
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supercapacitor electrode materials: nanostructures from 0 to 3 dimensions
Zenan
Yu
 ,
Laurene
Tetard
,
Lei
Zhai
and
Jayan
Thomas
*
,
Laurene
Tetard
,
Lei
Zhai
and
Jayan
Thomas
*
NanoScience Technology Center (NSTC), Department of Materials Science and Engineering, Department of Physics, Department of Chemistry, College of Optics and Photons, University of Central Florida, Orlando, FL 32826, USA. E-mail: Jayan.Thomas@ucf.edu; Fax: +1 407 882 2819; Tel: +1 407 882 0196
First published on 3rd December 2014
Abstract
Supercapacitors have drawn considerable attention in recent years due to their high specific power, long cycle life, and ability to bridge the power/energy gap between conventional capacitors and batteries/fuel cells. Nanostructured electrode materials have demonstrated superior electrochemical properties in producing high-performance supercapacitors. In this review article, we describe the recent progress and advances in designing nanostructured supercapacitor electrode materials based on various dimensions ranging from zero to three. We highlight the effect of nanostructures on the properties of supercapacitors including specific capacitance, rate capability and cycle stability, which may serve as a guideline for the next generation of supercapacitor electrode design.
 Zenan Yu | Zenan Yu is a Ph.D. student in the Department of Materials Science and Engineering at University of Central Florida. He received his B.S. degree in Electronic Science and Technology from Shanghai University in 2011. Under the supervision of Professor Jayan Thomas, his research interests mainly focus on the design, synthesis and applications of nanostructured materials for supercapacitors. |
 Laurene Tetard | Laurene Tetard is an assistant professor at the NanoScience Technology Center (NSTC) and Department of Physics. She received her B.S. and M.S. from the University of Burgundy in France and completed her Ph.D. with Professor Thundat at the University of Tennessee, Knoxville in 2010. She was a Eugene P. Wigner Fellow at the Oak Ridge National Laboratory from 2011 to 2013 before joined the University of Central Florida in 2013. Her current research interest includes innovative nanoscale characterization of energy-related materials including two dimensional materials and soft matter. |
 Lei Zhai | Lei Zhai is an associate professor at the NanoScience Technology Center (NSTC) and Department of Chemistry. He received his Ph.D. from Carnegie Mellon University in 2002 and worked as a postdoctoral research associated at Massachusetts Institute of Technology before he joined the University of Central Florida in 2005. He is a recipient of NSF CAREER Award and a Scialog Fellow of Research Corporation for Science Advancement. His research focuses on conjugated polymers and composites for energy conversion and storage, surface science and engineering, and polymer derived ceramics. |
 Jayan Thomas | Jayan Thomas is an assistant professor at the NanoScience Technology Center (NSTC), College of Optics and Photonics (CREOL) and College of Engineering and Computer Science at the University of Central Florida (UCF). After receiving Ph.D. from Cochin University of Science and Technology in India, he joined College of Optical Sciences, University of Arizona in 2001 as a research faculty. He moved to UCF in 2011 and is currently working on the development of supercapacitors, photorefractive polymers and solar cells. He is a recipient of NSF CAREER award, VEECO's 2010 best nanotechnology innovation award and WTN World Technology (Energy) Award-2014 sponsored by FORTUNE and TIME. |
Broader contextThe ever-growing global demand of energy together with the depletion of fossil fuels makes it critical to develop sustainable and renewable energy resources. Developing relevant energy storage systems (e.g. batteries and supercapacitors) is essential to utilizing sustainable and renewable energy resources. A supercapacitor is highly beneficial in storing renewable energy. For example, when light is not shining or wind is not blowing, the energy needs to be stored in devices like batteries and supercapacitors. Among the efforts of building efficient supercapacitors, electrode materials with rational nanostructured designs have offered major improvements in performance over the past several years. This review article is intended to examine recent progress in nanostructuring supercapacitor electrode materials, highlighting the fundamental understanding of the relationship between structural properties and electrochemical performances, as well as an outlook on the next generation of nanostructured supercapacitor electrodes design. |
1 Introduction
Before a ray of sun light peeked through the curtain and shined on John's face, everything was perfect for a quiet morning. Suddenly jumping off the bed, “Oh, my, I forgot to set the alarm last night.” He got dressed and rushed to his electric car; “LOW BATTERY” exclaimed John, “wish I could charge it up in a few minutes…” As a matter of fact, such energy storage technologies like supercapacitors which can charge very quickly have been around for decades, but their low energy densities have held them back in many such applications. The ever growing demand for energy urges researchers to look for new materials and processes to develop highly improved energy storage devices. Among energy storage systems, supercapacitors have drawn considerable attention during recent years due to their fast charge–discharge characteristics, high power density, and excellent cycle stability. Currently, supercapacitors are used along with batteries to provide the additional power required in many applications. However, they cannot be used as stand-alone units in these applications since their energy density is inferior to that of batteries. For example, commercially available supercapacitors can provide energy densities of less than 10 W h Kg−1 whereas Li-ion batteries can provide energy densities of more than 180 W h Kg−1.1,2 Nevertheless, the power density and cycle life of supercapacitors are far superior to those of Li-ion batteries. At present, the scientific community is striving to considerably improve the energy and power density of supercapacitors by developing new electrode materials and electrolytes, and ingenious device design.Achieving noteworthy improvements in supercapacitor performances by means of better electrode materials together with low-cost and environment-friendly production, requires a deep fundamental understanding of the charge storage mechanisms, transport pathways of electrons and ions, and electrochemically active sites. Advances in nanomaterials, ideal candidates for improved ion adsorption or faster surface redox reactions, have fostered extensive research efforts toward the enhancement of electrochemically active sites for charge transfer and controlled ionic and electronic transport at small diffusion length scales.
Supercapacitors are generally classified into two types based on their storage mechanism, the electrochemical double layer capacitors (EDLCs) and pseudocapacitors. EDLCs physically store charges via reversible ion adsorption at the electrode–electrolyte interface, while pseudocapacitors chemically store their charges via redox reaction at the vicinity (a few nanometers) of the surface. Typically, electrode materials for EDLCs are made of carbon-based materials (including activated carbon, carbon nanotubes, graphene, etc.), while that for pseudocapacitors are transition metal oxides (e.g. RuO2, MnO2, CoOx, NiO, Fe2O3, etc.) and conducting polymers (e.g. polypyrrole, polyaniline, poly(3,4-ethylenedioxythiophene), etc.).
The criteria for designing a high-performance supercapacitor electrode include high specific capacitance (energy stored per unit mass, volume, or area of active materials), large rate capability (capacitance retention at high scan rate or current density), and high cycle stability. In addition, the toxicity and cost of the active materials used in an electrode design should be taken into account as well. Therefore, in order to realize a high-performance supercapacitor electrode, factors that determine the specific capacitance, rate capability, and cycle stability need to be mentioned here. (1) Surface area: Since charges are stored on the surface of the supercapacitor electrodes, an electrode with a higher surface area leads to an improved specific capacitance. Nanostructuring of electrode materials is a feasible method to considerably improve the surface area of the electrodes. (2) Electronic and ionic conductivity: As specific capacitance and rate capability are considerably dependent on both electronic and ionic conductivity, a high electronic and ionic conductivity will help to maintain the rectangular nature of cyclic voltammetry (CV) curve and symmetricity of galvanostatic charging–discharging (GCD) curves. They also reduce the specific capacitance losses as scan rates/current densities are increased. Typical approaches to enhance the electronic conductivity include binder-free electrode design and nanostructured current collector design to provide efficient electron pathways for charge transport. To increase the ionic conductivity, precise control of pore size and prudent design strategies are used (e.g. more open structures for ion transport). (3) Mechanical and chemical stability: The cycle stability is greatly influenced by the mechanical and chemical stability of electrode materials during cycling. Phase change, dissolution, and side reaction of active materials are the major reasons for causing cycle instability. Sensible electrode surface protection may substantially boost the cycle stability. In addition to investigating new electrode materials, electrode design with nanostructures offers a promising avenue to fulfill high-performance supercapacitors. This is because the aforementioned factors can be effectively manipulated by a nanostructured design, which will be discussed in detail in the following sections.
Typically, nanomaterials can be classified into zero dimensional (0D), one dimensional (1D), two dimensional (2D) and three dimensional (3D) categories.3–5 Particles which are more or less spherical in shape like fullerenes, quantum dots, nano-onions, nanoparticles, etc. are considered as 0D. Nanostructures with 1D are interesting due to their dimensionality dependence on its functional properties. They include nanotubes, nanofibers, nanowires, nanopillars, nanoribbons and nanobelts. Typically, materials with a thickness of a few atomic layers and with the other two dimensions beyond the nanometric size range are considered as 2D materials. Graphene and many other layered van der Waals solids like MoS2, CaGe2 and CaSi2, fall under the 2D category. Materials with three dimensions beyond the nanometric size range but still preserve the advantages of nano size effect are regarded as 3D materials. Mesoporous carbon and graphene aerogel fall under 3D category.
Even though there are several reviews describing the material aspects of supercapacitor electrodes,2,6–9 a comprehensive review of 0D, 1D, 2D and 3D materials used only in supercapacitors is still lacking. Although Wu and co-authors have reported the dimensional study of electrodes for 0 to 3D,10 they only focused on carbon–metal oxide composite electrodes from 0 to 3D. This review covers a much broader range of electrode materials. In recent years, a range of materials configurations ranging from 0D particles to 3D nanostructured frameworks have been developed and tested in supercapacitors devices. This review is aiming to fill this gap and provide the readers a clear idea about the performance of these materials as electrodes in electrochemical supercapacitors. Fig. 1 summarized the key points of electrode materials from 0 to 3D and a detailed discussion is given in the following sections.
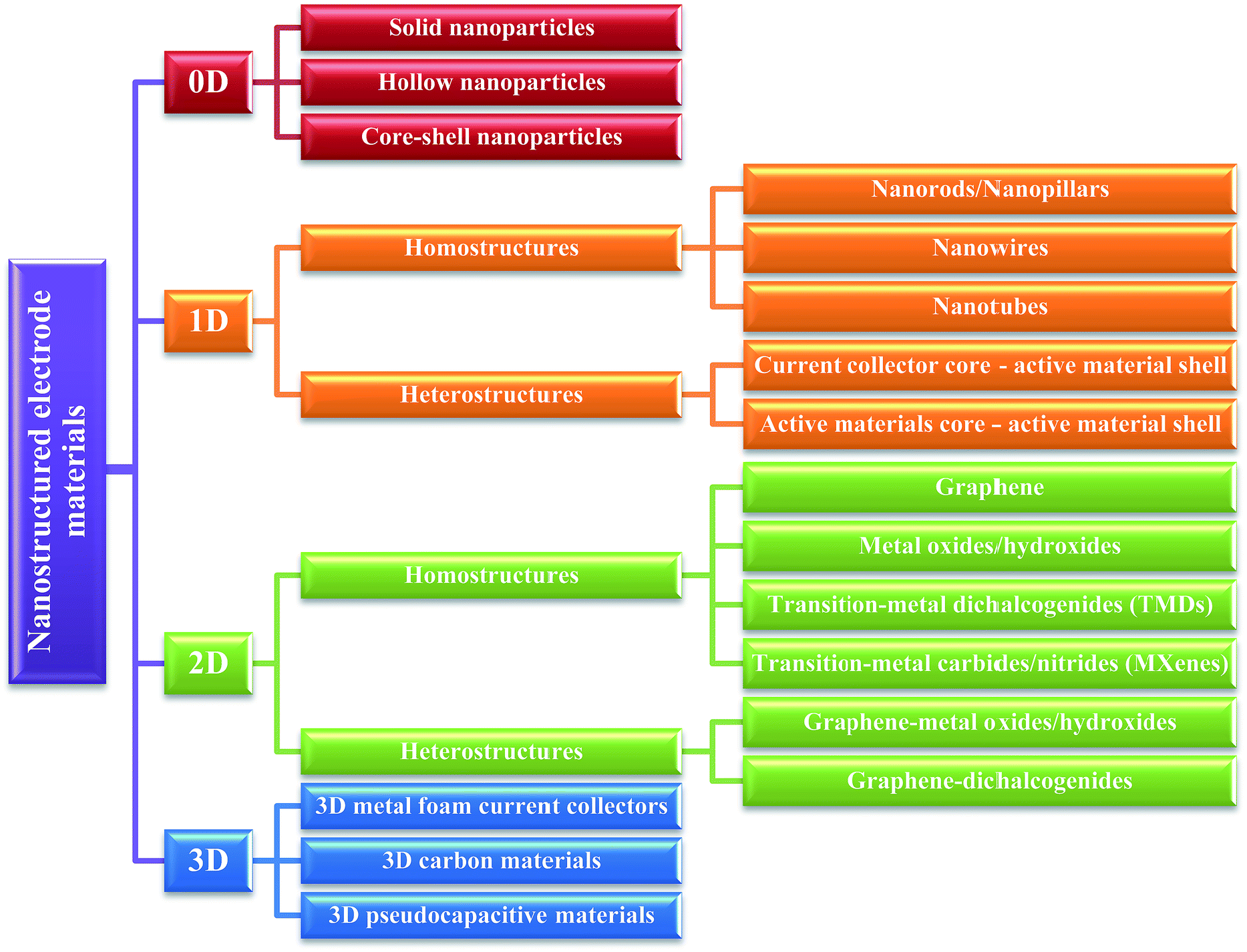 | ||
| Fig. 1 A schematic representation summarizing the key aspects of electrode materials from 0 to 3D. | ||
2 0D nanostructures
According to the strict definition, 0D nanostructures are spherical particles that have three dimensions constrained on the nanoscale (usually 1–100 nm). Nevertheless, particles with diameters up to 1 µm are usually regarded as nanoparticles as well, as the benefit provided by the effect of their small size has been observed for this size range. In this section, three subcategories of 0D nanostructures used as electrodes in supercapacitors are considered, viz., solid, hollow, and core–shell 0D nanostructures.2.1 Solid 0D nanostructures
As the foundation of 0D nanostructures, solid nanoparticles (nanospheres) are intensely investigated for designing supercapacitor electrodes. Carbon materials (e.g. activated carbon,11 carbon nanospheres,12 and mesoporous carbon13,14) and transition metal oxides (e.g. MnO2,15,16 NiO,17–19 Fe3O4,20etc.) have been widely fabricated into solid nanoparticles for electrode materials. Among carbon materials, activated carbons (ACs) are distinct due to their high surface area (up to 3000 m2 g−1) and economical fabrication process. In addition, ACs propose a wide range of pore size distribution including micropores (<2 nm), mesopores (2–50 nm), and macropores (>50 nm).21 ACs are generally produced by either physical or chemical activation of versatile carbonaceous precursors such as wood, coal, nutshell, and so forth. Larger surface area is usually expected to result in a higher specific capacitance. However, even with a surface area of up to 3000 m2 g−1, only small specific capacitance (<10 µF cm−2) could be obtained with ACs.1 This is due to the fact that specific capacitance is not only determined by surface area but also by other parameters such as pore size distribution, pore shape and structure, accessibility of the electrolyte, and electrical conductivity.6 Therefore, an ideal design of ACs should meet at least two requirements: (1) large surface area for the enhancement of electrochemically active sites and (2) suitable pore size and structure for the transportation of ions in the electrolyte. In addition to ACs, Gogotsi, Simon, and their co-workers have prepared onion-like carbon by annealing nanodiamond powder at 1800 °C.22,23 The carbon onions were found to possess a moderate specific surface area (∼500 m2 g−1) compared to ACs; however, the surface can be fully accessed by ions (Fig. 2). As a result, micro-supercapacitors based on these onion-like carbons showed ∼70% rate capability from 1 to 100 V s−1 and an excellent relaxation time constant (26 ms) compared to that of activated carbon (700 ms).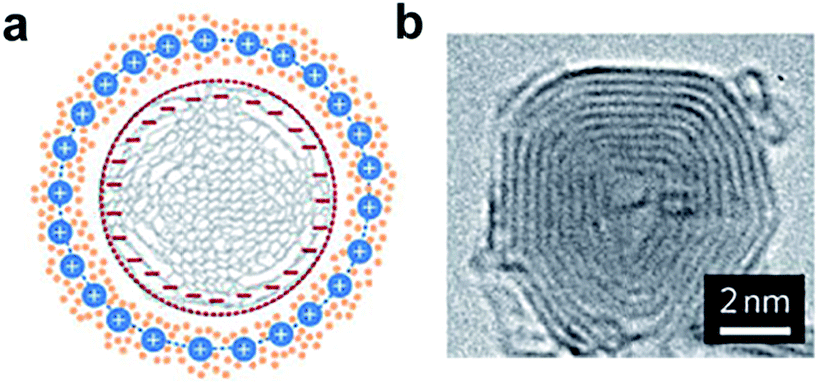 | ||
| Fig. 2 (a) Cross-section of a charged 0D onion-like carbon capacitor. (b) TEM image of a carbon onion with lattice spacing close to 0.35 nm. Reproduced with permission from ref. 23. Copyright 2010 Macmillan Publishers Limited. | ||
The above mentioned criteria for designing ideal ACs are also applicable to transition metal oxide nanoparticles in pseudocapacitors as they rely on fast surface redox reactions. Additionally, as most of the transition metal oxides suffer from poor electrical conductivity, enhancing the bulk electrical conductivity should also be taken into account. For example, Chen and co-workers have fabricated nanoporous metal/oxide hybrid supercapacitor electrodes in order to enhance the electrical conductivity of MnO2.24 In this method, conductive nanoporous gold was first produced by de-alloying Ag65Au35 using HNO3, followed by growing nanocrystalline MnO2 (with a grain size of ∼5 nm) into the nanopores. The resulting nanoporous Au/MnO2 supercapacitor electrode showed a high specific capacitance of ∼1145 F g−1 at 50 mV s−1, which can be attributed to nanoporous gold that allows easy and efficient access to both electrons and ions. Recent advances in using perovskite materials for solar energy harvesting have greatly stimulated the enthusiasm of researchers to exploit these materials for other potential fields. Lately, Stevenson, Johnston, and co-workers have prepared LaMnO3 nanoparticles via reverse-phase hydrolysis method as supercapacitor electrode material.25Fig. 3a shows an ideal LaMnO3 cubic perovskite structure, where lanthanum atom locates at body centered position, manganese atoms at cubic corner positions, and oxygen atoms at mid-edge positions respectively. The dark-field SEM image (Fig. 3b) presents the small (∼20–50 nm) nanocrystals of LaMnO3.09 that were sintered into agglomerated particles. They have found that oxygen intercalation has significant effect on LaMnO3 perovskite nanoparticles as supercapacitor electrodes. The specific capacitances of LaMnO3.09 and LaMnO2.91 reached 586.7 and 609.8 F g−1, respectively. Interestingly, the capacitance difference (∼4%) was found to be close to the oxygen content difference (∼6%), implying that the oxygen intercalation is the major charge storage mechanism of LaMnO3 perovskite supercapacitor electrodes. This assumption was further demonstrated by applying electrolyte with varying anion concentrations as well as different electrolyte types (i.e. neutral and alkaline electrolytes). As a result, perovskite-type materials with anion vacancies can be exploited to store charge through oxygen intercalation. Therefore, in addition to traditional solid 0D active materials (e.g. activated carbon), perovskite materials might open a new paradigm as supercapacitor electrode materials.
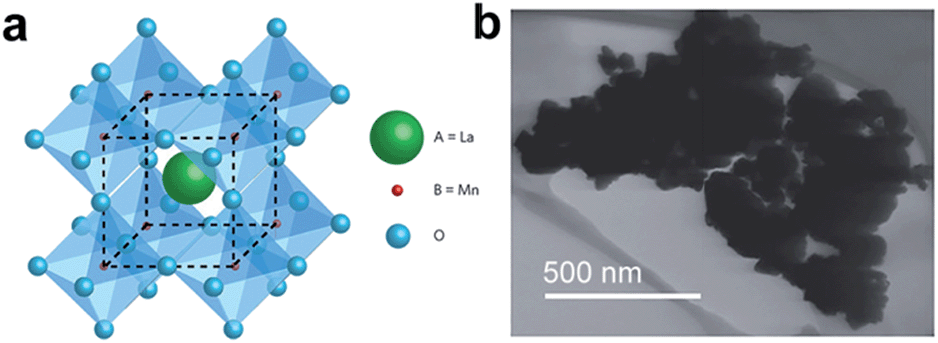 | ||
| Fig. 3 (a) Idealized cubic LaMnO3 perovskite structure. (b) Dark-field SEM image of LaMnO3.09 nanoparticles. Reproduced with permission from ref. 25. Copyright 2014 Macmillan Publishers Limited. | ||
2.2 Hollow 0D nanostructures
Hollow 0D nanostructures have been recognized as promising candidates for electrode design due to their peculiar properties such as low density, high surface-to-volume ratio and shortened pathways for transporting both mass and charges.26 Hollow 0D nanostructures are generally synthesized via three distinct approaches: hard templating, soft templating, and template-free methods. Among them, hard templating methods have been widely adopted because of their remarkable advantages in controlling the size, shape, and structure of the products.27 Typically, a single-shelled hollow nanosphere is created by coating a thin layer of precursor materials onto a spherical hard template (e.g. silica spheres, polystyrene colloid spheres, carbonaceous spheres, etc.), followed by removing the template either by calcination or by chemical etching. For example, Yang and co-workers reported the synthesis of hollow carbon nanospheres by carbonizing its hollow nanosphere precursor obtained through a silica sphere-assisted hard templating method.28 The resulting hollow carbon nanospheres showed a high surface area (1704 m2 g−1), large bimodal mesopores (6.4 and 3.1 nm), and large pore volumes (1.6 cm3 g−1). With such a design, a specific capacitance of 251 F g−1 at 50 mV s−1 was achieved for the hollow carbon nanospheres. Hollow 0D structures based on other electrode materials such as MnO2,29,30 NiO,31,32 NiS,33etc. have been investigated as well.In addition to single shelled hollow 0D nanostructures, multiple shelled samples have also shown fascinating characteristics (e.g. substantially improved surface-to-volume-ratio) for efficient electrode material design. Similarly, electrode materials like NiO,34 Co3O4,35 and Fe2O3,36 have been successfully fabricated into multi-shelled structures. For example, Zhang and co-workers reported the synthesis of NiO hollow nanospheres via layer-by-layer (LBL) self-assembly for supercapacitor application.34 In their method, the number of shells was directly determined by the cycles of immersion–precipitation process before removing the carbonaceous template via calcination (Fig. 4a). Single, double, and triple shelled NiO nanospheres were prepared by repeating the immersion–precipitation process for 0, 1, and 2 times (Fig. 4b and c). The double-shelled NiO nanospheres exhibited the highest surface area (92.00 m2 g−1), which ascribes to their more loosely-packed structure compared to their single and triple shelled counterparts. As a result, the double shelled NiO nanospheres showed the most enhancement in specific capacitance (612.5 F g−1). Since many other promising electrode materials have not been fabricated into 0D multi shelled structures, much effort still needs to be dedicated in the future.
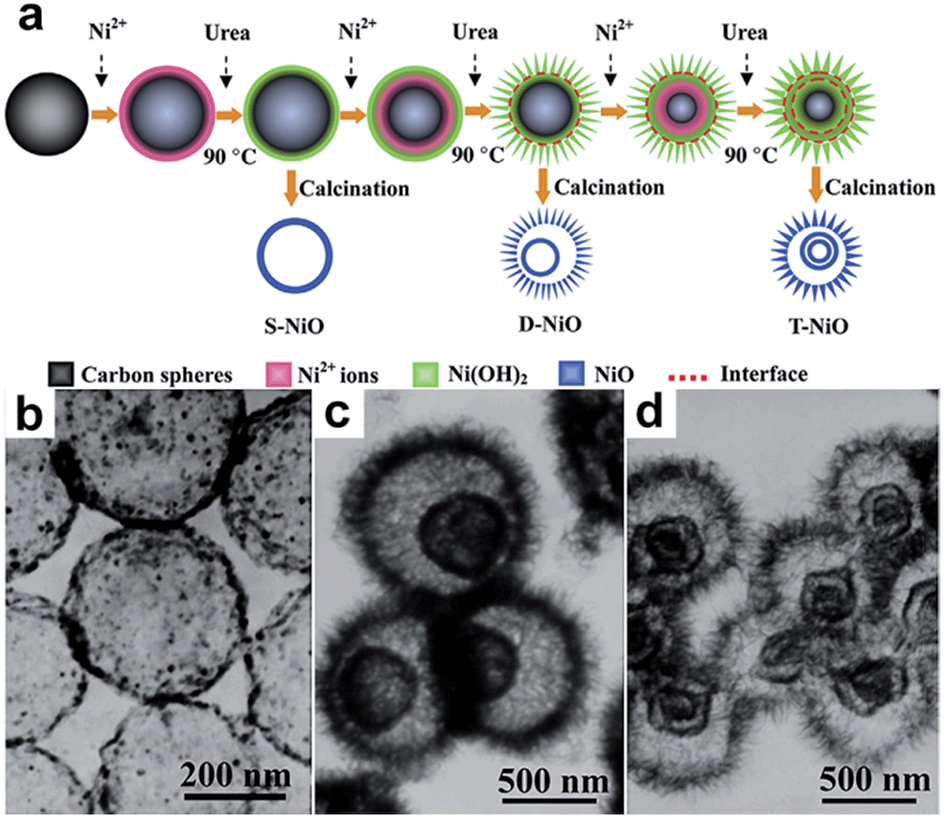 | ||
| Fig. 4 (a) Schematic of synthesizing multishelled NiO hollow nanospheres using LBL self-assembly. TEM images show NiO hollow nanospheres with (a) single-shell, (b) double-shells, and (c) triple-shells. Reproduced with permission from ref. 34. Copyright 2014 Elsevier. | ||
2.3 Core–shell 0D nanostructures
Core–shell 0D nanostructures often refer to a solid or hollow nanoparticle coated with a thin shell. Combining faradaic and non-faradaic materials into core–shell 0D nanostructures offer considerable advantages including enhanced electrical conductivity, less agglomeration, robust chemical and mechanical stability, etc.37–40 For example, Zhao and co-workers have grown conducting polymer (polyaniline (PANI)) on hollow carbon spheres as supercapacitor electrode materials.37 Hollow carbon spheres (HCS) with high specific area (2239 m2 g−1) were first prepared using ferrocene as the carbon precursor and silica spheres as the template. PANI was then conformally grown on the surface of the HCS by polymerizing aniline at room temperature for 12 h (Fig. 5a and b). HCS–PANI core–shell structures with an optimized content of PANI showed a specific capacitance of 525 F g−1, which is almost two times greater than that of pristine HCS (268 F g−1). However, the rate capability of HCS–PANI decayed with increasing PANI content, which can be attributed to the pore blockage due to the reduced accessibility of electrolyte ions. Zhao's group also synthesized core–shell nanoparticles with carbon core and MnO2 shell by direct redox reaction between graphitic hollow carbon spheres (GHCS) and KMnO4 solution at 70 °C (Fig. 5c and d).38 The as-prepared GHCS–MnO2 core–shell nanostructures showed a maximum specific capacitance of 190 F g−1 at 0.1 A g−1 with an optimized MnO2 content (64 wt%), more than two times higher than that of pristine GHCS (95 F g−1). Moreover, the GHCS–MnO2 core–shell nanostructures maintained 55% capacitance retention while current density increased from 0.1 to 10 A g−1, indicating a porous structure of MnO2 shell was formed to provide efficient diffusion pathway for electrolyte ions. | ||
| Fig. 5 (a) FESEM and (b) TEM images of HCS–PANI. (c) FESEM and (d) TEM images of GHCS–MnO2. Reproduced with permission from ref. 37 and 38. Copyright: 2010 American Chemical Society and 2012 The Royal Society of Chemistry. | ||
3 1D nanostructures
1D nanostructures are slivers of material with two dimensions constrained on nanoscale.41 They have been extensively studied for the design of electrode materials for supercapacitors due to their unique and superior chemical and physical properties.42–47 For instance, the longitudinal axis of 1D nanostructures can provide efficient transport pathway for both electrons and ions,48 while maintaining the similar benefits of 0D nanostructures in the two nanoscale dimensions. In this section, we place 1D nanostructures into two major categories: homostructures and heterostructures.3.1 1D homostructures
1D homostructures are structures with only one singular structure such as nanorods, nanowires, nanotubes, etc. They can be divided into three groups, namely, nanorods/nanopilars with aspect ratios (length/diameter) less than 10, nanowires with aspect ratios more than 10, and nanotubes with hollow interiors.49 The methods for synthesizing 1D homostructures are versatile and can generally be divided into two major types, template-assisted and template-free methods. For template-assisted methods, sacrificial templates (e.g. AAO templates) are commonly used to shape the initial structure, followed by the removal of the templates with an acidic or basic solution. For template-free methods, various approaches like hydrothermal synthesis, chemical vapor deposition (CVD), and electrodeposition are generally employed to directly grow 1D homostructures. Recently, vertical growth of active material on the conductive substrate either by a “bottom-up” or “top-down” method has become one of the most promising electrode design approaches. Such a design guarantees efficient transport of electrons between current collector and active material because of the binder-free design. It also enables fast transfer of ions between electrolyte and active materials because of the available void space between adjacent nanostructures.Tong and co-workers have reported oxygen-deficient Fe2O3 nanorods as supercapacitor electrode material.50 Nanorod-like FeOOH was first hydrothermally synthesized in an aqueous solution containing ferric chloride, sodium nitrate, and hydrogen chloride. Subsequently, the FeOOH nanorods were heated at high temperature in air and N2, separately, which resulted in pristine Fe2O3 nanorods and oxygen-deficient Fe2O3 nanorods. Interestingly, the oxygen-deficient Fe2O3 nanorods exhibited more than two times enhancement of the specific capacitance (64.5 F g−1) than that of pristine Fe2O3. This phenomenon may be attributed to the existence of oxygen vacancies which considerably enhance the donor density and in turn improve the conductivity and reactivity of Fe2O3 nanorods.
Recently, Thomas and co-workers reported a facile method to produce highly ordered carbon nanopillars as supercapacitor electrodes.51 They first developed a novel technique, called spin-on nanoimprinting (SNAP), to print highly ordered polymer nanostructures.51–54 In a typical SNAP process, a thin layer of polymer solution was spin-coated on an e-beam etched nanohole structured silicon mold, followed by curing and peeling off the polymer from the template (Fig. 6a). The SNAP technique was demonstrated to be simple, quick, and cost-effective since it requires just a spin coater and a pre-fabricated template. The entire printing process can be completed within a few minutes, but can be performed at a much faster pace if automated. Moreover, a large-scale nanostructure can be easily obtained by stitching several printed structures next to each other. Unlike commonly used sacrificial template (e.g. AAO template), this template can be re-used many times without any noticeable degradation of nanostructures since all the polymer nanopillars can easily be removed from the template. The carbon nanopillar arrays were obtained upon post stabilization and carbonization of the polymer nanostructures. Fig. 6b and c show the SEM images of printed polyacrylonitrile (PAN) and carbon nanopillar array. The diameter and height of each carbon nanopillar is ∼95 nm and ∼200 nm, respectively. Significantly, the specific capacitance of the carbon nanopillar arrays (3.4 mF cm−2) showed considerable enhancement compared to that of a planar carbon film (0.05 mF cm−2).
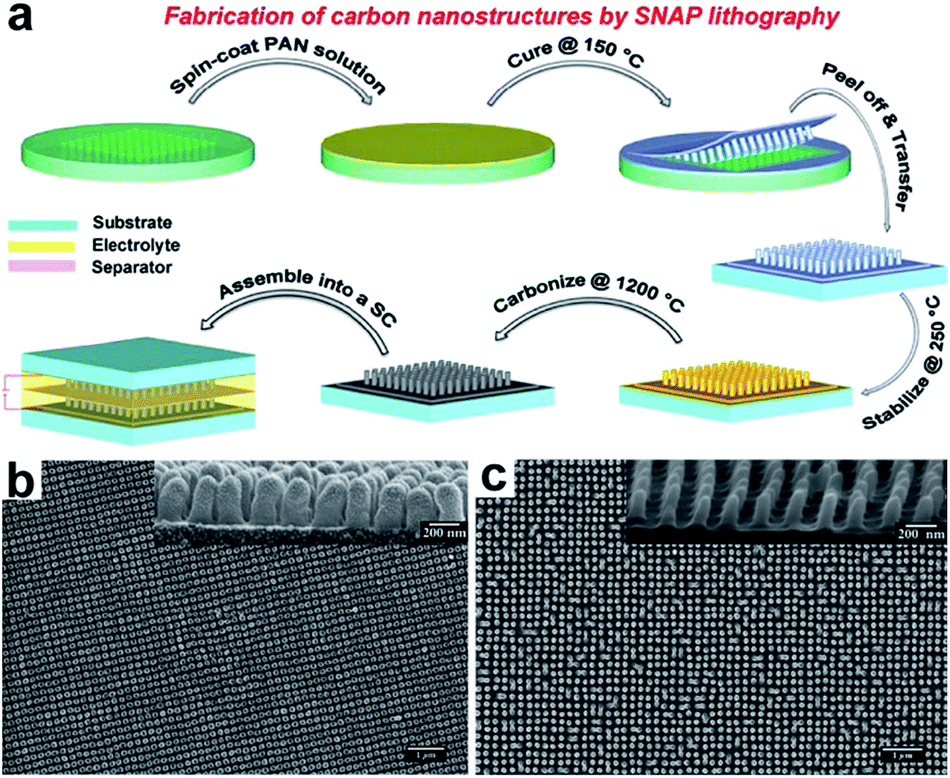 | ||
| Fig. 6 (a) Fabrication of carbon nanostructures by SNAP method. (b) SEM images of top view and side view (inset) of polymer nanopillars. (c) SEM images of top view and side view (inset) of carbon nanopillars. Reproduced with permission from ref. 51. Copyright 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Lately, a ternary metallic oxide, spinel nickel cobaltite (NiCo2O4), has attracted substantial attention due to its high conductivity (more than two orders of magnitude higher than single NiO or Co3O4) and multiple oxide states that enable rich redox reactions originating from both nickel and cobalt ions.57,58 For example, Lou and co-workers have successfully grown NiCo2O4 nanoneedle arrays directly on conductive substrates as supercapacitor electrodes.59 In their synthetic process, a conductive substrate was first immersed into a solution containing nickel nitrite, cobalt nitrite, ethanol, and water in an appropriate ratio and heated to 85 °C for 8 h, followed by annealing at 250 °C for 90 min. Remarkably, the NiCo2O4 nanoneedle arrays showed a capacitance of 1118.6 F g−1 and about 10.6% loss in capacitance after 2000 cycles.
The discovery of carbon nanotubes (CNTs) has led to several applications in energy storage field. CNTs as supercapacitor electrode materials, and especially vertically aligned CNTs directly grown on conductive substrates, have drawn remarkable attention due to their moderate to high surface area (120–500 m2 g−1), porous structure, superior electronic conductivity, and excellent mechanical and thermal stability. Ajayan and co-workers have grown aligned CNTs (400 µm long) on metal alloy (Inconel) using vapour-phase catalyst delivery.63 The CV curves showed rectangular and symmetric shape even at a scan rate of 1000 mV s−1, suggesting ideal capacitive behavior and low contact resistance between the CNTs and the substrate. The specific capacitance of a supercapacitor based on these vertically aligned CNT arrays was found to be 18 F g−1. Iijima and co-workers reported a method to prepare densely packed and aligned CNTs using zipping effect as supercapacitor electrode material.64 The specific capacitance of the aligned CNTs showed 80 F g−1, which is roughly twice the value of 45 F g−1 for the entangled CNTs. Recently, Kim and co-worker have grown vertically aligned CNTs directly on conductive carbon papers using an Al/Fe catalyst via water-assisted CVD.65 These aligned CNTs exhibited a specific capacitance of about 200 F g−1 at 20 A g−1.
3.2 1D heterostructures
Unlike homostructures, heterostructures usually consist of more than one component. One major advantage of using heterostructured electrode design is the synergic improvement of intrinsic properties of each component for better electrical conductivity, faster ionic transport, greater electrochemical reversibility and cycle stability, and improved mechanical stability. Owing to these advantages, 1D heterostructures are currently considered as one of the potential candidates for the next generation electrode designs.(1) Current collector core – active material shell. In this configuration, 1D nanostructured current collector core can provide shortened pathways for electron transport, which effectively enhances the available electrochemically active sites. Owing to the high electrical conductivity as well as excellent mechanical stability, metals have been selected as pivotal current collector candidates. When metals are forged into 1D nanostructures like nanowires or nanotubes, active materials can be deposited onto these 1D metal nanostructures and hence form core–shell 1D nanostructured electrode materials. There are several approaches to synthesize 1D metal nanostructures including template-assisted preparation,66–69 soft solution processing,49,70,71 vapor phase synthesis,72–74 and self-assembling.75,76 Among them, template-assisted preparation is the most intensively studied method for 1D metal nanostructure synthesis. Particularly, 1D metal nanostructures can be fulfilled by either shaping with the help of sacrificial templates (e.g. AAO) or directly coating active material on supercapacitive-inactive 1D nanostructures. For the former case, a pioneering work led by Teberna and co-workers reported Cu nanowire array prepared using AAO template as current collector, which was subsequently deposited with active materials (Fe3O4).69 Following a similar concept, other works have been demonstrated such as Ni@MnO2,77,78 Ni@NiO,79 Au@MnO2,80 Ni@Co3O4,81 Mn@MnO2,82etc. For the latter, since the inert core does not give any contribution to the electrochemical properties, the metal coated structure is still regarded as the current collector core.
There are several on-going efforts by Thomas's group to deposit metal on supercapacitive-inactive 1D nanostructures as current collector. Recently, they have demonstrated the synthesis of AuPd@MnO2 core–shell structures for supercapacitor application.52 AuPd was sputtered on PAN nanopillars prepared by SNAP technique, followed by electrochemical deposition of MnO2 (Fig. 7a). Fig. 7b and c shows the SEM image of the as-printed structures, suggesting a highly ordered nanopillar structure. These nanopillars have diameter of ∼140 nm, height of ∼250 nm, and centre-to-centre distance of ∼200 nm. As shown in Fig. 6d, MnO2 were conformally coated onto each pillar. A modelling of the 3D surface area enhancement indicated a ∼4.5 times surface area enhancement on the nanostructured region compared to a planar region (data based on the dimensions of nanopillars from SEM images). Accordingly, the specific capacitances of AuPd@MnO2 core–shell nanopillars (603 F g−1) consistently exhibited more than two-fold improvement compared to that of AuPd–MnO2 planar electrode, at various scan rates.
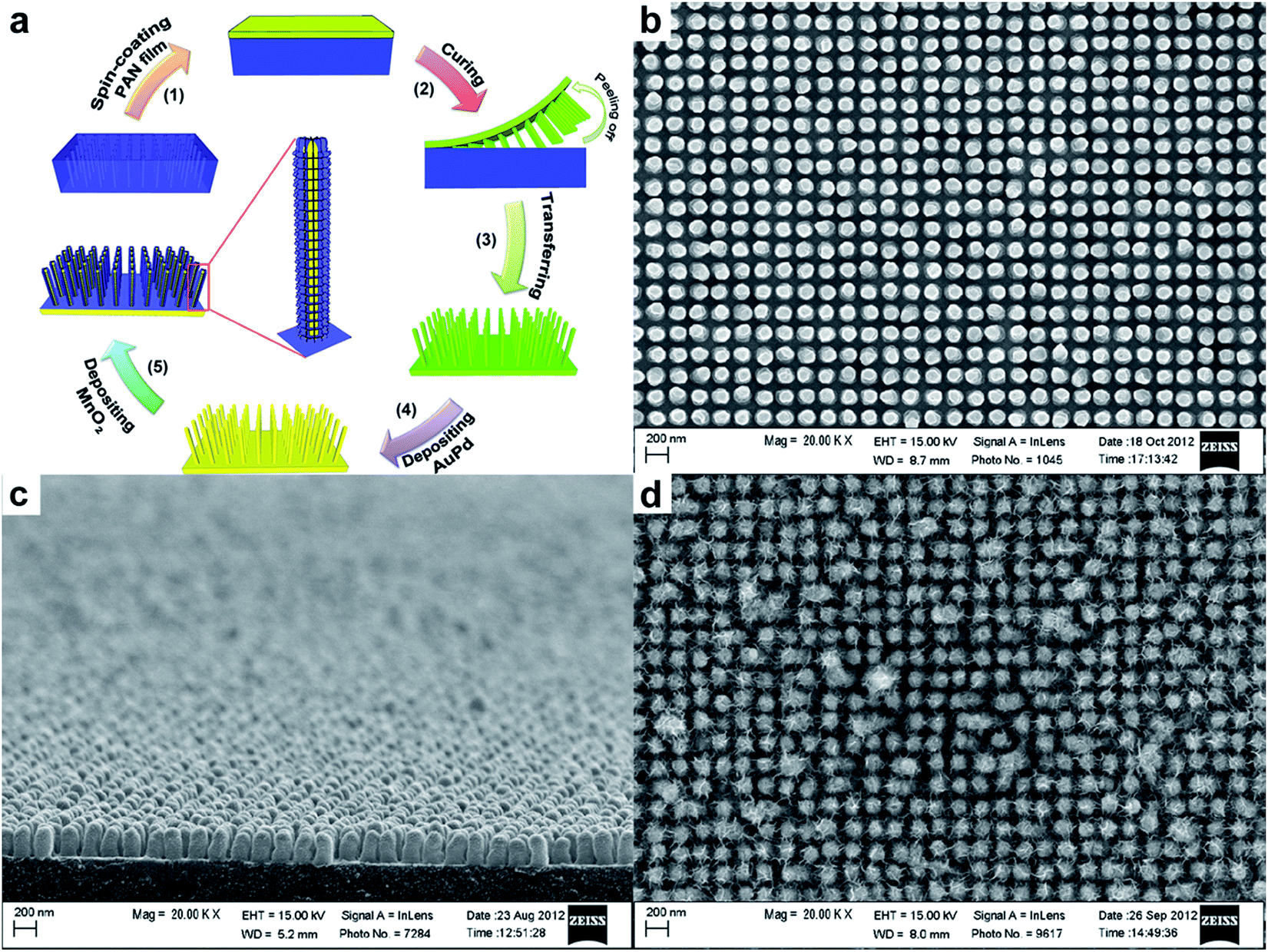 | ||
| Fig. 7 (a) Schematic illustration of the development of MnO2 coated electrodes. SEM images show top (a) and side (b) view of as-printed PAN nanostructures. (d) SEM image of electrochemically deposited MnO2 nanoparticulates on pillar nanoarrays. Reproduced with permission from ref. 52. Copyright 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Thomas and co-workers recently developed another strategy to prepare CuO@AuPd@MnO2 core–shell nanowhiskers (NWs) as supercapacitor electrodes.83 This was accomplished by sputtering AuPd on high aspect ratio CuO NWs (with diameter ∼100 nm and length ∼15 µm) that were obtained by simply heating a copper substrate in air at elevated temperature. MnO2 was then electrodeposited onto the CuO@AuPd structures hence producing CuO@AuPd@MnO2 core–shell NWs. Fig. 8a shows the schematic illustration of the fabrication process of CuO@AuPd@MnO2 core–shell NWs. Fig. 8b clearly presents that CuO NWs were grown uniformly on the Cu wire and Fig. 8c confirms that each CuO NW was conformally covered by MnO2. The as-prepared core–shell heterostructures showed excellent specific capacitance of 1376 F g−1 at a scan rate of 5 mV s−1. This may be attributed to two reasons: (1) the CuO nanowhiskers as scaffold provides substantially improved surface area (more than 2 orders of magnitude higher than a planar CuO substrate) for the electrode materials and (2) the core–shell heterostructures considerably enhance the ionic and electronic conductivity. Additionally, they have revealed that the CuO layer not only serves as high surface area scaffold but also works as an insulating layer to block charge transfer. As a result, they have successfully demonstrated that the inner copper wire can be used for electrical transmission as normal while an independent supercapacitor can be built on the outside by using overlapping layers. This led to the development of a dual-function (electrical conduction and energy storage) device.
 | ||
| Fig. 8 (a) Schematic illustration of the fabrication process of CuO@AuPd@MnO2 NWs. (b) SEM image shows that the copper wire is completely covered by CuO NWs. (c) SEM image of MnO2 that was uniformly electrodeposited onto NWs. Reproduced with permission from ref. 83. Copyright 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In addition to metals, other materials such as ZnO, SnO2, Zn2SnO4, and ITO have been fabricated into 1D nanostructures and served as current collector cores to deposit active material shells.84–91 To date, most 1D current collectors are either nanowires or nanorods, while reports about nantubular current collectors are relatively few. However, electrodes based on nanotubular current collectors show promising results since both inner and outer surfaces can be utilized for depositing active materials.
(2) Active material core – active material shell. Active material core – active material shell presents another promising combination of 1D core–shell heterostructures since both core and shell materials contribute to electrochemical performance. Generally, active materials used in supercapacitors can be classified into two groups: EDLC materials and pseudocapacitive materials. There can be four types of core–shell combinations, namely, EDLC material core – EDLC material shell, EDLC material core – pseudocapacitive material shell, pseudocapacitive material core – EDLC material shell, and pseudocapacitive material core – pseudocapacitive material shell. Among these categories, EDLC material core – pseudocapacitive material shell and pseudocapacitive material core – pseudocapacitive material shells are the two major players.
EDLC material core and pseudocapacitive material shell constitutes an appealing combination of core–shell design due to the following merits: (1) EDLC materials as core have better cycle stability due to their electrostatic charge and discharge storage mechanism (i.e. no phase changes), which makes them robust backbones even for large number of cycles; (2) EDLC core material also have better electrical conductivity compared to pseudocapacitive materials, which facilitates the transportation of electrons; (3) pseudocapacitive materials as shell can effectively offset the low capacitance contribution from EDLC materials core. In this regard, Zhai and co-workers have demonstrated the feasibility of EDLC material core and pseudocapacitive material shell such as CNT@PPy–MnO2,92 CNT@PEDOT–MnO2,93 and CNT@PANI–MnO2.94 In CNT@PPy–MnO2, multiwall CNTs were first well-dispersed and wrapped around using 18 wt% poly(4-styrenesulfonic acid) (PSS), which also provides attractive forces for pyrrole and metal ions upon negative charging. PPy–MnO2 composite shell was subsequently prepared by adding pyrrole and KMnO4 (Fig. 9). As a result, CNT@PPy–MnO2 core–shell nanowires showed ∼268 F g−1 specific capacitance, which is much higher than CNT@MnO2 (∼170 F g−1) and CNT@PPy (∼160 F g−1). Significantly, 93% rate capability of CNT@PPy–MnO2 was retained from 5 to 100 mV s−1, while only 35% and 72% were retained by CNT@MnO2 and CNT@PPy respectively in the same scan rate range when tested separately. Moreover, CNT@PPy–MnO2 exhibited very stable cycle life (10% reduction after 5000 cycles); in contrast, a specific capacitance reduction of 61% and 45% was observed under the same test conditions for CNT@MnO2 and CNT@PPy, respectively.
 | ||
| Fig. 9 High resolution TEM images of the MWCNT@PPy–MnO2 core–shell structure. Reproduced with permission from ref. 92. Copyright 2009 Elsevier. | ||
A similar strategy reported by Ajayan and co-workers is the combination of CNT core and MnO2 shell.95,96 First, MnO2 nanotubes were obtained by vacuum infiltrating manganese nitrate solution into an AAO template pre-electrodeposited with Au, followed by subsequent annealing in the air at 300 °C for 10 h. CNTs were then grown on the inner void left by the MnO2 shells by CVD process and hence obtained CNT@MnO2 1D core–shell heterostructure upon removing the AAO template. Significantly, the core–shell arrays showed almost two times enhancement of specific capacitance compared to pure MnO2. Moreover, no apparent structural change was detected even after 1000 cycles, indicating remarkable mechanical and cycle stability.
Pseudocapacitive material core – pseudocapacitive material shell presents another intriguing core–shell design because both core and shell materials have redox reactions during charge and discharge processes, which results in high specific capacitances. In this regard, Fan's group has synthesized versatile combinations of pseudocapacitive materials with 1D core–shell heterostructures including Co3O4@MnO2,97 CoO@NiHON,98 Co3O4@NiO,99 CoO@TiO2,100 and Co3O4@PEDOT–MnO2.101 Taking Co3O4@MnO2 core–shell nanowires as an example, Co3O4 nanowire core was first hydrothermally grown for 5 h, followed by coating a thin carbon layer and hydrothermal deposition of MnO2 from a KMnO4 solution at 160 °C for 1–5 h. The as-synthesized core–shell heterostructures presented a specific capacitance of 480 F g−1 at 2.67 A g−1 and excellent rate capability (56% retention with current density up to 44.7 mA cm−2).97 Following a similar concept, Liu and co-workers further improved the performance by employing NiCo2O4 instead of Co3O4 due to its better electronic conductivity (as mentioned earlier).102 Consequently, the final product of NiCo2O4@NixCo1−x(OH)2 core–shell nanowires considerably increased the specific capacitance up to 1500 F g−1 and improved the rate capability up to 67% retention at 90 mA cm−2.
It should be noted that some of the metal oxides, such as V2O5, MnO2 and Co3O4, suffer from dissolution issues due to phase changes upon cycling, which gradually erodes the quantity of electrochemically active materials, resulting in poor cycling and mechanical stability.103–105 Therefore, a rational design like a durable protective coating which can effectively inhibit the structural collapse or break-down of metal oxide core and alleviate the strain generated during cycling is essential. In this regard, conducting polymers are generally selected as promising protective shell materials due to their high stability, mechanical flexibility, and excellent electronic conductivity.106 For example, Wu and co-workers reported core–shell structure of PPy grown on V2O5 nanoribbon for supercapacitors.103 V2O5 nanoribbons were first prepared hydrothermally in a solution containing NH4VO3 and poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) copolymer at 120 °C. The as-prepared V2O5 nanoribbons were dispersed in water by using anionic surfactant. The anionic surfactant also provided electrostatic force to absorb pyrrole at a later stage. After adding pyrrole and FeCl3, a core–shell structured V2O5@PPy was obtained (Fig. 10a). Significantly, a supercapacitor assembled using V2O5@PPy as anode and activated carbon as cathode showed less than 5% capacitance fading after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, while one assembled under the same condition without PPy film on the V2O5 anode showed 17.5% capacitance loss. This result demonstrates the effectiveness of PPy shell in preventing the dissolution of V2O5 upon cycling (Fig. 10b). Furthermore, the electrolyte for PPy@V2O5 electrode after cycling showed transparent color, while yellow electrolyte was observed for the V2O5 electrode, indicating that vanadium ions diffuse into the electrolyte without a protective coating. Similarly, other works employing conducting polymers as protective shell, including, MnO2@PEDOT–PSS,107,108 Co3O4@PANI,109 MnO2@PPy,110,111 V2O5@PEDOT–MnO2,112etc. have been demonstrated. Further studies are needed to address the relationship between the thickness of protective coating and electrochemical performances (e.g. cycle stability and rate capability). This is because, even though the protective coating could effectively prevent the dissolution issue, it could, in turn, block the accessibility of ions if it is too thick, which impedes the rate capability.
000 cycles, while one assembled under the same condition without PPy film on the V2O5 anode showed 17.5% capacitance loss. This result demonstrates the effectiveness of PPy shell in preventing the dissolution of V2O5 upon cycling (Fig. 10b). Furthermore, the electrolyte for PPy@V2O5 electrode after cycling showed transparent color, while yellow electrolyte was observed for the V2O5 electrode, indicating that vanadium ions diffuse into the electrolyte without a protective coating. Similarly, other works employing conducting polymers as protective shell, including, MnO2@PEDOT–PSS,107,108 Co3O4@PANI,109 MnO2@PPy,110,111 V2O5@PEDOT–MnO2,112etc. have been demonstrated. Further studies are needed to address the relationship between the thickness of protective coating and electrochemical performances (e.g. cycle stability and rate capability). This is because, even though the protective coating could effectively prevent the dissolution issue, it could, in turn, block the accessibility of ions if it is too thick, which impedes the rate capability.
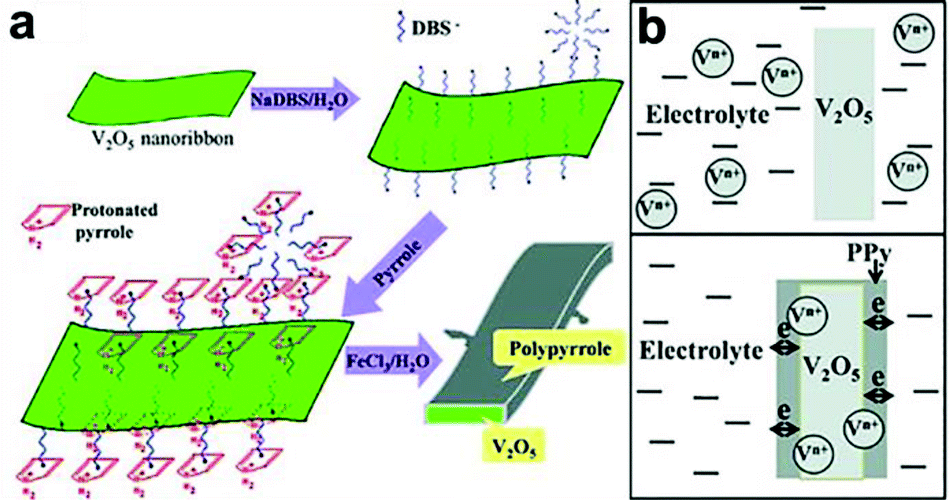 | ||
| Fig. 10 (a) Schematic shows the growth of PPy on V2O5 nanoribbon surface. (b) PPy shell helps the electronic transport and prevents the dissolution of vanadium in electrolyte. Reproduced with permission from ref. 103. Copyright 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
000 cycles. This impressive result may be attributed to the employment of CNT arrays, which provides not only high electronic and ionic conductivity but also sturdy mechanical stability. Another example reported by Lee and co-workers was the loading of well-dispersed MnO2 nanoparticles into PEDOT nanowires.114 PEDOT nanowires were first prepared by electrochemical deposition of PEDOT in AAO template, followed by removal of the template using NaOH solution. MnO2 nanoparticles were subsequently loaded into the PEDOT nanowires by soaking the PEDOT nanowires into KMnO4 solution. The MnO2/PEDOT composites delivered a specific capacitance of 250 F g−1 and an 80% rate capability was maintained from 5 to 25 mA cm−2.
4 2D nanostructures
Given the importance of properties such as diffusion ion length or contact area with electrolyte for utilization of active material in the performance of the electrochemical supercapacitors, the rise of 2D materials, and the fact that they exhibit only surface area (i.e. no bulk volume), has naturally attracted substantial interest and augmented a number of possible configurations of potential hybrid 2D configurations in the electrode material design. In addition, 2D materials exhibit unique properties such as mechanical stability, strength, flexibility, transparency, and chemical stability. However, in supercapacitors involving 2D structures, little is known of the unique behavior and properties most 2D materials and derived hybrid in aqueous or organic solvents that will exhibit. Since capacitance is highly dependent on the surface areas of the electrode materials, 2D materials are envisioned to bring a paradigm shift in the field of electrochemical capacitors.1154.1 2D homostructures
The most widely explored materials for 2D homostructured-electrode fall under the following three categories. The first one includes graphene-based electrodes, which have been the most extensively investigated since its emergence, and found to have extensive applications in EDLC. Secondly, electrodes made of metal oxides and hydroxides, which have also been widely studied as active materials for pseudocapacitors, will be discussed. Finally, recent developments in transition metal dichalcogenides (TMDs) including MoS2 and VS2 and transition metal carbides and/or nitrides (MXenes) will be overviewed.In addition, as discussed by Ruoff and co-workers in one of the first demonstrations of graphene as electrodes for supercapacitors, it eliminates the necessity of using conductive fillers to improve the electrode conductivity.125 It also offers the possibility to increase the electrode thickness while greatly improving the performance including a higher ratio of electrode material to collector/separator and higher energy densities. Moreover, large surface area of graphene is very attractive for supercapacitor electrode materials.125–127 The surface area of graphene is predicted to be as high as 2630 m2 g−1 and all the more exciting is the fact that it intrinsically consists of structured, accessible pores and interlayer spaces (i.e. better accessibility for ions).125,126 Thin films of graphene electrodes have been obtained by various methods aiming at large scale production, including CVD,128–130 electrostatic spray deposition,129 electrophoretic deposition,23,129 and ink-jet printing.94,129,131 Nevertheless, to date, chemical approaches to make graphene oxide (GO) are the most cost-effective ton-scale method to produce graphene-based electrode materials. However, despite higher production yield, additional treatments are required to overcome its low conductivity. Among various treatments, chemically modified graphene (CMG) and reduced graphene oxide (rGO) are of particular interest as they offer a scalable and cost-effective chemical approach and produce a material with conductivity close to that of graphene.116,125,132,133 As a result, rGO has been extensively used as a highly conductive electrode material and the mechanisms of reduction of GO becomes highly important in the development of 2D electrode materials for supercapacitors.
(a) Performance of chemically prepared rGO electrodes. A variety of reducing agents have been considered to restore the graphene network and its conductivity in the GO sheets.134–137 Hydrazine is the most commonly reported reducing agent for GO reduction. Ruoff and co-workers described the synthesis of CMG by reduction of suspended graphene oxide in water using hydrazine hydrate.125 The resulting sheets showed a surface area of 705 m2 g−1 and provided specific capacitances of 135 and 99 F g−1, respectively, in aqueous and organic electrolytes. Chen and co-workers discussed GO gas-based reduction by hydrazine at room temperature to lower agglomeration in the system.116,117 The electrode materials resulting from their protocol were shown to surpass CMG or CNT-based supercapacitors,125,138,139 with a maximum capacitance of about 205 F g−1, energy density of 28.5 W h kg−1 in an aqueous electrolyte solution and ∼90% specific capacitance retained after 1200 cycle tests. C–N and C–O bonds, creating numerous hydrophilic polar sites, were present in the material, and seemed to improve the wettability of the electrodes.116 Despite the seemingly high packing of the layers, the agglomeration was shown to be lower than with other protocols, allowing ions from the electrolyte to penetrate in-between layers and increase the capacitance of the system.
Other reducing agents including hydroquinone7,137 and NaBH4137,140,141 have also been used to reduce GO. Interestingly, activated graphene using KOH in GO, another form derived from graphene obtained by chemical activation of exfoliated GO prepared by Ruoff and co-workers, revealed large surface area (3100 m2 g−1), high specific capacitance (up to ∼170 F g−1) and great electrical conductivity.132 Despite the chemical processes involved in the formation of the activated layers, low oxygen and hydrogen content were found in the final product. Furthermore, ∼97% of the capacitance was retained after 10000 cycles.
(b) Performance of rGO electrodes prepared by physical methods. Vacuum low-temperature exfoliation and laser treatments are among the physical methods used for the reduction of GO in energy storage applications. Chen and co-workers have recently demonstrated that graphene flakes obtained as a result of low-temperature vacuum exfoliation demonstrated excellent energy storage performance.142 The devices were tested in aqueous and organic electrolytes and exhibited specific capacitance of 220 F g−1 and 120 F g−1, respectively.142
Thermal treatment of GO suspension from 150–200 °C has also been shown to produce rGO by removing the oxygen from GO thus enabling high conductivity up to 5230 S m−1.137 A specific capacitance of 122 F g−1 at 5 mA could be achieved after thermal treatment at 200 °C, corresponding to a charge–discharge rate of ∼1000 mA g−1. This constitutes an improvement of the specific capacitance of ∼20% by direct comparison with the performance of a supercapacitor prepared using rGO obtained with hydrazine,125 and compares well with other standard electrode-materials.143 The advantage of this approach is its potential for scalable “green” production of carbon-based supercapacitor electrode materials.
On the other hand, laser reduction of GO is also getting substantial attention,144–147 in part due to the ability to pattern rGO–GO structures with customized geometries in the same piece of GO, in plane or in 3D. For example, Ajayan and co-workers have developed a laser writing technique to directly convert GO to rGO (Fig. 11a).144 In their study, the structure of the laser induced rGO was found to be porous, due to the decomposition of functional groups and water during laser treatment. Due to the anisotropic nature of ion mobility and transport distances, the capacitance was found to be highly dependent on the geometry of the patterned structures. The highest area capacitance of 0.51 mF cm−2 was obtained for in-plane disc geometry with no electrolyte. Comparatively, an interdigitated rGO supercapacitor with an organic electrolyte and electrode of the same thickness exhibited a mean capacitance density of 0.4 mF cm−2.146 A 30 to 35% drop in capacitance was found after 10000 cycles, but the same performance could be improved by long exposure to ambient environment or aqueous electrolytes.144 In contrast, Kaner and co-workers found that the laser-treated GO obtained using a LightScribe CD/DVD optical drive can provide an impressive conductivity (∼1740 S m−1) (Fig. 11b).145 In addition, the nanosheets were found to be well aligned, with outstanding mechanical properties (only 1% change in conductivity after 1000 bending cycles). The formation of rGO–GO interfaces by laser reduction paves the way to in-plane supercapacitors with new configuration of ion percolation for higher performance.
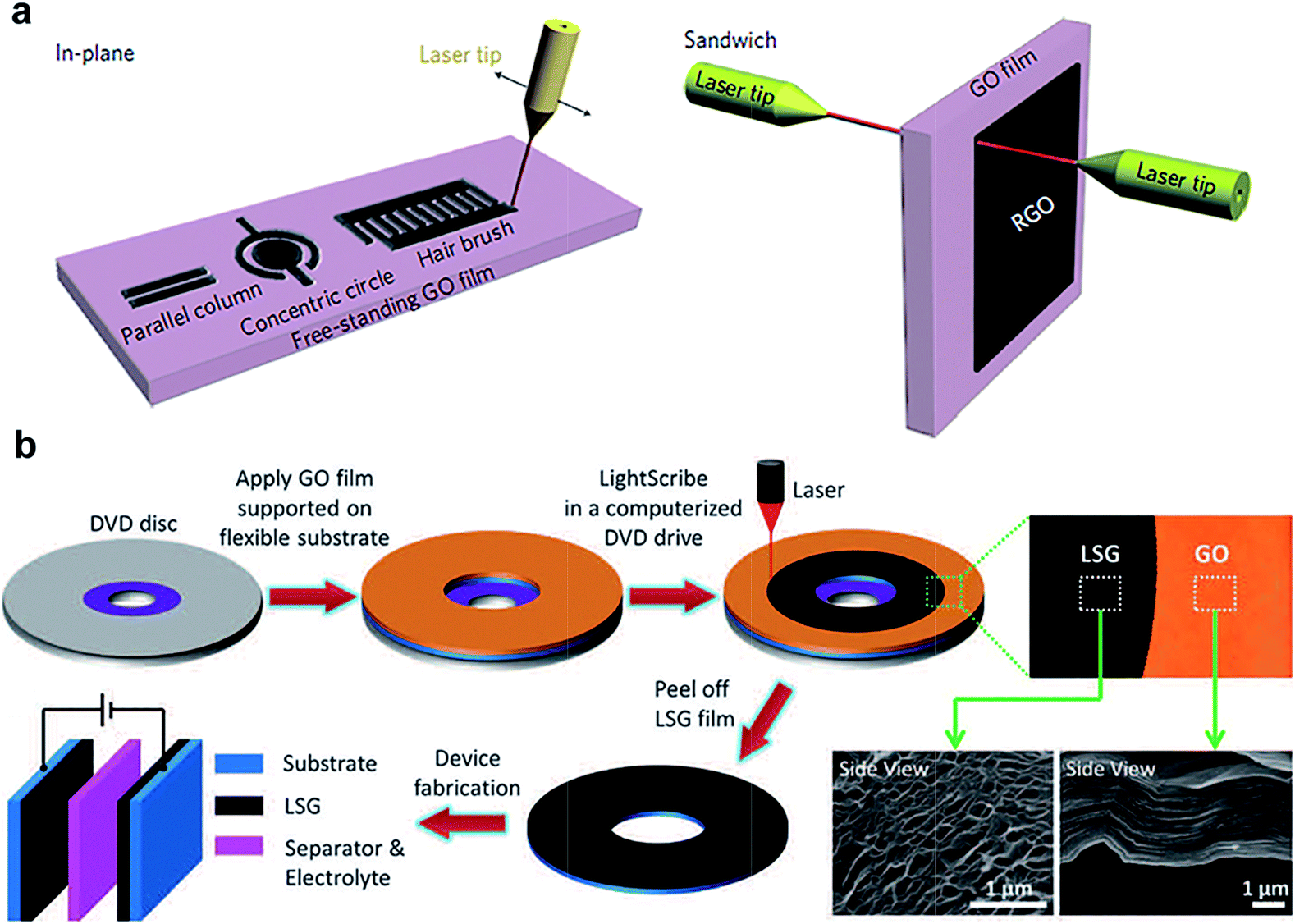 | ||
| Fig. 11 (a) rGO–GO–rGO in-plane and sandwich devices obtained by laser patterning. (b) Laser-scribed graphene-based supercapacitors. Reproduced with permission from ref. 144 and 145. Copyright: 2011 Macmillan Publishers Limited and 2012 American Association for the Advancement of Science. | ||
The laser reduction methods also addresses one of the several challenges that greatly impede the performance of the devices, in particular restacking of graphene nanosheets.120 This phenomenon is caused by van der Waals interaction between successive graphene layers.120 In fact this is one of many extensive research efforts that have been employed to develop protocols preventing graphene restacking. Curved graphene was also shown to improve the performance of supercapacitors by taking advantage of the maximum intrinsic surface capacitance and surface area.127 In addition, the formation of rGO–GO interfaces by laser reduction paves the way to in-plane supercapacitors with new configuration of ion percolation for higher performance. As demonstrated by Ajayan and co-workers (Fig. 12a),130 the in-plane design fosters ions to interact with all the graphene layers resulting in the maximum utilization of electrochemical surface area.
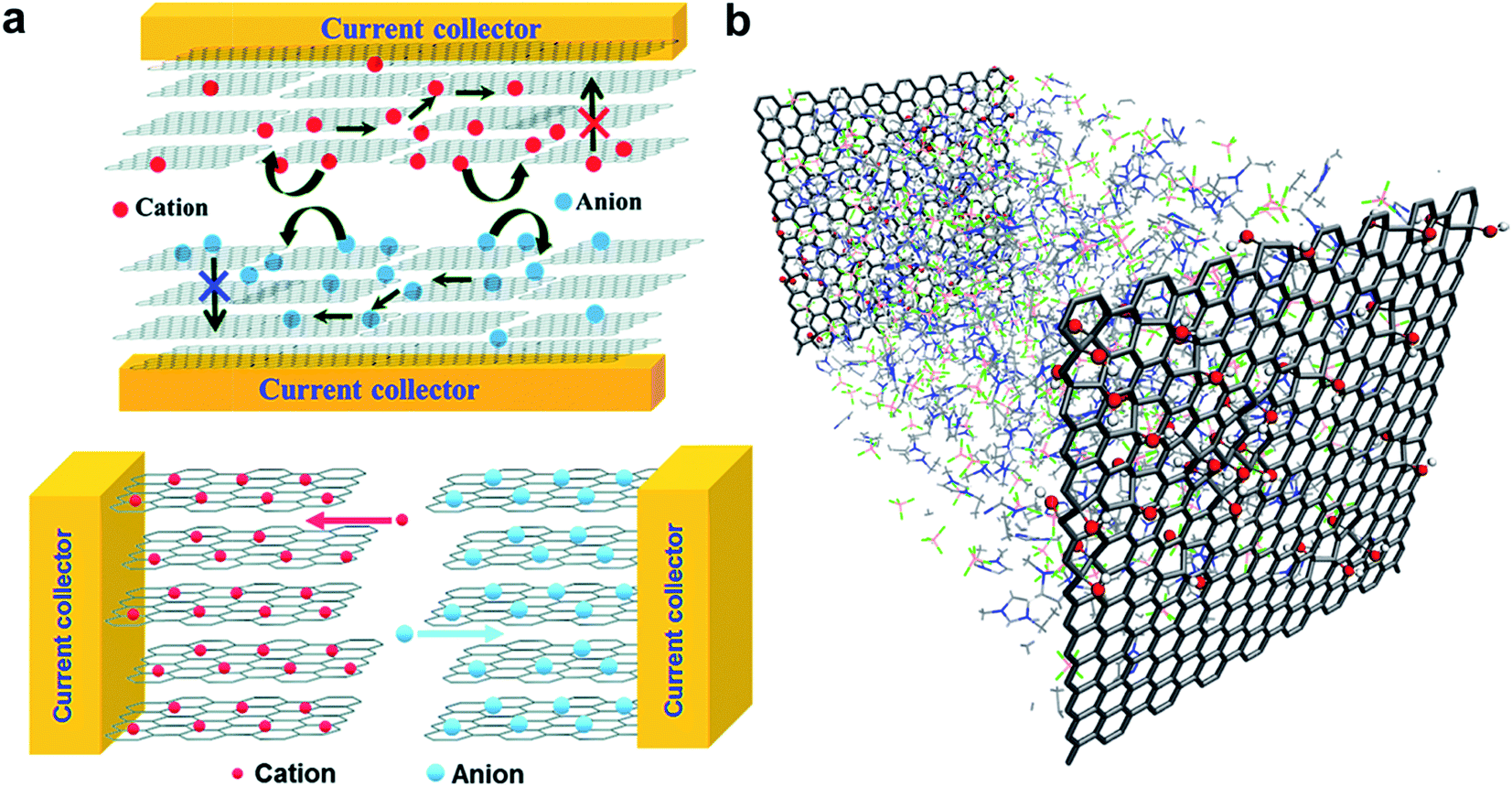 | ||
| Fig. 12 (a) Conventional stacked geometry used for carbon based electrode material (top) and in-plane supercapacitor configuration made of graphene electrodes (bottom). The schematic depicts the mechanisms of increase in electrolyte percolation for higher performance. (b) Representation of a supercapacitor model consisting of GO electrodes (10% oxidation) with pure EMI+BF4−. Reproduced with permission from ref. 130 and 133. Copyright: 2011 and 2014 American Chemical Society. | ||
Given the diversity of the results obtained with GO and reduced GO, a fundamental model describing the electrode polarizability, electrolyte dynamics, possible chemical processes, structural defects in the electrodes and pore characteristics would be of great interest to speed up the material discovery. However, this is a complex problem to model. Recently, Kim and co-workers considered the role of oxidation in molecular dynamics models (Fig. 12b) and demonstrated that the capacitance is highly dependent on the configuration of the oxidation,133 as it modifies the electrode surface accessibility of the ions. Although limited in the parameters considered for the calculations, the approach shows great promise to catalyse electrode optimizations.
Overall, the initial reports of graphene-based electrodes in electrochemical supercapacitors indicated specific capacitance of 117 F g−1 in aqueous H2SO4 with operating voltage to 1 V for electrode preparation by graphitic oxide exfoliation and by nanodiamond transformation.148 Specific capacitance of 135 F g−1 and voltage up to 1 V in aqueous media were obtained with CMG125 with energy density of about 32 W h g−1.148
2D MnO2 (up to 2 nm in thickness) has been considered for cathode electrodes. Using soft template methods, Kang and co-workers showed high specific capacitance as high as 774 F g−1 up to 10000 cycles.150 The MnO2 electrodes were used in aqueous Ca(NO3)2–SiO2 gel electrolyte. The electrodes were prepared using a soft template technique. MnO2 nanosheets were obtained by mixing sodium bis(2-ethylhexyl)sulfosuccinate and water to form a binary lamellar structure, to which KMnO4 is added. The dissolution of KMnO4 occurs only in the aqueous phase of the mixture and the growth of MnO2 is automatically initiated in the aqueous phase. Scan rates from 2 to 50 mV s−1 could be demonstrated. For RuO2 thin film electrodes, performances up to 730 F g−1 were demonstrated.151,152
Among TMDs being considered for electrode materials, MoS2 may be of special interest for EC, as its intrinsic conductivity was calculated to be higher than other similar metal oxides, while its theoretical capacity was predicted to surpass that of graphite.153,154 However, to date, the literature related to MoS2 for EC electrodes is rather limited.155,156 Soon and Loh carried out electrochemical impedance spectroscopy to characterize the performance of an arrangement of dense edge-oriented MoS2 films for micro-supercapacitor and found that the performance is similar to that of CNT electrodes.155,156 Other studies with various configuration of MoS2 lead to medium performance electrodes.102,111,157 More recently, flower-like MoS2 electrode material developed by Geng and co-workers exhibited a specific capacitance of 168 F g−1 (at 1 A g−1, 1 M KCl) and retained ∼93% capacitance after 3000 cycles.155
Among other TMDs with intrinsically high conductivity, VS2 represents a favorable alternative to graphene; further strengthened by the feasibility of exfoliation of layers after intercalation with NH3 (Fig. 13).158 Numerous tests were carried out on this new material. The conductivity remained constant throughout the 200 bending cycle test and is of great promise for flexible applications. The fabricated in plane device exhibited a specific capacitance ∼317 F cm−3, comparable to graphene based electrodes, with a good stability (90% after 1000 cycles).
 | ||
| Fig. 13 VS2 nanosheets for supercapacitor electrodes. S-V-S multilayers with intercalated NH3 molecules (a) is used to exfoliate thin layers of VS2 (b). The transfer is accomplished by vacuum filtration and the layers are transferred on mixed cellulose membranes (c). SEM (d) and HR-TEM (e) images of the multilayer with NH3. Reproduced with permission from ref. 158. Copyright 2011 American Chemical Society. | ||
MXenes derive from the family of layered hexagonal MAX phases, a group including more than 60 forms of ternary nitrides or carbides, where M represents an early transition metal (e.g. Ti, V, Cr, Nb, etc.), A stands for an A-group element (e.g. Al, Si, Sn, In, etc.), and X represents C and/or N. The synthesis of MXene involves removing the A of MAX compounds by selective etching (Fig. 14). While numerous MXenes were predicted theoretically,164,165 only about eleven MXenes are tested experimentally to date. Theoretically, it has been demonstrated that the surface termination (hydroxyl, fluorine) can be used to modify some of the MXene properties, such as their conductivity and bandgap.
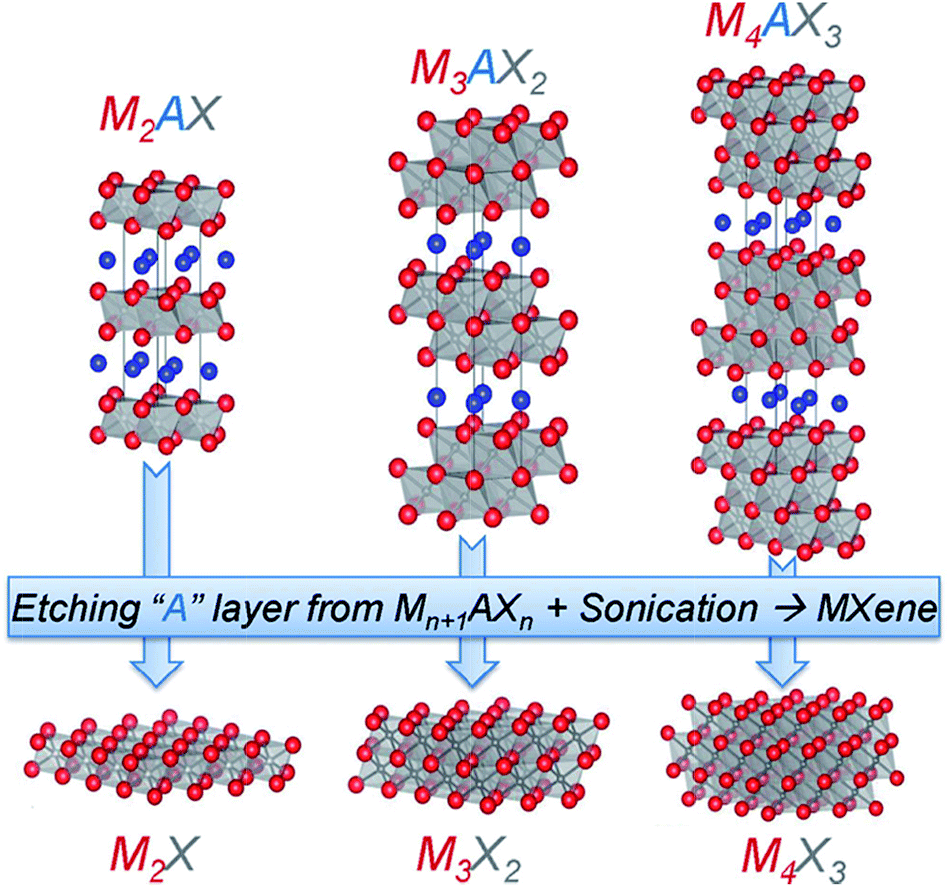 | ||
| Fig. 14 Schematic of the fabrication of MXene. Reproduced with permission from ref. 162. Copyright 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Among the reported studies, a MXene, Ti3C2Tx, was obtained by etching Ti3AlC2 by hydrofluoric acid (HF) at room temperature.161 A binder-free Ti3C2Tx MXene material with surface area of 98 m2 g−1 was obtained by delamination (single to few layers) and filtration of Ti3AlC2. It showed high flexibility and high volumetric capacitance (as high as 350 F cm−3) in aqueous electrolyte NaOH.159 To date, research towards electrode materials based on MXenes are limited; nevertheless, as a new promising 2D supercapacitor electrode material, much effort needs to be dedicated in this direction.
4.2 2D heterostructures
Regarding improvement of energy density in supercapacitors, making composites with transition–metal oxides and conducting polymers are being considered as they promise higher energy density than carbon materials. In addition, it has been shown that combining such high-energy metal oxides or conductive polymers with graphene based electrode materials can lead to high electrochemically accessible area. In addition, it leads to improvements in electrical conductivity, thermal stability and mechanical strength of the electrode, while minimizing the restacking of graphene layers.As a very high conductivity metal oxide, RuO2 has been known as a performing pseudocapacitive material.104,121,172,173 Recently, Dai and co-workers synthesized a capacitor-like RuO2–graphene (∼30 wt% graphene sheets) hybrid in solutions,121 which showed a specific capacitance of ∼370 F g−1 (at 2 mV s−1) and ∼280 F g−1 at a scan rate of 40 mV s−1 in 1 M KOH solution for a 0.2 V to −1.0 V voltage range. Another investigation of the performance of RuO2–rGO hybrid (Fig. 15) showed increasing capacitance with increasing Ru loading (at 16, 33 and 66 wt% Ru),174 with a specific capacitance of 357 F g−1 at a current density of 0.3 A g−1, which was attributed to the superior conductivity for rGO and an improved charge transport from RuO2 to the rGO layers. This is supported by structural comparison to materials made of rGO (Fig. 15a and b), and rGO–PANI (Fig. 15c and d). Ni(OH)2 nanoplates were also attached to graphene sheets to form a battery-like electrode material, and showed similar performance as RuO2–graphene structures.121 Finally pairing the two electrodes in an asymmetrical cell led to very attractive energy and power densities, suggesting that the performance of supercapacitors can be enhanced not only by hybrid electrode materials, but also by the combinations of electrodes with different properties.
 | ||
| Fig. 15 (a) SEM images of rGO electrode materials. TEM image of rGO (b), rGO–PANi (c and d) and rGO–RuO2 (e–f). Reproduced with permission from ref. 174. Copyright 2011 The Royal Society of Chemistry. | ||
Due to its great potential for enhanced supercapacitor performances, MnO2 in combination with graphene has received substantial interest.175–180 Wei and co-workers developed a facile method for graphene–MnO2 hybrid electrode material by microwave irradiation.179 With performance of 310 F g−1 at 2 mV s−1 (228 F g−1 at 500 mV s−1), MnO2–graphene surpasses pure graphene by a factor of three (104 and 103 F g−1, respectively). About 95% of capacitance retention was observed even after 15000 cycles. Another facile synthesis by Gao and co-workers showed high specific capacitance of 324 F g−1 in 1 M Na2SO4 with a retention of 97% after 1000 cycles.177 Other metal oxides such as Fe3O4,126 SnO2,181 and ZnO166 are also mixed with graphene to make supercapacitor electrode materials.
MoS2 has been combined with rGO to investigate the performance of hybrid energy storage devices. Covalent bonds occur between MoS2 and the oxygen atom in RGO, forming a Mo–O–C bond. The resulting hybrid 2D material is electroactive, and could be cycled in 1 M HClO4 between 0.25 V and up to 0.8 V, with energy density of 63 W h kg−1 and a specific capacitance decrease of 8% after 1000 cycles.182
WS2–rGO hybrids, synthesised by a simple hydrothermal method, were evaluated for their performance as supercapacitor material (Fig. 16).183 Comprehensive testing showed interesting specific capacitance of 350 F g−1 at a scan rate of 2 mV s−1 in 1 M Na2SO4, significantly higher than corresponding electrodes made of the bare materials measured at 130 and 70 F g−1. In addition, a high energy density (∼50 W h kg−1) and the capacitance remained almost the same after 1000 cycles in the hybrid structure, due to the faster transport of electrolyte ions in the presence of WS2.
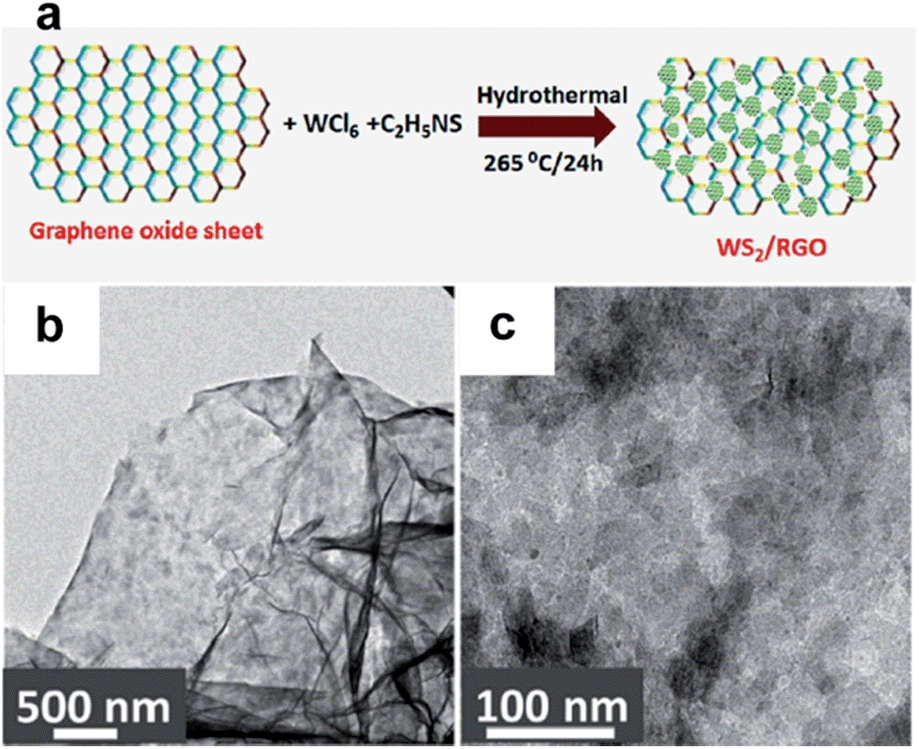 | ||
| Fig. 16 (a) Hydrothermal synthesis of the WS2–rGO materials. (b and c) TEM images of the resulting WS2–rGO electrode material. Reproduced with permission from ref. 183. Copyright 2013 American Chemical Society. | ||
5 3D nanostructures
3D porous structures of active materials provide large surface area, well-defined pathways to electrolyte access and mechanical stability for efficient supercapacitor electrodes. Generally, 3D electrodes are built either using metal foam as templates or organizing active materials into 3D nanostructures.5.1 Metal foams as 3D nanostructured current collectors
Metal foams such as nickel foam provide highly porous and conductive substrates for the deposition of active materials including graphene, CNTs, conjugated polymers and metal oxides to build supercapacitor electrodes.185–190 Compared with carbon based 3D substrates, metal foams have higher electrical conductivity and better mechanical properties but at the cost of larger density and lower specific surface area. 3D Co/Ni oxides with different morphologies were controllably synthesized and deposited on nickel foam substrates.186 The synthesis involves a co-electrodeposition of bimetallic (Ni, Co) hydroxide precursors on a nickel foam scaffold and a subsequent thermal transformation of the precursors to NixCo1−x oxides. The crystal structure, morphology and electrochemical performance of the 3D NixCo1−x oxides can be readily manipulated by simply varying the Co/Ni molar ratio in the electrodeposition electrolyte. With the increase in the Co/Ni molar ratio, the structure changes from a NiO crystal dominant structure to NiCo2O4 and finally to a Co3O4 dominant structure. The unique 3D network architecture provides large solid/liquid interfacial area and facilitates efficient electron and ion transport. The electrode prepared with Ni0.61Co0.39 oxide on Nickel foam showed a specific capacitance of 1523.0 F g−1 at 2 A g−1 current density and retained 95.30% of its specific capacitance at a current density of 30 A g−1 after 1000 cycles. This electrode was used as a positive electrode with activated carbon as a negative electrode to build an asymmetrical supercapacitor and delivered a prominent energy density of 36.46 W h kg−1 at a power density of 142 W kg−1.186Ozkan and co-workers used nickel foams to grow graphene and CNTs followed by a deposition of ruthenium oxide (RuO2) nanoparticles on CNTs (Fig. 17).185 The hierarchical and porous structures enable efficient access of electrolyte to the active materials (CNT–RuO2 network layer) and provide a conductive framework to charge transport attributed to the embedded CNTs. The composite had a high energy density of 39.28 W h kg−1 and power density of 128.01 kW kg−1 and showed an excellent cycle stability of 106% capacitance retention over 8100 cycles.
 | ||
| Fig. 17 (a) Schematic illustration of the preparation process of RGM nanostructure foam. SEM images of (b and c) as-grown GM foam, (d) lightly loaded RGM, and (e) heavily loaded RGM. Reproduced with permission from ref. 185. Copyright 2014 Macmillan Publishers Limited. | ||
Similarly, electrodes were prepared using well-aligned CoO nanowire arrays grown on 3D nickel foams with polypyrrole uniformly immobilized onto nanowire surface to utilize high electrochemical activities of CoO and polypyrrole as well as the short ion diffusion pathway in ordered mesoporous nanowires.190 Such synergy between materials and structures leads to a high specific capacitance of 2223 F g−1, good rate capability, and good cycle stability (99.8% capacitance retention after 2000 cycles). An asymmetrical supercapacitor device fabricated using the hybrid materials as the positive electrode and activated carbon film as the negative electrode achieved a maximum voltage of 1.8 V, high energy density (∼43.5 W h kg−1) and power density (∼5500 W kg−1 at 11.8 W h kg−1), and outstanding cycle stability (∼20000 times with 91.5% capacitance retention).
Mn/MnO2 core–shell 3D porous structures were fabricated from ordered polystyrene sphere templates. Mn was electrically deposited between polystyrene spheres from N-butyl-N-methyl pyrrolidinium TFSI ionic liquid solution of MnCl2. After removing polystyrene template using tetrahydrofuran, the 3D Mn template was anodized in a KCl aqueous solution to produce a Mn/MnO2 core–shell structure. Such core–shell porous structures contribute several positive factors including larger surface area, high electrical conductivity attributed to Mn and high capacitance of MnO2 for efficient supercapacitors. Especially, the electrolyte can easily access the active material, leading to high specific capacitance at high scan rate. The optimum specific capacitance of the electrode at 500 mV s−1 is 996 F g−1, maintaining 83% of the capacitance measured at 5 mV s−1.188 Composite supercapacitor electrodes based on metal foams have high electrical conductivity, good mechanical properties, and easy access of electrolyte to active materials, which leads to high specific capacitance and good cycle stability.
5.2 3D nanostructures based on carbon materials
Mesoporous carbon (MC) has been used as promising materials for supercapacitor electrode materials because of their large specific surface area and interconnected pores with tunable size. Since Ryoo and co-workers reported the fabrication of ordered MC from the ordered mesoporous silica molecular sieves as templates,191,192 much effort has been devoted to exploring the application of such materials as supercapacitor electrodes. The studies of utilizing MC as electrode materials for EDLCs suggest that the pore size and symmetry have profound effect on the performance by influencing the accessibility of electrolytes. The pore size distribution with best performance ranges from 0.6 to 3 nm depending on different types of electrolytes.193,194 While these EDLCs can reach the specific capacitance of 140 F g−1 which is much higher than those using commercial carbon, their performance is limited by the relatively low electrical conductivity.195Further improvement can be achieved by doping196 and depositing electrochemically pseudocapacitive materials such as metal oxides,197–199 PANI,200–204 and their mixtures205 onto MC templates. For example, nitrogen- and sulfur-codoped 3D cubic ordered MC (KNOMC) materials with controlled dopant content (10.0–4.6 atom % for nitrogen and 0.94–0.75 atom % for sulfur) have demonstrated a specific capacitance of 320 F g−1 because the incorporated S and N species function as pseudo-active materials.196 Metal oxides have also been added onto MC to increase the capacitance. For example, MnO2 nanoparticles were imbedded in the MC through a redox reaction between permanganate ions and carbons.199 The ordered mesoporous structures were retained through the control of the concentration of permanganate ion. The resulted composite structure showed a specific capacitance of 200 F g−1 with decent electrochemical stability and reversibility. Nanostructured PANI has been deposited on various MC substrates through chemical oxidative polymerization and it greatly increased the specific capacitance due to its high capacitance and electrical conductivity.206 Li and co-workers have deposited PANI needle-like structures on MC and studied the effect of the structure on electrochemical properties.202 It was found that the composite with 60% PANI deposition has the best performance (517 F g−1), suggesting that a bimodal pore structure with some open pores facilitates the ion diffusion and shorten the charge transfer distance. PANI–CMK-3–MnO2 ternary nanocomposites were built by co-deposition of conductive PANI and MnO2 on MC and further improved the specific capacitance to 695 F g−1.205 In such composite, MnO2 nanoparticles stabilizes the interaction between the quinoid ring of PANI and the CMK-3–MnO2, while the PANI nanolayer retards the dissolution of MnO2 nanoparticles during the charge–discharge process, leading to high cycle stability.
Theoretical studies207,208 and experimental investigation209 of the effect of pore size and distribution on ionic transport properties of porous structures suggest that the coexistence of macropore and mesopore would increase the efficiency of porous carbon as supercapacitor electrodes. Compared with MC, an ordered carbon structure with meso/macro/micropores can achieve a specific capacitance as high as 350 F g−1.209 Hierarchical carbon structures with a combination of meso/macro/micropores have been produced through various approaches,210–218 and used as electrode with and without metal oxides.219,220 These structures have demonstrated great potential in constructing high performance supercapacitors. Further improvement requires better control of pore size and distribution as well as increased electrical conductivity of porous carbon.
Taking advantages of extraordinary properties of CNTs and graphene including large specific surface area, high electrical conductivity and superior strength, assembling CNTs and graphene into 3D porous structures offers excellent opportunities for building high performance supercapacitors. 3D CNTs and graphene not only provide large surface area for double layer supercapacitors but also function as supports for metal oxides and conjugated polymers for pseudocapacitors. GO sheets dispersed in aqueous solutions offer promising materials for building 3D structures for graphene based supercapacitors. The GO can be reduced chemically or thermally to increase the electrical conductivity. To eliminate the aggregation of graphene sheets that limits the electron and ion transport, Huang and co-workers have transformed 2D graphene sheets into crumpled paper ball structures by isotropic capillary compression in rapidly evaporating droplets of GO dispersion using an aerosol spray pyrolysis setup.221Fig. 18 compares the microstructures of mechanically compressed pellets of stacked graphene materials of flat graphene sheets (a), heavily winkled sheets (b) and crumpled graphene balls (c) using surface and cross sectional SEM.222 The flat graphene sheets pack like a stack of paper with a smooth featureless surface and an ordered lamellar cross section (Fig. 18a). The pellets of wrinkled sheets (Fig. 18b) have uneven surfaces with many flattened islands in micrometers. The cross sectional SEM indicates a largely lamellar but significantly more disordered microstructure. For the crumpled balls (Fig. 18c), the morphology of individual particles in the compressed pellet was largely unchanged. Compared to flat or wrinkled sheets, the crumpled graphene balls have larger surface area and better defined voids for electron and ion transport. The electrodes based on crumpled graphene balls can deliver much higher specific capacitance (150 F g−1) and better rate capability. Furthermore, using flat graphene sheets as binder for the crumpled particles eliminates the use of insulating binder materials like PTFE colloids and further improve device performances.222
 | ||
| Fig. 18 Paper models and SEM images showing stacks of (a) flat graphene sheets, (b) heavily wrinkled sheets, and (c) crumpled graphene balls. Reproduced with permission from ref. 222. Copyright 2013 American Chemical Society. | ||
Graphene aerogel (GA) as electrode material for supercapacitor has drawn considerable attention lately due to its open 3D network structure that not only effectively reduces the aggregation of graphene nanosheets but also facilitates effective electrolyte ion transportation. As a result, more electrochemically active sites could be utilized compared to 2D graphene nanosheets. For example, Duan and co-workers have prepared a 3D holey graphene framework with a hierarchical porous structure by etching the GO sheets via H2O2 molecules.223 The resulting holey graphene framework showed a large specific surface area of 830 m2 g−1, which is significantly greater than that of non-holey one (∼260 m2 g−1). As a consequence, holey graphene framework electrodes delivered a specific capacitance of 310 F g−1 at 1 A g−1, while 208 F g−1 was achieved for non-holey graphene framework electrodes. It is to be noted that the electrical conductivity of GA is generally lower than that of pristine graphene due to the formation of defect sites during the oxidation–reduction process.224 It is advantageous to fabricate a GA with enhanced electrical conductivity. Recently, Thomas, Zhai, and co-workers have reported functionalized GA composites as supercapacitor electrodes with reduced electrical resistivity by incorporating palladium nanoparticles (Fig. 19a and b).225 The electrical resistivity of the functionalized GA (16 Ω cm) is substantially reduced compared to pristine GA (950 Ω cm). Consequently, the specific capacitance of the functionalized GA reached 175.8 F g−1, while only 51.9 F g−1 was obtained for the pristine GA.
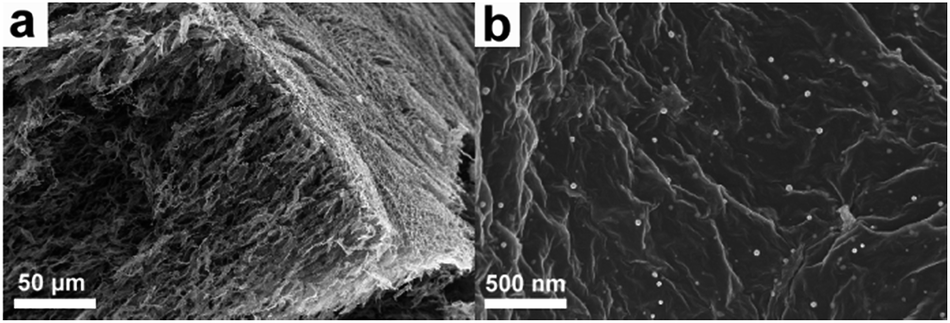 | ||
| Fig. 19 SEM images of (a) functionalized graphene aerogel and (b) graphene nanosheets uniformly loaded with metal nanoparticles. | ||
Similar to MC, high performance composite electrodes have been developed by doping graphene with nitrogen and boron226 and integrating pseudo-active materials on 3D CNT and graphene structures.227–239 3D macroporous electrodes of chemically modified graphene (CMG) were fabricated using polystyrene colloidal particles as a sacrificial template.227 A thin layer of MnO2 was deposited onto the porous electrodes to further improve the capacitance. The porous graphene structure facilitates fast ionic transport within the electrode while preserving decent electrical conductivity. Such combination grants the electrodes excellent electrochemical properties such as high specific capacitance (389 F g−1 at 1 A g−1) and 97.7% capacitance retention when the current increases to 35 A g−1. In addition, an asymmetrical supercapacitor assembled using MnO2/CMG composite electrode and a CMG electrode achieves an energy density of 44 W h kg−1, a power density of 25 kW kg−1, and excellent cycle life.
As discussed previously, ordered 3D porous structures with controlled pore size and distribution would improve the electrochemical properties. The concept of inherently nanoporous 3D pillared vertically aligned carbon nanotube (VA-CNT)–graphene hybrid architecture with a large surface area has been explored to develop efficient supercapacitors (Fig. 20a).240–242 Dai and co-workers prepared a VA-CNT–graphene hybrid by the intercalated growth of VA-CNTs into thermally expanded highly ordered pyrolytic graphite (Fig. 20b).241 The resulting VA-CNT–graphene hybridized with a nickel hydroxide coating also demonstrated a high specific capacitance with a notable charging-rate capability, as high as 1065 F g−1 (based on the active material) at a current density of 22.1 A g−1. Sun and co-worker have grown carbon nanotubes on graphene sheets through a facile one-pot pyrolysis strategy where cobalt ions and urea were mixed with GO followed by drying and heating the mixture at 900 °C. The length of the carbon nanotubes was tuned by adjusting the amount of precursors (Fig. 20c and d). The resulting graphene–CNT composites showed a high specific surface area of 903 m2 g−1 and maximum specific capacitance of 413 F g−1.242
 | ||
| Fig. 20 (a) Schematic diagram of a 3D pillared VACNT–graphene nanostructure. (b) A SEM image of the thermally expanded graphene layers intercalated with VACNTs for 10 minutes pyrolysis times. (c and d) SEM images of CNTs grown on graphene using urea as carbon sources. Reproduced with permission from ref. 240–242. Copyright: 2008, 2011, and 2014 American Chemical Society. | ||
As an example of depositing pseudocapacitive metal oxides on graphene/CNT scaffolds, 3D flower-like β-Ni(OH)2/GO/CNTs composites were prepared via a facile hydrothermal reaction in alkaline solutions. CNTs were introduced to the composite to improve the electrical conductivity. The electrochemical properties of the composites produced at different reaction time were evaluated to understand the effect of composite structures on energy storage efficiency. Hydrothermal reaction converted α-Ni(OH)2 to β-Ni(OH)2 (1 h reaction time) and further 3D β-Ni(OH)2 nanoflowers to stacked nanosheets (10 h). The electrochemical characterization of three different structures indicates that β-Ni(OH)2 composite prepared through one hour hydrothermal reaction has the best performance with a high specific capacitance of ∼1815 F g−1 based on Ni(OH)2 (nearly 96% of its theoretical pseudocapacitance) at 2 A g−1 and a good cycling performance of ∼97% capacitance retention after 2000 cycles at 10 A g−1, attributed to higher stability (vs. α-Ni(OH)2) and more efficient ion diffusion (vs. stacked β-Ni(OH)2).228
Zhai and co-workers have incorporated MnO2 into MWCNT 3D networks as supercapacitor electrodes. More importantly, they have discovered the relationship between MnO2 migration phenomenon and the scan rates. The structural evolution were examined by studying the structure and electrochemical properties of identical electrodes after 500 CV cycles at various scan rates.243 By using Electrochemical Quartz Crystal Microbalance (EQCM) to monitor the mass change of the electrode in situ and SEM to examine the electrodes after CV cycles, MnO2 was found to “migrate” from the MWCNTs network and “deposit” on the electrode surface upon cycling. The structural change mechanism contains the dissolution of MnO2 upon reduction, the diffusion of the reduced Mn species from the MWCNT network toward the electrolyte solution, and the deposition of MnO2 on the electrode surface upon oxidation.243 The structural change of MnO2 is highly dependent on the scan rate. For instance, the reduced Mn species have more diffusion time at a lower scan rate, while less reduced species are generated due to the lower associated current density and vice versa. A scan rate that might cause the greatest structural evolution of MnO2 is expected based on this contradictory effect. Fig. 21a shows the cross section of as-synthesized MWCNT/MnO2 composites sprayed on a graphite substrate (MCM0) as well as an X-ray line scan spectra of Mn element. The SEM and X-ray line scan spectral data clearly exhibit that MnO2 nanoflakes are uniformly embedded in the MWCNTs network as the active materials while the MWCNT network serves both as the support for MnO2 nanoflakes and current collector. In contrast, Fig. 21b–f shows the striking changes of electrode structures after 500 CV cycles, and the structural change varies at different scan rates. As a result, a higher specific capacitance could be achieved with a lower scan rate; however, the specific capacitance considerably drops upon cycling due to the dissolution of active material. Therefore, either developing a protective layer as mentioned in the 1D section or manipulating a suitable scan rate that induces the least structural changes while maintaining high specific capacitance is required.
 | ||
| Fig. 21 Cross-section morphologies and X-ray line scan spectra of Mn element in pristine composites (MCM0), and after 500 cycles at 20 mV s−1 (MCM20), 60 mV s−1 (MCM60), 100 mV s−1 (MCM100), 300 mV s−1 (MCM300), and 500 mV s−1 (MCM500). (a) MCM0, (b) MCM20, (c) MCM 60, (d) MCM100, (e) MCM300, and (f) MCM500. Reproduced with permission from ref. 232. Copyright 2012 Elsevier. | ||
5.3 3D nanostructures based on pseudocapacitive materials
Different nanostructures of pseudocapacitive materials offer intriguing composition for 3D supercapacitors. Various types of metal oxide nanostructures produced through sol–gel, solvothermal and other approaches have been evaluated electrochemically to understand the impact of their structures on the energy storage properties. NiO nanostructures with three distinct morphologies (flower-like, slice-like and particle-like) were fabricated by a sol–gel method. The BET specific surface area of nanoflower, nanoslice, and nanoparticle NiO was 159, 193, and 233 m2 g−1 and the average pore size was 16.7, 6.3, and 8.8 nm, respectively (Fig. 22).244 With the lowest surface area and largest pore size, the nanoflower NiO nanostructures formed a 3D network, offering the most efficient contact with and transport of the electrolyte. Therefore, this structure granted the best electrochemical properties among three different nanostructures. This study suggests that it is important to facilitate the transport of ions to active materials on the top of large surface area.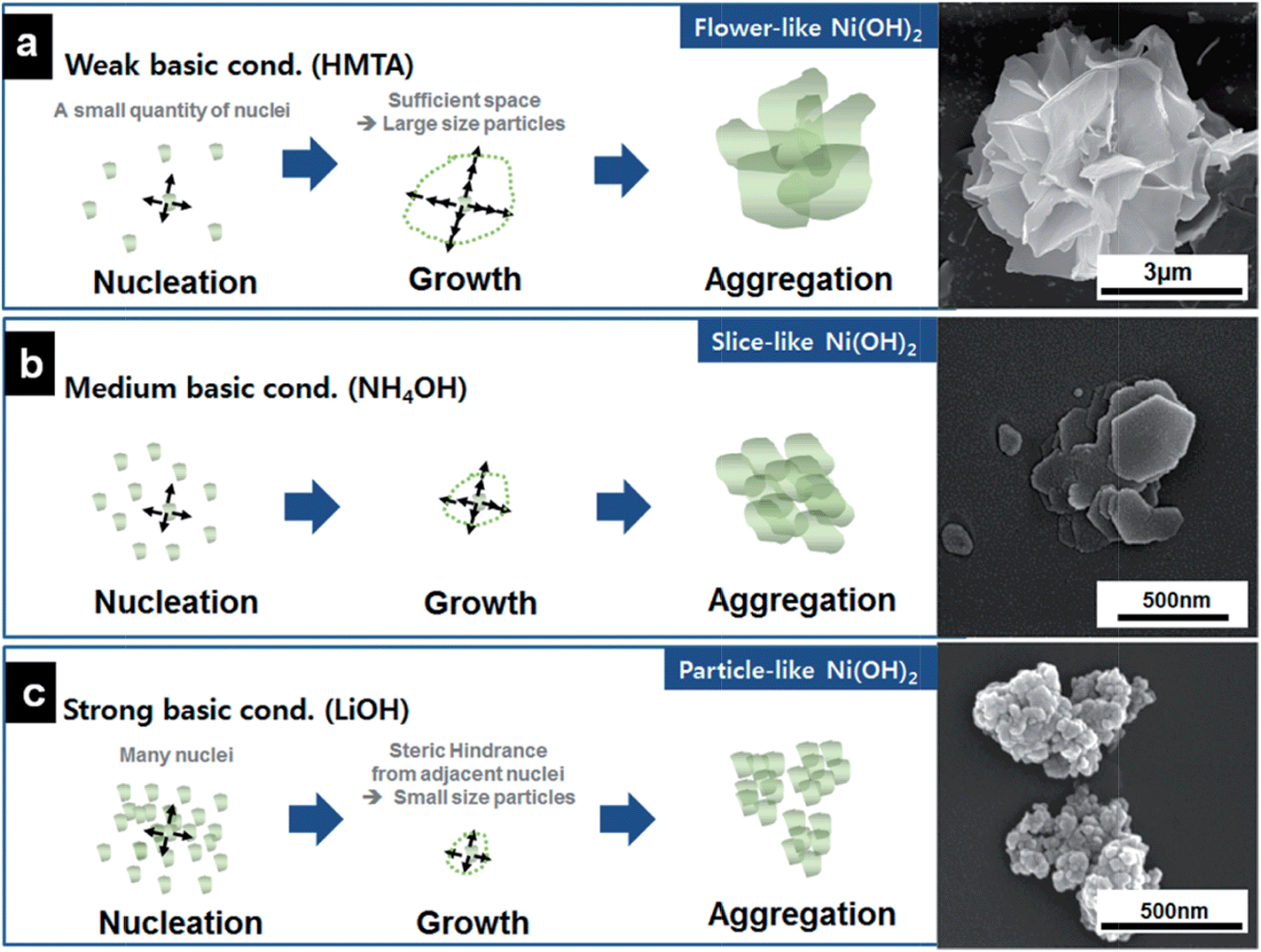 | ||
| Fig. 22 Schematic diagram showing the formation process of (a) flower-, (b) slice-, and (c) particle-like Ni(OH)2 under different pH conditions; corresponding SEM Images of as-prepared samples are in the Right Panel. Reproduced with permission from ref. 244. Copyright 2013 American Chemical Society. | ||
3D hierarchical Co3O4 twin-spheres with an urchin-like structure were produced by a solvothermal synthesis of cobalt carbonate hydroxide hydrate followed by calcination. The morphology of the precursors evolved from nanorods to sheaf-like bundles, to flower-like structures, to dumbbell-like particles, and eventually to twin-spheres through a three-stage self-assembly of nanorods. Such structure provided larger surface area and better accessibility of electrolytes compared to nanorods and nanodiscs of Co3O4. The specific capacitances of the urchin-like structure evaluated from the discharge curves were 781, 754, 700, 670, and 611 F g−1 at current densities of 0.5, 1, 2, 4, and 8 A g−1, respectively.245 Similar urchin-like MnO2 nanostructures were also produced through a hydrothermal method. It was found that the phase of the products transformed from α-MnO2 to ε-MnO2 when Al3+ was substituted by Fe3+ in the solution. At the same time, the morphology of the product was changed from urchin-like clusters to 3D clew-like structures.246 The 3D clew-like ε-MnO2 showed a specific capacitance of 120 F g−1 at the scan rate of 5 mV s−1.
Current research in this field focuses on the structural control of the products rather than achieving good electrochemical properties. These nanostructures, although present interesting shapes, have not demonstrated superior performance compared with pseudocapacitive materials deposited on porous carbons and metal foams. In order to advance the application of metal oxide nanostructures in supercapacitors, it is necessary to improve the electrical conductivity, electrolyte accessibility and cycle stability of the materials.
6 Conclusion and outlook
The quest for higher energy and power for supercapacitors to complement or compete with batteries has led to a major effort in developing advanced electrode materials. In this review article, we overviewed the fabrication and performance of 0–3 dimensional nanostructured electrode materials and discussed the impact of structures on the electrochemical properties. Table 1 exhibits a summary of the properties and characteristics of supercapacitor electrode materials based on various dimensional nanostructures.| Category | Subcategory | Selected examples | Electrochemical properties | Ref. |
|---|---|---|---|---|
| 0D | 0D solid nanostructures | Onion-like carbon | 0.9 mF cm−2; ∼70% rate capability from 1 to 100 V s−1; ∼100% capacitance retention after 10000 cycles |
23 |
| Porous Au/MnO2 hybrid electrode | 1145 F g−1 at 50 mV s−1; 80% capacitance retention after 500 cycles | 24 | ||
| LaMnO3 perovskite nanoparticles | 609.8 F g−1 at 2 mv s−1; 57% rate capability from 2 to 100 mV s−1 | 25 | ||
| 0D hollow nanostructures | Hollow carbon nanospheres | 251 F g−1 at 50 mV s−1; 77% rate capability from 50 to 500 mV s−1; 98% capacitance retention after 200 cycles | 28 | |
| NiO multi shelled nanospheres | 612.5 F g−1 at 0.5 A g−1; 83.1% rate capability from 0.5 to 3 A g−1; 90.1% capacitance retention after 1000 cycles | 34 | ||
| 0D core–shell nanostructures | HCS@PANI core–shell spheres | 525 F g−1 at 0.1 A g−1; 50% rate capability from 0.1 to 10 A g−1; 73% capacitance retention after 1000 cycles | 37 | |
| GHCS@MnO2 core–shell spheres | 190 F g−1 at 0.1 A g−1; 55% rate capability from 0.1 to 10 A g−1; 99% capacitance retention after 1000 cycles | 38 | ||
| 1D | 1D homostructures | Oxygen-deficient Fe2O3 nanorods | 64.5 F g−1 at 10 mV s−1; ∼35% rate capability from 10 to 400 mV s−1; 95.2% capacitance retention after 10000 cycles |
50 |
| Carbon nanopillars | 3.4 mF cm−2 at 100 mV s−1; 95% capacitance retention after 10000 cycles |
51 | ||
| PANI nanowires | 1142 F g−1 at 5 A g−1; 82% rate capability from 1 to 40 A g−1; 95% capacitance retention after 500 cycles | 55 | ||
| NiCo2O4 nanoneedle arrays | 1118.6 F g−1; 89.4% capacitance retention after 2000 cycles | 59 | ||
| MnO2 nanotube arrays | 320 F g−1 at 20 mV s−1; 70% rate capability from 1 to 10 A g−1; 81% capacitance retention after 2000 cycles | 60 | ||
| Vertically aligned CNTs | 200 F g−1 at 20 A g−1; 72% rate capability from 20 to 150 A g−1 | 65 | ||
| 1D heterostructures | AuPd@MnO2 core–shell nanopillars | 603 F g−1 at 5 mV s−1; 52% rate capability from 5 to 100 mV s−1; 93% capacitance retention after 5000 cycles | 52 | |
| CuO@AuPd@MnO2 core–shell nanowhiskers | 1376 F g−1 at 5 mV s−1; 58% rate capability from 5 to 100 mV s−1; 99% capacitance retention after 5000 cycles | 83 | ||
| CNT@PPy–MnO2 core–shell nanotubes | 268 F g−1; 93% rate capability from 5 to 100 mV s−1; 90% capacitance retention after 5000 cycles | 92 | ||
| Co3O4@MnO2 core–shell nanowires | 480 F g−1 at 2.67 A g−1; 56% rate capability from 4 to 44.7 mA cm−2; 97.3% capacitance retention after 5000 cycles | 97 | ||
| V2O5@PPy core–shell nanoribbons | 308 F g−1 at 100 mA g−1; 95% capacitance retention after 10000 cycles |
103 | ||
| 2D | 2D homostructures | Chemically modified graphene (CMG) | 135 F g−1 at 10 mA; 97% rate capability from 20 to 400 mV s−1 | 125 |
| Laser reduced graphite oxide | 0.51 mF cm−2 at 40 mV s−1; 70% capacitance retention after 10000 cycles |
144 | ||
| 2D MnO2 | 774 F g−1 at 0.1 A g−1; 63% rate capability from 0.1 to 1 A g−1; >100% capacitance retention after 10000 cycles |
150 | ||
| Flower-like MoS2 nanosheets | 168 F g−1 at 1 A g−1; 92.6% capacitance retention after 3000 cycles | 155 | ||
| VS2 nanosheets | 317 F cm−3; ∼100% capacitance retention after 1000 cycles | 158 | ||
| Ti3C2Tx (MXene) | 350 F cm−3 at 1 A g−1; ∼60% rate capability from 2 to 100 mV s−1; ∼100% capacitance retention after 10000 cycles |
159 | ||
| 2D heterostructures | RuO2–graphene hybrid | ∼370 F g−1 at 2 mV s−1; 76.3% rate capability from 2 to 40 mV s−1; 92% capacitance retention after 5000 cycles | 121 | |
| Graphene–MnO2 | 310 F g−1 at 2 mV s−1; 73.5% rate capability from 2 to 500 mV s−1; 95% capacitance retention after 15000 cycles |
179 | ||
| MoS2–rGO | 265 F g−1 at 10 mV s−1; 77.4% rate capability from 10 to 80 mV s−1; 92% capacitance retention after 1000 cycles | 182 | ||
| WS2–rGO | 350 F g−1 at 2 mV s−1; 42.8% rate capability from 2 to 200 mV s−1; ∼100% capacitance retention after 1000 cycles | 183 | ||
| 3D | 3D metal foam | Ni0.61Co0.39 oxide on nickel foam | 1523.0 F g−1 at 2 A g−1; 95.3% capacitance retention after 1000 cycles | 186 |
| CoO–PPy on 3D nickel foam | 2223 F g−1 at 1 mA cm−2; 29.1% rate capability from 1 to 50 mA cm−2; 99.8% capacitance retention after 2000 cycles | 190 | ||
| Mn/MnO2 core–shell 3D porous structure | ∼1200 F g−1 at 5 mV s−1; 83% rate capability from 5 to 500 mV s−1; 96% capacitance retention after 2000 cycles | 188 | ||
| 3D carbonaceous materials | Crumpled graphene balls | 150 F g−1 at 0.1 A g−1; 40% rate capability from 0.1 to 2.5 A g−1; ∼100% capacitance retention after 5000 cycles | 222 | |
| VA-CNT–graphene with Ni(OH)2 coating | 1065 F g−1 at 22.1 A g−1; 70% rate capability from 5 to 30 mV s−1; 96% capacitance retention after 20000 cycles |
241 | ||
| β-Ni(OH)2/GO/CNTs | 1815 F g−1 at 2 A g−1; 47.2% rate capability from 2 to 20 A g−1; 97% capacitance retention after 2000 cycles | 228 | ||
| 3D pseudocapacitive materials | 3D NiO nanoflowers | 480 F g−1 at 0.5 A g−1; 52.5% rate capability from 0.5 to 5 A g−1; ∼90% capacitance retention after 400 cycles | 244 | |
| 3D Co3O4 urchin-like nanostructures | 781 F g−1 at 0.5 A g−1; 78.2% rate capability from 0.5 to 8 A g−1; 97.8% capacitance retention after 1000 cycles | 245 |
For quite some time, 0D materials like activated carbon have been the work horse for developing supercapacitor electrodes. However, the necessity to add an insulating polymer binder to make the actual electrode diminishes its performance. The emergence of perovskites as active materials for solar cell applications has inspired the research of using them for supercapacitor applications. It has been observed that oxygen intercalation in perovskite-type materials can be used to store the charges and has a bright future as the potential material to improve the performance of supercapacitor electrodes. Among other 0D materials, single and multi-shelled hollow structures stand out because of their highly enhanced surface area. 1D nanostructure can also provide efficient transport pathways for both electrons and ions in the supercapacitor electrode. Particularly, binder-free and highly ordered designs can further improve the specific capacitance of the electrodes. Moreover, a rational 1D heterogeneous design helps to achieve enhanced rate capability and cycle stability.
Due to their high surface area, 2D materials offer a unique opportunity to surpass the current limitations of electrode materials. The great advantage of using 2D materials like graphene for supercapacitor application is its high conductivity and large surface area. In order to improve the energy density of pristine graphene-based devices, composites are made with 2D graphenes and conducting polymer or transition metal oxides. This resulted in improved devices with high thermal stability and mechanical strength with minimum restacking of 2D layers. However, stacking of these 2D layers to accomplish a considerable volume poses a major challenge. The availability of a large variety of van der Waals layered materials provides considerable opportunities to develop new 2D materials for future supercapacitor electrodes. Furthermore, MXene, as a new family of 2D materials, shows a great potential for supercapacitor electrode applications.
Building 3D structures with a controlled pore size distribution is very attractive as electrode materials. Utilizing 3D conductive materials as templates and depositing pseudocapacitive materials like conductive polymers and metal oxide on to these 3D structures is a very efficient method to construct high performing supercapacitor electrodes. Composite 3D metal foams are also very promising because of the high electrical conductivity and easy accessibility of electrolyte ions. Assembling prefabricated nanostructures into 3D structures is another very important direction to obtain highly efficient supercapacitor electrodes since nanostructures can be manipulated by controlling synthetic parameters. However, more effort needs to be focused on optimizing the interface between these nanostructures and charge collecting electrodes.
This review is focused on the supercapacitor electrode performance based on the dimensionality of the material used as the electrode. Due to the variability of conditions and parameters used to evaluate the materials, it is not easy to compare the performance of the electrodes with different dimensionality. Nevertheless, we can infer from this review that there are clear advantages and disadvantages for different dimensionalities. For example, it is possible to control the pore size of 0D carbon electrode to less than 10 nm to obtain better electrolyte accessibility than 1D materials, however, the conductivity of 0D materials remains low in the presence of an insulating polymer binder. In the case of 1D materials, it is possible to deposit active materials on a conductive nanowire, but precisely controlling the accessible space between high aspect ratio structures is not a simple task. A feasible future direction is the combination of different dimensionalities to generate a composite which can provide high conductivity, surface area, mechanical and performance stability and capacitance. 2D graphene made into aerogels (3D) with incorporated active nanoparticles like NiO (0D) is an example of such hybrid system. Such composite could also combine the properties of both EDLCs (long cycle life) and pseudocapacitive electrodes (better specific capacitance). Recently, there is a concerted effort to develop 2D materials beyond graphene using van der Waals solids. Many of these solids constitute promising materials for supercapacitor electrodes. It is expected that the combination of these nanomaterials leads to a new generation of supercapacitors electrodes with high energy and power densities in the near future.
Acknowledgements
J.T. acknowledges Florida Space Research Institute and Florida Space Grant Consortium for the financial support.Notes and references
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, Springer, New York, 1999 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- H. Gleiter, Acta Mater., 2000, 48, 1–29 CrossRef CAS.
- V. Pokropivny and V. Skorokhod, Mater. Sci. Eng., C, 2007, 27, 990–993 CrossRef CAS PubMed.
- J. N. Tiwari, R. N. Tiwari and K. S. Kim, Prog. Mater. Sci., 2012, 57, 724–803 CrossRef CAS PubMed.
- L. L. Zhang and X. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong, X. Li and L. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889–4899 CrossRef CAS PubMed.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5, 72–88 RSC.
- J. Gamby, P. Taberna, P. Simon, J. Fauvarque and M. Chesneau, J. Power Sources, 2001, 101, 109–116 CrossRef CAS.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- D.-W. Wang, F. Li, Z.-G. Chen, G. Q. Lu and H.-M. Cheng, Chem. Mater., 2008, 20, 7195–7200 CrossRef CAS.
- S. Prabaharan, R. Vimala and Z. Zainal, J. Power Sources, 2006, 161, 730–736 CrossRef CAS PubMed.
- L. Hu, W. Wang, J. Tu, J. Hou, H. Zhu and S. Jiao, J. Mater. Chem. A, 2013, 1, 5136–5141 CAS.
- M. Xue, Z. Xie, L. Zhang, X. Ma, X. Wu, Y. Guo, W. Song, Z. Li and T. Cao, Nanoscale, 2011, 3, 2703–2708 RSC.
- M. P. Yeager, D. Su, N. S. Marinković and X. Teng, J. Electrochem. Soc., 2012, 159, A1598–A1603 CrossRef CAS PubMed.
- Y.-Z. Zheng, H.-Y. Ding and M.-L. Zhang, Mater. Res. Bull., 2009, 44, 403–407 CrossRef CAS PubMed.
- Y. Chen, Z. Yu, Y. Chen, L. Luo and X. Wang, Materials for Renewable Energy & Environment (ICMREE), 2011 International Conference on, Shanghai, 2011 Search PubMed.
- X. Du, C. Wang, M. Chen, Y. Jiao and J. Wang, J. Phys. Chem. C, 2009, 113, 2643–2646 CAS.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- C. Portet, G. Yushin and Y. Gogotsi, Carbon, 2007, 45, 2511–2518 CrossRef CAS PubMed.
- D. Pech, M. Brunet, H. Durou, P. Huang, V. Mochalin, Y. Gogotsi, P.-L. Taberna and P. Simon, Nat. Nanotechnol., 2010, 5, 651–654 CrossRef CAS PubMed.
- X. Lang, A. Hirata, T. Fujita and M. Chen, Nat. Nanotechnol., 2011, 6, 232–236 CrossRef CAS PubMed.
- J. T. Mefford, W. G. Hardin, S. Dai, K. P. Johnston and K. J. Stevenson, Nat. Mater., 2014, 13, 726–732 CrossRef CAS PubMed.
- X. Lai, J. E. Halpert and D. Wang, Energy Environ. Sci., 2012, 5, 5604–5618 CAS.
- Z. Zhong, Y.-d. Yin, B. Gates and Y. Xia, Adv. Mater., 2000, 12, 206–209 CrossRef CAS.
- B. You, J. Yang, Y. Sun and Q. Su, Chem. Commun., 2011, 47, 12364–12366 RSC.
- X. Tang, Z.-h. Liu, C. Zhang, Z. Yang and Z. Wang, J. Power Sources, 2009, 193, 939–943 CrossRef CAS PubMed.
- S.-W. Bian, Y.-P. Zhao and C.-Y. Xian, Mater. Lett., 2013, 111, 75–77 CrossRef CAS PubMed.
- C.-Y. Cao, W. Guo, Z.-M. Cui, W.-G. Song and W. Cai, J. Mater. Chem., 2011, 21, 3204–3209 RSC.
- W. Yu, X. Jiang, S. Ding and B. Q. Li, J. Power Sources, 2014, 256, 440–448 CrossRef CAS PubMed.
- T. Zhu, Z. Wang, S. Ding, J. S. Chen and X. W. D. Lou, RSC Adv., 2011, 1, 397–400 RSC.
- Z. Yang, F. Xu, W. Zhang, Z. Mei, B. Pei and X. Zhu, J. Power Sources, 2014, 246, 24–31 CrossRef CAS PubMed.
- Y. Wang, A. Pan, Q. Zhu, Z. Nie, Y. Zhang, Y. Tang, S. Liang and G. Cao, J. Power Sources, 2014, 272, 107–112 CrossRef CAS PubMed.
- X. Lai, J. Li, B. A. Korgel, Z. Dong, Z. Li, F. Su, J. Du and D. Wang, Angew. Chem., Int. Ed., 2011, 50, 2738–2741 CrossRef CAS PubMed.
- Z. Lei, Z. Chen and X. Zhao, J. Phys. Chem. C, 2010, 114, 19867–19874 CAS.
- Z. Lei, J. Zhang and X. Zhao, J. Mater. Chem., 2012, 22, 153–160 RSC.
- L. Fan, L. Tang, H. Gong, Z. Yao and R. Guo, J. Mater. Chem., 2012, 22, 16376–16381 RSC.
- Z.-C. Yang, C.-H. Tang, H. Gong, X. Li and J. Wang, J. Power Sources, 2013, 240, 713–720 CrossRef CAS PubMed.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- X. Lu, G. Wang, T. Zhai, M. Yu, J. Gan, Y. Tong and Y. Li, Nano Lett., 2012, 12, 1690–1696 CrossRef CAS PubMed.
- X. Lu, D. Zheng, T. Zhai, Z. Liu, Y. Huang, S. Xie and Y. Tong, Energy Environ. Sci., 2011, 4, 2915–2921 CAS.
- X. Lu, G. Wang, T. Zhai, M. Yu, S. Xie, Y. Ling, C. Liang, Y. Tong and Y. Li, Nano Lett., 2012, 12, 5376–5381 CrossRef CAS PubMed.
- P. Yang, Y. Ding, Z. Lin, Z. Chen, Y. Li, P. Qiang, M. Ebrahimi, W. Mai, C. P. Wong and Z. L. Wang, Nano Lett., 2014, 14, 731–736 CrossRef CAS PubMed.
- Z. Yu, J. Moore, J. Lorenzo, C. Li and J. Thomas, ECS Trans., 2014, 61, 31–36 CrossRef PubMed.
- Z. Yu, J. Moore, B. Duong, C. Li and J. Thomas, ECS Trans., 2014, 61, 49–53 CrossRef PubMed.
- H. Wang, Q. Gao and L. Jiang, Small, 2011, 7, 2454–2459 CAS.
- J. Chen, B. J. Wiley and Y. Xia, Langmuir, 2007, 23, 4120–4129 CrossRef CAS PubMed.
- X. Lu, Y. Zeng, M. Yu, T. Zhai, C. Liang, S. Xie, M. S. Balogun and Y. Tong, Adv. Mater., 2014, 26, 3148–3155 CrossRef CAS PubMed.
- B. Duong, Z. Yu, P. Gangopadhyay, S. Seraphin, N. Peyghambarian and J. Thomas, Adv. Mater. Interfaces, 2014, 1, 1300014 Search PubMed.
- Z. Yu, B. Duong, D. Abbitt and J. Thomas, Adv. Mater., 2013, 25, 3302–3306 CrossRef CAS PubMed.
- Z. Yu, C. Li, D. Abbitt and J. Thomas, J. Mater. Chem. A, 2014, 2, 10923–10929 CAS.
- B. Duong, H. Khurshid, P. Gangopadhyay, J. Devkota, K. Stojak, H. Srikanth, L. Tetard, R. A. Norwood, N. Peyghambarian, M. H. Phan and J. Thomas, Small, 2014, 10, 2840–2848 CrossRef CAS PubMed.
- G.-Y. Zhao and H.-L. Li, Microporous Mesoporous Mater., 2008, 110, 590–594 CrossRef CAS PubMed.
- K. Wang, J. Huang and Z. Wei, J. Phys. Chem. C, 2010, 114, 8062–8067 CAS.
- M. R. Tarasevich and B. N. Efremov, in Electrodes of Conductive Metallic Oxides Part A, ed. S. Trasatti, Elsevier, USA, 1982, p. 227 Search PubMed.
- T. Y. Wei, C. H. Chen, H. C. Chien, S. Y. Lu and C. C. Hu, Adv. Mater., 2010, 22, 347–351 CrossRef CAS PubMed.
- G. Q. Zhang, H. B. Wu, H. E. Hoster, M. B. Chan-Park and X. W. D. Lou, Energy Environ. Sci., 2012, 5, 9453–9456 CAS.
- H. Xia, J. Feng, H. Wang, M. O. Lai and L. Lu, J. Power Sources, 2010, 195, 4410–4413 CrossRef CAS PubMed.
- C.-C. Hu, K.-H. Chang, M.-C. Lin and Y.-T. Wu, Nano Lett., 2006, 6, 2690–2695 CrossRef CAS PubMed.
- R. Liu, S. I. Cho and S. B. Lee, Nanotechnology, 2008, 19, 215710 CrossRef PubMed.
- S. Talapatra, S. Kar, S. Pal, R. Vajtai, L. Ci, P. Victor, M. Shaijumon, S. Kaur, O. Nalamasu and P. Ajayan, Nat. Nanotechnol., 2006, 1, 112–116 CrossRef CAS PubMed.
- D. N. Futaba, K. Hata, T. Yamada, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, H. Hatori, M. Yumura and S. Iijima, Nat. Mater., 2006, 5, 987–994 CrossRef CAS PubMed.
- B. Kim, H. Chung and W. Kim, J. Phys. Chem. C, 2010, 114, 15223–15227 CAS.
- C. R. Martin, Science, 1994, 266, 1961–1966 CAS.
- M. P. Zach, K. H. Ng and R. M. Penner, Science, 2000, 290, 2120–2123 CrossRef CAS.
- E. Menke, M. Thompson, C. Xiang, L. Yang and R. Penner, Nat. Mater., 2006, 5, 914–919 CrossRef CAS PubMed.
- P.-L. Taberna, S. Mitra, P. Poizot, P. Simon and J.-M. Tarascon, Nat. Mater., 2006, 5, 567–573 CrossRef CAS PubMed.
- Y. Sun, B. Gates, B. Mayers and Y. Xia, Nano Lett., 2002, 2, 165–168 CrossRef CAS.
- Y. Sun, Y. Yin, B. T. Mayers, T. Herricks and Y. Xia, Chem. Mater., 2002, 14, 4736–4745 CrossRef CAS.
- S. Vaddiraju, H. Chandrasekaran and M. K. Sunkara, J. Am. Chem. Soc., 2003, 125, 10792–10793 CrossRef CAS PubMed.
- H. Choi and S.-H. Park, J. Am. Chem. Soc., 2004, 126, 6248–6249 CrossRef CAS PubMed.
- J. Zhou, N. S. Xu, S. Z. Deng, J. Chen, J. C. She and Z. L. Wang, Adv. Mater., 2003, 15, 1835–1840 CrossRef CAS.
- N. Cordente, M. Respaud, F. Senocq, M.-J. Casanove, C. Amiens and B. Chaudret, Nano Lett., 2001, 1, 565–568 CrossRef CAS.
- C. Cheng, R. K. Gonela, Q. Gu and D. T. Haynie, Nano Lett., 2005, 5, 175–178 CrossRef CAS PubMed.
- Y. Lei, B. Daffos, P.-L. Taberna, P. Simon and F. Favier, Electrochim. Acta, 2010, 55, 7454–7459 CrossRef CAS PubMed.
- Z. Su, C. Yang, B. Xie, Z. Lin, Z. Zhang, J. Liu, B. Li, F. Kang and C. Wong, Energy Environ. Sci., 2014, 7, 2652–2659 CAS.
- J. YoungáKim, RSC Adv., 2012, 2, 8281–8285 RSC.
- W. Yan, J. Y. Kim, W. Xing, K. C. Donavan, T. Ayvazian and R. M. Penner, Chem. Mater., 2012, 24, 2382–2390 CrossRef CAS.
- D. Chao, X. Xia, C. Zhu, J. Wang, J. Liu, J. Lin, Z. Shen and H. J. Fan, Nanoscale, 2014, 6, 5691–5697 RSC.
- Q. Li, Z.-L. Wang, G.-R. Li, R. Guo, L.-X. Ding and Y.-X. Tong, Nano Lett., 2012, 12, 3803–3807 CrossRef CAS PubMed.
- Z. Yu and J. Thomas, Adv. Mater., 2014, 26, 4279–4285 CrossRef CAS PubMed.
- L. Bao, J. Zang and X. Li, Nano Lett., 2011, 11, 1215–1220 CrossRef CAS PubMed.
- P. Yang, X. Xiao, Y. Li, Y. Ding, P. Qiang, X. Tan, W. Mai, Z. Lin, W. Wu, T. Li, H. Jin, P. Liu, J. Zhou, C. P. Wong and Z. L. Wang, ACS Nano, 2013, 7, 2617–2626 CrossRef CAS PubMed.
- X. Sun, Q. Li, Y. Lü and Y. Mao, Chem. Commun., 2013, 49, 4456–4458 RSC.
- A. Sumboja, X. Wang, J. Yan and P. S. Lee, Electrochim. Acta, 2012, 65, 190–195 CrossRef CAS PubMed.
- G.-R. Li, Z.-L. Wang, F.-L. Zheng, Y.-N. Ou and Y.-X. Tong, J. Mater. Chem., 2011, 21, 4217–4221 RSC.
- J. Liu, C. Cheng, W. Zhou, H. Li and H. J. Fan, Chem. Commun., 2011, 47, 3436–3438 RSC.
- J. Yan, E. Khoo, A. Sumboja and P. S. Lee, ACS Nano, 2010, 4, 4247–4255 CrossRef CAS PubMed.
- J. Bae, M. K. Song, Y. J. Park, J. M. Kim, M. Liu and Z. L. Wang, Angew. Chem., Int. Ed., 2011, 50, 1683–1687 CrossRef CAS PubMed.
- R. K. Sharma, A. Karakoti, S. Seal and L. Zhai, J. Power Sources, 2010, 195, 1256–1262 CrossRef CAS PubMed.
- R. K. Sharma and L. Zhai, Electrochim. Acta, 2009, 54, 7148–7155 CrossRef CAS PubMed.
- Q. Li, J. Liu, J. Zou, A. Chunder, Y. Chen and L. Zhai, J. Power Sources, 2011, 196, 565–572 CrossRef CAS PubMed.
- A. L. M. Reddy, M. M. Shaijumon, S. R. Gowda and P. M. Ajayan, Nano Lett., 2009, 9, 1002–1006 CrossRef CAS PubMed.
- A. L. M. Reddy, M. M. Shaijumon, S. R. Gowda and P. M. Ajayan, J. Phys. Chem. C, 2009, 114, 658–663 Search PubMed.
- J. Liu, J. Jiang, C. Cheng, H. Li, J. Zhang, H. Gong and H. J. Fan, Adv. Mater., 2011, 23, 2076–2081 CrossRef CAS PubMed.
- C. Guan, J. Liu, C. Cheng, H. Li, X. Li, W. Zhou, H. Zhang and H. J. Fan, Energy Environ. Sci., 2011, 4, 4496–4499 CAS.
- X. Xia, J. Tu, Y. Zhang, X. Wang, C. Gu, X.-b. Zhao and H. J. Fan, ACS Nano, 2012, 6, 5531–5538 CrossRef CAS PubMed.
- C. Guan, X. Xia, N. Meng, Z. Zeng, X. Cao, C. Soci, H. Zhang and H. J. Fan, Energy Environ. Sci., 2012, 5, 9085–9090 CAS.
- X. Xia, D. Chao, Z. Fan, C. Guan, X. Cao, H. Zhang and H. J. Fan, Nano Lett., 2014, 14, 1651–1658 CrossRef CAS PubMed.
- L. Huang, D. Chen, Y. Ding, S. Feng, Z. L. Wang and M. Liu, Nano Lett., 2013, 13, 3135–3139 CrossRef CAS PubMed.
- Q. Qu, Y. Zhu, X. Gao and Y. Wu, Adv. Energy Mater., 2012, 2, 950–955 CrossRef CAS.
- Q. Li, J. M. Anderson, Y. Chen and L. Zhai, Electrochim. Acta, 2012, 59, 548–557 CrossRef CAS PubMed.
- G. Binotto, D. Larcher, A. Prakash, R. Herrera Urbina, M. Hegde and J.-M. Tarascon, Chem. Mater., 2007, 19, 3032–3040 CrossRef CAS.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS PubMed.
- R. Liu and S. B. Lee, J. Am. Chem. Soc., 2008, 130, 2942–2943 CrossRef CAS PubMed.
- J. Duay, E. Gillette, R. Liu and S. B. Lee, Phys. Chem. Chem. Phys., 2012, 14, 3329–3337 RSC.
- X. Xia, D. Chao, X. Qi, Q. Xiong, Y. Zhang, J. Tu, H. Zhang and H. J. Fan, Nano Lett., 2013, 13, 4562–4568 CrossRef CAS PubMed.
- J.-G. Wang, Y. Yang, Z.-h. Huang and F. Kang, Electrochim. Acta, 2014, 130, 642–649 CrossRef CAS PubMed.
- W. Yao, H. Zhou and Y. Lu, J. Power Sources, 2013, 241, 359–366 CrossRef CAS PubMed.
- L. Mai, F. Dong, X. Xu, Y. Luo, Q. An, Y. Zhao, J. Pan and J. Yang, Nano Lett., 2013, 13, 740–745 CrossRef CAS PubMed.
- H. Zhang, G. Cao, Z. Wang, Y. Yang, Z. Shi and Z. Gu, Nano Lett., 2008, 8, 2664–2668 CrossRef CAS PubMed.
- R. Liu, J. Duay and S. B. Lee, ACS Nano, 2010, 4, 4299–4307 CrossRef CAS PubMed.
- R. Mas-Balleste, C. Gomez-Navarro, J. Gomez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20–30 RSC.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CAS.
- D. Chen, L. Tang and J. Li, Chem. Soc. Rev., 2010, 39, 3157–3180 RSC.
- Y. Huang, J. Liang and Y. Chen, Small, 2012, 8, 1805–1834 CrossRef CAS PubMed.
- M. M. Shaijumon, F. S. Ou, L. Ci and P. M. Ajayan, Chem. Commun., 2008, 2373–2375 RSC.
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833–2838 CrossRef CAS PubMed.
- H. Wang, Y. Liang, T. Mirfakhrai, Z. Chen, H. Casalongue and H. Dai, Nano Res., 2011, 4, 729–736 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Falko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- J. Xia, F. Chen, J. Li and N. Tao, Nat. Nanotechnol., 2009, 4, 505–509 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- Q. Qu, S. Yang and X. Feng, Adv. Mater., 2011, 23, 5574–5580 CrossRef CAS PubMed.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- J. R. Miller, R. A. Outlaw and B. C. Holloway, Science, 2010, 329, 1637–1639 CrossRef CAS PubMed.
- M. Beidaghi and Y. Gogotsi, Energy Environ. Sci., 2014, 7, 867–884 CAS.
- J. J. Yoo, K. Balakrishnan, J. Huang, V. Meunier, B. G. Sumpter, A. Srivastava, M. Conway, A. L. Mohana Reddy, J. Yu, R. Vajtai and P. M. Ajayan, Nano Lett., 2011, 11, 1423–1427 CrossRef CAS PubMed.
- M. Kaempgen, C. K. Chan, J. Ma, Y. Cui and G. Gruner, Nano Lett., 2009, 9, 1872–1876 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- A. D. DeYoung, S.-W. Park, N. R. Dhumal, Y. Shim, Y. Jung and H. J. Kim, J. Phys. Chem. C, 2014, 118, 18472–18480 CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- S. Park, J. An, I. Jung, R. D. Piner, S. J. An, X. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS PubMed.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2009, 4, 25–29 CrossRef CAS PubMed.
- Y. Zhu, M. D. Stoller, W. Cai, A. Velamakanni, R. D. Piner, D. Chen and R. S. Ruoff, ACS Nano, 2010, 4, 1227–1233 CrossRef CAS PubMed.
- C. Niu, E. K. Sichel, R. Hoch, D. Moy and H. Tennent, Appl. Phys. Lett., 1997, 70, 1480–1482 CrossRef CAS PubMed.
- C. Du, J. Yeh and N. Pan, Nanotechnology, 2005, 16, 350 CrossRef CAS.
- H.-J. Shin, K. K. Kim, A. Benayad, S.-M. Yoon, H. K. Park, I.-S. Jung, M. H. Jin, H.-K. Jeong, J. M. Kim, J.-Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- W. Lv, D.-M. Tang, Y.-B. He, C.-H. You, Z.-Q. Shi, X.-C. Chen, C.-M. Chen, P.-X. Hou, C. Liu and Q.-H. Yang, ACS Nano, 2009, 3, 3730–3736 CrossRef CAS PubMed.
- A. Burke, Electrochim. Acta, 2007, 53, 1083–1091 CrossRef CAS PubMed.
- W. Gao, N. Singh, L. Song, Z. Liu, A. L. M. Reddy, L. Ci, R. Vajtai, Q. Zhang, B. Wei and P. M. Ajayan, Nat. Nanotechnol., 2011, 6, 496–500 CrossRef CAS PubMed.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 CrossRef CAS PubMed.
- Z. Wei, D. Wang, S. Kim, S.-Y. Kim, Y. Hu, M. K. Yakes, A. R. Laracuente, Z. Dai, S. R. Marder, C. Berger, W. P. King, W. A. de Heer, P. E. Sheehan and E. Riedo, Science, 2010, 328, 1373–1376 CrossRef CAS PubMed.
- Y. Zhang, L. Guo, S. Wei, Y. He, H. Xia, Q. Chen, H.-B. Sun and F.-S. Xiao, Nano Today, 2010, 5, 15–20 CrossRef CAS PubMed.
- S. R. C. Vivekchand, C. S. Rout, K. S. Subrahmanyam, A. Govindaraj and C. N. R. Rao, J. Chem. Sci., 2008, 120, 9–13 CrossRef CAS PubMed.
- C. D. Lokhande, D. P. Dubal and O.-S. Joo, Curr. Appl. Phys., 2011, 11, 255–270 CrossRef PubMed.
- S. Shi, C. Xu, C. Yang, Y. Chen, J. Liu and F. Kang, Sci. Rep., 2013, 3, 2598 Search PubMed.
- J. P. Zheng, P. J. Cygan and T. R. Jow, J. Electrochem. Soc., 1995, 142, 2699–2703 CrossRef CAS PubMed.
- T. P. Gujar, V. R. Shinde, C. D. Lokhande, W.-Y. Kim, K.-D. Jung and O.-S. Joo, Electrochem. Commun., 2007, 9, 504–510 CrossRef CAS PubMed.
- L. Cao, S. Yang, W. Gao, Z. Liu, Y. Gong, L. Ma, G. Shi, S. Lei, Y. Zhang, S. Zhang, R. Vajtai and P. M. Ajayan, Small, 2013, 9, 2905–2910 CrossRef CAS PubMed.
- B. Lei, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2014, 2, 3919–3925 CAS.
- X. Wang, J. Ding, S. Yao, X. Wu, Q. Feng, Z. Wang and B. Geng, J. Mater. Chem. A, 2014, 2, 15958–15963 CAS.
- J. M. Soon and K. P. Loh, Electrochem. Solid-State Lett., 2007, 10, A250–A254 CrossRef CAS PubMed.
- K. Krishnamoorthy, G. K. Veerasubramani, S. Radhakrishnan and S. J. Kim, Mater. Res. Bull., 2014, 50, 499–502 CrossRef CAS PubMed.
- J. Feng, X. Sun, C. Wu, L. Peng, C. Lin, S. Hu, J. Yang and Y. Xie, J. Am. Chem. Soc., 2011, 133, 17832–17838 CrossRef CAS PubMed.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
- M. D. Levi, M. R. Lukatskaya, S. Sigalov, M. Beidaghi, N. Shpigel, L. Daikhin, D. Aurbach, M. W. Barsoum and Y. Gogotsi, Adv. Energy Mater., 2014 DOI:10.1002/aenm.201400815.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- M. Kurtoglu, M. Naguib, Y. Gogotsi and M. W. Barsoum, MRS Commun., 2012, 2, 133–137 CrossRef CAS.
- M. Khazaei, M. Arai, T. Sasaki, C. Y. Chung, N. S. Venkataramanan, M. Estili, Y. Sakka and Y. Kawazoe, Adv. Funct. Mater., 2013, 23, 2185–2192 CrossRef CAS.
- Y. Zhang, H. Li, L. Pan, T. Lu and Z. Sun, J. Electroanal. Chem., 2009, 634, 68–71 CrossRef CAS PubMed.
- S. Yang, G. Cui, S. Pang, Q. Cao, U. Kolb, X. Feng, J. Maier and K. Müllen, ChemSusChem, 2010, 3, 236–239 CrossRef CAS PubMed.
- H. Wang, H. S. Casalongue, Y. Liang and H. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS PubMed.
- H. Wang, J. T. Robinson, G. Diankov and H. Dai, J. Am. Chem. Soc., 2010, 132, 3270–3271 CrossRef CAS PubMed.
- H. Wang, L.-F. Cui, Y. Yang, H. Sanchez Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS PubMed.
- Z.-S. Wu, D.-W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H.-M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS.
- H. Xia, Y. S. Meng, G. Yuan, C. Cui and L. Lu, Electrochem. Solid-State Lett., 2012, 15, A60–A63 CrossRef CAS PubMed.
- T.-S. Hyun, J.-E. Kang, H.-G. Kim, J.-M. Hong and I.-D. Kim, Electrochem. Solid-State Lett., 2009, 12, A225–A228 CrossRef CAS PubMed.
- J. Zhang, J. Jiang, H. Li and X. S. Zhao, Energy Environ. Sci., 2011, 4, 4009–4015 CAS.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L.-C. Qin, Carbon, 2011, 49, 2917–2925 CrossRef CAS PubMed.
- L. Peng, X. Peng, B. Liu, C. Wu, Y. Xie and G. Yu, Nano Lett., 2013, 13, 2151–2157 CrossRef CAS PubMed.
- Y. Qian, S. Lu and F. Gao, J. Mater. Sci., 2011, 46, 3517–3522 CrossRef CAS.
- Z.-S. Wu, W. Ren, D.-W. Wang, F. Li, B. Liu and H.-M. Cheng, ACS Nano, 2010, 4, 5835–5842 CrossRef CAS PubMed.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS PubMed.
- X. Zhao, L. Zhang, S. Murali, M. D. Stoller, Q. Zhang, Y. Zhu and R. S. Ruoff, ACS Nano, 2012, 6, 5404–5412 CrossRef CAS PubMed.
- L. Fenghua, S. Jiangfeng, Y. Huafeng, G. Shiyu, Z. Qixian, H. Dongxue, I. Ari and N. Li, Nanotechnology, 2009, 20, 455602 CrossRef PubMed.
- E. G. da Silveira Firmiano, A. C. Rabelo, C. J. Dalmaschio, A. N. Pinheiro, E. C. Pereira, W. H. Schreiner and E. R. Leite, Adv. Energy Mater., 2014, 4, 1301380 Search PubMed.
- S. Ratha and C. S. Rout, ACS Appl. Mater. Interfaces, 2013, 5, 11427–11433 CAS.
- S. Li, X. Wang, H. Xing and C. Shen, J. Micromech. Microeng., 2013, 23, 114013 CrossRef.
- W. Wang, S. Guo, I. Lee, K. Ahmed, J. Zhong, Z. Favors, F. Zaera, M. Ozkan and C. S. Ozkan, Sci. Rep., 2014, 4, 4452 Search PubMed.
- Y.-M. Wang, X. Zhang, C.-Y. Guo, Y.-Q. Zhao, C.-L. Xu and H.-L. Li, J. Mater. Chem. A, 2013, 1, 13290–13300 CAS.
- T. Zhai, F. Wang, M. Yu, S. Xie, C. Liang, C. Li, F. Xiao, R. Tang, Q. Wu, X. Lu and Y. Tong, Nanoscale, 2013, 5, 6790–6796 RSC.
- M.-J. Deng, P.-J. Ho, C.-Z. Song, S.-A. Chen, J.-F. Lee, J.-M. Chen and K.-T. Lu, Energy Environ. Sci., 2013, 6, 2178–2185 CAS.
- X. H. Xia, J. P. Tu, Y. Q. Zhang, Y. J. Mai, X. L. Wang, C. D. Gu and X. B. Zhao, J. Phys. Chem. C, 2011, 115, 22662–22668 CAS.
- C. Zhou, Y. Zhang, Y. Li and J. Liu, Nano Lett., 2013, 13, 2078–2085 CrossRef CAS PubMed.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS.
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677–681 CrossRef CAS.
- H. Hu, Z. Zhao, W. Wan, Y. Gogotsi and J. Qiu, Adv. Mater., 2013, 25, 2219–2223 CrossRef CAS PubMed.
- W. Xing, S. Z. Qiao, R. G. Ding, F. Li, G. Q. Lu, Z. F. Yan and H. M. Cheng, Carbon, 2006, 44, 216–224 CrossRef CAS PubMed.
- M. Inagaki, H. Konno and O. Tanaike, J. Power Sources, 2010, 195, 7880–7903 CrossRef CAS PubMed.
- D. Zhang, L. Zheng, Y. Ma, L. Lei, Q. Li, Y. Li, H. Luo, H. Feng and Y. Hao, ACS Appl. Mater. Interfaces, 2014, 6, 2657–2665 CAS.
- L. Wang, Y. Zheng, S. Chen, Y. Ye, F. Xu, H. Tan, Z. Li, H. Hou and Y. Song, Electrochim. Acta, 2014, 135, 380–387 CrossRef CAS PubMed.
- H. Jiang, J. Ma and C. Li, Adv. Mater., 2012, 24, 4197–4202 CrossRef CAS.
- X. Dong, W. Shen, J. Gu, L. Xiong, Y. Zhu, H. Li and J. Shi, J. Phys. Chem. B, 2006, 110, 6015–6019 CrossRef CAS PubMed.
- Y. Yan, Q. Cheng, G. Wang and C. Li, J. Power Sources, 2011, 196, 7835–7840 CrossRef CAS PubMed.
- Y. G. Wang, H. Q. Li and Y. Y. Xia, Adv. Mater., 2006, 18, 2619–2623 CrossRef CAS.
- Y. Yan, Q. Cheng, Z. Zhu, V. Pavlinek, P. Saha and C. Li, J. Power Sources, 2013, 240, 544–550 CrossRef CAS PubMed.
- Z. Lei, X. Sun, H. Wang, Z. Liu and X. S. Zhao, ACS Appl. Mater. Interfaces, 2013, 5, 7501–7508 CAS.
- W.-X. Liu, N. Liu, H.-H. Song and X.-H. Chen, Carbon, 2011, 49, 4610 CrossRef CAS PubMed.
- Y. Yan, Q. Cheng, V. Pavlinek, P. Saha and C. Li, Electrochim. Acta, 2012, 71, 27–32 CrossRef CAS PubMed.
- D. Li, J. Huang and R. B. Kaner, Acc. Chem. Res., 2008, 42, 135–145 CrossRef PubMed.
- H.-K. Song, Y.-H. Jung, K.-H. Lee and L. H. Dao, Electrochim. Acta, 1999, 44, 3513–3519 CrossRef CAS.
- G.-J. Lee and S.-I. Pyun, Langmuir, 2006, 22, 10659–10665 CrossRef CAS PubMed.
- H. Yamada, H. Nakamura, F. Nakahara, I. Moriguchi and T. Kudo, J. Phys. Chem. C, 2006, 111, 227–233 CrossRef.
- A. J. Amali, J.-K. Sun and Q. Xu, Chem. Commun., 2014, 50, 1519–1522 RSC.
- S. Kubo, R. J. White, K. Tauer and M.-M. Titirici, Chem. Mater., 2013, 25, 4781–4790 CrossRef CAS.
- Q. Long, W. Chen, H. Xu, X. Xiong, Y. Jiang, F. Zou, X. Hu, Y. Xin, Z. Zhang and Y. Huang, Energy Environ. Sci., 2013, 6, 2497–2504 CAS.
- M. Oschatz, L. Borchardt, K. Pinkert, S. Thieme, M. R. Lohe, C. Hoffmann, M. Benusch, F. M. Wisser, C. Ziegler, L. Giebeler, M. H. Ruemmeli, J. Eckert, A. Eychmueller and S. Kaskel, Adv. Energy Mater., 2014, 4, 1300645 Search PubMed.
- Q. Zhao, X. Wang, C. Wu, J. Liu, H. Wang, J. Gao, Y. Zhang and H. Shu, J. Power Sources, 2014, 254, 10–17 CrossRef CAS PubMed.
- D.-W. Wang, F. Li, M. Liu, G. Q. Lu and H.-M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373–376 CrossRef CAS PubMed.
- S. Dutta, A. Bhaumik and K. C. W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 CAS.
- B. Xu, S. Hou, G. Cao, M. Chu and Y. Yang, RSC Adv., 2013, 3, 17500–17506 RSC.
- R. Liu, J. Duay and S. B. Lee, Chem. Commun., 2011, 47, 1384–1404 RSC.
- D. Lan, Y. Chen, P. Chen, X. Chen, X. Wu, X. Pu, Y. Zeng and Z. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 11839–11845 CAS.
- C. Yuan, J. Li, L. Hou, J. Lin, X. Zhang and S. Xiong, J. Mater. Chem. A, 2013, 1, 11145–11151 CAS.
- J. Luo, H. D. Jang, T. Sun, L. Xiao, Z. He, A. P. Katsoulidis, M. G. Kanatzidis, J. M. Gibson and J. Huang, ACS Nano, 2011, 5, 8943–8949 CrossRef CAS PubMed.
- J. Luo, H. D. Jang and J. Huang, ACS Nano, 2013, 7, 1464–1471 CrossRef CAS PubMed.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- Z. Yu, M. McInnis, J. Calderon, S. Seal, L. Zhai and J. Thomas, Nano Energy DOI:10.1016/j.nanoen.2014.11.030.
- Z.-S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Muellen, Adv. Mater., 2012, 24, 5130–5135 CrossRef CAS PubMed.
- B. G. Choi, M. H. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- X. Ma, J. Liu, C. Liang, X. Gong and R. Che, J. Mater. Chem. A, 2014, 2, 12692–12696 CAS.
- X. Jiang, X. Yang, Y. Zhu, J. Shen, K. Fan and C. Li, J. Power Sources, 2013, 237, 178–186 CrossRef CAS PubMed.
- X. Zhu, P. Zhang, S. Xu, X. Yan and Q. Xue, ACS Appl. Mater. Interfaces, 2014, 6, 11665–11674 CAS.
- S. Wu, W. Chen and L. Yan, J. Mater. Chem. A, 2014, 2, 2765–2772 CAS.
- J. T. Zhang, S. Liu, G. L. Pan, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2014, 2, 1524–1529 CAS.
- Q. Wu, Y. Xu, Z. Yao, A. Liu and G. Shi, ACS Nano, 2010, 4, 1963–1970 CrossRef CAS PubMed.
- J. Xu, K. Wang, S.-Z. Zu, B.-H. Han and Z. Wei, ACS Nano, 2010, 4, 5019–5026 CrossRef CAS PubMed.
- X. Yu, B. Lu and Z. Xu, Adv. Mater., 2014, 26, 1044–1051 CrossRef CAS PubMed.
- F. Zhang, D. Zhu, X. a. Chen, X. Xu, Z. Yang, C. Zou, K. Yang and S. Huang, Phys. Chem. Chem. Phys., 2014, 16, 4186–4192 RSC.
- X. Xie, C. Zhang, M.-B. Wu, Y. Tao, W. Lv and Q.-H. Yang, Chem. Commun., 2013, 49, 11092–11094 RSC.
- L. Zhang, F. Zhang, X. Yang, G. Long, Y. Wu, T. Zhang, K. Leng, Y. Huang, Y. Ma, A. Yu and Y. Chen, Sci. Rep., 2013, 3, 1408 Search PubMed.
- Y. Shao, H. Wang, Q. Zhang and Y. Li, J. Mater. Chem. C, 2013, 1, 1245–1251 RSC.
- G. K. Dimitrakakis, E. Tylianakis and G. E. Froudakis, Nano Lett., 2008, 8, 3166–3170 CrossRef CAS PubMed.
- F. Du, D. Yu, L. Dai, S. Ganguli, V. Varshney and A. K. Roy, Chem. Mater., 2011, 23, 4810–4816 CrossRef CAS.
- Z.-Y. Yang, Y.-F. Zhao, Q.-Q. Xiao, Y.-X. Zhang, L. Jing, Y.-M. Yan and K.-N. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 8497–8504 CAS.
- Q. Li, J. M. Anderson, Y. Chen and L. Zhai, Electrochim. Acta, 2012, 59, 548–557 CrossRef CAS PubMed.
- S.-I. Kim, J.-S. Lee, H.-J. Ahn, H.-K. Song and J.-H. Jang, ACS Appl. Mater. Interfaces, 2013, 5, 1596–1603 CAS.
- Y. Xiao, S. Liu, F. Li, A. Zhang, J. Zhao, S. Fang and D. Jia, Adv. Funct. Mater., 2012, 22, 4052–4059 CrossRef CAS.
- P. Yu, X. Zhang, D. Wang, L. Wang and Y. Ma, Cryst. Growth Des., 2009, 9, 528–533 CAS.
| This journal is © The Royal Society of Chemistry 2015 |