
Synthesis of high quality alkyl naphthenic kerosene by reacting an oil refinery with a biomass refinery stream†
Karen S.
Arias
a,
Maria J.
Climent
a,
Avelino
Corma
*ab and
Sara
Iborra
a
aInstituto de Tecnología Química (UPV-CSIC), Universitat Politécnica de València, Avda dels Tarongers s/n, 46022, Valencia, Spain. E-mail: acorma@itq.upv.es; Fax: +34 963877809
bKing Fahd University of Petroleum and Minerals, P. O. Box 989, Dhahran 31261, Saudi Arabia
First published on 6th November 2014
Abstract
Alkylation of aromatics with HMF is a new route for the synthesis of biofuels. Alkylation of toluene with HMF has been studied in the presence of large pore (HBeta, USY and Mordenite), delaminated zeolites as well as on mesoporous aluminosilicates. In all cases a mixture of monoalkylated products of 5-(o-, m- and p-methyl)benzylfuran-2-carbaldehyde and OBMF coming from self etherification of HMF were obtained. Large pore 3D (USY) and especially 2D (ITQ-2) zeolites are active and selective catalysts for this transformation. The alkylation reaction was extended successfully to other substituted benzenes as well as to a heavy reformate mixture as source aromatic compounds, achieving 91% yield of alkylated products with 93% selectivity. Further hydrodeoxygenation of alkylated compounds in a fixed bed continuous reactor was performed using Pt/C and Pt/TiO2 as catalysts allowing to obtain a hydrocarbon mixture containing alkylcyclohexane compounds that can be used as high quality kerosene.
Broader contextIn recent time, the production of fuels from biomass resources has emerged as a promising alternative to fossil fuels. The research interest for biofuels has shifted from the first generation biofuels, derived from plant sugars and oils, to the second generation which are produced from biopolymers such as lignocellulosic biomass, with the predominant monomers being hexoses and pentoses. However, the difficulty of transforming that biomass into biofuels is due to the great difference in the molecular composition between the biofuels and the liquid fuels required today. Therefore an interesting approach to produce advanced biofuels from biomass is through the transformation of a set of biomass derivative molecules, the so-called platform molecules. However, platform molecules are highly oxygenated products and their conversion into liquid hydrocarbon fuels requires the adjustment of the molecular weight via C–C coupling reactions (e.g. aldol-condensation, ketonization, and oligomerization) of reactive intermediates in combination with oxygen removal reactions. In this work we present the alkylation of aromatics with a platform molecule, 5-hydroxymethylfurfural, using large pore 3D (USY) and especially 2D (ITQ-2) zeolites as catalysts followed by hydrodeoxygenation as a new route for the production of high quality alkyl naphthenic kerosene. |
1. Introduction
In the last decade, policies related to sustainability and reduction of carbon dioxide emissions, have stimulated research to develop efficient processes for the transformation of renewable biomass into fuels and chemicals,1,2 with lignocellulosic biomass being the most appropriated feedstock since it does not compete with food supplies. The production of fuels and chemicals from this renewable resource involves different direct routes such as direct pyrolysis or catalytic pyrolysis of the lignocelluloses,3 hydrothermal treatment under pressure and temperature,4 and gasification to produce synthetic gas.There is another way to process the biomass that requires separation of its components into lignin, cellulose and hemicellulose.5 This route opens the possibility for hydrolysis of cellulosic and hemicellulosic components to produce C5 and C6 sugars, which are converted into fuels and chemicals by either fermentation or chemical reactions. An interesting approach to convert biomass into fuels and chemicals is through the transformation of the so called platform molecules.6 Among them, 5-hydroxymethylfurfural (HMF) coming from dehydration of hexoses (glucose and fructose) appears as one of the most promising platform molecules due to its chemical versatility, which allows us to convert HMF into a wide variety of high value chemicals and alternative biofuels.1,2,7
HMF is produced by acid dehydration of C6 carbohydrates such as fructose and glucose or polymeric carbohydrates such as starch, inulin, cellulose and raw biomass, with D-fructose being the feed of choice. While aqueous processes appear to be the most convenient from an environmental point of view, they are not efficient since under acidic aqueous conditions, polymerization and hydrolysis of HMF to levulinic and formic acid occur giving a low yield of HMF.8 In order to overcome HMF degradation and to optimize the HMF yield, a variety of homogeneous and heterogeneous acid catalysts, solvents and biphasic reaction systems for the production of HMF have been used with varying success rates, and they have been extensively reviewed recently.9
In the non-aqueous systems, the hydrolysis of HMF can be suppressed, however the cross-polymerization reaction also occurs. The use of high boiling point polar solvents such as DMSO and DMF, habitually allows obtaining high yields of HMF (90%),10 however the extraction of HMF from these solvents is rather complicated. An interesting approach for the production of HMF, which could be applied at a large scale, is the use of solvent/aqueous biphasic systems since the extraction of HMF from the aqueous phase avoids its degradation.11 However, the key issue of these biphasic systems is the high extraction efficiency of the solvent and its recyclability.
Despite the attractive functionality of HMF and the great efforts for finding an optimized method of HMF production, HMF was not industrially manufactured until very recently.12 However, a few months ago, AVA Biochem announced “the first commercial-scale production of HMF from biomass” at its Biochem-1 facility.13 The process, which is based on a hydrothermal carbonization of lignocelluloses feedstock (HTC),14 will produce, in the first phase, up to 20 tonnes of HMF per year in various levels of purity (up to 99%).
Several platform molecules can be transformed into liquid hydrocarbon fuels by catalytic routes involving deoxygenation combined with C–C coupling reactions.2,15 For instance levulinic acid (LA) can be converted into liquid hydrocarbon fuels via gamma valerolactone through two routes. One of them involves the ring opening of GVL into pentenoic acid isomers which can be subsequently decarboxylated over a SiO2/Al2O3 catalyst producing an equimolar mixture of butenes and CO2. The oligomerization of butenes over an acidic catalyst produces alkenes which can be used as a jet fuel upon hydrogenation.16 However the oligomerization products of molecular size in the range of C8 are not suitable for diesel formulation and therefore the diesel yield is reduced to 20%. Another route to produce liquid alkanes from levulinic acid is via production of pentanoic acid through ring-opening/hydrogenation of GVL on a bifunctional (acid–metal) catalyst. Ketonization of two molecules of pentanoic acid can be performed over a variety of heterogeneous catalysts such as zirconium oxide, zirconium–cerium mixed oxide, cerium oxide, alumina, silica or MgO17–19 yielding 5-nonanone along with CO2 and water. 5-Nonanone can be upgraded to liquid alkanes, through its hydrogenation/dehydration to n-nonane using bifunctional catalysts such as Pt/C or Pt/TiO217 and Pt/Nb2O5 or Pt/Al2O3.18
Kunkes et al.20 have developed a process to produce higher alkanes starting from polyols, such as sorbitol, which consist of a two-step cascade process that combines oxygen removal and C–C coupling reactions. Polyols are firstly deoxygenated over a Pt–Re/C catalyst giving a mixture of organic compounds in the C4–C6 range which contains acids, alcohols, ketones and heterocycles. In a subsequent step, this mixture is converted into higher hydrocarbons through different C–C coupling reactions such as aldol or ketonization processes. However, a drawback of the process is the high cost of the catalyst.
Starting from different furan derived platform molecules such as furfural, 5-hydroxymethylfurfural and 2-methylfuran different strategies involving C–C bond formation reactions have been developed in order to produce liquid biofuels. For instance, Corma et al.21–23 have demonstrated the hydroxyalkylation/alkylation of 2-methylfuran just with itself or with other aldehydes or ketones in the presence of an acid catalyst (such as sulphuric acid or solid acid catalysts). A C–C bond formation occurs to obtain intermediate oxygenated molecules, which are converted subsequently into alkanes with excellent diesel properties23 through a hydrodeoxygenation step. Dumesic et al.24,25 have proposed as a carbon–carbon forming reaction, the aldol condensation of furfurals (furfural and 5-hydroxymethyl furfural) with ketones (such as acetone) to produce the mono and bis-aldol condensate products, which by subsequent hydrodeoxygenation generate linear alkanes. However, this cross-aldol condensation also involves the self condensation of the ketone leading to lower selectivity to the desired compound. To overcome this problem an alternative is to perform firstly the hydrogenation of the furan ring of HMF and furfural to give 5-hydroxymethyltetrahydrofurfural and tetrahydrofurfural which after self condensation and hydrogenation/dehydration process yield C12 and C10 alkanes respectively.26
Another strategy to obtain diesel fuel precursors from furan derived platform molecules, such as HMF, is the Friedel–Crafts alkylation of arenes (derived from oil or from lignin) with HMF. This reaction has been mainly performed using FeCl3 as the Lewis acid catalyst. For instance, Iovel et al.27 performed the alkylation of o-xylene with HMF in the presence of FeCl3 (10 mol%) at 80 °C yielding a 37% mixture of 4- and 3-substituted-xylenes (62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :38) after 24 h reaction time. More recently Zhou et al.28 performed the Friedel–Crafts alkylation between HMF and mesitylene in nitromethane as the solvent. When using FeCl3 (10 mol%) as the catalyst, 94% yield of mesitylmethylfurfural was obtained after 1 h, while with p-toluenesulfonic acid (10 mol%) only 76% yield was achieved after 2 h. Additionally the authors reported that formic acid can act as the solvent and catalyst for the one pot process involving the dehydration of fructose or glucose to HMF that, by alkylation of mesitylene gives mesitylmethylfurfural with reasonable yields (20–70%). While the above procedures are interesting, they present some drawbacks for the synthesis of 5-benzyl HMF derivatives, i.e., use of volatile and toxic solvents such as nitromethane and the presence of homogeneous Lewis or Brønsted catalysts which require a neutralization step, avoiding the recovery and reuse of the catalyst. Therefore, the development of new catalytic processes capable of producing alkylated HMF derivatives by more sustainable methods with heterogeneous acid catalysts and avoiding the use of solvents is of much interest. Following this, Onorato et al.29 have recently used poly(3,4-ethylenedioxythiophene) (PEDOT+) salts as acid catalysts to perform the dehydration of different ketohexoses to HMF. The authors found that when the reaction is performed using fructose at reflux of toluene a mixture of ortho and para 5-(methyl)benzylfuran-2-carbaldehyde in 84% yield was obtained and similar results were obtained when reacting HMF with toluene. However, in both processes high substrate/catalyst (1:1) molar ratios as well as long reaction times (20 h) were required.
:38) after 24 h reaction time. More recently Zhou et al.28 performed the Friedel–Crafts alkylation between HMF and mesitylene in nitromethane as the solvent. When using FeCl3 (10 mol%) as the catalyst, 94% yield of mesitylmethylfurfural was obtained after 1 h, while with p-toluenesulfonic acid (10 mol%) only 76% yield was achieved after 2 h. Additionally the authors reported that formic acid can act as the solvent and catalyst for the one pot process involving the dehydration of fructose or glucose to HMF that, by alkylation of mesitylene gives mesitylmethylfurfural with reasonable yields (20–70%). While the above procedures are interesting, they present some drawbacks for the synthesis of 5-benzyl HMF derivatives, i.e., use of volatile and toxic solvents such as nitromethane and the presence of homogeneous Lewis or Brønsted catalysts which require a neutralization step, avoiding the recovery and reuse of the catalyst. Therefore, the development of new catalytic processes capable of producing alkylated HMF derivatives by more sustainable methods with heterogeneous acid catalysts and avoiding the use of solvents is of much interest. Following this, Onorato et al.29 have recently used poly(3,4-ethylenedioxythiophene) (PEDOT+) salts as acid catalysts to perform the dehydration of different ketohexoses to HMF. The authors found that when the reaction is performed using fructose at reflux of toluene a mixture of ortho and para 5-(methyl)benzylfuran-2-carbaldehyde in 84% yield was obtained and similar results were obtained when reacting HMF with toluene. However, in both processes high substrate/catalyst (1:1) molar ratios as well as long reaction times (20 h) were required.
While the alkylation of aromatic compounds by alkyl and benzyl halides, alcohols and alkenes on different solid acid catalysts has been reported,30 structured solid acid catalysts with good accessibility for larger molecules can present an opportunity for these types of reactions. Therefore we present here the conversion of HMF into 5-benzyl HMF derivatives on large pore and delaminated zeolites as well as on mesoporous aluminosilicates. There is no doubt that if one could design a solid catalyst that would avoid competitive reactions such polyalkylations and self etherification of HMF, while maximizing alkylation of aromatics with HMF, new opportunities will be opened for the synthesis of biofuels and chemicals. It should be noted that while the methods reported above produce linear or branched alkanes, the production and hydrodeoxygenation of 5-benzyl HMF derivatives, besides to be a selective and environmental friendly process, produces alkyl cycloalkanes which have high densities and volumetric heating values due to the robust ring strain. This variety of compounds distributed over the diesel and jet range can be added to the conventional fuels to increase the density or volumetric heating of the fuel.
2. Results and discussion
2.1 Alkylation of toluene with HMF in the presence of acid catalysts
Two different routes allow to obtain 5-benzyl HMF derivatives starting from HMF: Friedel–Crafts alkylation of aromatic compounds with 5-chloromethyl-2-furfural in the presence of an excess of Lewis acid catalysts (see Scheme 1a), and by the direct reaction of HMF in the presence of Brønsted acids (see Scheme 1b). When homogeneous Lewis or Brønsted acids are used in a conventional alkylation reaction large amounts of residues such as salts or polyalkylated compounds are generated. Moreover when using halides such as 5-chloromethyl-2-furfural, an additional halogenation reaction is required. This step could be avoided if solid acid catalysts could carry out the direct alkylation of aromatic compounds and HMF with high conversion and selectivities. Among solid acids, zeolites may offer interesting opportunities for these type of reactions since they have been applied successfully to a large number of alkylation processes.31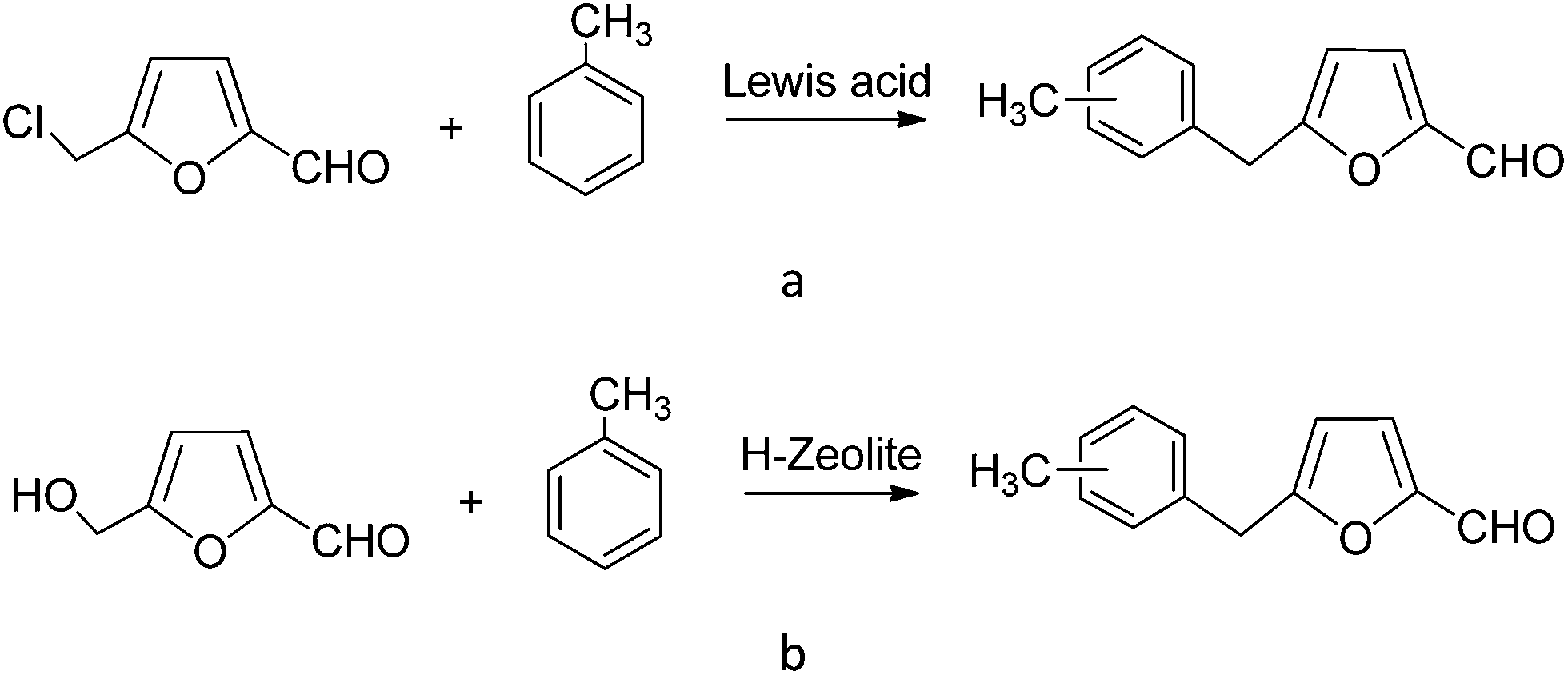 | ||
| Scheme 1 Synthesis of alkylated products of toluene from 5-chloromethyl-2-furfural (a) and from HMF (b). | ||
Considering the size of the reaction products, suitable zeolites for the alkylation of aromatics with HMF will require a pore–structure system that allows faster diffusion of products out of the pores to avoid multiple alkylation reactions and catalyst deactivation. Then, two dimensional (2D) or layered zeolites, as well as structured mesoporous materials can be an adequate choice. On the other hand, since different isomers, i.e. meta, ortho and para can be obtained when alkylating alkylaromatics with HMF, pore dimensions and topology should have an impact on product distribution. Based on the above premises, we have selected as the starting reference three protonic large pore zeolites with different pore topologies and close pore diameters (∼0.7 nm): HMordenite, with a system of unidimensional twelve ring pores (12R); HBeta zeolite with a system of tridirectional 12R pore without cavities and finally a tridirectional 12R pore zeolite that contains cavities (faujasite Y zeolite). The main characteristics of the zeolites used are given in the experimental section (Table 11).
Toluene was used first as a model aromatic molecule for studying the alkylation reaction with HMF. Then, when, the reaction between HMF and toluene was carried with HBeta zeolite (Si/Al ratio of 12) at 115 °C the corresponding monoalkylated products, a mixture of 5-(o-, m- and p-methyl)benzylfuran-2-carbaldehyde were obtained with 54% yield after 8 h (see Fig. 1 and Table 1). Moreover, 5,5′-(oxy-bis(methylene))bis-2-furfural (OBMF) coming from the self etherification of HMF (Scheme 2) was also obtained in 15% yield, while polyalkylated, oligomers, and compounds coming from the hydroxyalkylation of the aromatic ring were not detected in the reaction media. No products were detected when the reaction was carried out in the absence of the catalyst.
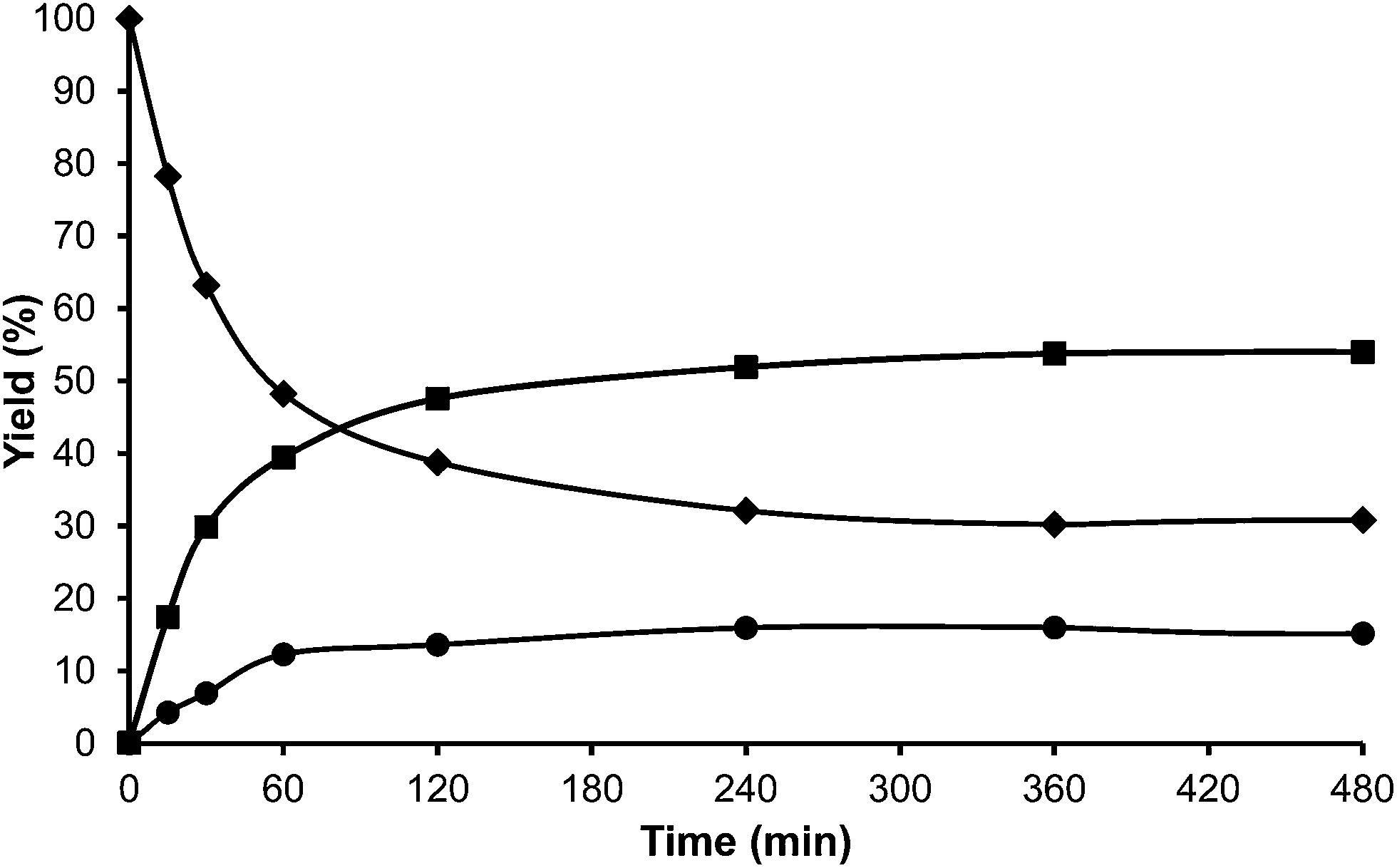 | ||
| Fig. 1 Kinetic curves of alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of HBeta (25 wt% HMF) at reflux temperature under a nitrogen atmosphere: HMF (♦), 5-alkylated products: ortho, meta, para isomers (■) and OBMF (●). | ||
| Catalyst (Si/Al) | r oAlk 105 mol min−1 | r oOBMF 105 mol min−1 | Time (h) | Conv. (%) | Yield (%) OBMF | Yield (%) alk | Sel. (%) alk | Sel. (%) alk (o:p:m) |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: HMF (0.5 mmol), toluene (25 ml), catalysts 15.75 mg (25 wt%), reflux temperature, nitrogen atmosphere. b 12 mg of PTSA was added. | ||||||||
| Mordenite (10) | 0.16 | 0.14 | 8 | 20 | 9 | 11 | 55 | 27:73:0 |
| USY-720 (12.2) | 2.56 | 0.57 | 6 | 100 | 5 | 95 | 95 | 46:52:2 |
| HBeta (12.5) | 0.57 | 0.14 | 6 | 69 | 15 | 54 | 78 | 24:72:4 |
| ITQ-2 (15) | 1.54 | — | 8 | 99 | 1 | 98 | 99 | 22:76:2 |
| MCM-41 (12) | 0.97 | 0.38 | 8 | 99 | 15 | 84 | 85 | 40:56:4 |
| PTSAb | 1.01 | 6.1 | 8 | 100 | 14 | 86 | 86 | 45:48:7 |
| 24 | 100 | 5 | 95 | 95 | 45:48:7 |
|||
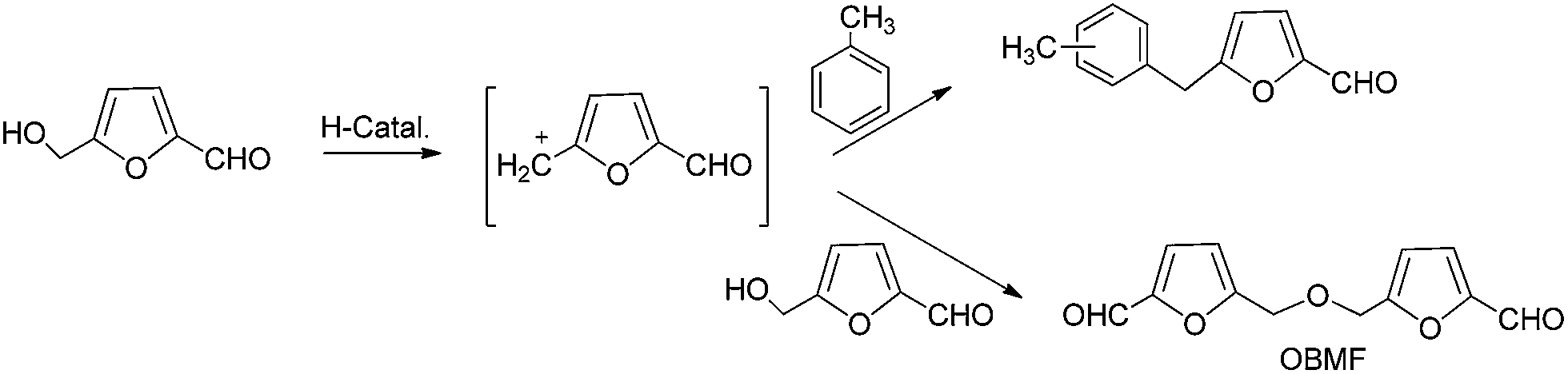 | ||
| Scheme 2 Products obtained (5-alkylated and OBMF) in the alkylation reaction of toluene with HMF. | ||
The formation of products could be explained by considering that the hydroxylmethyl group of HMF reacts on the acid sites to form the methylfurfuryl cation, which can react with the aromatic ring giving 5-benzylfuran-2-carbaldehyde. On the other hand the methylfurfuryl cation can also react with the hydroxyl group of another HMF molecule giving the corresponding ether (OBMF) (Scheme 2).
The yields of 5-alkylated-HMF and the OBMF products were plotted versus reaction time, and both appear as primary products (Fig. 1). As can be seen there, the initial reaction rate for the formation of 5-alkylated products is faster than the formation of OBMF, indicating that under our reaction conditions the alkylation reaction is preferred with respect to the self etherification of HMF. The result is not surprising considering that we work under a large excess of the aromatic compound. Despite this, the kinetic curves in Fig. 1 show that the reaction stops before complete conversion indicating deactivation of the catalysts. This can be attributed to the formation of polyalkylated or, in general, to the formation of bulky products which can remain strongly adsorbed inside the micropores of the Beta zeolite and which can hardly diffuse out, blocking active sites or even pores. To check this hypothesis we have determined the amount of organic products remaining adsorbed on the zeolite after the reaction by TG-MS, and the “coked” sample was subjected to Soxhlet extraction with dichloromethane. By doing this we obtained 4.4 mg of organic (composed of toluene, OBMF and alkylated compounds) on the solid that corresponds to 28 wt% with respect to the catalyst weight. Moreover, nitrogen adsorption was performed on the used catalyst after extraction with dichloromethane, and an important decrease of the pore volume of the catalyst after the reaction was observed going from 0.18 cm3 g−1 for the fresh sample to 0.08 cm3 g−1 for the used zeolite. The micropore volume decrease confirms that there is an important reduction of the internal pore volume by pore blocking with reaction products.
For comparison purposes, the alkylation of HMF was performed in the presence of p-toluenesulfonic acid (PTSA) as the catalyst and the results are shown in Table 1 and Fig. 2. As can be seen, PTSA promotes the self etherification of HMF at a much higher rate than alkylation, achieving a maximum concentration of OBMF that then decreases with time. In this case, the strong acidity of the homogeneous catalyst and the absence of diffusional limitations favour the alkylation of toluene by the OBMF achieving a final selectivity to the alkylated compounds of 95% after 24 h reaction time.
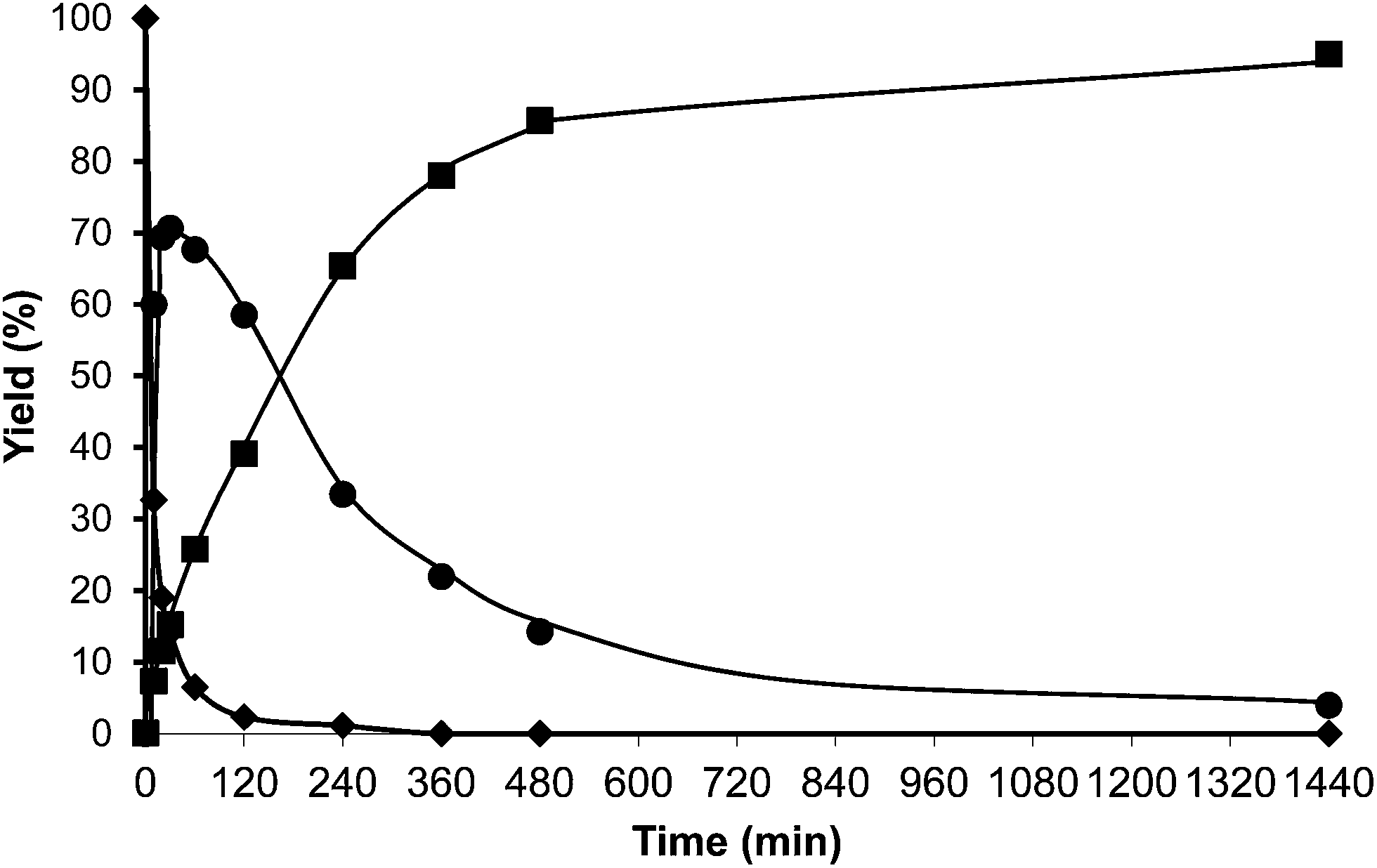 | ||
| Fig. 2 Results of alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of p-toluenesulfonic acid (18 wt% HMF) at reflux temperature under a nitrogen atmosphere: HMF (♦), 5-alkylated products: ortho, meta, para isomers (■) and OBMF (●). | ||
With respect to selectivity at the different regioisomers (see Fig. 3), the ratio of para/ortho isomers observed for the homogeneous catalyst, was approximately 1.2, which is similar to the value reported for the alkylation of toluene with benzyl alcohol using Nafion-H as the catalyst.32 However, Beta zeolite was much more selective to the para-isomer (less sterically impeded) than p-toluenesulfonic or Nafion, with a para to ortho ratio of 3.0, with little selectivity to the electronically disfavored meta-isomer. It should be noticed that the para to ortho ratio decreases with reaction time (not showed), probably indicating a larger contribution of the external surface of the zeolite when the pores of the catalyst become blocked.
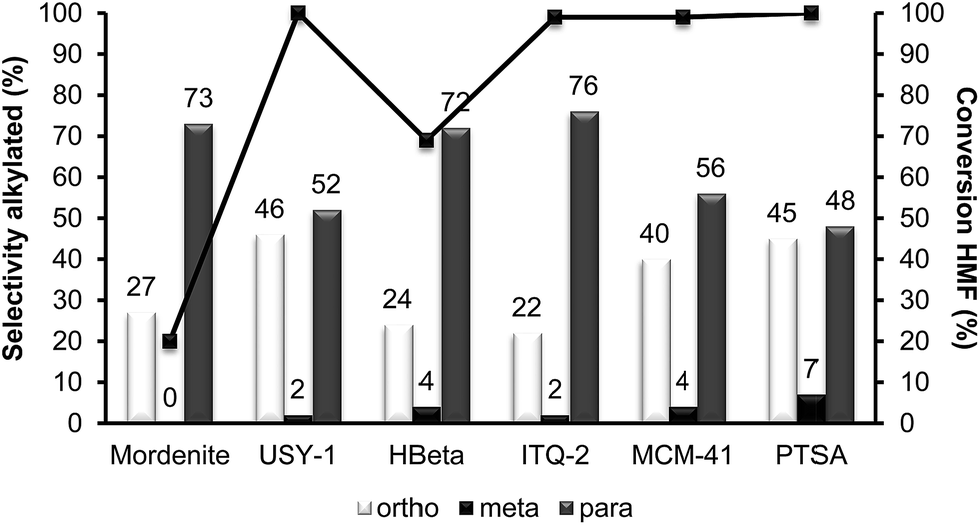 | ||
| Fig. 3 Results of HMF conversion and selectivity to ortho, meta and para alkylated isomers obtained in the alkylation of toluene with HMF over different acid catalysts after 8 h of reaction. | ||
At this point we thought that perhaps HMordenite could yield a higher para/ortho ratio, although we were since the first moment concerned about the potential faster deactivation of the unidimensional zeolite by pore blocking. Indeed, a much faster deactivation was observed in the case of the HMordenite with respect to HBeta, together with a much higher selectivity to OBMF (see Fig. 4 and Table 1). Meanwhile, no benefit on the para to ortho ratio was obtained. After these results the HMordenite was already rejected as a potential catalyst for the alkylation of aromatics with HMF.
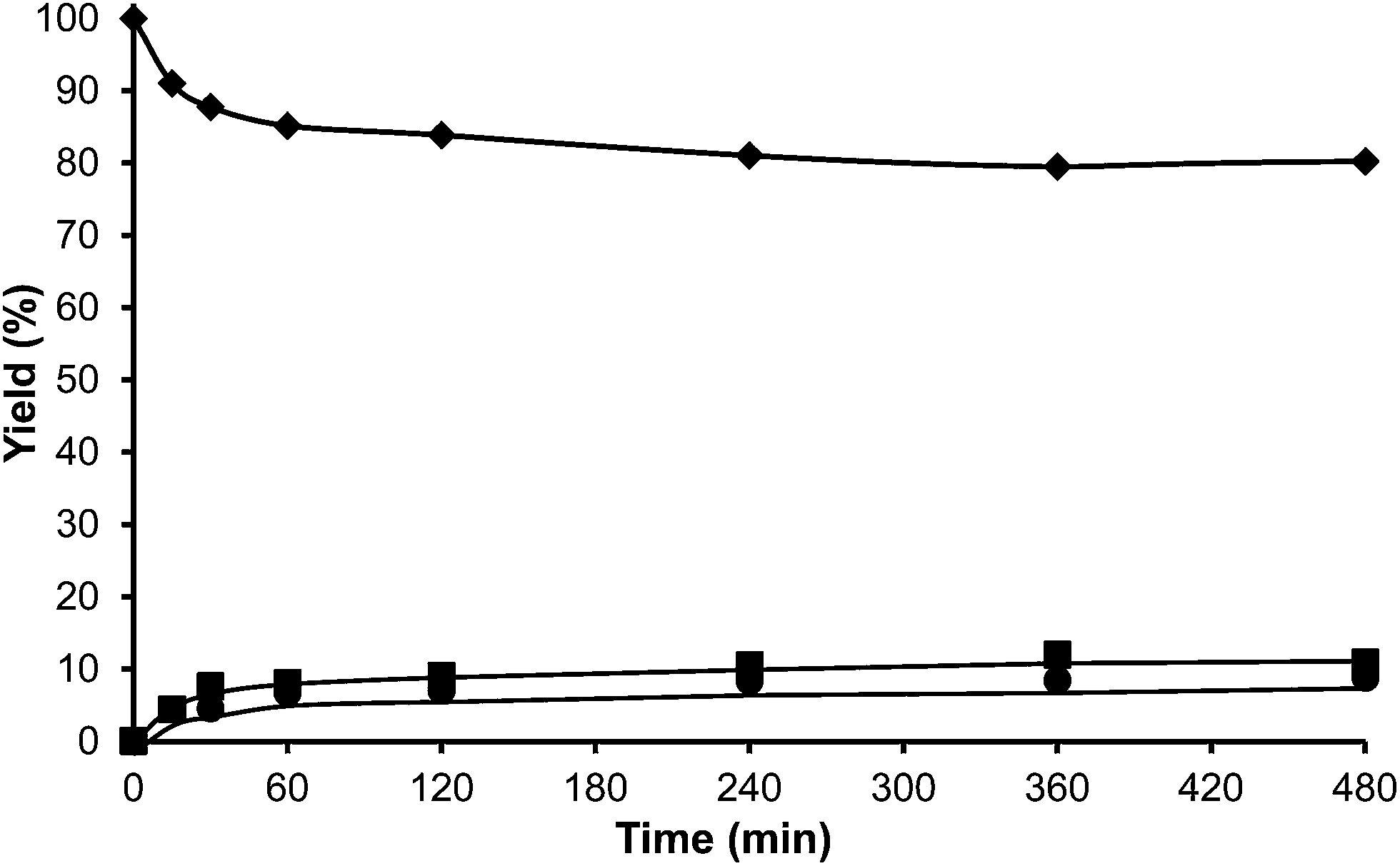 | ||
| Fig. 4 Kinetic curves of alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of Mordenite (25 wt% HMF) at reflux temperature under a nitrogen atmosphere: HMF (♦), 5-alkylated products: ortho, meta, para isomers (■) and OBMF (●). | ||
The following step in the study was to use a tridimensional zeolite with larger pores than HBeta, and ultra stable Y zeolites USY samples were selected as alkylation catalysts. These types of zeolites have shown to be active and stable for the alkylation of aromatic molecules with alcohols.33 Indeed, we have found that when toluene and HMF were reacted in the presence of a USY zeolite (USY-720) with a unit cell size of 24.32 Å, equivalent to a framework Si/Al ratio of 19, the results in Fig. 5 show a high selectivity to the desired alkylated product with low selectivity to the undesired OBMF ether.
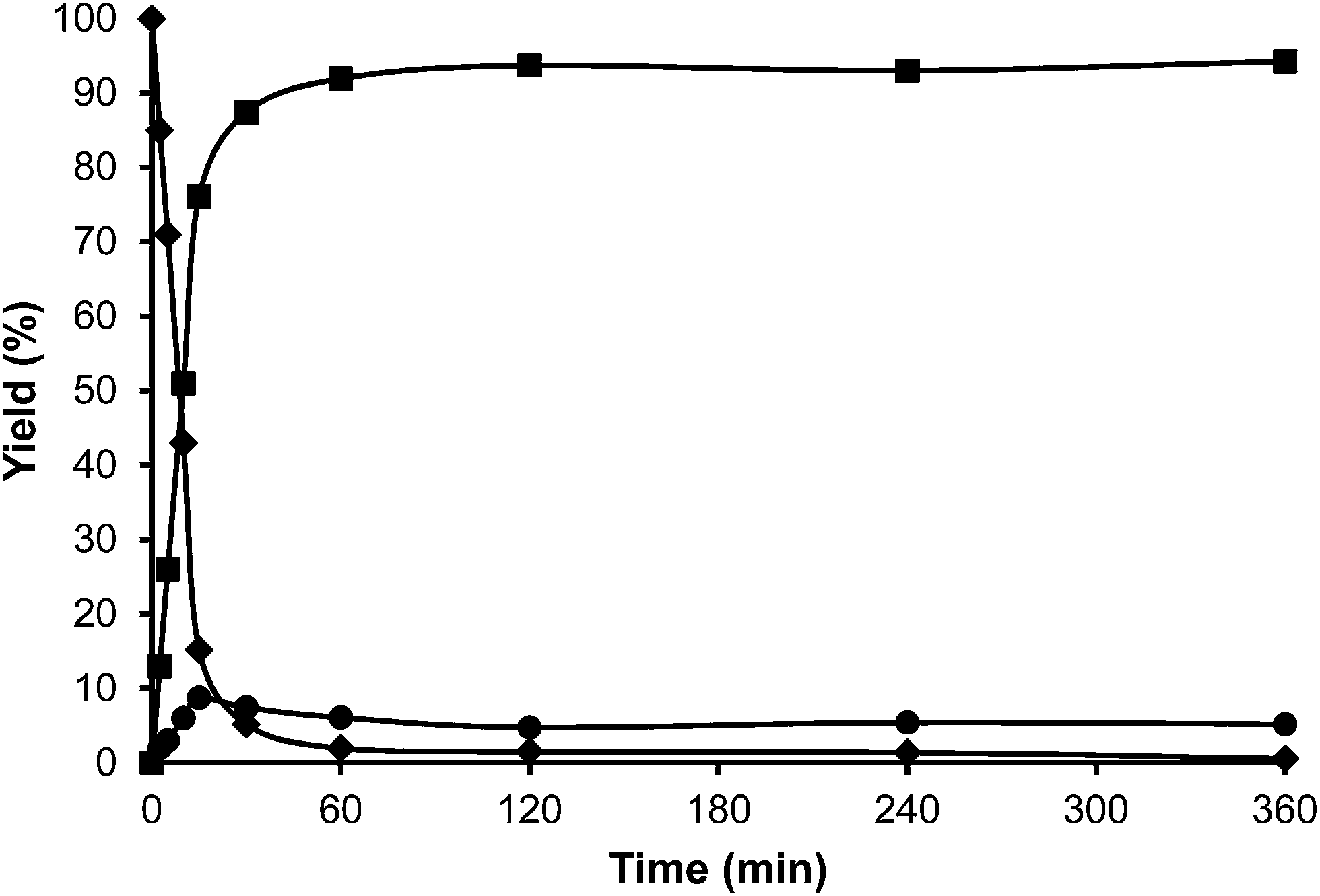 | ||
| Fig. 5 Kinetic curves of alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of USY-720 (25 wt% MF) at reflux temperature under a nitrogen atmosphere: HMF (♦), 5-alkylated products: ortho, meta, para isomers (■) and OBMF (●). | ||
Interestingly, 100% conversion is achieved very fast, suggesting not only a high activity but also probably a slower rate of deactivation. In an analogous way, as was done previously with HBeta, the amount of organic remaining and the micropore volume of USY zeolite after the reaction and Soxhlet extraction were measured. The results presented in Table 2 show that the amount of remaining organic and the percentage of the micropore volume lost follow the order Beta > USY > Mordenite.
| Catalyst (Si/Al) | Organic material (%) | V micro lost (%) |
|---|---|---|
| Mordenite (10) | 10 | 17 |
| USY-720 (12.2) | 16 | 41 |
| HBeta (12.5) | 28 | 55 |
The organic remaining on the USY-720 zeolite was burned off by calcination at 450 °C during 3 h, and the catalyst was reused again. The kinetic curves in Fig. 6a–b compare the results with fresh Beta and USY-720 and the corresponding regenerated catalysts. It can be seen that the activity can be perfectly restored by calcination of the catalysts in air. However, when the para to ortho ratio is considered (see Fig. 3) it is possible to see that owing to the large cavities (∼1.2 nm) present in the USY zeolite the ratio is practically the same than the one obtained with PTSA in which no geometrical restrictions from the pore exist. In other words, USY-720 is a very active and selective zeolite that allows alkylation to occur freely within the cavities, while products can diffuse out.
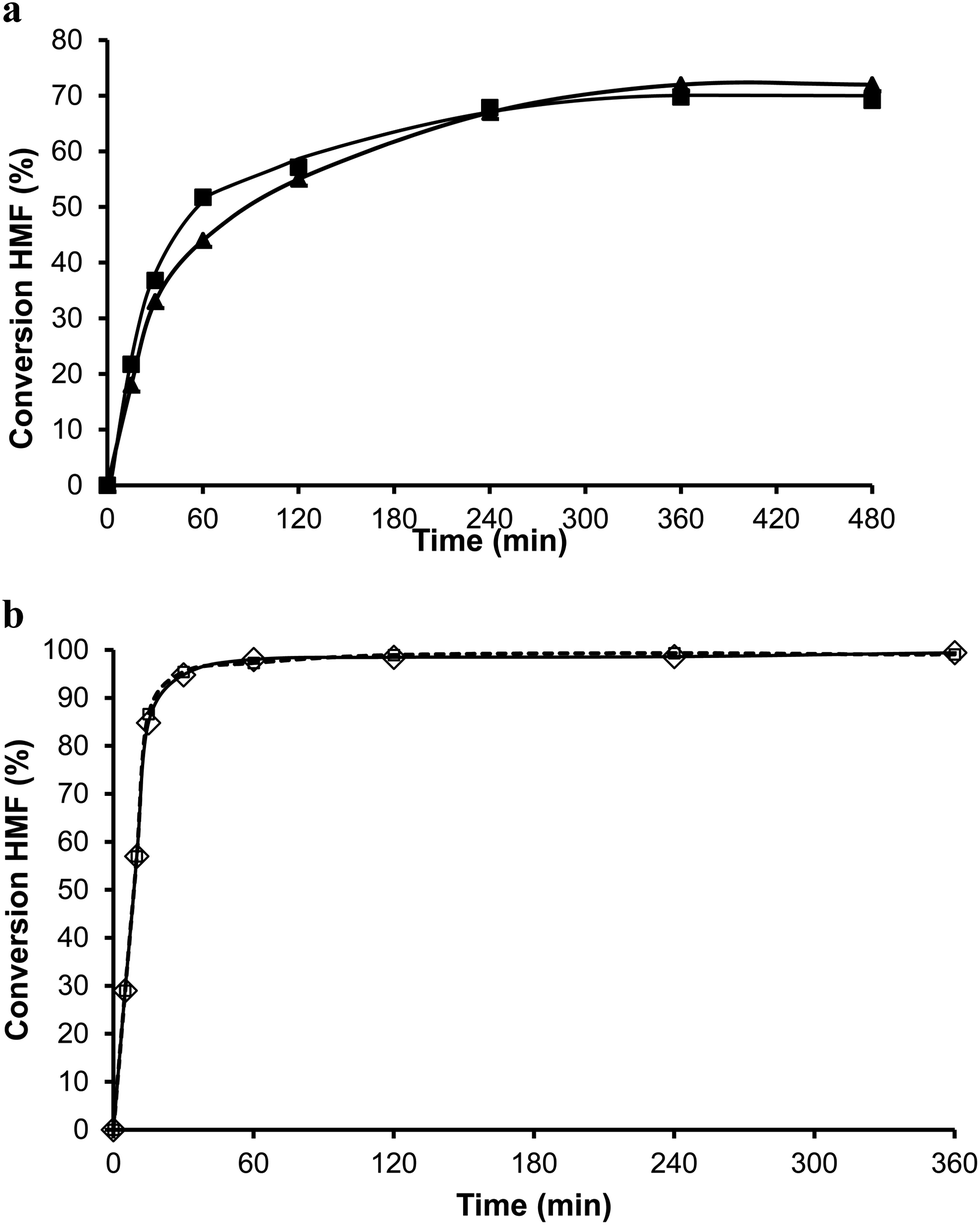 | ||
| Fig. 6 (a) Conversion versus time plot for the alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of Beta 1st cycle (■) and 2nd cycle (after calcination) (▲) (25 wt% HMF) at reflux temperature and nitrogen atmosphere. (b) Conversion versus time plot for the alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of USY-720. 1st cycle (–□–) and 2nd cycle (after calcination) (◊) (25 wt% HMF) at reflux temperature under a nitrogen atmosphere. | ||
Since the pore dimensions and topology of the Y zeolite seem to be convenient, we have explored the implications of the framework composition/acidity (framework Si/Al ratio), as well as the influence of the mesoporosity of different ultrastable Y zeolites (USY) on the catalyst activity, selectivity and catalyst decay.
In order to check the influence of the framework Si/Al (FAL), four commercial USY samples with different Al contents: USY-500, USY-712, USY-720 and USY-760 and similar crystallite size (see Table S1 in the ESI†) were selected. Measurements of acidity and physico-chemical characteristics are presented in Tables 3 and 4.
| Catalyst | Si/Al | 150 °C | 250 °C | 350 °C | |||
|---|---|---|---|---|---|---|---|
| Brønsted | Lewis | Brønsted | Lewis | Brønsted | Lewis | ||
| a Acidity μmol pyridine per gram catalyst, calculated using extinction coefficients given in ref. 46. | |||||||
| USY-300 | 2.6 | 65 | 106 | 31 | 83 | 3 | 57 |
| USY-500 | 2.7 | 111 | 56 | 92 | 44 | 52 | 33 |
| USY-712 | 5.6 | 58 | 25 | 49 | 19 | 31 | 14 |
| USY-720 | 12.2 | 79 | 16 | 66 | 13 | 41 | 10 |
| USY-760 | 27 | 24 | 11 | 21 | 10 | 6 | 7 |
| USY-MY | 4.3 | 88 | 49 | 64 | 34 | 27 | 25 |
| USY-HMY | 5.3 | 34 | 25 | 29 | 21 | 19 | 22 |
| Catalyst | Unit cell size (Å) | Crys.a (%) | Si/Al (ICP) | Si/Al (framew.)b | Al per unit cell | BET (m2 g−1) | V micro (cm3 g−1) | V meso (cm3 g−1) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | FAL | EFAL | ||||||||
| a Referred to as NaY CBV100. b Framework Si/Al ratio, determined by means of Breck–Flanigen47 or Fichtner-Schmittler48 equations for zeolites with Si/Al ratios below and above 3, respectively. c Calculated from the t-plot. FAL = framework Al, EFAL = extra framework Al. | ||||||||||
| USY-500 | 24.56 | 100 | 2.7 | 3.5 | 51.9 | 42.5 | 9.4 | 609 | 0.27 | 0.07 |
| USY-712 | 24.36 | 100 | 5.6 | 12.4 | 29 | 14.3 | 15 | 593 | 0.25 | 0.13 |
| USY-720 | 24.32 | 99 | 12.2 | 18.6 | 14.5 | 9.8 | 4.7 | 603 | 0.27 | 0.13 |
| USY-760 | 24.26 | 36 | 27 | 63 | 6.9 | 3 | 3.9 | 509 | 0.15 | 0.20 |
The alkylation of toluene with HMF was performed with these samples and the initial rate of alkylation versus Si/Al framework (FAL) is displayed in Fig. 7. As can be seen, maximum activity was found at the framework Si/Al ratio between 18.6 and 63, indicating that the amount of framework Al plays an important role in the catalytic activity. In fact, it is known that by increasing the Si/Al ratio, the hydrophobic character of a zeolite increases while the number of potential acid sites decreases. In this case it is shown that an optimum in adsorption properties, acidity and catalytic activity exists for a framework Si/Al value of ∼19 that corresponds to 10 Al per unit cell (see Tables 3–5).
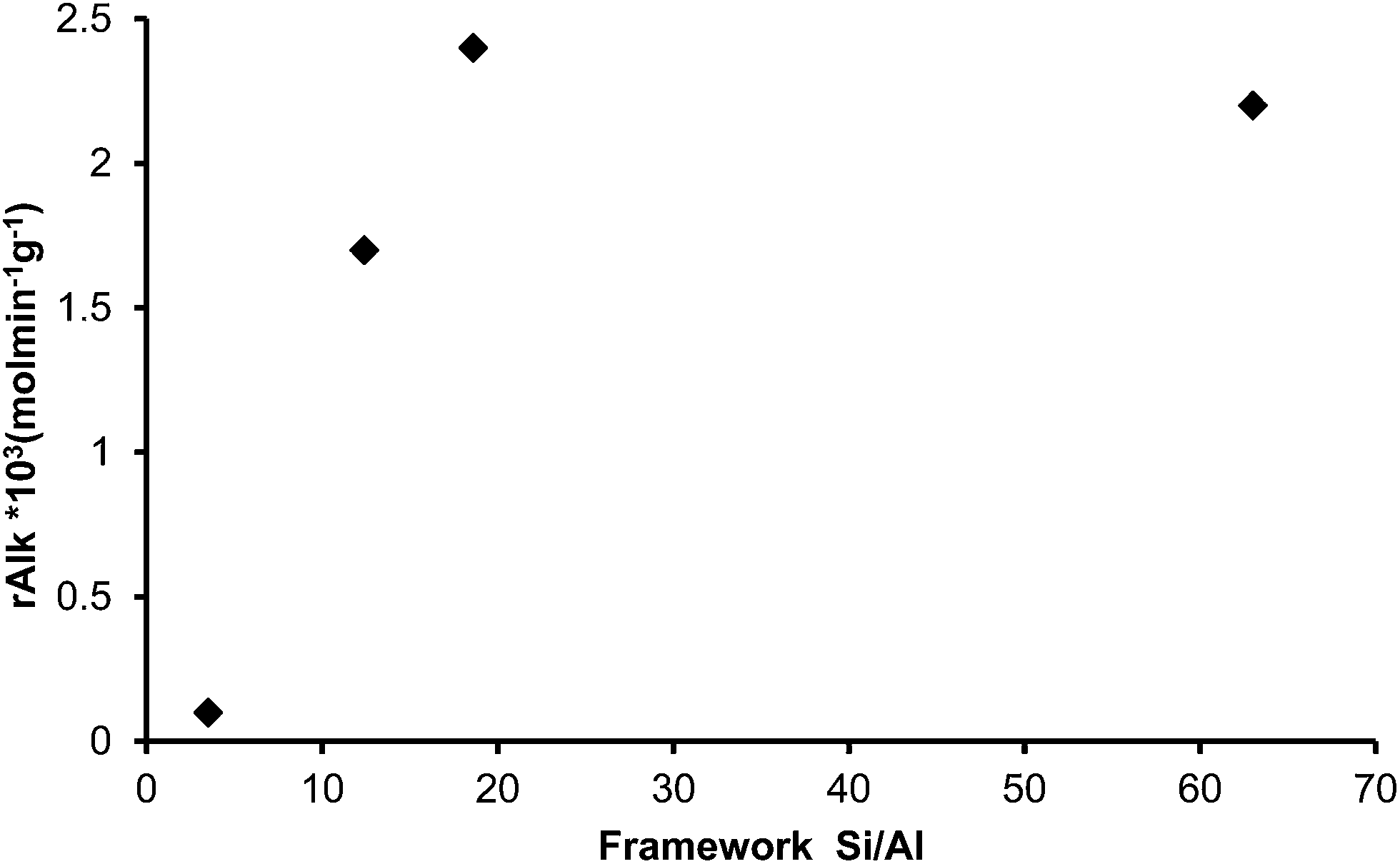 | ||
| Fig. 7 Rate of appearance of alkylated compounds versus FAL content of different USY samples. | ||
| Catalyst | Si/Al(UCS) FAL | Si/Al (ICP) | r oAlk 103 (mol min−1 g−1) | Conv.a HMF (%) | Yield alk (%) | Select. (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: HMF (0.5 mmol), toluene (25 ml), catalysts 15.75 mg (25 wt%), refluxed temperature, nitrogen atmosphere after 8 h of reaction. | ||||||
| USY-500 | 3.5 | 2.7 | 0.1 | 15 | 9 | 60 |
| USY-712 | 12.4 | 5.6 | 1.7 | 99 | 91 | 92 |
| USY-720 | 18.6 | 12.2 | 2.4 | 100 | 95 | 95 |
| USY-760 | 63.0 | 27.0 | 2.2 | 99 | 91 | 92 |
Additionally, the influence of the framework Si/Al ratio on deactivation is shown in Fig. 8. As can be observed, the deactivation rate follows the order USY-500 > USY-712 > USY-760 > USY-720, showing also in this case the existence of an optimum for catalyst deactivation for the zeolite with ∼10 framework Al per unit cell.
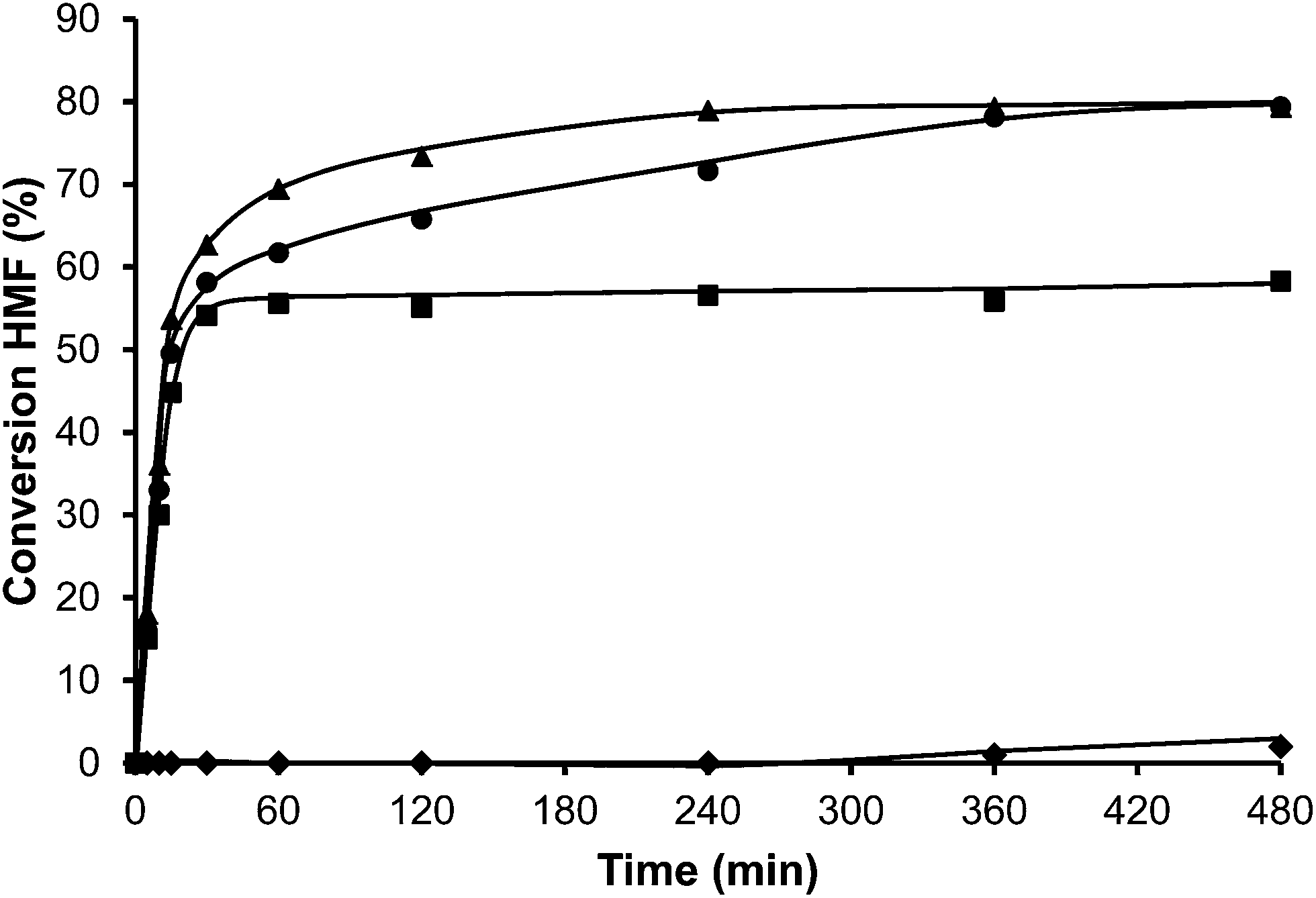 | ||
| Fig. 8 Conversion versus time plot for the alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of different USY samples (10 wt% HMF) at reflux temperature under a nitrogen atmosphere: USY-500 (♦), USY-712 (■), USY-720 (▲) and USY-760 (●). | ||
With this value and considering a statistical distribution of Al, the optimum will correspond to a sample in where most of the framework Al will be isolated sites, i.e. Al with no Al in the next nearest neighbor positions.34
| Sample | UCS (Å) | Cryst.a (%) | Si/Al (ICP) | Si/Al(UCS) FALb | %Na2O | BET (m2 g−1) | V micro (cm3 g−1) | V meso (cm3 g−1) |
|---|---|---|---|---|---|---|---|---|
| a Referred to as NaY CBV100. b Determined by means of Breck–Flanigen47 or Fichtner-Schmittler48 equations for zeolites with Si/Al ratios below and above 3, respectively. c Calculated from the t-plot. d Calculated from BJH correlation. | ||||||||
| USY-300 | 24.64 | 100 | 2.6 | 2.7 | 2.8 | 925 | 0.30 | 0.04 |
| USY-MY | 24.60 | 58 | 4.3 | 3.7 | 1.4 | 617 | 0.16 | 0.22 |
| USY-HMY | 24.47 | 28 | 5.3 | 6.2 | <0.1 | 449 | 0.09 | 0.24 |
The alkylation of toluene with HMF was performed using USY-300 and USY-MY samples. Fig. 9, shows that the activity of USY-300 zeolite is very low which can be attributed to the high sodium content. However in contrast, the mesoporous USY-MY sample showed higher catalytic activity than the precursor sample (USY-300) (Fig. 10), achieving around 40% of HMF conversion after 8 h. When the activity of theUSY-MY sample is compared with that exhibited by a sample (USY-500) with a similar unit cell size value, i.e. similar framework aluminum content but higher acidity it is possible to see that the latter exhibits considerably lower activity (see Table 3), achieving only 15% conversion of HMF after 8 h reaction time. These results clearly indicate that the generation of intracrystalline mesopores in the USY zeolite has a very positive effect on the catalytic activity for the alkylation of toluene with HMF.
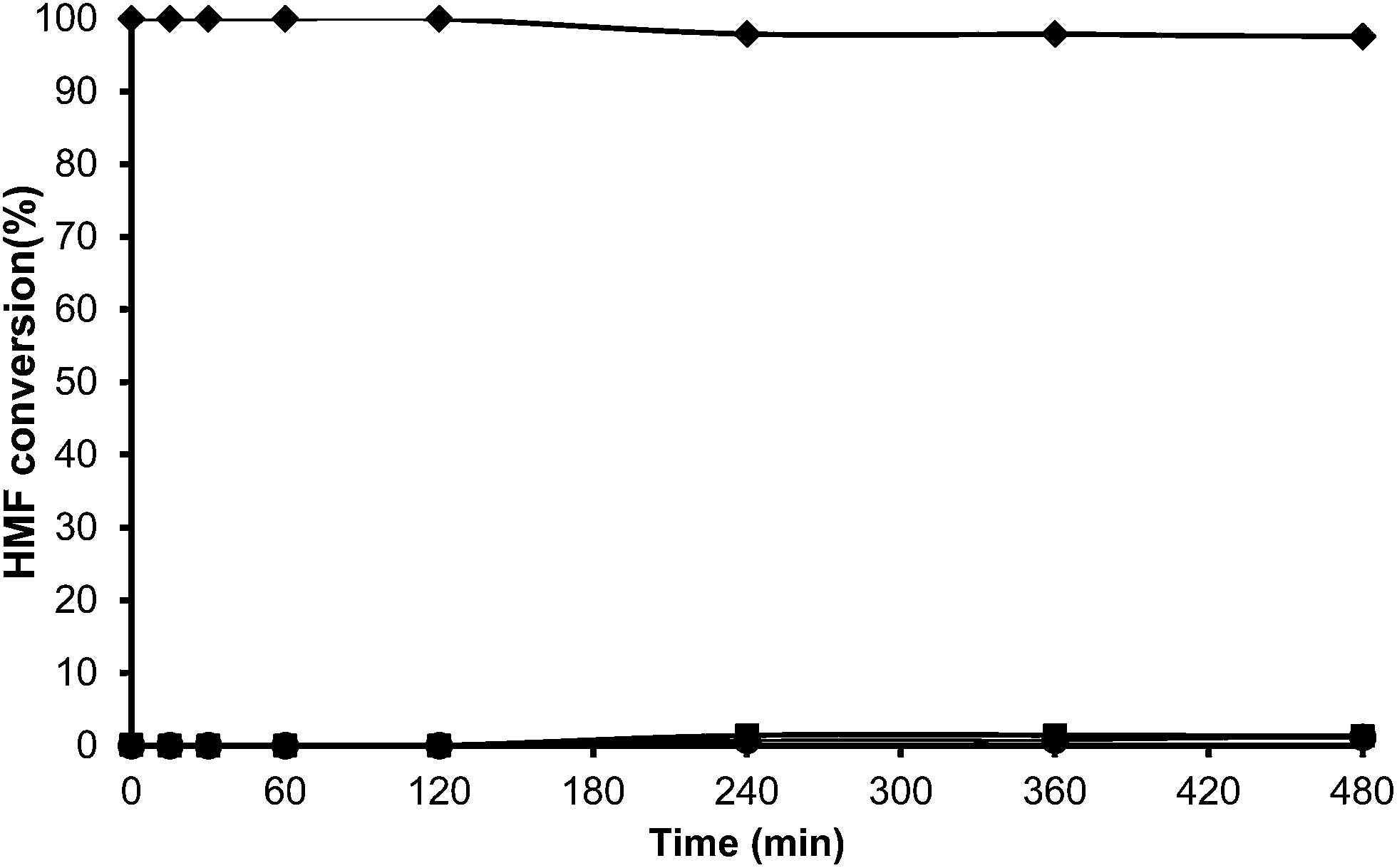 | ||
| Fig. 9 Kinetic curves of alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of USY-300 (25 wt% HMF) at reflux temperature under a nitrogen atmosphere: HMF (♦), 5-alkylated products: ortho, meta, para isomers (■) and OBMF (●). | ||
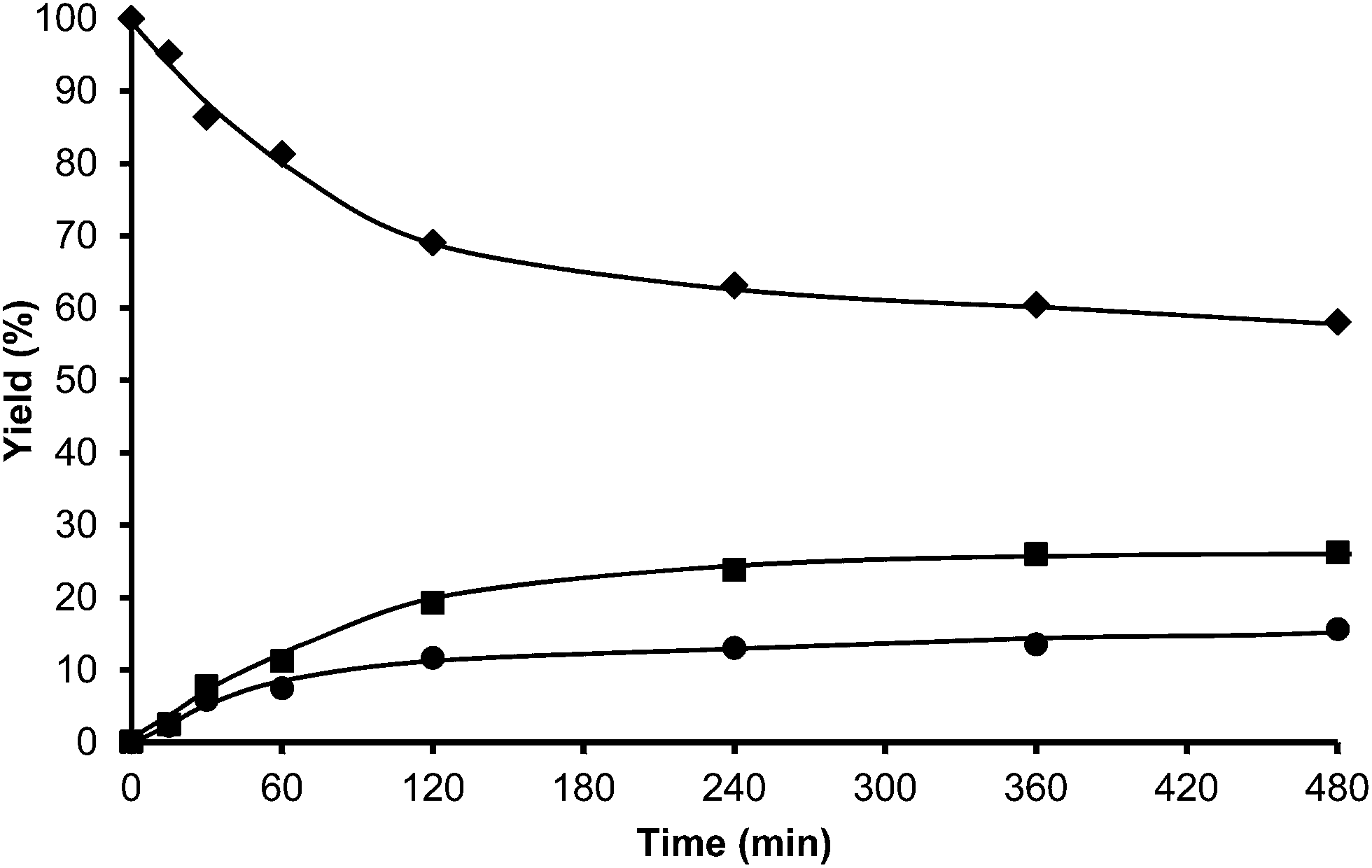 | ||
| Fig. 10 Kinetic curves of alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of mesoporous USY-MY (25 wt% HMF) at reflux temperature under a nitrogen atmosphere: HMF (♦), 5-alkylated products: ortho, meta, para isomers (■) and OBMF (●). | ||
Additionally, we reduced the Na content of the USY-MY by a conventional ammonium exchange procedure followed by calcination in order to study the influence of increasing acidity on the mesoporous sample. The ammonium exchanged sample (USY-HMY) obtained, had a Na2O content below 0.1%. As can be seen in Table 6, the mesoporosity of this material is preserved, however a loss of micropore volume and framework aluminum, with a considerable decrease in acidity is evidenced. Unfortunately, these changes affect significantly the catalytic activity and in fact, only 10% of conversion of HMF was achieved after 8 h reaction time (see Fig. 11).
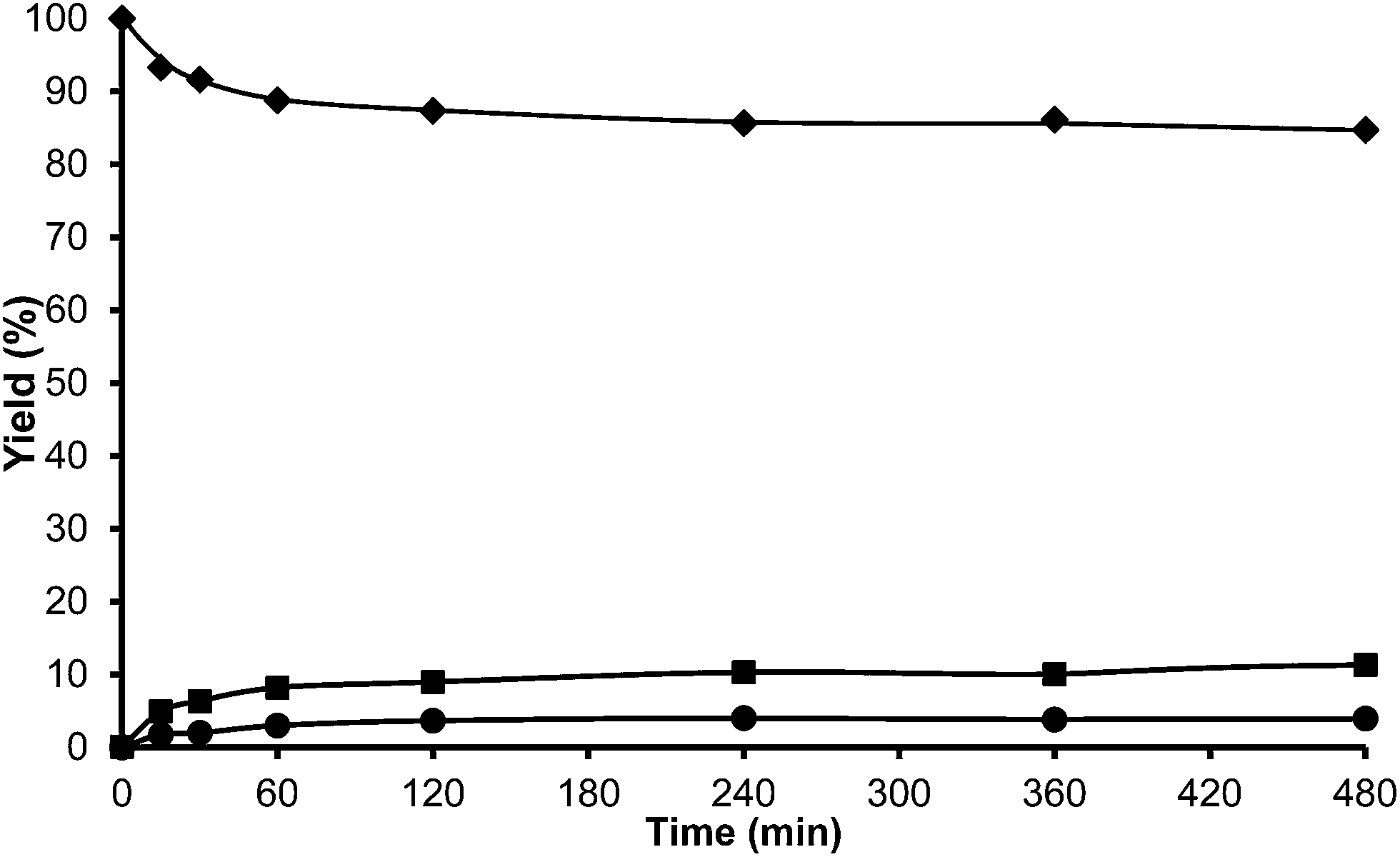 | ||
| Fig. 11 Kinetic curves of alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of USY-HMY (25 wt% HMF) at reflux temperature under a nitrogen atmosphere: HMF (♦), 5-alkylated products: ortho, meta, para isomers (■) and OBMF (●). | ||
Since diffusional effects are important for the alkylation of toluene with HMF, we have also considered the possibility of using a two dimensional layered zeolite, where most of the surface can be considered as the external surface (ITQ-2). The catalytic results obtained with this type of material will be described below.
In the case of the laminar precursor of MWW, the 2D layered zeolite ITQ-2 was prepared where the 12R hemicavities similar to cups are exposed to the exterior ready to adsorb-react-desorb without requiring diffusion through micropores42 (see Fig. 12 and S1 ESI†). Since this material with external surface areas larger than 600 m2 g−1 was shown to be more active for alkylation of biphenyl with propene than the corresponding 3D MWW zeolite,43 we have studied here, the catalytic behavior of ITQ-2 for the alkylation of toluene with HMF. The results in Fig. 12 and Table 1 show that this is an active zeolite with a very low initial rate of OBMF formation. Moreover, it is observed that the OBMF formed also reacts with toluene giving the alkylated compounds with, a final selectivity of 99% at 99% of HMF conversion (see Fig. 12). No deactivation can be inferred from the kinetic curves. However since batch reactors are not the best to discuss catalyst decay from the kinetic curves the used ITQ-2 was tested again after the reaction and Soxhlet extraction. The results in Fig. 13 and 14 indicate that ITQ-2, as it occurs with USY-720, strongly deactivates during the reaction, reaching only a 10% of the initial reaction rate. In any case, the initial activity of ITQ-2 and USY-720, catalysts can be restored after calcination at 450 °C for 3 h.
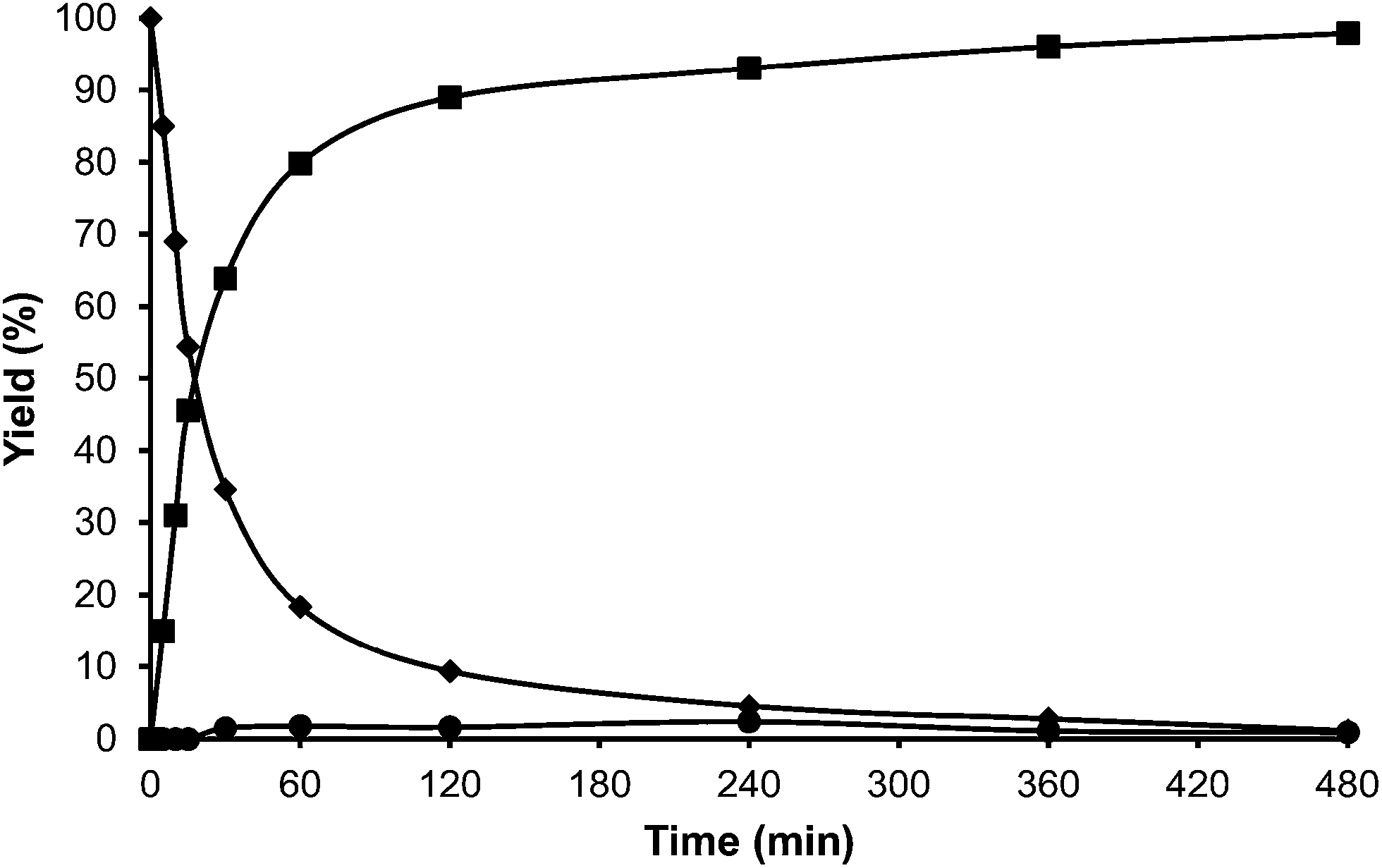 | ||
| Fig. 12 Kinetic curves of alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of ITQ-2 (25 wt% HMF) at reflux temperature and nitrogen atmosphere: HMF (♦), 5-alkylated products: ortho, meta, para isomers (■) and OBMF (●). | ||
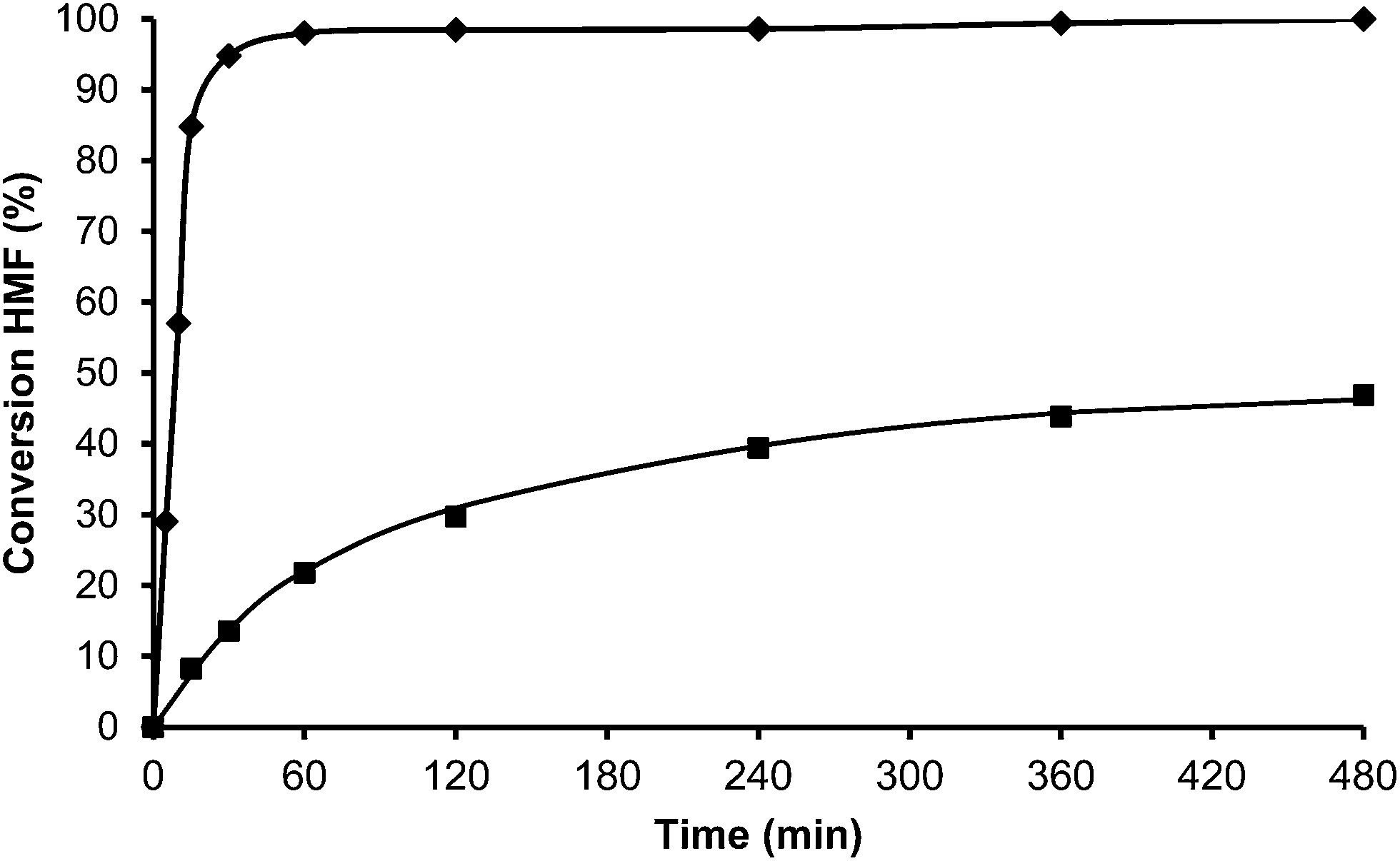 | ||
| Fig. 13 Conversion versus time plot for alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of USY-720 1st cycle (♦) and 2nd cycle (after Soxhlet extraction) (■) (25 wt% HMF) at reflux temperature under a nitrogen atmosphere. | ||
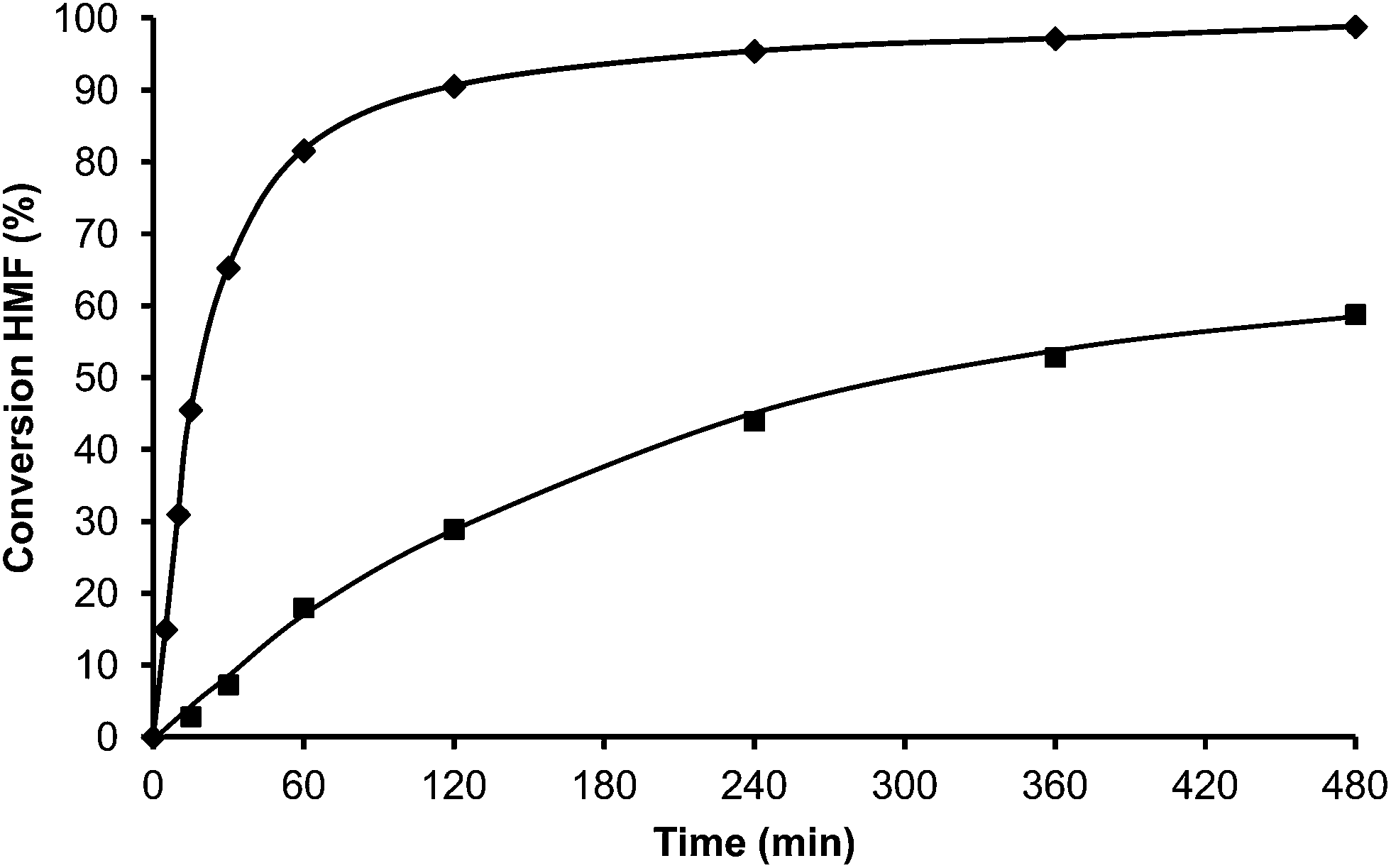 | ||
| Fig. 14 Conversion versus time plot for alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of ITQ-2 1st cycle (♦) and 2nd cycle (after Soxhlet extraction) (■) (25 wt% HMF) at reflux temperature under a nitrogen atmosphere. | ||
Interestingly, while the alkylation isomer distribution for USY-720 was similar to that of PTSA, ITQ-2 clearly gives the highest para/ortho ratio among the zeolites studied (see Fig. 3), indicating the key role played by the external “cup” like hemicavities (see Fig. 1 ESI†) during the catalytic reaction. It appears then, that the excellent catalytic behavior of ITQ-2 is due to the presence of zeolitic type of acidity together with a well structured very high external surface area formed by cup like hemicavities. These cups are not connected with the microporous channel system, and allow the reaction and desorption of the products. Notice that the ITQ-2 retains the initial activity and selectivity after four reaction–regeneration cycles (see Fig. 15).
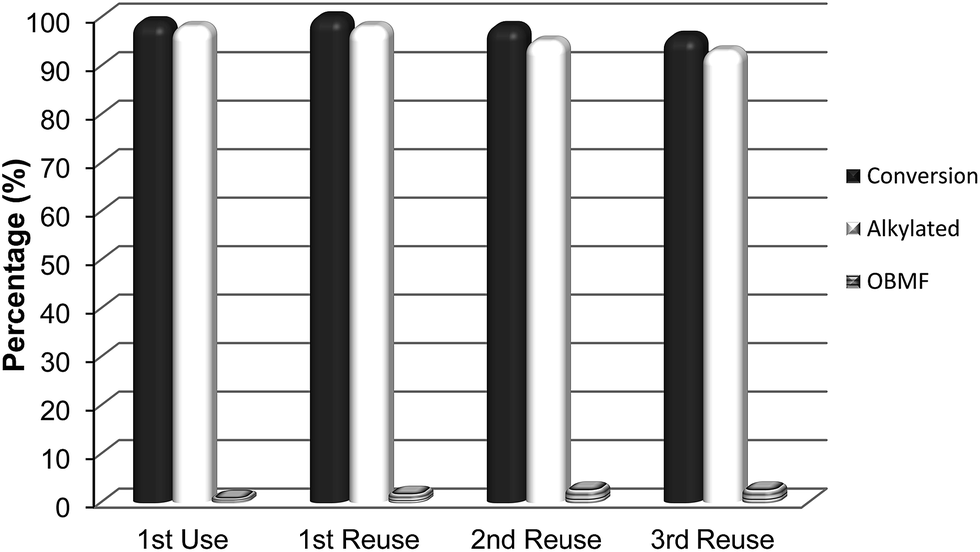 | ||
| Fig. 15 Reuse of the ITQ-2 zeolite. Conversion and selectivity after 8 h reaction time. | ||
Since acid strength, diffusion and desorption are key catalyst variables, we decided to study finally the catalytic behavior of structured mesoporous materials, and more specifically of the MCM-41 material. The acidity shown by this material is weaker than that of zeolites and closer to the acidities of amorphous silica-alumina. However, the fact that it presents regular pores with approximately 3.5 nm diameter should allow fast desorption and diffusion of reactants and products. The results in Fig. 16 and Table 1 show that while the activity of MCM-41 is reasonable, its selectivity for the formation of the undesired OBMF is the highest. These results are probably due to the lower acidity of the MCM-41 as demonstrated by pyridine adsorption–desorption. With respect to the para/ortho ratio, it is much lower than for ITQ-2.
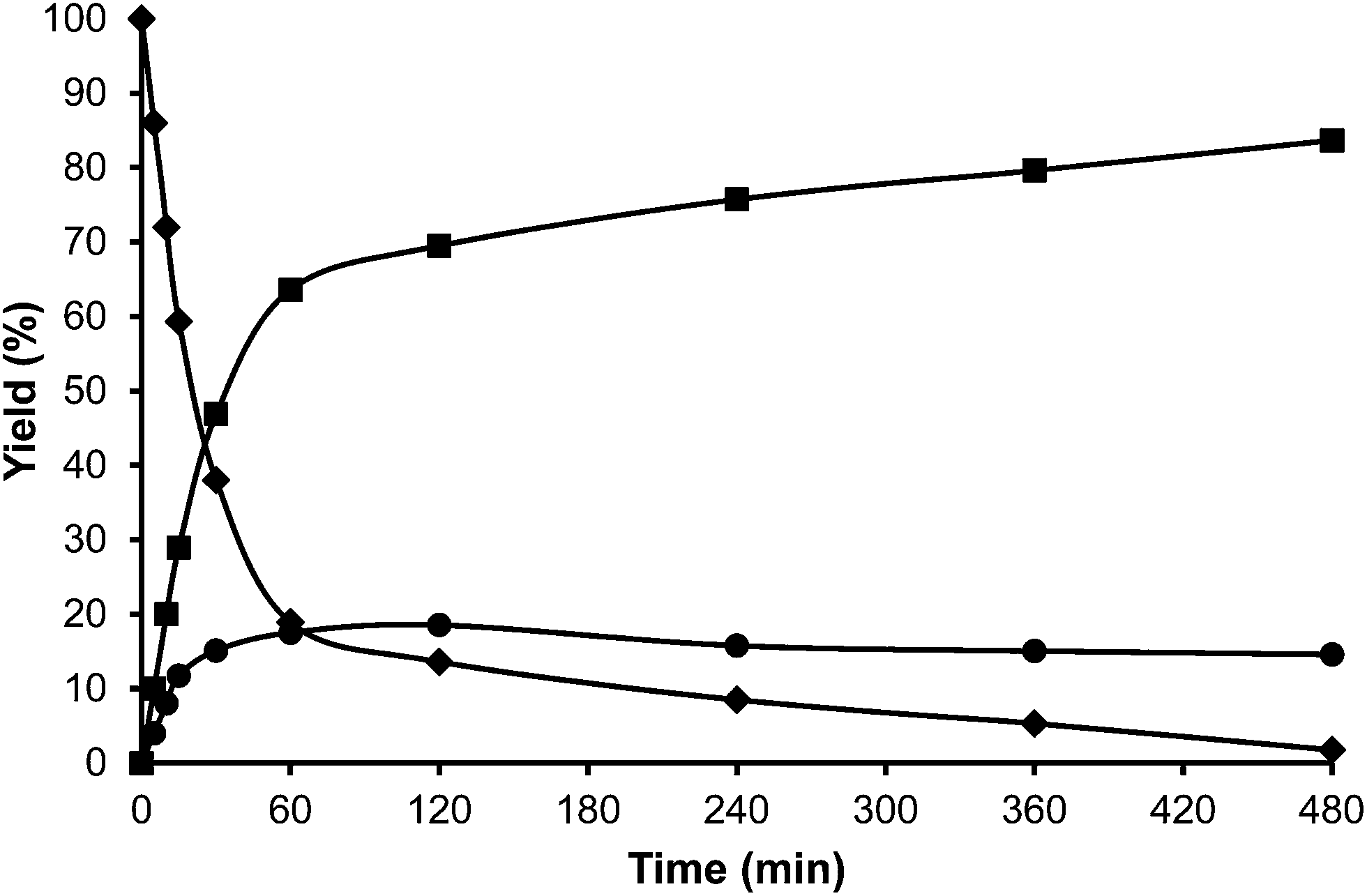 | ||
| Fig. 16 Kinetic curves of alkylation of toluene (25 ml) with HMF (0.5 mmol) in the presence of MCM-41 (25 wt% HMF) at reflux temperature and nitrogen atmosphere: HMF (♦), 5-alkylated products: ortho, meta, para isomers (■) and OBMF (●). | ||
| Toluene/HMF (mol mol−1) | Tol (ml) | T (°C) | Time (h) | Yield (%) alk. | Yield (%) OBMF | Selectivity (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: HMF (0.5 mmol), ITQ-2(15) (15.75 mg, 25 wt%), 115 °C, N2. b 30 wt% ITQ-2 (15) with respect to HMF. c 50 wt% ITQ-2 (15) with respect to HMF. | ||||||
| 47 | 2.5 | 115 | 2 | 61 | 29 | 66 |
| 8 | 68 | 28 | 69 | |||
| 47 | 2.5 | 150 | 2 | 70 | 22 | 76 |
| 8 | 83 | 14 | 85 | |||
| 47 | 2.5 | 170 | 2 | 79 | 13 | 86 |
| 8 | 90 | 10 | 90 | |||
| 94 | 5 | 115 | 2 | 70 | 21 | 77 |
| 8 | 83 | 17 | 83 | |||
| 94b | 5 | 115 | 2 | 79 | 18 | 81 |
| 8 | 87 | 13 | 87 | |||
| 94c | 5 | 115 | 2 | 85 | 15 | 85 |
| 8 | 96 | 4 | 96 | |||
| 94 | 5 | 150 | 2 | 85 | 11 | 89 |
| 8 | 96 | 4 | 96 | |||
| 189 | 10 | 115 | 2 | 74 | 14 | 84 |
| 8 | 80 | 15 | 82 | |||
| 472 | 25 | 115 | 2 | 89 | 2 | 98 |
| 8 | 98 | 1 | 99 | |||
| Aromatic compound | t (h) | HMF conv (%) | Yield (%) alkylated | Selec. (%) alkylated |
|---|---|---|---|---|
| a Reaction conditions: HMF (0.5 mmol), aromatic hydrocarbon 25 ml, ITQ-2(15) (25 wt%), 115 °C. | ||||
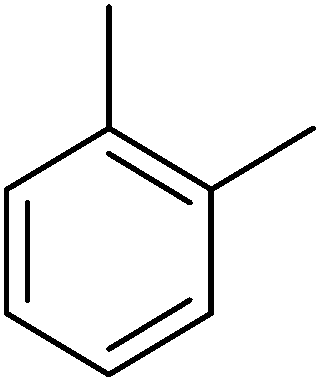
|
2 | 100 | 99 | 99 |
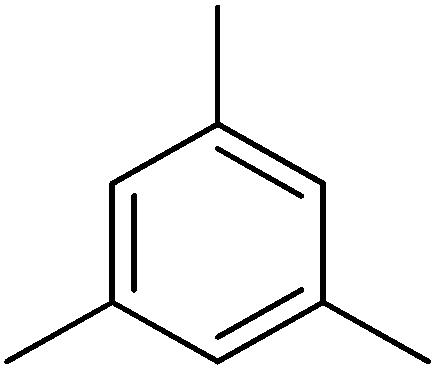
|
6 | 55 | 37 | 67 |
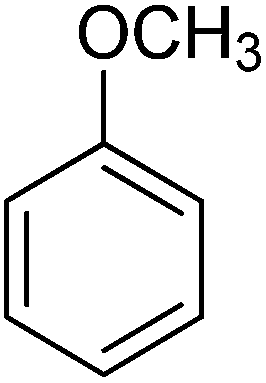
|
24 | 80 | 77 | 96 |
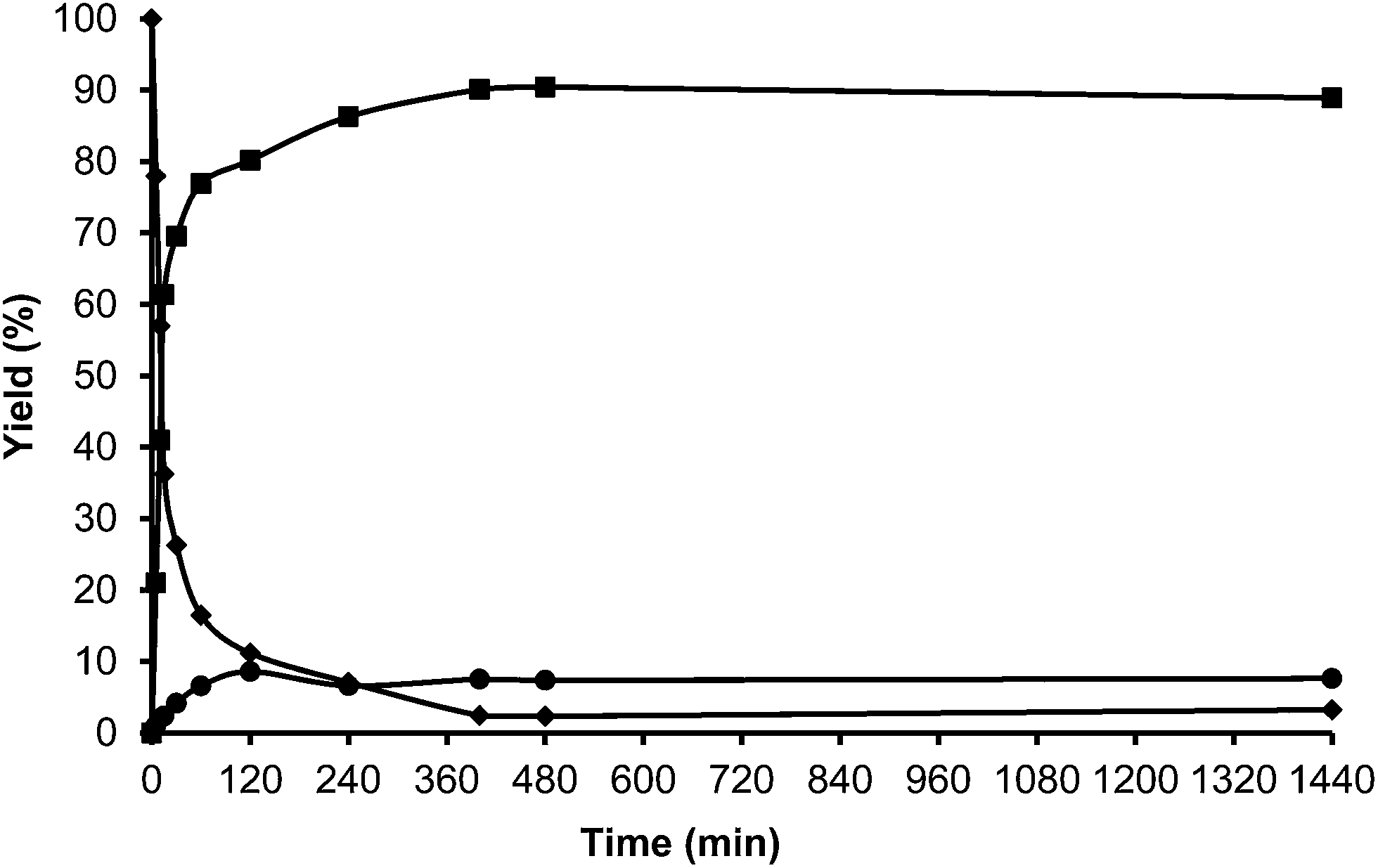 | ||
| Fig. 17 Kinetic curves of alkylation of a heavy reformate mixture (5 ml: 1,2,3-trimethylbenzenes (54 v/v%), 3-ethyl-toluene (33.4 v/v%), n-propylbenzene (6 v/v%) and o-xylene (6.6 v/v%)) with HMF (0.5 mmol) in the presence of ITQ-2 (25 wt% HMF) at 150 °C and nitrogen atmosphere: HMF (♦), 5-alkylated products: ortho, meta, para isomers (■) and OBMF (●). | ||
2.2 Hydrodeoxygenation of 5-(o-, m- and p-methyl)benzylfuran-2-carbaldehyde
Finally we have studied the possibility of transforming 5-(o-, m- and p-methyl)benzylfuran-2-carbaldehyde, an oxygenated alkylated compound derived from biomass, into a mixture of hydrocarbons and hydrophobic molecules that can be blended with kerosene or diesel. Thus, the hydrodeoxygenation of the alkylated compound was performed in a fixed bed continuous reactor introducing the feed without any solvent after pretreatment at 65 °C. We have selected a physical mixture of platinum supported on carbon and on titanium oxide as the catalyst to carry out the hydrodeoxygenation process. It has been proposed that platinum metal is a good candidate for hydrodeoxygenation and in the presence of titania the carbonyl groups are activated favoring the hydrogenation by platinum.23 Thus, a mixture of Pt/C–Pt/TiO2 was prepared as the catalyst and the reaction was performed at a hydrogen pressure of 40 bar and 350 °C, with a flow rate of 450 ml min−1 of hydrogen. After 1.67 h time on stream, the liquids were separated into an aqueous phase and a transparent light yellow organic phase. GC and GC-MS analysis of the organic phase was used to identify the main products (Table 9). As can be seen the highest selectivity was to saturated hydrocarbons (77.49%), followed by mono-aromatic hydrocarbons (16.82%), di-aromatic and triaromatic hydrocarbons (<5%) and polar compounds (0.21%). Thus, the main product obtained, 1-hexyl-4-methylcyclohexane is the C13 alkanes generated by hydrogenation of the unsaturated double C–C bond, ring opening and complete hydrogenolysis of all carbon–oxygen bonds as can be seen in Scheme 3. The second most abundant, a C12 alkane molecule (see Scheme 3), results from a decarbonylation reaction. A very low amount (2.61%) of hydrocarbon products with >C13 atoms were also detected indicating that carbon–carbon bond formation occurred, though at little extension, under our reaction conditions.| Organic compounds | Percentages (wt%) | |
|---|---|---|
| Saturated | 77.49 | |
| Naphthenes C13 | 37.58 | |
| Naphthenes C12 | 0.76 | |
| Polynaphthenes | 10.44 | |
| n-Hexane | 2.48 | |
| Naphthene C7 | 13.92 | |
| Others saturates (C9H18, C11H22, C13H28, >C14) | 12.30 | |
| Mono-aromatics | 16.82 | |
| Toluene | 6.31 | |
| Mono-aromatics C13 | 8.83 | |
| Mono-aromatics C12 | 0.26 | |
| Others aromatics (C8–C11, C14–C18) | 2.61 | |
| Di-aromatics | 2.70 | |
| Tri-aromatics | 1.59 | |
| Polars | 0.21 | |
| 100.00 | ||
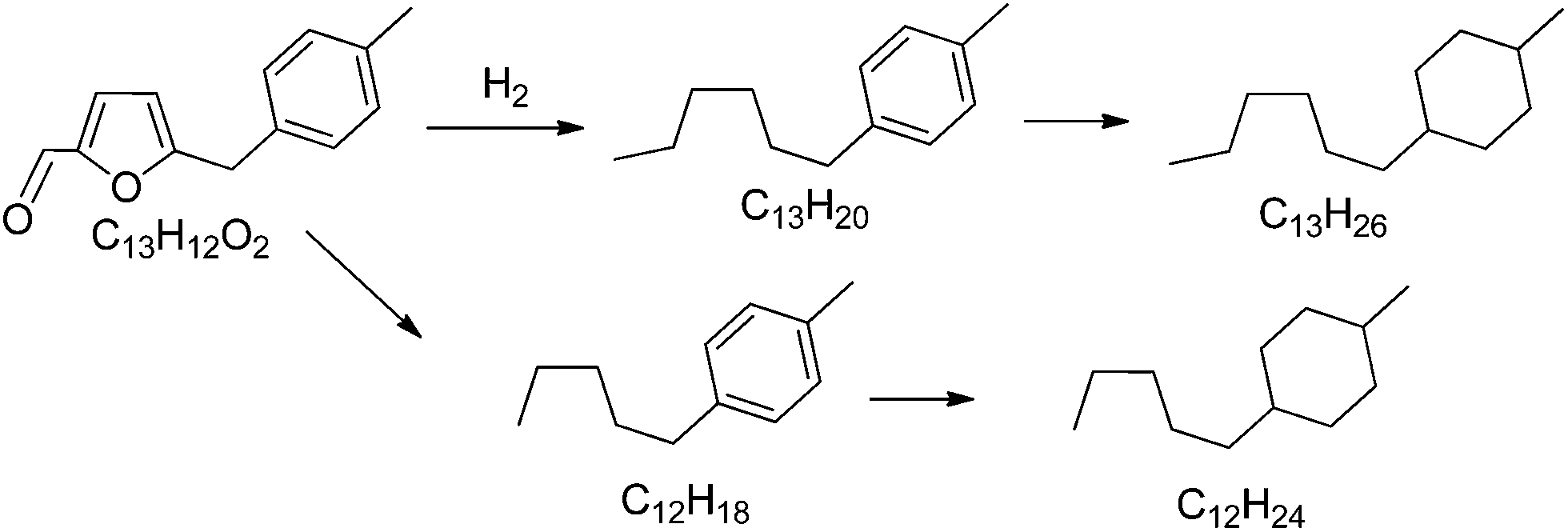 | ||
| Scheme 3 Main detected products after hydrodeoxygenation of 5-(o-, m- and p-methyl)benzylfuran-2-carbaldehyde in the presence of Pt/C and Pt/TiO2 catalysts. | ||
The gas phase products (4.1% yield), included C1 to C6 alkanes, traces of propylene, CO and CO2.
The simulated distillation of the organic phase (see Fig. 18) showed that most of the products are in the kerosene range (200–300 °C) (Table 10). One can expect that a kerosene, like the one produced here containing a large amount of alkylcyclohexane products, with high energy density and low smoke point, should be a high quality kerosene.
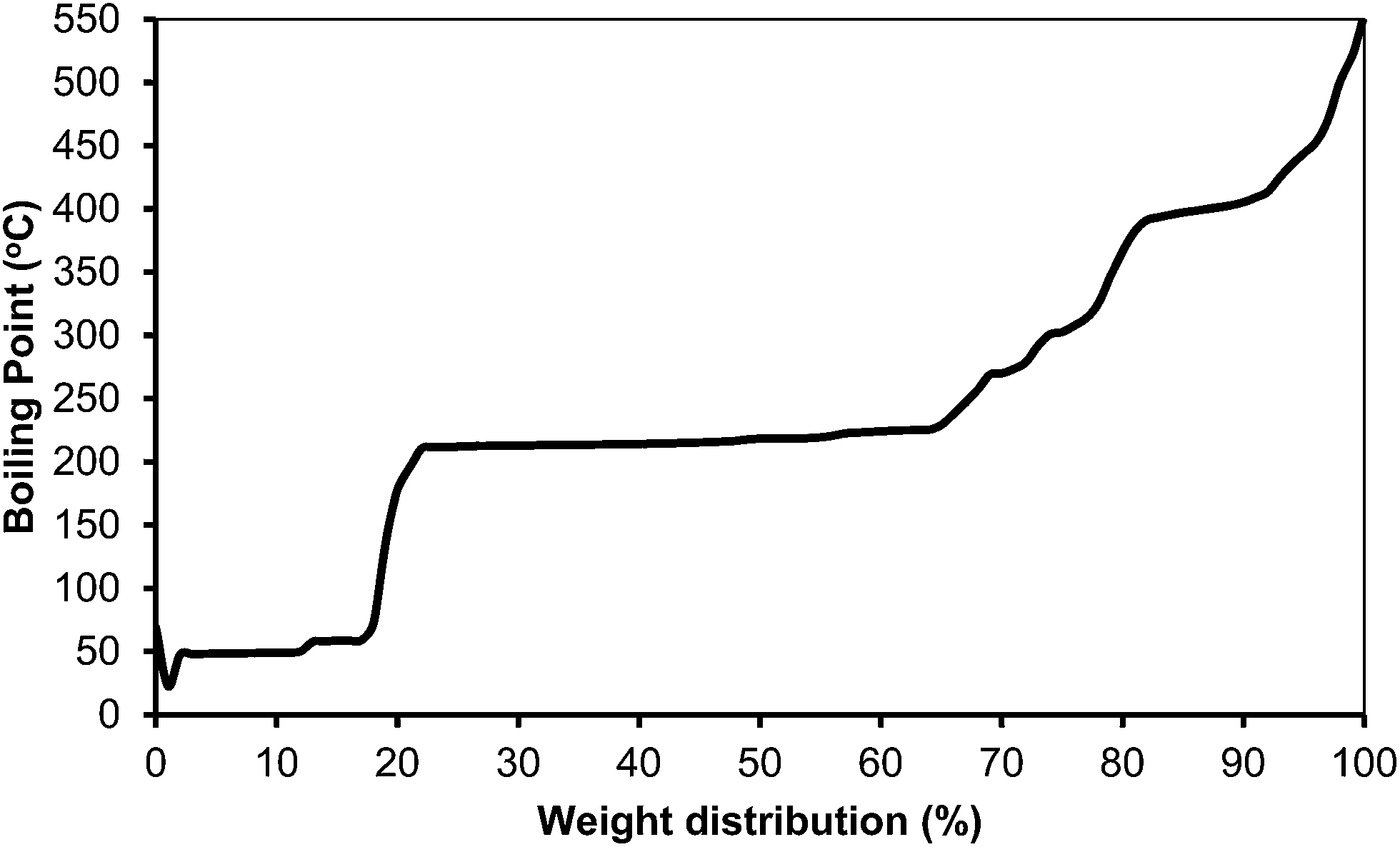 | ||
| Fig. 18 Simulated distillation for the product mixture obtained when 5-(o-, m- and p-methyl)benzylfuran-2-carbaldehyde was passed over Pt/C and Pt/TiO2 in a fixed bed reactor on the basis of a kerosene boiling point range from 200 to 300 °C. | ||
| T (°C) | Fraction | Percentage (wt%) |
|---|---|---|
| 150.8 | Light gasoline | 19.25 |
| 216.1 | Heavy gasoline | 27.84 |
| 359 | Kerosene | 32.54 |
| 482 | HCO (gasoil) | 17.78 |
| 1000 | Slurry | 2.63 |
| Catalyst | BET (m2 g−1) | Pore vola. (cm3 g−1) | 150 °C | 250 °C | 350 °C | |||
|---|---|---|---|---|---|---|---|---|
| Brønsted | Lewis | Brønsted | Lewis | Brønsted | Lewis | |||
| a Acidity μmol pyridine per gram catalyst at different temperatures, calculated using extinction coefficients given in ref. 46. | ||||||||
| HBeta (12.5) | 602 | 0.36 | 65 | 69 | 58 | 56 | 25 | 29 |
| USY-720 (12.2) | 780 | 0.49 | 79 | 16 | 66 | 13 | 41 | 10 |
| ITQ-2 (15) | 643 | 0.53 | 57 | 27 | 37 | 18 | 16 | 13 |
| MCM-41 (15) | 1100 | 0.94 | 19 | 62 | 5 | 46 | 4 | 34 |
| Mord. (10) | 550 | 0.40 | 67 | 25 | 54 | 25 | 29 | 28 |
With respect to the gasoline obtained, which corresponds to 19 wt%, it has an excellent Research (RON) and Octane number (MON) of 93 and 88 respectively according to the PIONA analysis.44 We have performed a detailed analysis of the products boiling in the gasoline range (up to 200 °C) (Fig. S2†), and the main products are given in Scheme 4, where we can see there that the most of the products up to C10 (200 °C B.P.) are alkyl cycloalkanes coming from cracking-hydrogenation of the products formed by alkylation of toluene with HMF. The cracking will probably occur on some acid sites of the catalyst support (titania and carbon) though the occurrence of some additional hydrogenolysis on the metal cannot be neglected. Note that toluene is also formed supporting the idea that some cracking takes place during the hydrogenation (Scheme 4).
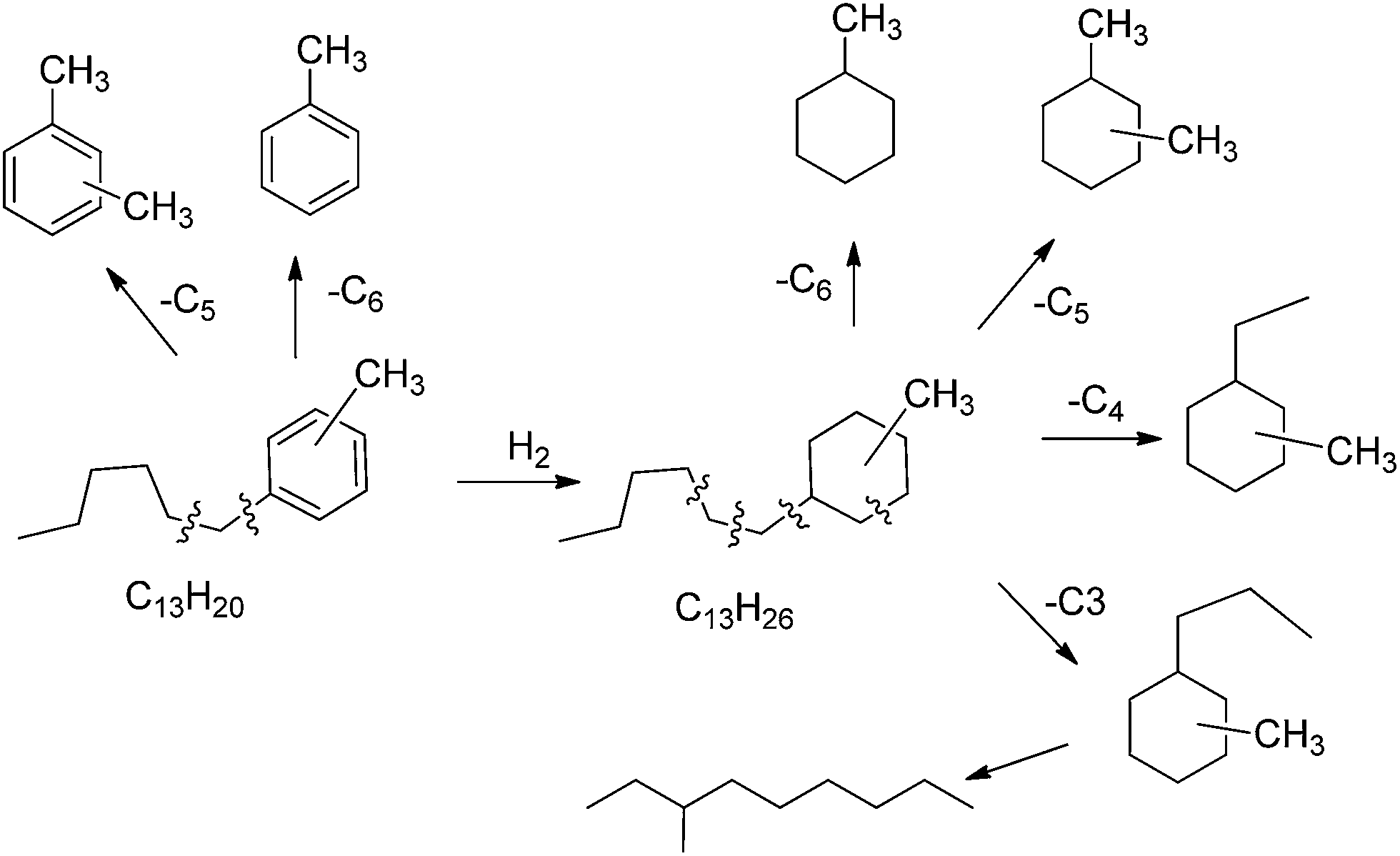 | ||
| Scheme 4 Main detected products in the range C6–C10 after hydrodeoxygenation of 5-(o-, m- and p-methyl)benzylfuran-2-carbaldehyde in the presence of Pt/C and Pt/TiO2 catalysts. | ||
3. Conclusions
We have showed that is possible to alkylate aromatics with HMF with very good activity and selectivities using large pore 3D and 2D zeolites. The 2D delaminated zeolite ITQ-2 is an excellent shape selective catalyst for alkylation of aromatics with HMF that can be subjected to multiple reaction–regeneration cycles. This catalyst can be used to alkylate heavy reformate fractions with HMF with very good yields to alkylated products, which after hydrodeoxygenation yield excellent kerosene. The process shown here allows reacting feeds rich in aromatics coming from refineries or produced from lignin, with HMF to yield high quality kerosene.4. Experimental part
4.1 Materials and catalyst
5-Hydroxymethyl-2-furfural (99%), p-methoxybenzene, toluene, o-dimethylbenzene, mesitylene, nonane, and p-toluenesulfonic acid were acquired from Sigma-Aldrich and all the solvents were used without drying.HBeta (CP811) (Si/Al = 12.5), Mordenite (CBV 20A) (Si/Al = 10), and USY (CBV 500, CBV 712, CBV 720, and CBV 760) (Si/Al = 2.7, 5.7, 12.2, and 27 respectively) zeolites were purchased from PQ Zeolites B. V. and were calcined before use at 580 °C for 3 h. The following catalysts were prepared according to the literature: MCM-41 (Si/Al = 15).45 The 2D layered ITQ-2 (Si/Al = 15) catalyst was prepared by expansion and subsequent exfoliation of the corresponding laminar precursors of the MWW structure by following ref. 39.
IR spectra were obtained on a Nicolet 750 FTIR spectrophotometer, using wafers of 10 mg cm2 treated under vacuum (104 to 105 Pa) at 400 °C overnight. After equilibration, the samples were degassed for 1 h at increasing temperatures (150, 250, and 350 °C). After each desorption step, the spectrum was recorded at room temperature and the background was subtracted.
Surface area measurements were carried out with a Micrometrics ASAP 2000 apparatus following the BET procedure by means of nitrogen and argon adsorption at 77 and 85 K, respectively. Thermogravimetric analyses (TGA) were performed with a Netzsch STA 409 EP thermal analyzer with about 20 mg of sample and a heating rate of 10 °C min−1 in air flow. The physicochemical characteristics of the different samples studied are presented in Table 1.
The metal catalysts (Pt/C and Pt/TiO2) were prepared by the incipient wetness impregnation procedure. The desired amount of metal precursor (hexachloroplatinic acid) was dissolved in water. The carbon and metal oxide support were impregnated as pellets (particle size of 0.425 to 0.850 mm) and dried at 100 °C overnight.
4.2 Alkylation reactions
5-(o-, m and p-Methyl)benzylfuran-2-carbaldehyde. 1H NMR (CDCl3): δ 9.53 (s, 2H), 7.11–7.32 (m, 8H), 6.16 (d, J = 2.8 Hz, 1H), 6.12 (d, J = 2.5 Hz, 1H), 4.08 (s, 1H), 4.02 (s, 1H), 2.34 (s, 3H), 2.28 (s, 3H); 13C NMR (CDCl3): δ 177.7, 177.2, 162.4, 161.8, 157.2, 152.17, 148.9, 136.7, 134.4, 130.6, 129.8, 129.6, 129.5, 128.8, 128.7, 128.5, 127.8, 127.4, 126.4, 125.9, 123.1, 121.8, 111.8, 109.9, 109.7, 109.6, 34.8, 34.5, 32.7, 21.3, 21, 19.4. GC-MS: m/z 200, 185, 171, 128, 115, 91, 77.
5-(p-Methyl)benzylfuran-2-carbaldehyde. 1H NMR (CDCl3): δ 9.55 (s, 1H), 7.11–7.18 (m, 5H), 6.19 (d, J = 2.8 Hz, 1H), 6.18 (d, J = 2.5 Hz, 1H), 4.04 (s, 2H), 2.35 (s, 3H), 2.28 (s, 3H); 13C NMR (CDCl3): δ 177.2, 162.4, 152.2, 136.7, 133, 129.5, 128.8, 123, 109.6, 34.5, 21. GC-MS: m/z 200, 185, 171, 128, 115, 91, 77.
5-(2,3-and 3,5-Dimethyl)benzylfuran-2-carbaldehyde. 1H NMR (300 MHZ, CDCl3): δ 9.44 (s, 2H), 6.85–7.11 (m, 6H), 6.10 (d, J = 2.8 Hz, 1H), 5.95 (d, J = 2.5 Hz, 1H), 3.99 (s, 1H), 3.9 (s, 1H), 2.21 (s, 3H), 2.16 (s, 3H), 2.09 (3H); 13C NMR (75 MHz, CDCl3) δ 177.24, 162.58, 162.27, 152.15, 137.37, 136.99, 135.34, 135.08, 134.27, 133.48, 130.16, 129.99, 129.15, 127.87, 126.24, 125.79, 123.15, 109.73, 109.62, 34.46, 33.35, 20.65, 19.72, 19.35, 15.34. GC-MS: m/z 214, 199, 185, 142, 115.
5-(o and p-Methoxy)benzylfuran-2-carbaldehyde. 1H NMR (300 MHz, CDCl3) δ 9.44 (s, 2H), 7.16–6.74 (m, 8H), 6.08 (d, J = 2.7 Hz), 6.04 (d, J = 2.6 Hz), 3.71 (s, 1H), 3.69 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 177.15, 158.23, 134.59, 130.45, 130.20, 129.94, 129.66, 129.44, 128.53, 128.13, 124.67, 120.69, 120.65, 114.21, 113.91, 113.82, 113.72, 113.63, 110.64, 109.56, 108.71, 106.44, 55.40, 55.29, 55.23, 55.13, 49.29, 34.05, 33.69, 29.20. GC-MS: m/z 216, 187, 144, 115.
5-(Mesityl)benzylfuran-2-carbaldehyde. 1H NMR (300 MHz, CDCl3) δ 9.45 (s, 1H), 7.04–7.03 (d, J = 3 Hz, 1H), 6.82 (s, 2H), 5.84–5.83 (d, J = 3 Hz, 1H), 3.97 (s, 2H), 2.21 (s, 3H), 2.19 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 177.07, 161.94, 152.11, 136.82, 136.65, 129.68, 129.13, 109.09, 28.65, 20.88, 19.93. GC-MS: m/z 228, 182, 143, 91.
5,5-Oxy(bis-methylene)-2-furaldehyde. 1H NMR (300 MHz, CDCl3): δ 9.56 (s, 2H), 7.15–7.14 (d, J = 3 Hz, 1H), 6.50–6.49 (d, J = 3 Hz, 1H), 4.56 (s, 4H). 13C NMR (75 MHz, CDCl3) δ 177.7, 157.21, 152.81, 121.77, 111.83, 64.63. GC-MS: m/z 234, 206, 125, 109, 81.
4.3 Hydrodeoxygenation process
A physical mixture of 5.34 g of 3 wt% Pt on active carbon and 1.34 g of 3 wt% Pt on TiO2, was placed in the stainless steel tubular reactor with an inner diameter of 7.7 mm and a length of 38 cm. Before the reaction the metal catalyst was activated (reduced) applying a hydrogen pressure of 40 bar and a hydrogen flow of 450 ml min−1 at 400 °C during 1 h.The reactor pressure was controlled with a Swagelok back-pressure regulator. Hydrogen gas flow to the reactor was controlled using a Bronkhorst EL-FLOW select mass flow controller. A mixture of 5-(o-, m- and p-methyl)benzylfuran-2-carbaldehyde (10.09 g) was pumped into the reactor with a Gilson 305 pump equipped with a HPLC pump head and mixed with the hydrogen flow before entering the reactor and a rate of 0.1 ml min−1 with the same hydrogen flow and pressure as during the activation step. The reaction temperature was 65 °C at the entry of the reactor with a gradient to the middle of the reactor of 120 °C and 350 °C in the second half of the reactor.
Two liquid phases (aqueous and organic) were collected in two tanks at room temperature at the exit of the reactor with a total weight of 8.8 g, the aqueous phase 15% and the organic phase 85%.
Gaseous samples were taken after the back-pressure regulation (4%) and analyzed by GC. The mass balance was 90.6%.
4.4 Simulated distillation
The organic phase was analyzed by simulated distillation using the ASTM D2887 method in order to study the boiling range distribution of different product samples at different temperatures using a BRUKER 450 gas chromatograph. A capillary column CP SimDis (10 m × 0.53 mm diameter × 2.65 μm) was used to separate hydrocarbons present in different product samples based on the order of their boiling range. To identify different boiling fractions, a calibration curve was used which was obtained by chromatographic analysis of a mixture of known boiling point compounds. A flame ionization detector (FID) was used to detect different boiling ranges using helium as a carrier gas at a flow rate of 30 ml min−1. The hydrogen and air flow were maintained at 35 and 400 ml min−1, respectively. The detector temperature and final oven temperature were maintained at 375 and 380 °C, respectively.Blobs (peaks) were classified as C9 to C31 saturated (linear, naphthenes, polynaphthenes), as low weight hydrocarbons (C4 to C8), monoaromatics, diaromatics, triaromatics and polar compounds.
Gas samples from hydrodecarboxylation were analyzed with a Varian 3800 GC equipped with three detectors.
Two detectors used were thermal conductivity detectors (TCD) for the analysis of H2 (2.5 m molecular sieves 13X column). The third detector was a flame ionization detector (FID) for the analysis of C1 to C6 hydrocarbons separated on a 50 m Plot/Al2O3 column.
Acknowledgements
Financial support by Consolider-Ingenio 2010 (project MULTICAT), Spanish MICINN Project CTQ-2011-27550), Generalitat Valenciana (Prometeo program) and Program Severo Ochoa are gratefully acknowledged. This work was supported by Consolider. KSA is grateful to ITQ for a doctoral grant.References
- (a) G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed; (b) J. Holmgren, C. Gosling, R. Mariangeli, T. Marker, G. Faraci and C. Perego, Hydrocarbon Proc., 2007, 86, 67 CAS.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516 RSC.
- Y. T. Cheng, J. Jae, S. Jian, F. Wei and G. W. Huber, Angew. Chem., Int. Ed., 2012, 51, 1387 CrossRef CAS PubMed.
- P. Gullon, A. Romani, C. Vila, G. Garrote and J. C. Parajo, Biofuels, Bioprod. Biorefin., 2012, 6(2), 219 CrossRef CAS.
- P. Dominguez de Maria, J. Chem. Technol. Biotechnol., 2014, 89, 11 CrossRef CAS.
- (a) T. Werpy and G. R. Petersen, Top Value Added Chemicals from Biomass. Volume I. Results of Screening for Potential Candidates from Sugars and Synthesis Gas, U.S.D. Energy, 2004 Search PubMed; (b) J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- (a) M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520 RSC; (b) A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754 RSC.
- B. F. M. Kuster and L. M. Tebbens, Carbohydr. Res., 1977, 54, 159 CAS.
- (a) X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1 CrossRef CAS PubMed; (b) R. J. van Putten, J. C. van der Waaal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499 CrossRef CAS PubMed; (c) S. P. Teong, G. Yi and Y. Zhang, Green Chem., 2014, 16, 2015 RSC.
- Y. Nakamura and S. Morikawa, Bull. Chem. Soc. Jpn., 1980, 53, 3705 CrossRef CAS.
- (a) Y. Roman-Leshkow, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933 CrossRef PubMed; (b) B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24 RSC.
- S. Frenzel, S. Peters, T. Rose and M. Kunz, Industrial Sucrose, in Sustainable Solutions for Modern Economies, ed. R. Höfer, RSC Green Chemistry, No 4, RSC Publ., Cambridge, 2009, pp. 264–299 Search PubMed.
- Biochem AVA (2014) First Industrial Production for Renewable 5-HMF, http://www.ava-biochem.com/media/downloads-EN/press-releases/First-Industrial-Production-For-Renewable-5-HMF.pdf, accessed April 16, 2014.
- J. Vyskocil and A. Kruse, DE102011053034, 2011.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS PubMed.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110 CrossRef CAS PubMed.
- A. Corma, M. Renz and C. Schaverien, ChemSusChem, 2008, 1, 739 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184 CrossRef CAS PubMed.
- A. Pulido, B. Oliver-Tomas, M. Renz, M. Boronat and A. Corma, ChemSusChem, 2013, 6, 141 CrossRef CAS PubMed.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gartner and J. A. Dumesic, Science, 2008, 322, 417 CrossRef CAS PubMed.
- A. Corma, O. de la Torre, M. Renz and N. Villandier, Angew. Chem., Int. Ed., 2011, 50, 2375 CrossRef CAS PubMed.
- A. Corma, O. de la Torre and M. Renz, ChemSusChem, 2011, 4, 1574 CrossRef CAS PubMed.
- A. Corma, O. de la Torre and M. Renz, Energy Environ. Sci., 2012, 5, 6328 CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446 CrossRef CAS PubMed.
- C. J. Barrett, J. N. Chheda, G. W. Huber and J. A. Dumesic, Appl. Catal., B, 2006, 66, 111 CrossRef CAS PubMed.
- J. Dumesic, G. W. Huber, N. J. Chheda and C. J. Barret, WO Pat. 2007103858, 2007.
- I. Iovel, K. Mertins, J. Kischel, A. Zapf and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 3913 CrossRef CAS PubMed.
- X. Zhou and T. Rauchfuss, ChemSusChem, 2013, 6, 383 CrossRef CAS PubMed.
- A. Onorato, C. Pavlik, M. A. Invernale, I. D. Berghorn, G. A. Sotzing, M. D. Morton and M. B. Smith, Carbohydr. Res., 2011, 346, 1662 CrossRef CAS PubMed.
- (a) A. M. F. Bidart, A. P. S. Borges, L. Nogueira, E. R. Lachter and C. J. A. Mota, Catal. Lett., 2001, 75, 155 CrossRef CAS; (b) K. Okumura, K. Nishigaki and M. Niwa, Microporous Mesoporous Mater., 2001, 44–45, 509 CrossRef CAS; (c) P. Kalita, N. M. Gupta and R. Kumar, J. Catal., 2007, 245, 338 CrossRef CAS PubMed; (d) Y. Sun and R. Prins, Appl. Catal., A, 2008, 336, 11 CrossRef CAS PubMed.
- (a) M. J. Climent, A. Corma, H. Garcia and J. Primo, Appl. Catal, 1989, 51, 113 CrossRef CAS; (b) S. Al-Khattaf, M. A. Ali and J. Cejka, Recent Development in Transformation of Aromatic Hydrocarbons over Zeolites, in Zeolites and Catalysis, ed. J. Cejka, A. Corma and S. Zones, Wiley-VCH, Weinheim, 2010, vol. 2, pp. 623–648 Search PubMed; (c) D. Verboekend and J. Perez-Ramirez, Catal. Sci. Technol., 2011, 1, 879 RSC; (d) R. Gounder and E. Iglesia, Chem. Commun., 2013, 49, 3491 RSC.
- T. Yamato, C. Hidezhima, G. K. S. Prakash and G. Olah, J. Org. Chem., 1991, 56, 2089 CrossRef CAS.
- (a) A. Corma, C. Zicovich-Wilson and P. Viruela, J. Phys. Org. Chem., 1994, 7, 364 CrossRef CAS; (b) S. Van der Beken, E. Dejaegere, K. A. Tehrani, J. S. Paul, P. A. Jacobs, G. V. Baron and J. F. M Denayer, J. Catal., 2005, 235, 128 CrossRef CAS PubMed.
- L. A. Pine, P. J. Maher and W. A. Wacher, J. Catal., 1984, 85, 466 CrossRef CAS.
- D. Verboekend, G. Vile and J. Perez-Ramirez, Adv. Funct. Mater., 2012, 22, 916 CrossRef CAS.
- J. Klinowski, J. M. Thomas, C. A. Fyfe and G. C. Gobbi, Nature, 1982, 296, 533 CrossRef CAS.
- M. E. Leonowicz, J. A. Lawton, S. L. Lawton and M. K. Rubin, Science, 1994, 264, 1910 CAS.
- (a) F. Cavani, V. Arrigoni, R. Ghezzi and G. Bellusi, CA Pat. 2030067, 1991; (b) A. Corma, V. Martínez-Soria and E. Schnoeveld, J. Catal., 2000, 192, 163 CrossRef CAS.
- A. Corma, V. Fornes, S. B. Pergher, T. Maesen and J. G. Buglass, Nature, 1998, 396, 353 CrossRef CAS PubMed.
- S. Zanardi, A. Alberti, G. Cruciani, A. Corma, V. Fornes and M. Brunelli, Angew. Chem., Int. Ed., 2004, 43, 4933 CrossRef CAS PubMed.
- A. Corma, U. Diaz, T. García, G. Sastre and A. Velty, J. Am. Chem. Soc., 2010, 132, 15011 CrossRef CAS PubMed.
- U. Diaz, V. Fornes and A. Corma, Microporous Mesoporous Mater., 2006, 90, 73 CrossRef CAS PubMed.
- J. Aguilar, S. B. C. Pergher, C. Detoni, A. Corma, F. V. Melo and E. Sastre, Catal. Today, 2008, 133–135, 667 CrossRef CAS PubMed.
- H. J Lugo, G. Ragone and J. Zambrano, Ind. Eng. Chem. Res., 1999, 38, 2171 CrossRef.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartulli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347 CrossRef CAS.
- D. W. Breck and E. Flanigen, Molecular Sieves, Society of Chemical Industry, 1968, p. 47 Search PubMed.
- H. Fichtner-Schmittler, U. Lohse, G. Engelhardt and V. Patzelova, Cryst. Res. Technol., 1984, 19, K1 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ee03194f |
| This journal is © The Royal Society of Chemistry 2015 |