
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen-tolerant proton reduction catalysis: much O2 about nothing?†
David W.
Wakerley
and
Erwin
Reisner
*
Christian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk; Web: http://www-reisner.ch.cam.ac.uk/
First published on 29th May 2015
Abstract
Proton reduction catalysts are an integral component of artificial photosynthetic systems for the production of H2. This perspective covers such catalysts with respect to their tolerance towards the potential catalyst inhibitor O2. O2 is abundant in our atmosphere and generated as a by-product during the water splitting process, therefore maintaining proton reduction activity in the presence of O2 is important for the widespread production of H2. This perspective article summarises viable strategies for avoiding the adverse effects of aerobic environments to encourage their adoption and improvement in future research. H2-evolving enzymatic systems, molecular synthetic catalysts and catalytic surfaces are discussed with respect to their interaction with O2 and analytical techniques through which O2-tolerant catalysts can be studied are described.
Broader contextThe generation of hydrogen from water is a potential approach to develop a clean and renewable fuel. This process is carried out by proton reduction catalysts and currently research is focussed on the development of efficient and robust catalytic species. Application of the water-splitting process will be carried out on a large scale, not restricted to the laboratory, and as such it is necessary to consider how O2 in our atmosphere or produced as a side product from water splitting would interact with such an arrangement. O2 is an inhibitor of a number of catalytic processes and therefore designing strategies to avoid O2 inhibition is crucial in the production of viable proton reduction systems. |
1. Introduction
The large scale production of H2 through artificial photosynthesis stands as an aspiring goal of contemporary science.1–3 Chemical-energy storage through water splitting generates both H2 and O2 and relies on efficient reduction and oxidation catalysts, respectively [reaction (1)].| H2O → H2 + ½ O2 ΔE0 = −1.23 V | (1) |
One issue that remains relatively underexplored is the impact of O2 on synthetic proton-reducing systems. Less than a decade ago it seemed common sense that synthetic molecular H2-evolving catalysts would operate poorly under air due to the propensity of O2 to irreversibly damage a catalytic structure during turnover. As a result, research was carried out under inert atmospheres of N2 or Ar. Given that the end goal for a proton reduction catalyst would be its widespread use in a H2-fuelled economy, any observable O2-sensitivity would seriously impair its practicality. Adding to this, stringent anaerobic conditions are costly to maintain on an industrial scale. Developing catalysts that could operate under O2 consequently stood as a major challenge for H2 production research,5,6 yet recent publications have demonstrated that avoiding the inhibiting effects of O2 may be more manageable than first imagined and O2-tolerant proton reduction is now a fast-developing field.
Exposure of a proton reduction catalyst to O2 in a water splitting system, particularly over prolonged periods of time, is almost unavoidable. Fig. 1a shows a standard electrolyser/photoelectrochemical (PEC) cell, which contains an O2 evolving anode and a H2 producing cathode separated by a proton exchange membrane to prevent crossover of the evolved gaseous products.7 Interaction between O2 and the proton reducing cathode can still occur through O2 leakage from the atmosphere into the electrochemical cell or from the anodic chamber after membrane degradation.8,9 Another configuration is the ‘artificial leaf’,10,11 a simplification of which can be seen in Fig. 1b. The cathode and anode are attached on opposing sides of a photovoltaic layer that drives catalysis and some exposure of the proton reduction catalyst to O2 is inherent in the system's design. Photocatalytic water-splitting particles are also a promising route to full water splitting, see Fig. 1c.12,13 H2 and O2 are produced on the same or a neighbouring light-absorbing particle, which is often loaded with a catalyst to enhance catalysis. The close proximity of O2 and H2 evolution sites makes interaction between catalyst and O2 inevitable without additional protection of the catalyst.
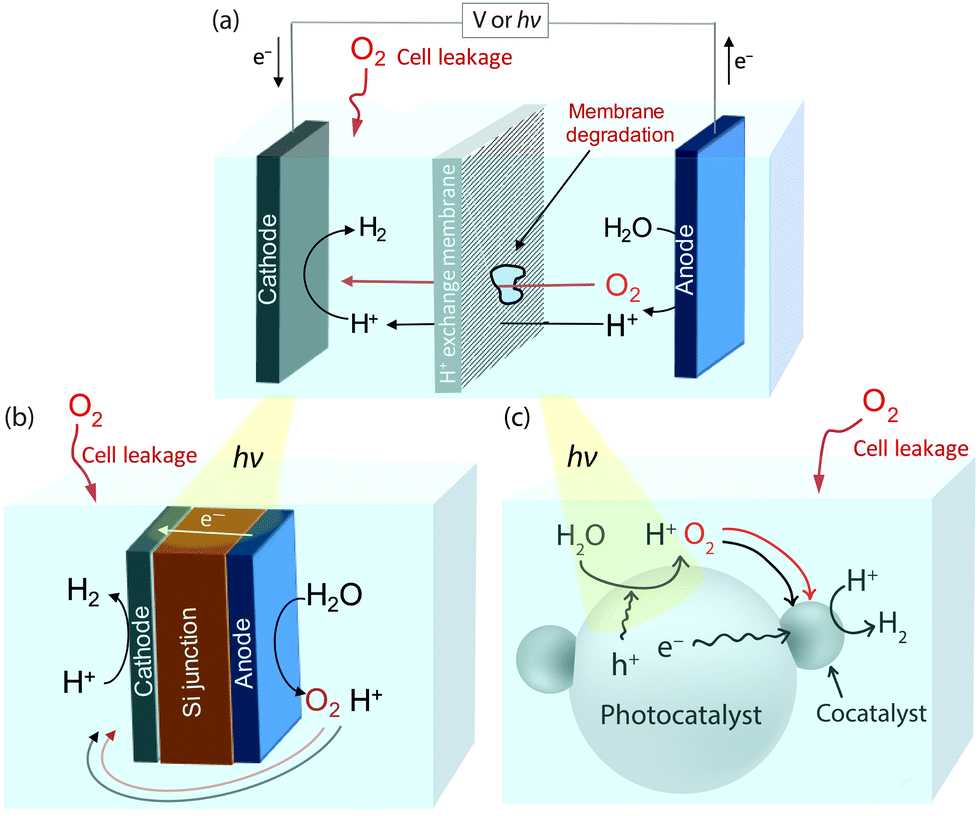 | ||
| Fig. 1 Potential routes through which a proton reducing catalyst could be exposed to O2 in (a) a standard electrolysis/PEC cell, (b) an artificial leaf and (c) photocatalytic water-splitting particles. | ||
Contemporary research has started to cover the concept of O2-tolerant H2 generation to realise systems in which the presence of O2 is inconsequential. This field is still in its infancy, nonetheless the reported O2-tolerant systems present innovative routes to efficient, aerobic proton reduction. Broadly speaking the current examples fall into one of three areas of catalyst: proton reducing enzymes (hydrogenases),14 molecular complexes5 and catalytic surfaces.15,16
In this perspective, each of these examples will be discussed to encourage a holistic development of O2-tolerant catalyst systems. A discussion of the electrochemical/spectroscopic study of O2-tolerance is also provided to highlight key techniques that will be vital for fully understanding the effects of O2 on a proton reduction system.
2. Oxygen in a proton reducing system
Proton reduction is a pH dependent redox process that has a formal redox potential, E0′, of 0 − (pH × 59) mV vs. the normal hydrogen electrode (NHE) (25 °C). Applied potentials more negative than E0′ are needed to drive H2 evolution and under aerobic conditions it is necessary to consider the effect such potentials have on O2. In a pH 7 solution there are a number of potential O2 reduction reactions that could occur, many of which form reactive oxygen species (ROS):17Water formation:
| O2 + 4H+ + 4e− → 2H2O 7E0′ = +0.82 V | (2) |
| O2 + 2H+ + 2e− → H2O2 7E0′ = +0.28 V | (3) |
| O2 + e− → O2˙− 7E0′ = −0.33 V | (4) |
| H2O2 + H+ + e− → HO˙ + H2O 7E0′ = +0.38 V | (5) |
| H2O2 + 2H+ + 2e− → 2H2O 7E0′ = +1.35 V | (6) |
| HO˙ + H+ + e− → H2O 7E0′ = +2.32 V | (7) |
| O2˙− + 2H+ + e− → H2O2 7E0′ = +0.89 V | (8) |
| 2H+ + 2e− → H2 7E0′ = –0.41 V | (9) |
Direct O2 reduction to water through reaction (2) forms the most thermodynamically stable product, but the process is kinetically slow due to the high dissociation energy of the dioxygen bond,18 which has a considerable thermodynamic barrier of 498 kJ mol−1. The reduction also requires 4e− and 4H+ and therefore, with the exception of a few highly active catalytic sites, it is much more likely that incomplete O2 reduction occurs to form H2O2, O2˙− or ˙OH if sufficiently reducing conditions are available [reactions (3) to (5)]. These species can subsequently be reduced to water in a multi-step reaction sequence [reactions (6) to (8)].
Each of the O2-reduction reactions (2) to (8) occurs at a less negative potential than the proton reduction reaction (9), which implies that any system capable of reducing protons will have sufficient driving force for O2 reduction to either generate water or ROS. It should be noted that photochemical systems may also generate reactive singlet O2 (1O2) through triplet–triplet annihilation. The interaction of a H2 evolving catalyst with O2 has two potential outcomes: O2-tolerant proton reduction or inhibited catalysis due to O2-sensitivity (Fig. 2).
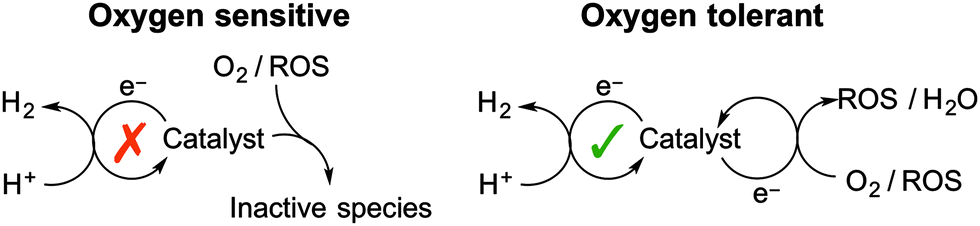 | ||
| Fig. 2 Two routes through which O2 can affect catalytic proton reduction. | ||
Oxygen-sensitive catalyst
O2-sensitive proton reduction catalysts undergo a critical drop in H2 production activity in the presence of O2. In this case the catalyst is susceptible to deactivation by reaction with O2 or with the ROS produced in reactions (3)–(5) or (8). The reducing sites at which O2 or ROS attack are typically essential to proton reduction activity and therefore the catalyst is irreversibly inhibited.O2-sensitive catalysts require a defensive approach to overcome irreversible O2 inhibition (see below). This involves protecting a catalyst from exposure to O2/ROS in order to generate a locally anaerobic environment.
Oxygen-tolerant catalyst
O2-tolerance is a term used to describe a catalyst that maintains a degree of activity in the presence of O2. In this case the catalyst is able to reduce the incoming O2 or ROS without being irreversibly damaged. Proton reduction is therefore in competition with O2 reduction and H2 is often produced at a decreased rate and efficiency under aerobic conditions.The reduction of O2 by O2-tolerant catalysts can be seen as an offensive approach to prevent O2-inhibition. The catalyst is able to remove O2 as a threat and allows H2 evolution to continue. Designing a proton reduction catalyst capable of reducing O2 and ROS to harmless by-products is an elegant strategy to realise aerobic proton reduction. O2-tolerance can be enhanced further through design of a catalyst that has favourable kinetics for proton reduction over O2 reduction.
3. Analytical techniques to study oxygen tolerance
Studying the O2 tolerance of a proton reducing species is a relatively new line of research and as such, routine analytical techniques are not commonplace in most laboratories. Currently, electrochemistry offers the simplest and most effective approach. Analysis of currents stemming from a catalyst and quantification of the H2 produced can be used to calculate turnover frequencies (TOFs),19 turnover numbers (TONs) and determine redox processes under O2.20 These techniques can be applied across all types of H2-evolving catalysts.Cyclic voltammetry (CV) offers a fast method to study redox changes and catalytic currents. CV analysis starts from a catalytically-inert potential and scans to a more negative potential at which clear proton reduction currents are observable. The onset of proton reduction and size of the reduction wave, along with Tafel slope analysis, provide a measure of a catalyst's activity. The first step in the study of O2 tolerance is to establish whether this activity changes under aerobic conditions. If a catalyst is O2 sensitive, a CV in air will result in a significant drop in proton reduction current, whereas little change in the proton reduction wave indicates O2-tolerant catalysis. An O2-tolerant catalyst may also display an O2 reduction wave, demonstrating simultaneous proton/O2 reduction. O2 tolerance is visible on a Pt electrode, where an O2 reduction wave (onset +0.5 V vs. NHE) can be observed under an O2 atmosphere, whilst the proton reduction wave (onset around −0.4 V) is maintained (Fig. 3). CV only gives an indication of O2-tolerance on a short time-scale, and analysis must therefore be supplemented with other techniques.
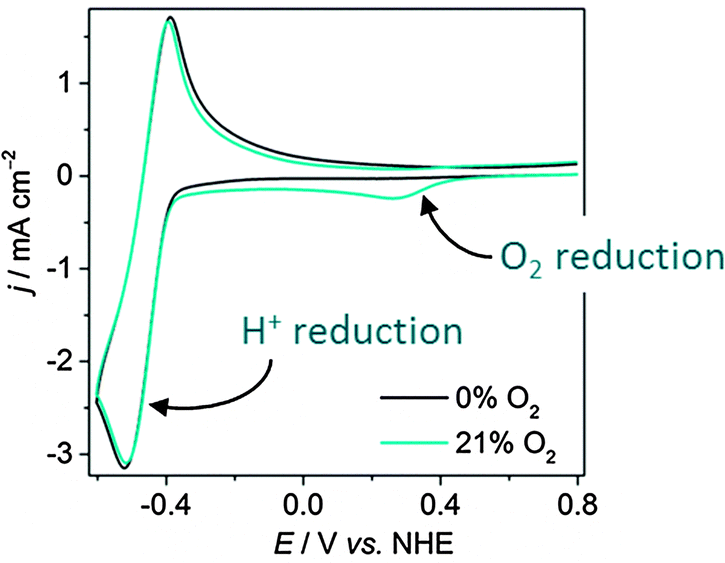 | ||
| Fig. 3 Cyclic voltammograms on a Pt disk electrode in phosphate buffer (pH 7, 0.1 M) under aerobic and anaerobic conditions under N2 at a scan rate of 50 mV s−1 at room temperature.21 The anodic wave can be attributed to the oxidation of H2 generated during the cathodic scan. | ||
Controlled potential electrolysis (CPE) is another vital tool in the study of proton reduction catalysis. In this process a constant potential is applied to a catalyst, allowing measurable quantities of H2 to build up that can be quantified through techniques such as gas chromatography. Confirming that H2 has been produced under aerobic conditions is of paramount importance, as otherwise it is not clear if an observed current stems from H2 evolution or O2/ROS reduction. Quantification of H2 also allows the Faradaic efficiency (FE) to be calculated. FE is a measure of the electrons used vs. the H2 produced and would be 100% if all electrons were consumed for proton reduction. Quantification of the H2 produced and FE from CPE under aerobic and anaerobic atmospheres gives a clear indication of a catalyst's O2 tolerance and selectivity for proton reduction over O2 reduction. CPE is also necessary to establish long-term catalytic stability under O2, as inhibition may occur over prolonged O2/ROS exposure. Such experiments may be further extended to include the effect of varying levels of O2 on catalysis.
Interaction between photocatalysts and O2 may also be studied using surface photovoltage spectroscopy. This technique monitors the contact potential difference as a function of photon energy in order to determine the surface states and energy necessary for O2 reduction on a given substrate.22
At present, analysis of O2-tolerance is confined to measuring the H2 produced by a catalyst with and without O2, however this should be coupled with analysis of the formed ROS to gain a complete appreciation of the catalyst's aerobic activity. Rotating ring-disk electrochemistry is one of the most common methods of ROS detection, which can distinguish the production of H2O2vs. H2O. This technique requires a disk electrode, consisting of the catalyst to be studied, encircled by an electrode ring, which is typically Pt. When this electrode is rotated there is laminar flow of solution from the central disk to the outer ring electrode.20 By holding the ring at oxidizing potentials with a bipotentiostat, it is possible to detect products from O2 and H+ reduction through their unique redox potentials. This technique can be used to monitor the production of H2O2 or H2,23 which can determine the degree of selectivity and O2-tolerance of a given proton reduction catalyst.24
A range of electrochemical sensors can similarly be implemented to detect the formation of ROS. Detection of O2˙− has been achieved by a number of protein-based electrodes, such as those loaded with superoxide dismutase25–27 or cytochrome c28,29 and more recently, protein-free detectors have been utilised.30–32 Similarly H2O2 can be detected through attachment of horseradish peroxidase,33 cytochrome c34 or CuS35 to an electrode. This subject has recently been reviewed.36
ROS detection can also be achieved through the measurement of a unique spectroscopic signal, such as the UV peak of H2O237 and mass-spectrometry allows the quantification of 18O2 reduction to H218O. Alternatively, spectroscopic probes can be used, which can specifically determine nM concentrations of a given ROS.38 Spectroscopic probing of the catalyst during proton reduction is equally important in order to visualise the structural and electronic changes that lead to O2-sensitivity and tolerance. Through such analysis a complete appreciation for ROS/H2 formed at a given applied potential vs. current expended can be realised, allowing conclusions concerning the interaction of the catalyst with O2 to be drawn.
4. Oxygen-tolerant hydrogenases
Hydrogenases are nature's H2-cycling catalysts and display a high ‘per active site’ activity with TOFs up to 104 s−1, rivalling that of Pt.39,40 These enzymes consist of well-suited structures to undertake proton reduction/H2 oxidation and as such have received much attention.14 [NiFe] and [FeFe] hydrogenases, categorised according to their active site composition, are the two classes of hydrogenases capable of proton reduction to H2. In each hydrogenase the active metal ions are ligated by CN−, CO and cysteine ligands and are typically connected to the protein exterior via iron–sulphur clusters. The disadvantages to the use of hydrogenases include difficult and costly purification, fragility, a large catalyst footprint (high ‘volume per active site’ ratio) and an infamous sensitivity to small quantities of O2.Hydrogenase interaction with O2 is a considerably well-established area of research and may be instrumental in engineering O2-tolerant synthetic systems.41 In-depth electrochemical and spectroscopic studies have illustrated the route to O2 inhibition across a range of hydrogenases and this work has been reviewed a number of times.14,42 As such this perspective will only briefly summarise the interaction between hydrogenases and O2 and instead focus on emerging strategies to shield the enzyme from aerobic atmospheres.
Both classes of hydrogenase consist of a range of subclasses and the O2 susceptibility of each depends to some extent on the environment in which the enzyme functions biologically. Generally, both the [NiFe] and [FeFe] hydrogenases are inhibited by O2 due to their interaction with ROS. Upon exposure of a [FeFe] hydrogenase to air, the active site, known as the H-cluster, is believed to form a ROS, which oxidises its proximal [4Fe–4S] cluster and prevents electron transfer through the enzyme to the active site.44 [NiFe] hydrogenases deactivate through the reduction of O2 to form an oxidised and paramagnetic ‘unready’ Ni-A state of the active site that is slow to reactivate45 (see Fig. 4a). The exact form of this state is debated, but crystallographic studies have suggested that a hydroperoxo ligand is ligated to the Ni ion as a result of incomplete O2 reduction.46
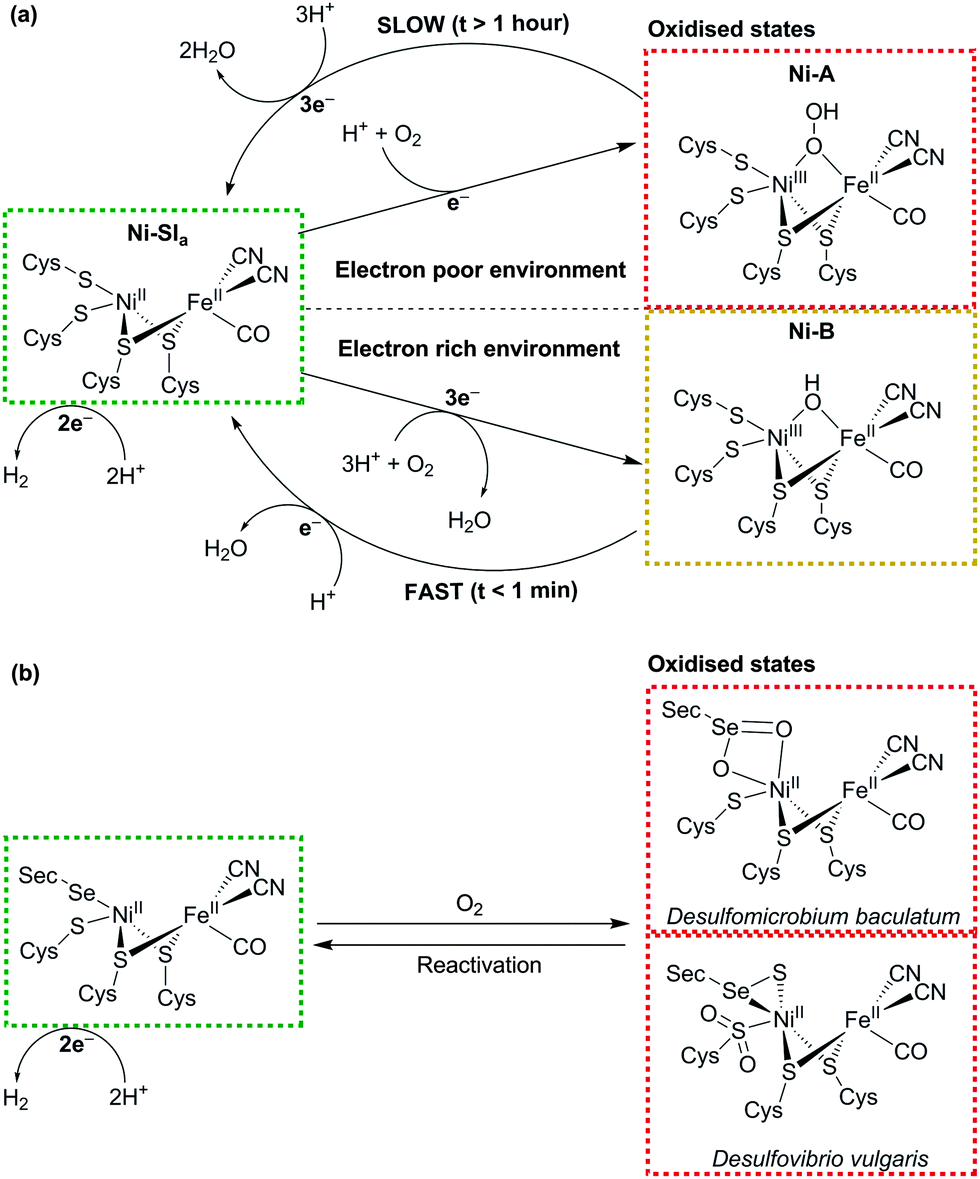 | ||
| Fig. 4 (a) Schematic representation of the formation and recovery of the oxidised Ni-A and Ni-B states in the [NiFe] hydrogenase active site (adapted from ref. 43). (b) Active site of the [NiFeSe] hydrogenase and two reported oxidised structures from Desulfomicrobium baculatum (Ox4B state) and Desulfovibrio vulgaris (conformer I). | ||
The concept of O2-tolerant H2 oxidation has become an exciting branch of research, in particular for the membrane-bound [NiFe] hydrogenase from Ralstonia eutropha, which can oxidise H2 under atmospheric levels of O2.47–49 O2-tolerant hydrogenases are more likely to form a paramagnetic Ni-B (or ‘ready’) state upon exposure to O2, as a result of more complete O2 reduction to form a bridging hydroxo ligand.46 The route to their tolerance is believed to originate from six cysteine residues surrounding the unique proximal [4Fe–3S] cluster next to the enzyme's active site.50 The cysteines facilitate structural changes that allow the cluster to transfer two electrons within a small potential range.51,52 When O2 enters the active site, one electron from the reduced Ni and two from the proximal [4Fe–3S] cluster allow the hydrogenase to consistently form the Ni-B state (Fig. 4a), which very quickly reactivates (t < 1 min). Recent evidence has suggested that conversion from Ni-A to Ni-B may occur through the oxygenation of one of the bridging S-atoms.53 Despite promising O2-tolerance, this exceptional type of [NiFe] hydrogenase is biased towards H2 oxidation over proton reduction and is inhibited by H2.42
The [NiFeSe] hydrogenase is a subclass of the [NiFe] hydrogenase that is highly active for proton reduction in the presence of H2 and illustrates a promising degree of tolerance to O2.14 [NiFeSe] hydrogenases contain a ligated selenocysteine moiety in place of one of the terminal cysteines of the conventional [NiFe] enzyme (Fig. 4). O2 exposure of the enzyme does not form substantial quantities of Ni-A/Ni-B states as a paramagnetic NiIII is not observed.54 The major products from oxidation of two [NiFeSe] hydrogenases are presented in Fig. 4b. The active site from Desulfomicrobium baculatum when crystallised aerobically contains an oxidised selenocysteine moiety (referred to as Ox4B)54 and the Desulfovibrio vulgaris species, when purified and crystallised aerobically, contains an oxidised Se and doubly-oxidised S (referred to as conformer I).55,56 The chemical role of selenocysteine in protecting the hydrogenase from oxidative damage is currently under investigation,57 but it has been shown that the [NiFeSe] hydrogenase is able to reactivate faster under anaerobic conditions after O2-exposure in comparison to the O2-sensitive [NiFe] species.58 The O2 tolerance may be a result of the easier redox chemistry of Se compared to S.59
Due to the extreme O2 sensitivity of many hydrogenases, engineering the enzymes to reduce protons and O2 simultaneously is a significant challenge,60,61 and currently more practicable approaches to aerobic H2-evolution involve shielding the enzyme from exposure to O2. This involves a ‘retrofitted’ O2-defending shield that reduces O2 before it can have adverse effects on enzyme activity. To date, ‘shields’ have been predominantly based on photochemical systems that remove O2 from a system during irradiation.
In 2009 we reported that Desulfomicrobium baculatum [NiFeSe] hydrogenase attached to a Ru-sensitised TiO2 nanoparticle was able to produce H2 photocatalytically in a N2 purged vial outside a glovebox.64 Although this sacrificial photosystem sustains H2 generation under traces of O2, it cannot maintain photo-H2 production activity under atmospheric O2 levels due to the lack of efficient O2 shielding and presumably enzyme-damaging ROS formation on irradiated TiO2 in the presence of O2 (see Section 5).
Peters and coworkers showed in 2012 that a [NiFe] hydrogenase from Thiocapsa roseopersicina covalently linked to a Ru dye was able to photocatalytically reduce protons under aerobic conditions in the presence of the soluble redox mediator methyl viologen (MV) and a sacrificial electron donor.65 Under an aerobic atmosphere and an initial lag period, where presumably dissolved O2 was photo-reduced, this system generated H2 at 11% of the initial rate observed under pseudo-inert conditions. An analogous system that used a Ru dye, which was not linked to the enzyme, showed no activity under air. It was therefore concluded that by attaching the Ru dye to the hydrogenase a local concentration of reduced MV was generated around the hydrogenase, which reduced O2 before it reached the enzyme and partially shielded it from inhibition.
Another example of O2-shielding came in 2013,62 when we reported photocatalytic H2 production with a Desulfomicrobium baculatum [NiFeSe] hydrogenase and the organic dye eosin Y in the presence of a sacrificial electron donor (Fig. 5a). The photoactivity of this mediator-free system was tested under increasing concentrations of O2 and it was able to maintain a notable degree of photocatalytic activity. Even under 21% O2, 10% of the enzyme's activity (corresponding to a TOF of 1.5 s−1) was sustained relative to the anaerobic experiment, without the observation of a significant lag phase to start H2 production. Excited eosin Y promotes proton reduction, reduction of O2 and conversion of O2 to 1O2.66 The O2-tolerance of the system may therefore stem from the photo-reduction of O2 and fast formation of 1O2 by the dye, which presumably reacts with eosin Y or the electron donor to create an anaerobic environment (Fig. 5a).
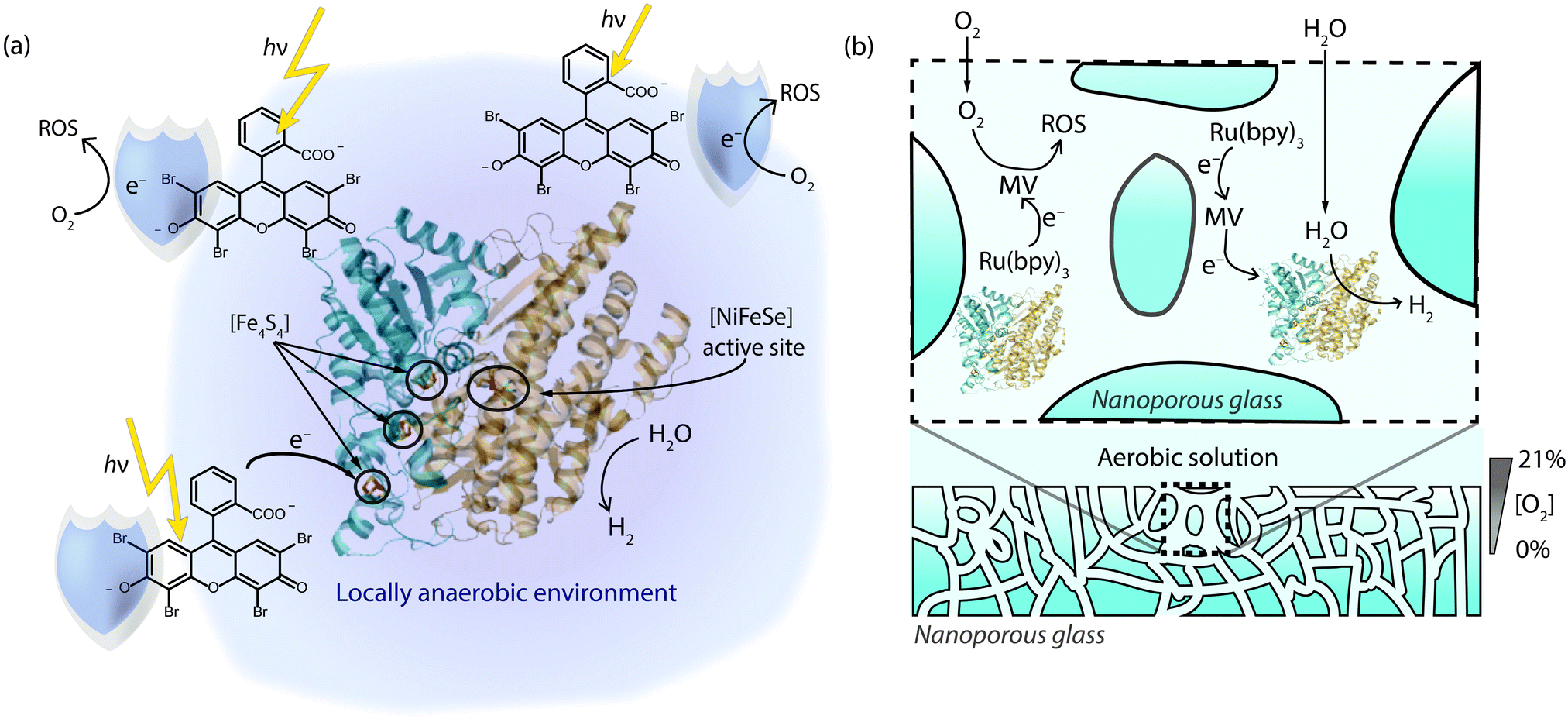 | ||
| Fig. 5 (a) Photo-excited eosin Y as a shield to protect a [NiFeSe] hydrogenase.62 (b) O2-shielding strategy based on a multi-component system consisting of a Ru dye, methyl viologen as soluble redox mediator and a hydrogenase in nanoporous glass. Reduced methyl viologen is generated upon photo-excitation of the dye and used to reduce the hydrogenase and quench O2 inside the pores to produce an anaerobic environment.63 The sacrificial electron donor used to quench the dye omitted for clarity in (a) and (b). | ||
The concept of shielding has been extended by Dewa and coworkers in 2014 through the implementation of porous enzyme-immobilising frameworks.63 In this case, a nanoporous glass plate was soaked in a tris(bipyridine)rutheniumII dye, MV and a [NiFe] hydrogenase from Desulfovibrio vulgaris. The nanoporous framework consisted of 50 nm channels that directed diffusion of O2 into the structure. The MV reduced O2 in the channels as it entered the glass during irradiation, producing a shielded pathway that allowed protons to reach the hydrogenase but not O2 (Fig. 5b). The glass framework thereby allowed sacrificial H2 evolution to be powered photocatalytically through the Ru dye. The system was able to generate H2 at photocatalytic rates as high as 7.9 s−1 per enzyme, with a TON of 130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 over 12 hours under aerobic atmospheres.
000 over 12 hours under aerobic atmospheres.
Shielding strategies have also been applied to H2 oxidising systems. Redox active polymers containing viologen moieties are capable of simultaneously immobilising and protecting hydrogenases during H2 oxidation,67,68 and 3D porous carbon electrodes loaded with hydrogenase have sustained H2 oxidation activity by favouring the effusion of H2 over O2.69 These approaches could also be employed for H2 evolving systems.
Despite being complex and multifaceted, the interaction between hydrogenases and O2 is generally thoroughly investigated. Yet there is currently enormous scope for the development of improved O2 shielding systems and scaffolds to protect the enzyme and allow the use of more O2-sensitive hydrogenases in less stringent environments. Future work should remove redox mediators and sacrificial agents from these systems and focus on constructing O2 shields on hydrogenase-modified electrodes to retroactively produce O2-tolerant hydrogenase systems.
5. Oxygen-tolerant molecular synthetic catalysts
Synthetic molecular catalysts are discrete transition metal complexes consisting of metal/ligand combinations designed to promote proton reduction.4,70 Study of their activity is normally restricted to the homogeneous phase, containing the dissolved catalyst and an electron source, which is typically an electrode, a dye with a sacrificial electron donor or a strong chemical reducing agent. Recent examples have shown innovative rational design71–75 and the field has been reviewed numerous times.5,76 These catalysts do not typically exhibit TONs or TOFs comparable to hydrogenases and the most active solid-state catalysts, but offer a defined catalytic site that can be easily manipulated and used to establish functionality and mechanisms that are essential for efficient proton reduction activity.Molecular catalysts are often inspired by the active site of hydrogenases and are frequently referred to as ‘artificial hydrogenases’ accordingly.77 Due to the low tolerance of hydrogenases towards O2, for a long time molecular catalysts were assumed to be unusable under aerobic conditions,5 however it is becoming increasingly apparent that molecular synthetic catalysts do not necessarily exhibit the debilitating O2-sensitivity of the enzymes they mimic.
Our group reported the first full study of O2-tolerant proton reduction with a synthetic molecular complex.78 The study used a water-soluble [Et3NH][CoIIICl(dimethylglyoximato)2(pyridyl-4-hydrophosphonate)] catalyst (Fig. 6 shows fully protonated complex 1A) and explored changes in activity under varying levels of O2. CVs of the catalyst were undertaken under N2, O2 and CO (Fig. 7).79 Catalytic currents were seen under N2 and O2 (Fig. 7a) but not CO, a known catalyst inhibitor (Fig. 7b). The large difference in proton reduction current between the CO-inhibited CV and the aerobic CV illustrates the O2-tolerant activity of the complex. Evidence of O2 reduction was also visible as the non-catalytic CoII/CoIII oxidation wave from the cobaloxime was not seen under aerobic conditions and the size of the CoIII/CoII wave increased, indicating competitive O2 reduction by the cobaloxime in the CoII oxidation state (Fig. 7a).
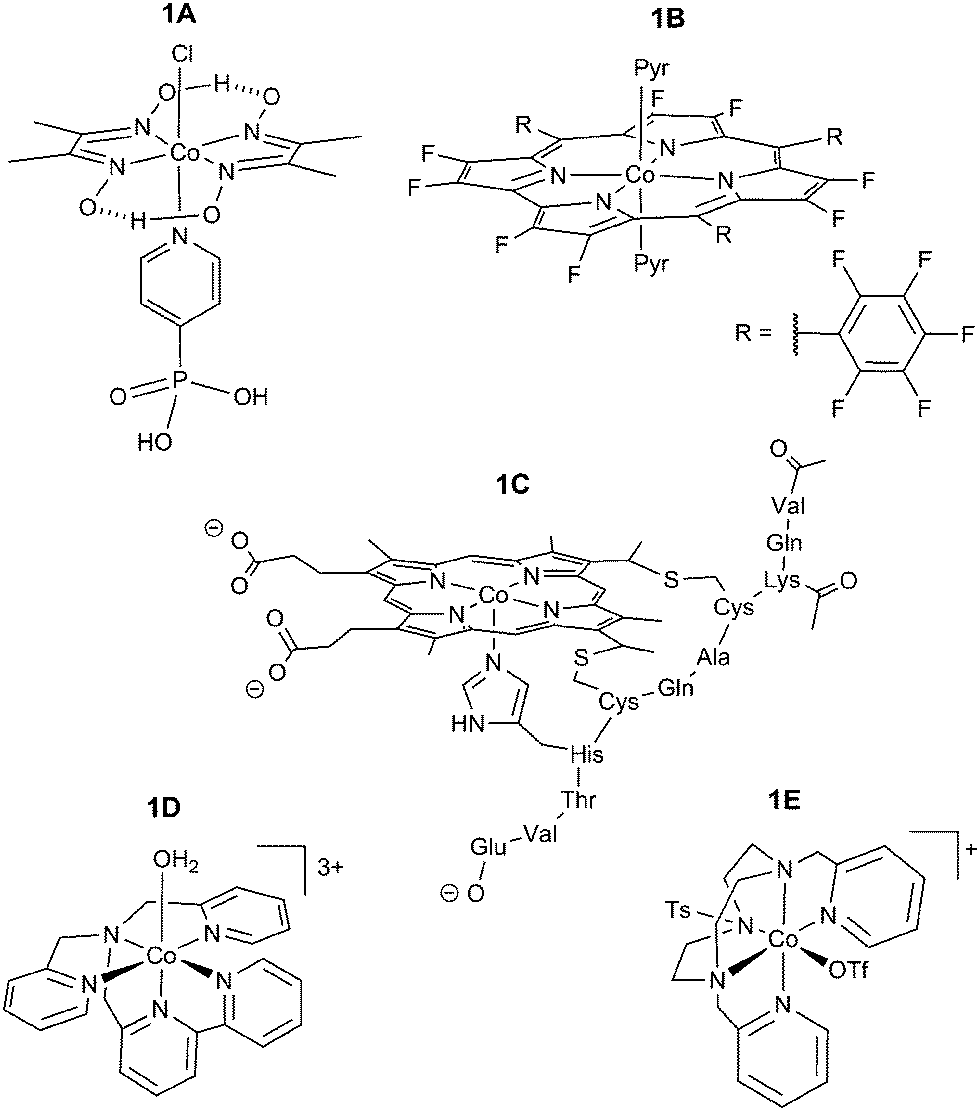 | ||
| Fig. 6 Currently known Co-based O2-tolerant molecular proton reduction catalysts. 1A: water-soluble cobaloxime;781B: fluorinated Co corrole;801C: acetylated Co microperoxidase-11;811D/1E: Co polypyridyl catalysts.82,83 | ||
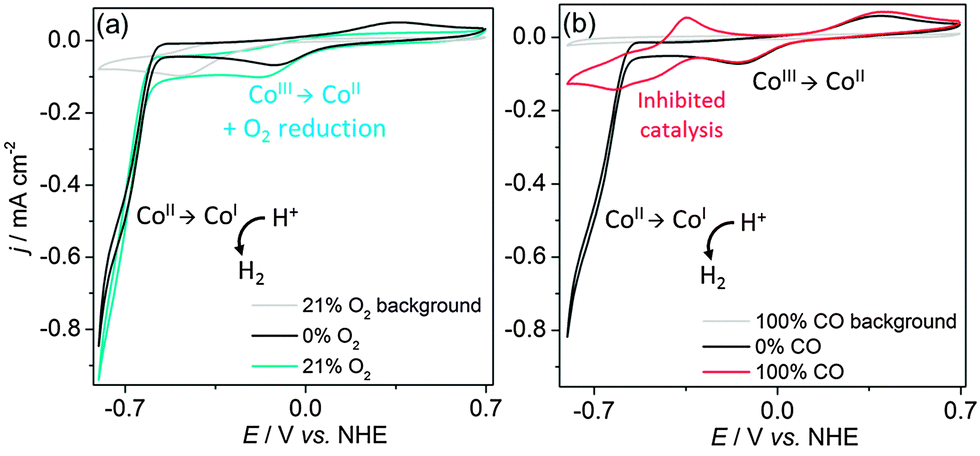 | ||
| Fig. 7 CVs of 1A (1 mM) in 0.1 M triethanolamine/Na2SO4 at pH 7 under atmospheres of (a) N2 and air and (b) N2 and CO. Scan rate was 100 mV s−1 on a glassy carbon working electrode. Taken from ref. 79. | ||
Subsequent CPE of this complex under inert and aerobic conditions at Eappl = −0.7 V vs. NHE (0.29 V overpotential) showed that substantial H2 production activity remained in the presence of O2. After re-purging the aerobic catalyst solution with N2 and repeating CPE, the cobaloxime regained 100% of its initial activity, suggesting the drop in activity under air was a result of competitive O2 reduction by the cobaloxime and not O2 sensitivity.
Photochemical experiments supported this result. Catalysis was driven photochemically using either a heterogeneous Ru-photosensitised TiO2 nanoparticle system or a homogeneous dye, eosin Y, and the evolved H2 was measured under increasing concentrations of O2. Under 21% O2, 71% of the original H2 evolution activity was measured in the homogenous system and 17% was maintained in the colloidal system, which illustrated the O2 tolerance of the cobaloxime complex. Subsequent experiments with other cobaloxime variants have shown similar levels of O2 tolerance.24,84
It should be noted that the degree of O2 tolerance exhibited by 1A varied depending on the electron source and as such the dye or electrode and the correspondingly applied potential to the catalyst must be considered when studying molecular systems under O2. Most commonly used electrodes are capable of reducing O2 to some extent and any currents stemming from a homogeneous catalyst must be deconvoluted from this background electrode activity. CVs of glassy carbon in air show a wave at −0.5 V vs. NHE in pH 7 solution (Fig. 7a, background) and FEs of a catalyst will typically be significantly less than the expected 100% for the same reason.79 The photosensitiser will also react with O2 during catalysis, lowering the rate of electron transfer to the catalyst and producing ROS. Organic dyes, such as fluorescein, rose bengal and eosin Y are common photosensitisers due to their appealing lack of precious metal centre, however under O2 they are a source of 1O2,66 which will rapidly react with catalyst ligands. Ruthenium polypyridine dyes are similarly quenched by O2.85 These dyes can be coupled to TiO2 to assist in charge separation, however the TiO2 is capable of producing ROS in the form of O2˙− and OOH− during irradiation.86 The low activity of the heterogeneous TiO2-based system that drove photocatalysis of 1A could be a result of O2˙− formation with concomitant desorption or decomposition of the Ru dye or catalyst.87
Following on from the cobaloxime system, a Co corrole catalyst synthesised by the Dey group demonstrated similar levels of O2 tolerance in 2013 (1B, Fig. 6).80 The study used a fluorinated macrocycle to decrease the overpotential needed for proton reduction and catalytic activity was established using a rotating ring-disk electrode consisting of the complex immobilised on an edge plane graphitic electrode with a Pt ring. Rotating ring-disk experiments were carried out in the presence of O2, allowing the authors to analyse the O2 reduction by the Co corrole through oxidation of the generated H2O2. This demonstrated the real time reduction of protons to H2 under aerobic conditions by the catalyst and CPE gave a FE of 52% under air after 10 hours of electrolysis in 0.5 M H2SO4. The O2 tolerance of the Co corrole stems from its ability to reduce O2 without deactivation, which had been reported previously.88
Bren and coworkers demonstrated in 2014 that an acetylated Co microperoxidase-11 complex (1C, Fig. 6) was O2 tolerant.81 This catalyst has a macrocyclic centre similar to that of 1B and showed a high FE of 85% when CPE was carried out over 4 hours in a pH 7 solution (13% lower than the equivalent experiment under N2). The high FE seen in this case may be a result of the large applied overpotential (850 mV), making the barrier of proton reduction over O2 reduction less significant. In such a case the relative concentrations of protons over O2 would determine catalyst selectivity. At room temperature the concentration of O2 is 0.3 mM under aerobic conditions89 with a diffusion coefficient of 2 × 10−5 cm2 s−1,90 and is therefore outmatched by the highly available and faster diffusing protons.
Cobalt polypyridyl catalysts have also demonstrated a degree of tolerance to O2. These catalysts typically show high stability towards deactivation and a number of structural variants have been synthesised.91,92 [Co(N,N-bis(2-pyridinylmethyl)-2,2′-bipyridine-6-methanamine)(OH2)][PF6]3 ([Co(DPA-Bpy)(OH2)][PF6]3) (1D, Fig. 6) is an O2-tolerant Co polypyridyl complex published by Zhao and coworkers.82 Using a [Ru(bpy)3]2+ photosensitiser in the presence of ascorbic acid as a sacrificial electron donor, the catalyst retained 40% of its activity in the presence of air, however this was not explored in more detail. This has been followed up by Lloret-Fillol and coworkers who used a 1,4-di(picolyl)-7-(p-toluenesulfonyl)-1,4,7-triazacyclononane (Py2TStacn) ligand to form a Co complex capable of generating H2 under O2 (1E, Fig. 6).83 In this case 25% of catalytic activity was maintained under air using a molecular Ir photosensitiser.
The O2-tolerant catalysts discussed thus far have a similar structure, consisting of N-ligating ligands to a Co centre. Proton reduction in such species is thought to occur through CoII/CoI intermediates to form a CoIII–H.82,93,94 The hydridic intermediate may then reduce a proton to form H2 or be further reduced to CoII–H, which evolves H2 (Fig. 8). Each of the reduced Co centres could also be active for O2 reduction95,96 (Fig. 8) and there is precedent for the formation of H2O2 by cobaloximes24,97 and H2O by Co corroles.88 Proficient reduction of O2 and ROS to harmless species by these catalysts may explain their limited deactivation in a similar manner to O2-tolerant hydrogenases. The catalytic core of these complexes is also comparable to Vitamin B12 and parallels can be drawn between the H2 production and O2 reduction activity of these species.96 Comparison of these complexes to biological structures will be useful in understanding the effects of O2 inhibition in both classes of catalyst.
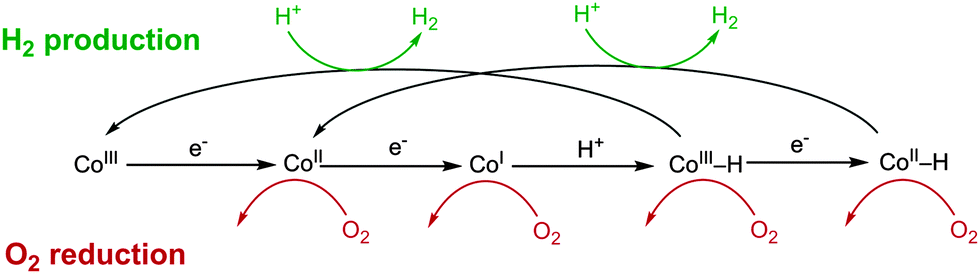 | ||
| Fig. 8 The proposed mechanism for heterolytic H2 evolution from Co complexes 1A–E and the potential O2 reduction reactions that could be carried out at the reduced intermediates. Adapted from ref. 98. | ||
It is important for the study of O2-tolerant molecular complexes to move away from the Co–N based scaffold and branch out into different ligand structures and metal centres to establish other functionalities insensitive to deactivation. A recent study of O2 tolerance with a Ni bis(diphosphine) catalyst (1F, Fig. 9) was consequently carried out by our group.79 The cyclic phosphine ligand-set coordinated to Ni contains pendant amines, which serve as proton relays that has led to high activity in organic and aqueous solution.72,75 CV of this hydrogenase-inspired catalyst showed little difference between anaerobic and aerobic conditions, however CPE at −0.4 V vs. NHE (0.13 V overpotential) at pH 4.5 produced 1.05 μmol of H2 (72% FE) under N2, but no H2 under 21% O2, indicating a high degree of O2-sensitivity.79 In its native Ni2+ oxidation state this catalyst is air stable, suggesting that a reduced form of the catalyst is susceptible to reaction with ROS/O2. The inactivation has been assigned to oxidation of the phosphine ligands to phosphine oxides during turnover under O2 (Fig. 9), which show no proton reduction activity. This effect has been observed when using compounds with similar composition as O2 reduction catalysts.99
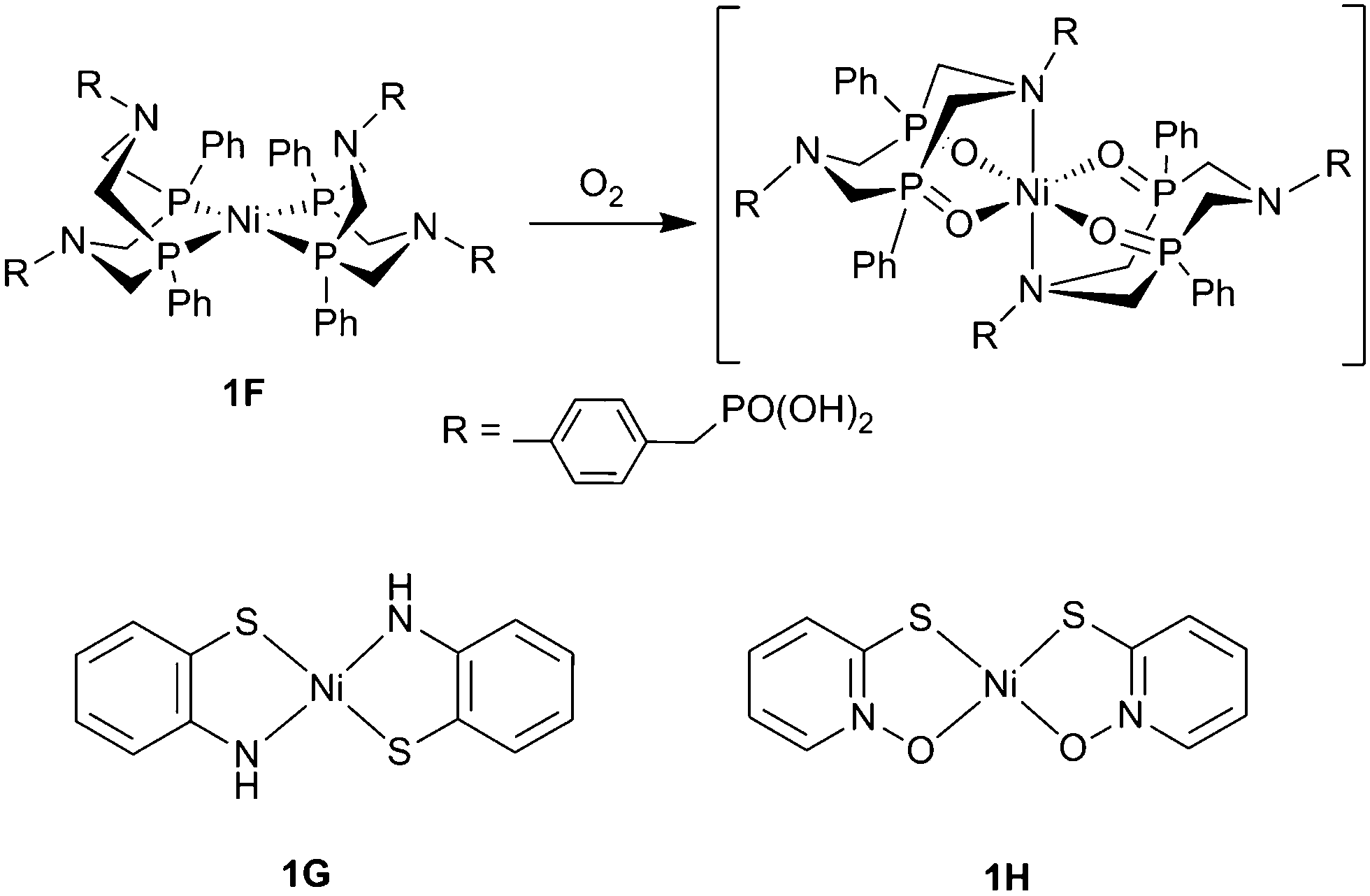 | ||
| Fig. 9 The O2-sensitive Ni bis(diphospine) complex, 1F, and the proposed route of inhibition. Complexes 1G and 1H are O2 tolerant square planar Ni complexes.100 | ||
Recently two square planar Ni thiolate-containing complexes have shown a high degree of O2 tolerance. These simple structures are notable for their high stability and in a recent report Eisenberg and coworkers showed that catalysts 1G and 1H (Fig. 9) exhibited TONs of 62000 and 80000, respectively, over 40 h CPE in aerobic solutions.100 CVs of the catalysts were identical under Ar or air and CPE showed a 15–18% drop in FE between inert and aerobic conditions (93 to 78% for 1G and 98 to 80% for 1H). The high FE suggests that these catalysts are robust in air, which may be related to the high overpotential applied (between 700–800 mV), much like catalyst 1C.
To gauge the current state of O2-tolerant molecular proton reduction catalysts, all examples known to us and their catalytic properties are summarised in Tables 1 and 2. In an ideal situation, H2 would be produced at mild overpotentials, with the same rate and efficiency regardless of whether O2 is present. This is not yet the case, however, examples continue to push the boundaries of what was previously thought possible and it appears that this could be realised within the next few years.
| Complex | Catalyst/electrode material | TOF under anaerobic/aerobic atm. (h−1) | pH | Over-potential (mV) | FE under anaerobic/aerobic atm. | Ref. |
|---|---|---|---|---|---|---|
| 1A | Cobaloxime/glassy carbon | 3.68/0.83 | 7 | 290 | 67/10 to 43% | 78 and 79 |
| 1B | Co corrole/graphite | N/A | 0 | 800 | N/A/52% | 80 |
| 1C | Acetylated Co microperoxidase-11/Hg pool | 6250/4750 | 7 | 850 | 98/85% | 81 |
| 1G | [Ni(2-aminobenzenethiolate)2]/glassy carbon | N/A/1550 | 7 | 800 | 93/78% | 100 |
| 1H | [Ni(2-pyridinethiolate-N-oxide)2]/glassy carbon | N/A/2000 | 7 | 780 | 98/80% | 100 |
| Complex | Catalyst/photosensitiser | TOF under anaerobic/aerobic atm. (h−1) | % Activity in aerobic atm. (%) | pH | λ of light | Ref. |
|---|---|---|---|---|---|---|
| 1A | Cobaloxime/TiO2-tris(bipyridine)Ru | 15/2.6 | 17 | 7 | λ > 420 nm | 78 |
| 1A | Cobaloxime/eosin Y | 62.0/44.2 | 71 | 7 | λ > 420 nm | 78 |
| 1D | [Co(DPA-Bpy)(OH2)][PF6]3/tris(bipyridine)Ru | N/A | 40 | 4 | 450 nm | 82 |
| 1E | [Co(CF3SO3)(Py2TStacn)][CF3SO3]/bis(2-phenylpyridine)(bipyridine)Ir | 147/44 | 30 | N/A | 447 nm | 83 |
There are many other known molecular catalysts that should be studied under O2 to establish a clear trend between catalyst structure and O2-tolerant proton reduction. It is also important that O2-tolerance studies are carried out in aqueous solution, rather than commonly used organic solvents as the solubility and behaviour of O2 in these environments is drastically different (O2 solubility in acetonitrile = 8.1 mM at 25 °C).101 Computational studies have begun to establish the effects of O2 on a molecular catalyst structure,102 but further expansion and comparison to experimental data is required. Future investigation must also include the study of ROS intermediates and their interaction with metal complexes to establish the O2 reduction tendencies of the O2-tolerant vs. the O2-sensitive catalysts. Nevertheless, at present it would seem that choosing a molecular catalyst capable of both catalytic O2 and proton reduction is the most viable strategy to attain an O2-tolerant molecular system.
6. Oxygen-tolerant catalytic surfaces
‘Catalytic surfaces’ is a broad term that we apply to heterogeneous surfaces, nanoparticles and immobilised assemblies in this perspective. Given their generally high stability and amenability to widespread use, such surfaces have been able to produce large amounts of H2 at rates rivalling those of enzymatic systems and many new examples have recently emerged.15,103 The wide scope for structural and geometric modification through methods such as doping, nanostructuring or controlled deposition of multifunctional layers has allowed rational surface design to maximise catalytic turnover and stability.12,104,105 Their use includes a few disadvantages however, as they have generally low ‘per atom activity’ and ascertaining the exact nature of the catalytically active site and mechanism can be difficult.Heterogeneous surfaces are considerably less sensitive to O2 than molecular complexes and hydrogenases (presumably due to the absence of fragile organic ligand frameworks) and many proton reducing surfaces are active O2 reduction catalysts.106,107 New developments in this field are instead focused on increasing catalytic selectivity for H2 evolution over O2 reduction in order to maximise efficiency.
Surface engineering to exclude O2 diffusion to the active catalyst seeks to defend catalytic surfaces from O2 entirely. One example of O2 exclusion has been presented by Domen and coworkers on a photocatalytic water-splitting particle consisting of a (Ga1−xZnx)(N1−xOx) photocatalyst loaded with Rh. O2 is particularly problematic in these systems as the Rh is able to catalyse the H2 and O2-consuming back reaction of water splitting (the reverse of reaction 1).13 It was found that the back reaction could be completely prevented through the use of a Cr2O3 layer. When the Rh cocatalyst was coated with Cr2O3 the water-splitting activity was greatly enhanced as the Cr2O3 blocked O2 from diffusing to the Rh surface (Fig. 10a).108,109 This effect was confirmed through a voltammetric study of a Cr2O3-coated Rh electrode, which showed complete loss of the O2 reduction wave on Rh.110 Proton reduction activity still remained and was only slightly diminished as a result of the Cr2O3 layer blocking some catalytic sites on the Rh. This was confirmed through infrared spectroscopy, which illustrated that protons were able to penetrate the Cr2O3 to reach a catalytic Pt surface.
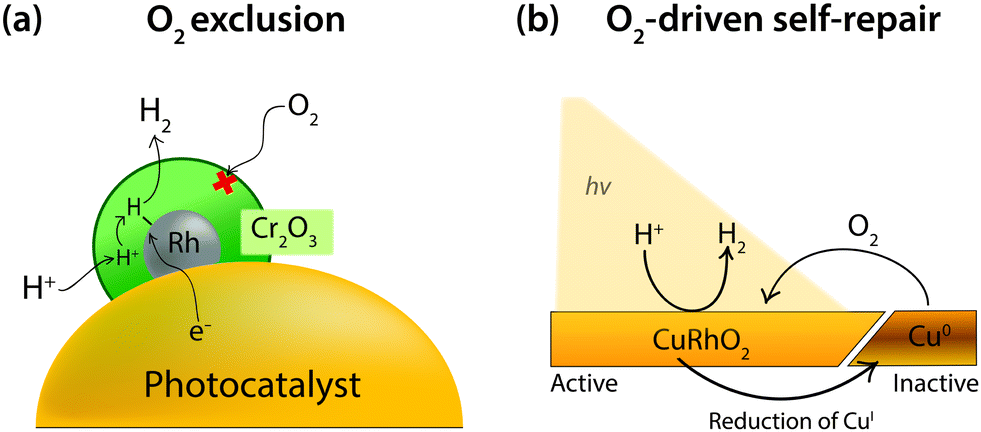 | ||
| Fig. 10 (a) Schematic representation of O2 exclusion by a Cr2O3 layer loaded on a Rh cocatalyst for photocatalytic H2 production.110 (b) Illustration of O2-driven self-repair after photocorrosion of a CuRhO2 electrode to form inactive Cu0.111 | ||
A similar strategy has been utilised by Dey and coworkers using ammonium tetrathiomolybdate (ATM),112 a reagent commonly used as a precursor to H2-evolving MoSx. It was proposed that the ATM formed a layer on Au that could shuttle protons, whilst preventing access of O2 to catalytically active sites. CV of an ATM-Au electrode showed no O2 reduction wave and CPE with 180 mV applied overpotential under air gave a high FE of 89% for proton reduction over 10 hours. The oxygen tolerance of the MoSx archetype is believed to originate from the S ligand, which plays a key role in the proton reduction mechanism.103
A number of other surface coatings have been able to prevent O2 reduction at photocatalyst surfaces, such as: lanthanide oxide layers based on La, Pr, Sm, Gd, and Dy on Rh loaded (Ga1−xZnx)(N1−xOx);113 amorphous Si and Ti oxyhydroxides on perovskite-type oxynitride, LaMgxTa1−xO1+3xN2−3x (x ≥ 1/3);114 surface-corroded Ti4+-doped Fe2O3;115 electrodeposited amorphous TiO2 on W-doped BiVO4;116 NiO-loaded on NaTaO3117 and cocatalysts of Au or RuO2.12,118 O2-excluding SiO2 layers for electrocatalytic CO2 reduction have also emerged119 and the presence of Li+ counter ions over K+ or Na+ has been shown to assist in the preclusion of O2 reduction.120
Other strategies to prevent a catalyst from O2 interaction may be achievable through O2-impermeable polymers. Research in this field is well-established due to its amenability to industrial applications, such as O2-impermeable packaging materials. A number of polymer layers are generally impermeable to O2 and thin coatings of metal oxides such as ZnO/SiOx and Al can lower the O2 permeability further.121
Preventing O2 reduction can also be achieved through use of selective catalysts. Takanabe and coworkers have synthesised tungsten carbide nanoparticle cocatalysts that illustrate an affinity for proton reduction over O2 reduction catalysis.122 Loading the nanoparticles onto a Na-doped SrTiO3 photocatalyst increased H2-evolution activity and prevented O2 reduction, which led to the UV light-driven production of stoichiometric quantities of H2 and O2 through water splitting.
Alternatively, O2 in solution can be used to maintain a catalytic structure through O2-driven self-repair. This has been demonstrated by Bocarsly and coworkers using a delafossite CuRhO2 structured electrode that functions most effectively under air (Fig. 10b).111 O2-driven self-repair is a form of O2 tolerance that reduces O2 to regenerate the active catalytic material. CuRhO2 is a photocathode for proton reduction at an applied bias of −0.7 V vs. NHE in 1 M NaOH. Under inert atmospheres the surface is active for 3 hours of photoelectrolysis, whereas in an aerobic atmosphere the activity remained constant over 8 hours. The increased stability in the presence of O2 was proven via X-ray photoelectron spectroscopy to be a result of regeneration of CuI by dissolved O2, which precluded the accumulation of Cu0 deposits on the surface. The material had a lowered FE compared to surfaces under inert atmospheres, at 80%, however this number is respectable in such challenging conditions and the lost efficiency is merely a result of the O2 reduction necessary for electrode regeneration.
In a similar example to the delafossite electrode above, a CuFeO2 electrode presented by Choi and coworkers was more stable in the presence of O2.123 The surface was able to produce H2 under visible light with a very large applied bias of −1.4 V vs. NHE in O2-saturated 1 M NaOH. The electrode had a photon to current ratio of 2.2% under Ar saturated and 3.7% under O2 saturated solutions suggesting that the electrode was less selective towards H2 evolution than CuRhO2. This has since been followed up by the Sivula group who described a sol–gel technique to fabricate a similar electrode,124 which was further doped with O2 to improve performance.
Heterogeneous, proton-reducing surfaces offer the most simple and robust strategies to achieve O2-tolerant H2 evolution. The use of O2-excluding layers is particularly interesting as the approach is also amenable to the systems discussed in Sections 4 and 5 of this perspective. It should be noted that it is still rare for H2 evolution activity to be studied under aerobic conditions and more studies of the presented strategies in the presence of O2 are therefore necessary.
7. Conclusion and future outlook
This perspective describes the state-of-the-art for the rapidly developing field of O2-tolerant proton reduction catalysis. Each of the catalytic classes discussed in Sections 4 to 6 demonstrate distinct approaches to achieve aerobic proton reduction, which revolve around either a defensive or an offensive strategy (Fig. 11). Future advances will surely involve a combined use of such techniques across enzymatic, molecular and surface-based catalysts, which we hope to bring together in this work.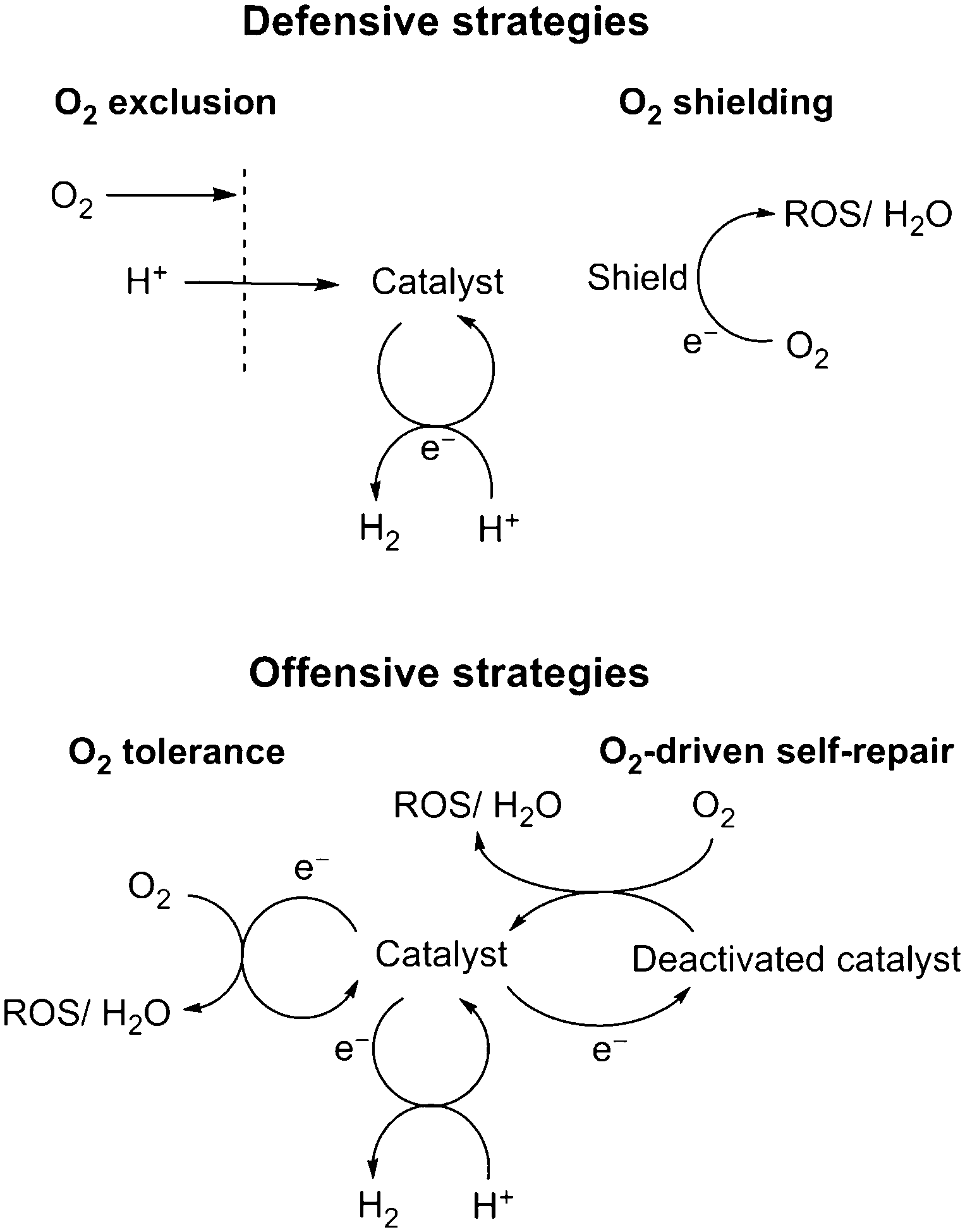 | ||
| Fig. 11 A summary of the offensive/defensive strategies used to evolve H2 in the presence of O2. | ||
Defensive methods to preclude O2 inhibition will allow the use of O2-sensitive catalysts under less stringent conditions. The use of O2 shields offers a simple and effective approach to remove O2, but such systems do not ensure complete elimination of O2 from a system and greatly lower catalytic efficiency. O2-exclusion layers are in theory a more effective route for O2-sensitive systems as they generate an anaerobic environment for catalysis without reducing the overall efficiency. These would be particularly useful for highly O2-sensitive catalysts, such as hydrogenases.
Offensive techniques utilise the catalytic centre to remove O2 from solution without damaging the catalyst and will be much simpler to utilise on a large scale. O2 tolerance has been identified in a number of catalysts and although not formally tested, is presumably present in a number of other species. O2 tolerance results in a lowered efficiency for proton reduction and decreasing the catalytic affinity for O2 reduction is therefore the predominant issue to be solved. O2-tolerant systems can be further optimised through combination with defensive strategies, such as O2-exclusion layers. Alternatively O2 can be used to improve the stability of reductively corroded catalysts through O2-driven self-repair, taking advantage of oxidising aerobic atmospheres. This has proven particularly useful for delafossite structured catalysts and may also prove effective for other catalysts that decompose in inert atmospheres.
To make further progress in this field it is important that O2 inhibition becomes a more common test of a proton reduction system. A tolerance to O2 is an excellent trait for a catalyst to exhibit and should be reported alongside other catalytic properties. Establishing the impact of O2 is simple; a catalyst's interaction with O2 can be studied with an extra electrolysis or photolysis experiment under aerobic conditions rather than an inert atmosphere.
More in depth studies of O2-tolerant catalyst systems should also become commonplace. Future studies would benefit from the use of rotating ring-disk electrodes and quantification of the produced ROS to help gain a better understanding of catalytic behaviour and deactivation pathways under air. Appreciating the factors that contribute to proton reduction inhibition by O2 should then pave the way for water splitting systems capable of functioning flawlessly under aerobic conditions. Whether such a system would be best implemented with an enzymatic, molecular or surface-based catalyst is yet to be determined, however the chemical strategies used to avoid O2 inhibition can mutually benefit the field as a whole.
The strategies considered in this perspective are also applicable to the production of other renewable fuels. Catalytic processes, such as CO2 reduction, offer alternate routes to artificial photosynthesis and would similarly benefit from O2-tolerant catalysts (for high aerobic stability) in combination with O2-exclusion strategies (for high efficiency). There are also other inhibitors to investigate, such as CO, which is formed in synthesis gas producing systems or through unwanted side reactions (e.g. in formic acid decomposition), the impact of which is seldom explored.79 Understanding inhibition across a range of inhibitors and catalytic processes will have the dual benefit of increasing our understanding of catalytic active sites and increasing the viability of each system to more widespread production of sustainable, pollution-free fuel.
Note added after first publication
This article replaces the version published on the 29th of May 2015, which contained an error in reaction (1).Acknowledgements
Financial support from the EPSRC (EP/H00338X/2), the Christian Doppler Research Association (Austrian Federal Ministry of Science, Research and Economy and National Foundation for Research, Technology and Development), and the OMV Group is gratefully acknowledged. Further thanks are extended to Mr Timothy E. Rosser, Dr Chong-Yong Lee and Dr Hyun S. Park for useful comments and fruitful discussions.Notes and references
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS PubMed.
- C.-Y. Lin, D. Mersch, D. A. Jefferson and E. Reisner, Chem. Sci., 2014, 5, 4906–4913 RSC.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- W. T. Eckenhoff and R. Eisenberg, Dalton Trans., 2012, 41, 13004–13021 RSC.
- F. E. Osterloh, J. Phys. Chem. Lett., 2014, 5, 2510–2511 CrossRef CAS.
- C.-Y. Lin, Y.-H. Lai, D. Mersch and E. Reisner, Chem. Sci., 2012, 3, 3482–3487 RSC.
- L. Ghassemzadeh, K.-D. Kreuer, J. Maier and K. Müller, J. Phys. Chem. C, 2010, 114, 14635–14645 CAS.
- M. Danilczuk, F. D. Coms and S. Schlick, J. Phys. Chem. B, 2009, 113, 8031–8042 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- R. E. Rocheleau, E. L. Miller and A. Misra, Energy Fuels, 1998, 12, 3–10 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- P. C. K. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- P. M. Wood, Biochem. J., 1988, 253, 287–289 CAS.
- A. A. Gewirth and M. S. Thorum, Inorg. Chem., 2010, 49, 3557–3566 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- Electrochemical measurements were carried out on a potentiostat (Ivium) with a Ag/AgCl reference electrode (sat. KCl, BASi) and a platinum-mesh counter electrode. Potentials were converted to NHE through the relationship: E (V) vs. NHE = E (V) vs. Ag/AgCl + 0.197 V.
- J. Zhao and F. E. Osterloh, J. Phys. Chem. Lett., 2014, 5, 782–786 CrossRef CAS.
- E. Yeager, Electrochim. Acta, 1984, 29, 1527–1537 CrossRef CAS.
- N. Kaeffer, A. Morozan and V. Artero, J. Phys. Chem. B, 2015 DOI:10.1021/acs.jpcb.5b03136.
- Y. Tian, L. Mao, T. Okajima and T. Ohsaka, Anal. Chem., 2002, 74, 2428–2434 CrossRef CAS.
- L. Campanella, G. Favero and M. Tomassetti, Anal. Lett., 1999, 32, 2559–2581 CrossRef CAS PubMed.
- M. I. Song, F. F. Bier and F. W. Scheller, Bioelectrochem. Bioenerg., 1995, 38, 419–422 CrossRef CAS.
- K. Tammeveski, T. T. Tenno, A. A. Mashirin, E. W. Hillhouse, P. Manning and C. J. McNeil, Free Radical Biol. Med., 1998, 25, 973–978 CrossRef CAS.
- W. Scheller, W. Jin, E. Ehrentreich-Förster, B. Ge, F. Lisdat, R. Büttemeier, U. Wollenberger and F. W. Scheller, Electroanalysis, 1999, 11, 703–706 CrossRef CAS.
- H. Flamm, J. Kieninger, A. Weltin and G. A. Urban, Biosens. Bioelectron., 2015, 65, 354–359 CrossRef CAS PubMed.
- X. J. Chen, A. C. West, D. M. Cropek and S. Banta, Anal. Chem., 2008, 80, 9622–9629 CrossRef CAS PubMed.
- S. Madhurantakam, S. Selvaraj, N. Nesakumar, S. Sethuraman, J. B. B. Rayappan and U. M. Krishnan, Biosens. Bioelectron., 2014, 59, 134–139 CrossRef CAS PubMed.
- Q. Zhang, Y. Qiao, L. Zhang, S. Wu, H. Zhou, J. Xu and X.-M. Song, Electroanalysis, 2011, 23, 900–906 CrossRef CAS PubMed.
- C. Xiang, Y. Zou, L.-X. Sun and F. Xu, Electrochem. Commun., 2008, 10, 38–41 CrossRef CAS PubMed.
- J. Bai and X. Jiang, Anal. Chem., 2013, 85, 8095–8101 CrossRef CAS PubMed.
- C. Calas-Blanchard, G. Catanante and T. Noguer, Electroanalysis, 2014, 26, 1277–1286 CrossRef CAS PubMed.
- J. M. Burns, W. J. Cooper, J. L. Ferry, D. W. King, B. P. DiMento, K. McNeill, C. J. Miller, W. L. Miller, B. M. Peake, S. A. Rusak, A. L. Rose and T. D. Waite, Aquat. Sci., 2012, 74, 683–734 CrossRef CAS.
- G. Bartosz, Clin. Chim. Acta, 2006, 368, 53–76 CrossRef CAS PubMed.
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed.
- A. K. Jones, E. Sillery, S. P. J. Albracht and F. A. Armstrong, Chem. Commun., 2002, 866–867 RSC.
- M.-E. Pandelia, H. Ogata and W. Lubitz, ChemPhysChem, 2010, 11, 1127–1140 CrossRef CAS PubMed.
- B. Friedrich, J. Fritsch and O. Lenz, Curr. Opin. Biotechnol., 2011, 22, 358–364 CrossRef CAS PubMed.
- F. A. Armstrong, N. A. Belsey, J. A. Cracknell, G. Goldet, A. Parkin, E. Reisner, K. A. Vincent and A. F. Wait, Chem. Soc. Rev., 2009, 38, 36–51 RSC.
- S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17331–17336 CrossRef CAS PubMed.
- H. Ogata, S. Hirota, A. Nakahara, H. Komori, N. Shibata, T. Kato, K. Kano and Y. Higuchi, Structure, 2005, 13, 1635–1642 CrossRef CAS PubMed.
- A. Volbeda, L. Martin, C. Cavazza, M. Matho, B. W. Faber, W. Roseboom, S. P. J. Albracht, E. Garcin, M. Rousset and J. C. Fontecilla-Camps, J. Biol. Inorg. Chem., 2005, 10, 239–249 CrossRef CAS PubMed.
- L. Lauterbach and O. Lenz, J. Am. Chem. Soc., 2013, 135, 17897–17905 CrossRef CAS PubMed.
- T. Burgdorf, O. Lenz, T. Buhrke, E. van der Linden, A. K. Jones, S. P. J. Albracht and B. Friedrich, J. Mol. Microbiol. Biotechnol., 2005, 10, 181–196 CrossRef CAS PubMed.
- G. Goldet, A. F. Wait, J. A. Cracknell, K. A. Vincent, M. Ludwig, O. Lenz, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 11106–11113 CrossRef CAS PubMed.
- J. Fritsch, P. Scheerer, S. Frielingsdorf, S. Kroschinsky, B. Friedrich, O. Lenz and C. M. T. Spahn, Nature, 2011, 479, 249–252 CrossRef CAS PubMed.
- A. Volbeda, P. Amara, C. Darnault, J.-M. Mouesca, A. Parkin, M. M. Roessler, F. A. Armstrong and J. C. Fontecilla-Camps, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5305–5310 CrossRef CAS PubMed.
- Y. Shomura, K.-S. Yoon, H. Nishihara and Y. Higuchi, Nature, 2011, 479, 253–256 CrossRef CAS PubMed.
- M. Horch, L. Lauterbach, M. A. Mroginski, P. Hildebrandt, O. Lenz and I. Zebger, J. Am. Chem. Soc., 2015, 137, 2555–2564 CrossRef CAS PubMed.
- A. Volbeda, P. Amara, M. Iannello, A. L. De Lacey, C. Cavazza and J. C. Fontecilla-Camps, Chem. Commun., 2013, 49, 7061–7063 RSC.
- M. C. Marques, R. Coelho, I. A. C. Pereira and P. M. Matias, Int. J. Hydrogen Energy, 2013, 38, 8664–8682 CrossRef CAS PubMed.
- M. C. Marques, R. Coelho, A. L. De Lacey, I. A. C. Pereira and P. M. Matias, J. Mol. Biol., 2010, 396, 893–907 CrossRef CAS PubMed.
- C. Wombwell and E. Reisner, Chem. – Eur. J., 2015, 21, 8096–8104 CrossRef CAS PubMed.
- M. Teixeira, G. Fauque, I. Moura, P. A. Lespinat, Y. Berlier, B. Prickril, H. D. Peck Jr., A. V. Xavier, J. Le Gall and J. J. G. Moura, Eur. J. Biochem., 1987, 167, 47–58 CrossRef CAS PubMed.
- O. Skaff, D. I. Pattison, P. E. Morgan, R. Bachana, V. K. Jain, K. I. Priyadarsini and M. J. Davies, Biochem. J., 2012, 441, 305–316 CrossRef CAS PubMed.
- V. Radu, S. Frielingsdorf, S. D. Evans, O. Lenz and L. J. C. Jeuken, J. Am. Chem. Soc., 2014, 136, 8512–8515 CrossRef CAS PubMed.
- S. V. Hexter, F. Grey, T. Happe, V. Climent and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11516–11521 CrossRef CAS PubMed.
- T. Sakai, D. Mersch and E. Reisner, Angew. Chem., Int. Ed., 2013, 52, 12313–12316 CrossRef CAS PubMed.
- T. Noji, M. Kondo, T. Jin, T. Yazawa, H. Osuka, Y. Higuchi, M. Nango, S. Itoh and T. Dewa, J. Phys. Chem. Lett., 2014, 5, 2402–2407 CrossRef CAS.
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed.
- O. A. Zadvornyy, J. E. Lucon, R. Gerlach, N. A. Zorin, T. Douglas, T. E. Elgren and J. W. Peters, J. Inorg. Biochem., 2012, 106, 151–155 CrossRef CAS PubMed.
- A. P. Gerola, J. Semensato, D. S. Pellosi, V. R. Batistela, B. R. Rabello, N. Hioka and W. Caetano, J. Photochem. Photobiol., A, 2012, 232, 14–21 CrossRef CAS PubMed.
- S. V. Morozov, O. G. Voronin, E. E. Karyakina, N. A. Zorin, S. Cosnier and A. A. Karyakin, Electrochem. Commun., 2006, 8, 851–854 CrossRef CAS PubMed.
- N. Plumeré, O. Rüdiger, A. A. Oughli, R. Williams, J. Vivekananthan, S. Pöller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef PubMed.
- L. Xu and F. A. Armstrong, RSC Adv., 2015, 5, 3649–3656 RSC.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698–702 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. Rakowski DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 15368–15371 CrossRef CAS PubMed.
- Z. Han, W. R. McNamara, M.-S. Eum, P. L. Holland and R. Eisenberg, Angew. Chem., Int. Ed., 2012, 51, 1667–1670 CrossRef CAS PubMed.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS PubMed.
- T. S. Teets and D. G. Nocera, Chem. Commun., 2011, 47, 9268–9274 RSC.
- G. Caserta, S. Roy, M. Atta, V. Artero and M. Fontecave, Curr. Opin. Chem. Biol., 2015, 25, 36–47 CrossRef CAS PubMed.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS PubMed.
- D. W. Wakerley, M. A. Gross and E. Reisner, Chem. Commun., 2014, 50, 15995–15998 RSC.
- B. Mondal, K. Sengupta, A. Rana, A. Mahammed, M. Botoshansky, S. G. Dey, Z. Gross and A. Dey, Inorg. Chem., 2013, 52, 3381–3387 CrossRef CAS PubMed.
- J. G. Kleingardner, B. Kandemir and K. L. Bren, J. Am. Chem. Soc., 2014, 136, 4–7 CrossRef CAS PubMed.
- W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. N. Ma, H. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 5941–5944 CrossRef CAS PubMed.
- A. Call, Z. Codolà, F. Acuña-Parés and J. Lloret-Fillol, Chem. – Eur. J., 2014, 20, 6171–6183 CrossRef CAS PubMed.
- D. W. Wakerley and E. Reisner, Phys. Chem. Chem. Phys., 2014, 16, 5739–5746 RSC.
- S. Ji, W. Wu, W. Wu, P. Song, K. Han, Z. Wang, S. Liu, H. Guo and J. Zhao, J. Mater. Chem., 2010, 20, 1953–1963 RSC.
- Y.-F. Li and A. Selloni, J. Am. Chem. Soc., 2013, 135, 9195–9199 CrossRef CAS PubMed.
- K. Hanson, M. K. Brennaman, H. Luo, C. R. K. Glasson, J. J. Concepcion, W. Song and T. J. Meyer, ACS Appl. Mater. Interfaces, 2012, 4, 1462–1469 CAS.
- A. Schechter, M. Stanevsky, A. Mahammed and Z. Gross, Inorg. Chem., 2012, 51, 22–24 CrossRef CAS PubMed.
- R. Sander, Atmos. Chem. Phys. Discuss., 2014, 14, 29615–30521 Search PubMed.
- P. Han and D. M. Bartels, J. Phys. Chem., 1996, 100, 5597–5602 CrossRef CAS.
- Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212–9215 CrossRef CAS PubMed.
- R. S. Khnayzer, V. S. Thoi, M. Nippe, A. E. King, J. W. Jurss, K. A. El Roz, J. R. Long, C. J. Chang and F. N. Castellano, Energy Environ. Sci., 2014, 7, 1477–1488 CAS.
- M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786–4795 CrossRef CAS PubMed.
- R. M. Kellett and T. G. Spiro, Inorg. Chem., 1985, 24, 2373–2377 CrossRef CAS.
- G. N. Schrauzer and L. P. Lee, J. Am. Chem. Soc., 1970, 92, 1551–1557 CrossRef CAS.
- G. N. Schrauzer, Angew. Chem., Int. Ed., 1976, 15, 417–426 CrossRef CAS PubMed.
- M. Shamsipur, A. Salimi, H. Haddadzadeh and M. F. Mousavi, J. Electroanal. Chem., 2001, 517, 37–44 CrossRef CAS.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- J. Y. Yang, R. M. Bullock, W. G. Dougherty, W. S. Kassel, B. Twamley, D. L. DuBois and M. Rakowski DuBois, Dalton Trans., 2010, 39, 3001–3010 RSC.
- A. Das, Z. Han, W. W. Brennessel, P. L. Holland and R. Eisenberg, ACS Catal., 2015, 5, 1397–1406 CrossRef CAS.
- J. M. Achord and C. L. Hussey, Anal. Chem., 1980, 52, 601–602 CrossRef CAS.
- P. H.-L. Sit, R. Car, M. H. Cohen and A. Selloni, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2017–2022 CrossRef CAS PubMed.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878–3888 CAS.
- M. P. Soriaga, J. H. Baricuatro, K. D. Cummins, Y.-G. Kim, F. H. Saadi, G. Sun, C. C. L. McCrory, J. R. McKone, J. M. Velazquez, I. M. Ferrer, A. I. Carim, A. Javier, B. Chmielowiec, D. C. Lacy, J. M. Gregoire, J. Sanabria-Chinchilla, X. Amashukeli, W. J. Royea, B. S. Brunschwig, J. C. Hemminger, N. S. Lewis and J. L. Stickney, Surf. Sci., 2015, 631, 285–294 CrossRef CAS PubMed.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- T. Wang, D. Gao, J. Zhuo, Z. Zhu, P. Papakonstantinou, Y. Li and M. Li, Chem. – Eur. J., 2013, 19, 11939–11948 CrossRef CAS PubMed.
- Y. Wang, E. Laborda, K. Tschulik, C. Damm, A. Molina and R. G. Compton, Nanoscale, 2014, 6, 11024–11030 RSC.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 7554–7560 CAS.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151–10157 CAS.
- J. Gu, Y. Yan, J. W. Krizan, Q. D. Gibson, Z. M. Detweiler, R. J. Cava and A. B. Bocarsly, J. Am. Chem. Soc., 2014, 136, 830–833 CrossRef CAS PubMed.
- S. Chatterjee, K. Sengupta, S. Dey and A. Dey, Inorg. Chem., 2013, 52, 14168–14177 CrossRef CAS PubMed.
- M. Yoshida, K. Maeda, D. Lu, J. Kubota and K. Domen, J. Phys. Chem. C, 2013, 117, 14000–14006 CAS.
- C. Pan, T. Takata, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, Angew. Chem., Int. Ed., 2015, 54, 2955–2959 CrossRef CAS PubMed.
- D. Cao, W. Luo, J. Feng, X. Zhao, Z. Li and Z. Zou, Energy Environ. Sci., 2014, 7, 752–759 CAS.
- D. Eisenberg, H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2014, 136, 14011–14014 CrossRef CAS PubMed.
- Y. Matsumoto, U. Unal, N. Tanaka, A. Kudo and H. Kato, J. Solid State Chem., 2004, 177, 4205–4212 CrossRef CAS PubMed.
- A. Iwase, H. Kato and A. Kudo, Catal. Lett., 2006, 108, 7–10 CrossRef CAS PubMed.
- G. Yuan, A. Agiral, N. Pellet, W. Kim and H. Frei, Faraday Discuss., 2014, 176, 233–249 RSC.
- C. Ding, X. Zhou, J. Shi, P. Yan, Z. Wang, G. Liu and C. Li, J. Phys. Chem. B, 2015, 119, 3560–3566 CrossRef CAS PubMed.
- Y. Leterrier, Prog. Mater. Sci., 2003, 48, 1–55 CrossRef CAS.
- A. T. Garcia-Esparza, D. Cha, Y. Ou, J. Kubota, K. Domen and K. Takanabe, ChemSusChem, 2013, 6, 168–181 CrossRef CAS PubMed.
- C. G. Read, Y. Park and K.-S. Choi, J. Phys. Chem. Lett., 2012, 3, 1872–1876 CrossRef CAS.
- M. S. Prévot, N. Guijarro and K. Sivula, ChemSusChem, 2015, 8, 1359–1367 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data used to prepare Fig. 3. See DOI: 10.1039/c5ee01167a |
| This journal is © The Royal Society of Chemistry 2015 |