Ar-H), 4.06 (sep, 3JHH = 6.8 Hz, 2 H Dipp-CH), 3.79 (sep, 3JHH = 7.0 Hz, 2 H Dipp-CH) 3.57–3.34 (m, 4 H, CH2(CH2NDipp), 2.65–2.51 (m, 1 H, HCH(CH2NDipp)2), 1.87–1.79 (m, 1 H, HCH(CH2NDipp)2), 1.47 (d, 3JHH = 6.8 Hz, 6 H, Dipp-CH3), 1.33 (d, 3JHH = 7.0 Hz, 6 H, Dipp-CH3), 1.29 (d, 3JHH = 6.8 Hz, 6 H, Dipp-CH3), 1.21 (d, 3JHH = 7.0 Hz, 6 H, Dipp-CH3), 0.30 (s with 119Sn and 117Sn satellites 2JH(117)Sn = 74.9 Hz 2JH(119)Sn = 78.2 Hz, 3 H, Sn-CH3). 13C NMR (75.4 MHz, 25 °C, C6D6): δ (ppm) 149.7 (Ar-C), 147.8 (Ar-C), 145.3 (Ar-C), 126.5 (Ar-C), 125.2 (Ar-C), 123.5 (Ar-C), 57.3 (CH2), 37.0 (CH2) 28.4 28.0 27.0 26.7 25.6, 23.7 (iPr-C), 2.2 (Sn-CH3). 119Sn NMR (111.9 MHz, 25 °C, C6D6): δ (ppm) −185. Mass spectrometry (E.I.): m/z 654 (M+), 639 (M+ − CH3), 527 (M+ − I), 512 (M+ − CH3 and I) all with correct isotope distributions.
[Sn(Me)I(L3)] (14).
Method B: The diamine (L3)H2 (0.965 g, 1.8 mmol) was dilithiated and reacted with SnCl2 (0.342 g, 1.8 mmol) according to method B. MeI (0.46 cm3, 7.2 mmol) was added and colourless crystals were isolated after storage at −20 °C (0.532 g, 0.67 mmol, 37% overall yield). 1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 8.04 (m, 2H, Ar-H), 7.17–6.97 (m, 12 H, Ar-H), 6.84 (m, 2H, Ar-H), 4.16 (sep, 3JHH = 6.8 Hz, 4H, Dipp-CH), 1.27 (d, 3JHH = 6.8 Hz, 12H, Dipp-CH3), 0.99 (s, with 119Sn and 117Sn satellites 2JH(119)Sn = 74.6 Hz, 2JH(117)Sn = 71.6 Hz, Sn-CH3), 0.78 (d, 3JHH = 6.8 Hz). 13C NMR (75.5 MHz, 25 °C, C6D6): δ (ppm) 147.1, 140.7, 137.5, 135.9, 135.2, 134.2, 129.8, 127.4, 127.3, 124.9, 124.1 (collection of Ar-C), 28.2 25.4 24.9 (iPr-C), 16.1 (Sn-CH3). 119Sn NMR (111.9 MHz, 25 °C, C6D6): δ (ppm) −129. 29Si NMR (59.6 MHz, 25 °C, C6D6): δ (ppm) −17. Elemental analysis calculated for C37H47IN2SiSn (%): C 56.01 H 5.97 N 3.53. Found (%): C 56.92 H 6.41 N 3.76.
[Sn(Me)I(L4)] (15).
Method A: 9 (0.380 g, 0.70 mmol) and MeI (0.17 cm3, 2.8 mmol) were reacted together. Colourless crystals were isolated after storage at −20 °C overnight (0.30 g, 0.44 mmol, 62%). 1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 7.18–7.19 (m, Ar-H), 3.96 (sep, 3JHH = 7.0 Hz, 2H Dipp-CH), 3.61 (sep, 3JHH = 7.0 Hz, 2H, Dipp-CH), 3.44 (m, 4H, O-CH2), 3.36 (m, 2H, N-C(H)H), 3.05 (m, 2H, N-C(H)H), 1.44 (d, 3JHH = 7.0 Hz, Dipp-CH3), 1.33 (d, 3JHH = 7.0 Hz, Dipp-CH3), 1.31 (d, 3JHH = 7.0 Hz, Dipp-CH3), 1.22 (d, 3JHH = 7.0 Hz, Dipp-CH3), 0.56 (very br. s, Sn-CH3). 13C NMR (75.5 MHz, 25 °C, C6D6): δ (ppm) 149.9 (Ar-C), 148.5 (Ar-Cortho), 146.9 (Ar-Cortho), 126.6 (Ar-Cpara), 124.9 (Ar-Cmeta), 124.0 (Ar-Cmeta), 71.2 (O-CH2), 52.3 (N-CH2), 28.9 (Dipp-CH3), 28.7 (Dipp-CH3), 26.3 (Dipp-CH3), 26.2 (Dipp-CH3), 25.5 (Dipp-CH), 24.1 (Dipp-CH), 1.7 (Sn-CH3). 119Sn NMR (111.9 MHz, 25 °C, C6D6): δ (ppm) −220 (very br. s). Elemental analysis calculated for C29H45IN2OSn (%): C 50.98 H 6.64 N 4.10. Found (%): C 51.18 H 7.03 N 4.13.
General method for the formation of tin triflates
The tin iodide (1 equiv.) was dissolved in CH2Cl2 or toluene (10 cm3), cooled to −78 °C and AgOTf (1 equiv.) was added. The reaction was stirred for 16 h, filtered and then the product was crystallised from the minimum volume of toluene or ether.
[Sn(Me)OTf(L1)] (16).
12 (0.50 g, 0.78 mmol) in CH2Cl2 (20 cm3) was reacted with AgOTf (0.201 g, 0.78 mmol). The reaction mixture was evaporated to dryness and extracted with CH2Cl2 (20 cm3), filtered and the product crystallised from Et2O as colourless crystals at room temperature (0.217 g, 0.328 mmol, 42%). 1H NMR (300 MHz, 25 °C, CD2Cl2): δ (ppm) 7.17–7.13 (m, 6H, Ar-H), 3.85 (sep, 3JHH = 7.0, 2H, Dipp-CH), 3.60 (sep, 3JHH = 6.8 Hz, 2H, Dipp-CH), 3.55 (m, 2H, C(H)HN), 3.37 (m, 2H, C(H)HN), 1.29 (overlapping d, Dipp-CH3), 1.25 (s, Sn-CH3). 119Sn NMR (111.9 MHz, 25 °C, CD2Cl2): δ (ppm) −155. 19F NMR (282.7 MHz, 25 °C, CD2Cl2): δ (ppm) −78.0. cf. AgOTf 19F NMR (282.7 MHz, 25 °C, CD2Cl2): δ (ppm) −111.8 ppm.
[Sn(Me)OTf(L2)] (17).
13 (0.217 g, 0.332 mmol) in toluene (10 cm3) was reacted with AgOTf (85 mg, 0.33 mmol) and colourless crystals were grown from toluene at −20 °C for 3 days (0.128 g, 0.19 mmol, 57%). 1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 7.23–7.04 (m, 6H, Ar-H), 3.95 (sep, 3JHH = 6.6 Hz, 2H, Dipp-CH3), 3.46 (sep, 3JHH = 6.6 Hz, 2H, Dipp-CH3), 3.28–3.19 (m, 4H, CH2N), 2.32–2.27 (m, 1H, NCH2C(H)HCH2N), 1.71–1.65 (m, 1H, NCH2C(H)HCH2N), 1.55 (d, 3JHH = 6.6 Hz, 6H, Dipp-CH3), 1.33 (d, 3JHH = 6.6 Hz, 6H, Dipp-CH3), 1.28 (d, 3JHH = 6.6 Hz, 6H, Dipp-CH3), 1.13 (d, 3JHH = 6.6 Hz, 6H, Dipp-CH3), 0.41 (s with 119Sn and 117Sn satellites 2JH(119)Sn = 86.9 Hz, 2JH(117)Sn = 82.9 Hz, 3 H, Sn-CH3). 13C NMR (75.4 MHz, 25 °C, C6D6): δ (ppm) 149.7, 146.6, 143.9, 127.4, 125.6, 123.8 (collection of Ar-C), 119.9 (q, CF3), 57.8 (CH2N), 32.8 (NCH2CH2CH2N), 29.2 (Dipp-CH), 28.0 (Dipp-CH), 26.9 (Dipp-CH3), 26.1 (Dipp-CH3), 25.1 (Dipp-CH3), 24.4 (Dipp-CH3), −3.0 (s with 119Sn and 117Sn satellites 1JC(119)Sn = 653 Hz, 1JC(117)Sn = 624 Hz, Sn-CH3). 119Sn NMR (111.9 MHz, 25 °C, C6D6): δ (ppm) −95. 19F NMR (282.7 MHz, 25 °C, C6D6): δ (ppm) −77.1. Elemental analysis calculated for (%): C 51.57 H 6.42 N 4.15. Found (%): C 52.89 H 6.56 N 4.22.
[Sn(Me)OTf(L3)] (18).
14 (0.258 g, 0.325 mmol) in toluene (10 cm3) was reacted with AgOTf (84 mg, 0.325 mmol) and colourless crystals were obtained from toluene after storage at −20 °C (77 mg). Although the molecular structure of the desired product was determined, multi-nuclear NMR spectroscopy showed that there were multiple products formed in the reaction that could not be separated.
[Sn(Me)OTf(L4)] (19).
15 (0.210 g, 0.307 mmol) in toluene (10 cm3) at −78 °C was reacted with AgOTf (79 mg, 0.307 mmol) and colourless crystals were obtained from toluene after storage at −20 °C (89 mg, 0.126 mmol, 41%). 1H NMR (300 MHz, 25 °C, C6D6): δ (ppm) 7.19–7.05 (m, 6H, Ar-H), 3.91 (app. sep, 3JHH = 6.6 and 6.8 Hz, 2H, Dipp-CH), 3.78 (br. s, 2H, CH2), 3.42 (br. m, 4H, Dipp-CH and CH2), 3.18 (br. m, 2H, CH2), 2.99 (m, 2H, CH2), 1.39 (d, 3JHH = 6.6 Hz, 12H, Dipp-CH3), 1.23 (br. d, 3JHH ≈ 6.2 Hz, 6H, Dipp-CH3), 1.18 (d, 3JHH = 6.8 Hz, 6H, Dipp-CH3), −0.18 (br. s, 3H, Sn-CH3). 13C NMR (75.4 MHz, 25 °C, C6D6): δ (ppm) 149.4, 147.8, 144.1, 127.2, 125.1, 123.8 (collection of Ar-C), 69.6 (bs, OCH2), 50.9 (NCH2), 28.9 (Dipp-CH), 28.0 (Dipp-CH), 26.4 (Dipp-CH3), 26.2 (Dipp-CH3), 24.3 (Dipp-CH3), 24.0 (Dipp-CH3). 119Sn NMR (111.9 MHz, 25 °C, C6D6): δ (ppm) −152. 19F NMR (282.7 MHz, 25 °C, C6D6): δ (ppm) −76.8. Elemental analysis calculated for C30H45F3N2O4SSn (%): C 51.08 H 6.43 N 3.97. Found (%): C 51.08 H 6.38 N 4.19.
X-ray crystallographic studies
Crystals suitable for X-ray crystallographic analysis were grown from saturated n-hexane solution (3, 4, 5monoclinic, 10–13, 15), saturated toluene solution (1, 2, 5triclinic, 14, 17–19) or saturated diethyl ether solution (16). The crystals were covered in an inert oil and then transferred to the cold stream of the diffractometer. Experiments were performed using either a Bruker-AXS SMART, SMART-APEX three-circle diffractometer48 using Mo-Kα radiation (λ = 0.71073 Å). Intensities were integrated49 from several series of exposures, each exposure covering 0.3° in ω. Data for 18 were collected using Cu-Kα radiation (λ = 1.54184 Å) on an Oxford diffraction Gemini diffractometer. Absorption corrections were applied based on multiple and symmetry-equivalent measurements.50 The structures were solved by direct methods and refined by least-squares on weighted F2 values for all reflections.51 All non-hydrogen atoms were assigned anisotropic displacement parameters and refined without positional constraints. Hydrogen atoms were constrained to ideal geometries and refined with fixed isotropic displacement parameters except for the N–H hydrogen atoms in 5monoclinic and 5triclinic which were located in the Fourier difference map and were refined with fixed isotropic parameters. Refinement proceeded smoothly to give the residuals shown in Table ESI 1† and final R1 values (I > 2σ) range from 1.9 to 9.0%. Complex neutral-atom scattering factors were used.52 The structure of 5monoclinic showed disorder of one carbon atom in the ligand back bone that was refined over two sites with site occupancy factors of 0.51 and 0.49. The structures of 5triclinic and 13 showed disorder in one isopropyl group which were modelled over two sites with sites occupancy factors of 0.94:0.06 and 0.52:0.48. 15 showed disorder in the iodide and methyl groups with site occupancy factors of 0.85/0.15. CCDC reference numbers: 1027507–1027522.
Conclusions
Dimethyltindiamide compounds with chelating ligands are readily synthesised using a salt metathesis strategy from the dilithiated diamide ligand precursors and Me2SnCl2. Attempts to make dichlorotindiamide compounds with chelating amide ligands showed an unexpected dependence on the ligand with the C2 linked diamine only yielding a trichlorotinamide/amine compound via both an oxidative synthetic route from an N-heterocyclic stannylene or from reaction of the diamine with SnCl4 and NEt3. However, reaction of the C3 linked diamine with SnCl4 and NEt3 furnished the desired dichlorotindiamide. Saturated N-heterocyclic stannylenes were found to readily undergo oxidative addition reactions with S and Se (E) yielding chalcogen-bridged four-membered Sn2E2 ring systems, and with MeI to give crystalline tin(IV) products. These compounds were found to be suitable starting materials for further reactivity as demonstrated by their reaction with AgOTf to generate the corresponding tin triflates.
Acknowledgements
The authors would like to thank the University of Bristol for funding. S.M.M. thanks Heriot-Watt University, The Royal Society (Research grant: RG130436) and the EPSRC (First grant: EP/M004767/1) for current funding.
Notes and references
- R. W. Chorley, P. B. Hitchock, B. S. Jolly, M. F. Lappert and G. A. Lawless, J. Chem. Soc., Chem. Commun., 1991, 1302 RSC.
- M. F. Lappert, Main Group Met. Chem., 1994, 17, 183 CrossRef CAS PubMed.
- B. Wrackmeyer, G. Kehr, H. Zhou and S. Ali, Inorg. Chim. Acta, 1992, 197, 129 CrossRef CAS.
- B. Wrackmeyer, W. Milius, H. E. Maisel, H. Vollrath and M. Herberhold, Z. Anorg. Allg. Chem., 2003, 629, 1169 CrossRef CAS PubMed.
- C. Lehnert, J. Wagler, E. Kroke and G. Roewer, Khim. Geterotsikl. Soedin., 2006, 1845 Search PubMed.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354 CrossRef CAS PubMed.
- M. F. Lappert, M. C. Misra, M. Onyszchuk, R. S. Rowe, P. P. Power and M. J. Slade, J. Organomet. Chem., 1987, 330, 31 CrossRef CAS.
- M. Veith and A. Muller, J. Organomet. Chem., 1988, 342, 295 CrossRef CAS.
- C. Eaborn, M. S. Hill, P. B. Hitchcock, D. Patel, J. D. Smith and S. B. Zhang, Organometallics, 2000, 19, 49 CrossRef CAS.
- A. Kavara, K. D. Cousineau, A. D. Rohr, J. W. Kampf and M. A. B. Holl, Organometallics, 2008, 27, 1041 CrossRef CAS.
- J. D. Cotton, P. J. Davidson and M. F. Lappert, J. Chem. Soc., Dalton Trans., 1976, 2275 RSC.
- H. Jing, S. K. Edulji, J. M. Gibbs, C. L. Stern, H. Zhou and S. T. Nguyen, Inorg. Chem., 2004, 43, 4315 CrossRef CAS PubMed.
- J. Jastrzebski, P. A. Vanderschaaf, J. Boersma, G. Vankoten, M. Dewit, Y. F. Wang, D. Heijdenrijk and C. H. Stam, J. Organomet. Chem., 1991, 407, 301 CrossRef CAS.
- N. Tokitoh and R. Okazaki, Coord. Chem. Rev., 2000, 210, 251 CrossRef CAS.
- M. Saito, N. Tokitoh and R. Okazaki, J. Am. Chem. Soc., 2004, 126, 15572 CrossRef CAS PubMed.
- M. Saito, N. Tokitoh and R. Okazaki, J. Am. Chem. Soc., 1997, 119, 11124 CrossRef CAS.
- T. Tajima, N. Takeda, T. Sasamori and N. Tokitoh, Organometallics, 2006, 25, 3552 CrossRef CAS.
- P. P. Power, Chem. Rev., 1999, 99, 3463 CrossRef CAS PubMed.
- T. Chivers and D. J. Eisler, Angew. Chem., Int. Ed., 2004, 43, 6686 CrossRef CAS PubMed.
- P. B. Hitchcock, J. Hu, M. F. Lappert and J. R. Severn, Dalton Trans., 2004, 4193 RSC.
- M. C. Kuchta and G. Parkin, Coord. Chem. Rev., 1998, 176, 323 CrossRef CAS.
- Y. L. Zhou and D. S. Richeson, J. Am. Chem. Soc., 1996, 118, 10850 CrossRef CAS.
- S. G. Hickey, C. Waurisch, B. Rellinghaus and A. Eychmueller, J. Am. Chem. Soc., 2008, 130, 14978 CrossRef CAS PubMed.
- P. B. Hitchcock, M. F. Lappert, L. J. M. Pierssens, A. V. Protchenko and P. G. H. Uiterweerd, Dalton Trans., 2009, 4578 RSC.
- P. B. Hitchcock, E. Jang and M. F. Lappert, J. Chem. Soc., Dalton Trans., 1995, 3179 RSC.
- M. Veith, O. Recktenwald and E. Humpfer, Z. Naturforsch., B: Chem. Sci., 1978, 33, 14 Search PubMed.
- M. Veith, M. Notzel, L. Stahl and V. Huch, Z. Anorg. Allg. Chem., 1994, 620, 1264 CrossRef CAS PubMed.
- S. M. Mansell, C. A. Russell and D. F. Wass, Inorg. Chem., 2008, 47, 11367 CrossRef CAS PubMed.
- J. P. H. Charmant, M. F. Haddow, E. Hahn, D. Heitmann, R. Fröhlich, S. M. Mansell, C. A. Russell and D. F. Wass, Dalton Trans., 2008, 6055 RSC.
- T. Gans-Eichler, D. Gudat and M. Nieger, Angew. Chem., Int. Ed., 2002, 41, 1888 CrossRef CAS.
- T. Gans-Eichler, D. Gudat, K. Nattinen and M. Nieger, Chem. – Eur. J., 2006, 12, 1162 CrossRef CAS PubMed.
- S. M. Mansell, R. H. Herber, I. Nowik, D. H. Ross, C. A. Russell and D. F. Wass, Inorg. Chem., 2011, 50, 2252 CrossRef CAS PubMed.
- A. D. Dilman and S. L. Ioffe, Chem. Rev., 2003, 103, 733 CrossRef CAS PubMed.
- G. A. Lawrance, Chem. Rev., 1986, 86, 17 CrossRef CAS.
- J. Beckmann, Appl. Organomet. Chem., 2005, 19, 494 CrossRef CAS PubMed.
- H. Nakazawa, M. Kishishita, T. Ishiyama, T. Mizuta and K. Miyoshi, J. Organomet. Chem., 2001, 617, 453 CrossRef.
- H. V. R. Dias and W. C. Jin, Inorg. Chem., 2000, 39, 815 CrossRef CAS.
- Y. Q. Ding, H. W. Roesky, M. Noltemeyer, H. G. Schmidt and P. P. Power, Organometallics, 2001, 20, 1190 CrossRef CAS.
- A. E. Ayers and H. V. R. Dias, Inorg. Chem., 2002, 41, 3259 CrossRef CAS PubMed.
- P. B. Hitchcock, M. F. Lappert, G. A. Lawless, G. M. de Lima and L. J. M. Pierssens, J. Organomet. Chem., 2000, 601, 142 CrossRef CAS.
- M. Veith, B. W. Royan and V. Huch, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 79, 25 CrossRef CAS.
- O. Hiemisch, D. Henschel, P. G. Jones and A. Blaschette, Z. Anorg. Allg. Chem., 1997, 623, 147 CrossRef CAS PubMed.
- D. A. Evans, D. W. C. MacMillan and K. R. Campos, J. Am. Chem. Soc., 1997, 119, 10859 CrossRef CAS.
- M. Westerhausen and W. Schwarz, Main Group Met. Chem., 1997, 20, 351 CrossRef CAS.
- A. Bartole-Scott, R. Resendes, F. Jakle, A. J. Lough and I. Manners, Organometallics, 2004, 23, 6116 CrossRef CAS.
- A. Asadi, A. G. Avent, C. Eaborn, M. S. Hill, P. B. Hitchcock, M. M. Meehan and J. D. Smith, Organometallics, 2002, 21, 2183 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Vanrijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
-
SMART diffractometer control software, Bruker Analytical X-ray Instruments Inc., Madison, WI, 1998 Search PubMed.
-
SAINT integration software, Siemens Analytical X-ray Instruments Inc., Madison, WI, 1994 Search PubMed.
-
G. M. Sheldrick, SADABS: A program for absorption correction with the Siemens SMART system, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
-
E. Prince, International Tables for Crystallography, Mathematical, Physical and Chemical Tables, Wiley, 2004,
vol. C Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A table containing additional crystallographic information and molecular structures of compounds 1–5 and 10–19. CCDC 1027507–1027522. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01022e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a