
Bioelectrochemical systems for nitrogen removal and recovery from wastewater
M.
Rodríguez Arredondo
ab,
P.
Kuntke
*a,
A. W.
Jeremiasse
c,
T. H. J. A.
Sleutels
a,
C. J. N.
Buisman
ab and
A.
ter Heijne
b
aWetsus, Centre of Excellence for Sustainable Water Technology, Oostergoweg 7, 8911 MA Leeuwarden, The Netherlands. E-mail: philipp.kuntke@wetsus.nl; Fax: +31 (0)58 284 30 01; Tel: +31 (0)58 284 30 00
bSub-Department of Environmental Technology, Wageningen University, Bornse Weilanden 9, P.O. Box 17, 6700 AA Wageningen, The Netherlands
cMAGNETO Special Anodes B.V., Calandstraat 109, 3125 BA, Schiedam, The Netherlands
First published on 5th November 2014
Abstract
Removal of nitrogen compounds from wastewater is essential to prevent pollution of receiving water bodies (i.e. eutrophication). Conventional nitrogen removal technologies are energy intensive, representing one of the major costs in wastewater treatment plants. For that reason, innovations in nitrogen removal from wastewater focus on the reduction of energy use. Bioelectrochemical systems (BESs) have gained attention as an alternative to treat wastewater while recovering energy and/or chemicals. The combination of electrodes and microorganisms has led to several methods to remove or recover nitrogen from wastewater via oxidation reactions, reduction reactions and/or transport across an ion exchange membrane. In this study, we give an overview of nitrogen removal and recovery mechanisms in BESs based on state-of-the-art research. Moreover, we show an economic and energy analysis of ammonium recovery in BESs and compare it with existing nitrogen removal technologies. We present an estimation of the conditions needed to achieve maximum nitrogen recovery in both a microbial fuel cell (MFC) and a microbial electrolysis cell (MEC). This analysis allows for a better understanding of the limitations and key factors to take into account for the design and operation of MFCs and MECs. Finally, we address the main challenges to overcome in order to scale up and put the technology in practice. Overall, the revenues from removal and recovery of nitrogen, together with the production of electricity in an MFC or hydrogen in an MEC, make ammonium recovery in BESs a promising concept.
Water impactNitrogen is an essential element for life. It is applied in reactive form (NH3 or NO3−) as a fertilizer on soils for growth of agricultural crops. The accumulation of reactive nitrogen in the environment causes environmental problems such as eutrophication and acidification. To decrease the accumulation of nitrogen in the environment, it needs to be removed from wastewaters before discharge into receiving water bodies. Classically, this is done using a combined aerobic and anaerobic treatment process in wastewater treatment plants (WWTPs). This process requires energy for aeration, which can constitute up to 60% of the total energy consumption of a WWTP, while nitrogen is lost. This review gives an overview of the innovative nitrogen removal and recovery technologies based on bioelectrochemical systems (BESs). We show that BESs enable energy-efficient nitrogen removal and even recovery of useful nitrogen (ammonia) from wastewaters. |
1 Introduction
Population growth has dramatically accelerated,1 fueled by developments during and following the industrial revolution. As of 2012, over 7 billion people live on earth.2 As a result, the impact of human activities on the environment has reached a critical point, not just for humanity.3 Three out of nine planetary boundaries – proposed thresholds for a ‘safe operation space of humanity’ – have been breached, namely climate change, biodiversity loss and nitrogen cycle.3Nitrogen is one of the elements essential for life. Most of the nitrogen is present in the atmosphere as inert N2 gas, where it represents approximately 78% of the present gases. In its inert form, nitrogen is not available to most living organisms and needs to be converted to more reactive forms like nitrate (NO3−), nitrite (NO2−), ammonium (NH4+), and ammonia (NH3). Natural nitrogen fixation of inert N2 to reactive NH3 is carried out by some prokaryotes (e.g. Azotobacter, Clostridium, cyanobacteria, Rhizobium) using the nitrogenase enzyme.4 This conversion is essential to provide the more reactive forms of nitrogen, as a nutrient for other living organisms.
Nowadays, humanity severely interferes with the natural nitrogen cycle to provide fertilizers for agriculture. Large amounts (120–160 Mt per year) of inert N2 are transformed into NH3via the Haber–Bosch process,3,5 a value exceeding the total amount of N2 fixed annually via natural terrestrial processes.3 While this conversion of N2 into reactive NH3 is essential for fertilizer production and to increase food production, a large part of this ‘newly produced’ nitrogen eventually ends up in the environment,6 where it accumulates and leads to pollution of waterways, soils and atmosphere (acidification). One of the strategies to mitigate the effects of nitrogen on the environment is, amongst others, removal of nitrogen from domestic wastewater. In the current practice of wastewater treatment, alternating aerobic and anaerobic treatments are required to remove the dissolved nitrogen and to convert it to inert N2 gas. The disadvantage of aerobic treatment is the considerable amount of energy required for aeration. Innovations in nitrogen removal from wastewater therefore focus on the reduction of energy use.
Bioelectrochemical systems (BESs) offer clean and CO2 neutral recovery of energy from wastewaters. The core of BESs is electrochemically active microorganisms, which can exchange electrons between electrodes and organic substances to convert chemical energy into electrical energy or the other way around.7–12 This variety of conversions has led to research in many areas: from wastewater treatment for electricity production, and the production of chemicals, to conversion of electricity into methane or organic compounds. Alternatively, BESs have been proposed as a new treatment technology to remove nitrogen from wastewaters while producing electricity.13,14 Different types of nitrogen-based reactions have been reported both at the anode and at the cathode. Recently, some of these reactions have been addressed in a study on nutrient removal and recovery in BESs by Kelly and He (2014).15
The objective of our study is to give an overview of the state-of-the-art research on nitrogen removal or recovery in BESs and to compare these new processes to existing nitrogen removal technologies. Our economic analysis, that builds on the previous work by Sleutels et al. (2012),16 shows that ammonium recovery from urine is a promising concept for economical application of BESs, mainly because of the high revenues generated from removal and recovery of nitrogen.
2 The influence of humans on the nitrogen cycle: biological, chemical, and physical processes
The Haber–Bosch process is the primary human-driven industrial conversion process of inert N2 to NH3. In this conversion, nitrogen gas reacts with hydrogen gas (H2) according to (reaction (1), Fig. 1):| N2 + 3H2 → 2NH3 | (1) |
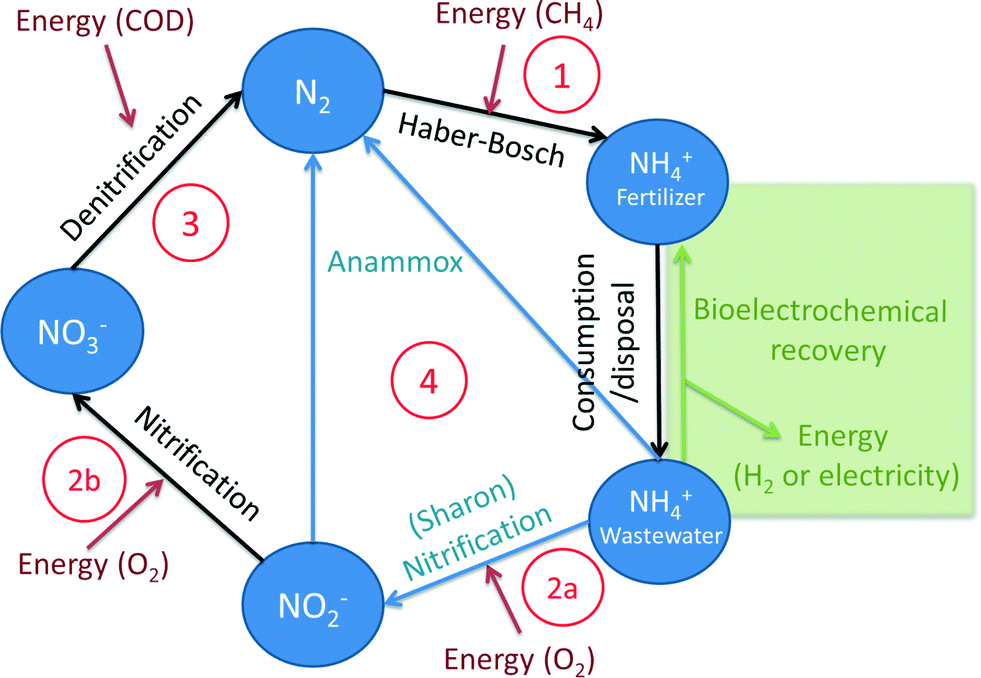 | ||
| Fig. 1 The nitrogen cycle in fertilizer production and wastewater treatment processes. | ||
The H2 required for the Haber–Bosch process is produced via steam reforming of natural gas. Most of the industrially produced ammonia (~80 Mt N per year) is used as a fertilizer in agriculture to enhance food production.3,5 Part of the ammonium is taken up by the crops, but another part ends up in the environment, where they increase the risk of eutrophication of receiving water bodies, and adds to the pollution of the atmosphere.
Because of these adverse effects on the environment, reactive nitrogen compounds in wastewater were considered as pollutants, and the importance of removing or recovering nitrogen compounds from wastewater was recognized.17 In wastewater, nitrogen is mostly present in the form of ammonium, and removal of ammonium in wastewater treatment plants mainly occurs via a two-step process.18 During the first step, nitrification under aerobic conditions, ammonia is oxidized to nitrite by Nitrosomonas, and subsequently, nitrite is oxidized to nitrate by Nitrobacter according to (reaction (2), Fig. 1):
 | (2) |
So for each mole of ammonium, 2 moles of oxygen are required for full oxidation to NO3−. During the second step, denitrification under anoxic conditions, nitrate is reduced to N2 gas, for example, by Paracoccus denitrificans, according to (reaction (3), Fig. 1):
| 2NO3− + COD → N2 + CO2 + 2OH− + H2O | (3) |
The produced N2 gas is released (or recycled) to the atmosphere.
The disadvantage of the conventional nitrification/denitrification reaction is that it requires considerable amounts of energy, because the wastewater needs to be aerated to supply oxygen for the conversion of ammonium to nitrate, and a supply of electrons for denitrification in the form of COD (e.g. methanol) is required. The advantage of nitrification/denitrification is that nitrogen can be removed at low concentrations.
Anammox (anaerobic ammonium oxidation) has been proposed as a more energy-efficient alternative to the conventional nitrification and denitrification process. Anammox relies on the biological conversion of ammonium and nitrite to N2 gas by specialized bacteria (planctomycete-like).19 First, part of the ammonium needs to be partially oxidized to nitrite in a pretreatment step. Therefore, Anammox is combined with, for example, the SHARON (Single reactor system for High-rate Ammonium Removal Over Nitrite)20,21 process. SHARON is a biological process in which ammonium is oxidized to nitrite (instead of a complete oxidation to nitrate). The oxidation to nitrite is possible due to the specific growth rates of nitrite-oxidizing bacteria, ammonium-oxidizing bacteria and the applied loading rate of the reactor. Another approach is the so-called CANON (Completely Autotrophic Nitrogen removal Over Nitrite) process, where the aerobic and anaerobic ammonium oxidizers symbiotically coexist. In the CANON process, it is possible to perform aerobic and anaerobic ammonium oxidation simultaneously. Therefore, ammonium-oxidizing and Anammox bacteria cooperate.22 The overall Anammox process can be described by (reaction (4), Fig. 1):
 | (4) |
Because only half of the ammonium needs to be oxidized to nitrite and no addition of COD is required, the energy input for the SHARON/Anammox process is considerably lower than the energy input for conventional nitrification/denitrification.19,21
A recent study23 reports a new process for the removal of nitrogen from wastewater: Coupled Aerobic–anoxic Nitrous Decomposition Operation (CANDO). CANDO consists of three steps: (1) partial nitrification of NH4+ to NO2− (by the SHARON process), (2) partial anoxic reduction of NO2− to N2O and (3) decomposition or combustion of N2O to nitrogen, oxygen and energy.23 Even though pilot-scale studies are needed, this process has the potential to lower oxygen demand and sludge production, as well as recover energy from nitrogen.
Recently, bioelectrochemical systems (BESs) have been investigated as an alternative to the conventional wastewater treatment processes, such as organic matter and nitrogen removal. A BES is an electrochemical cell which uses microorganisms to catalyse one or more reactions taking place on the electrodes. At the anode, anaerobic microorganisms can oxidize biodegradable organic matter to carbon dioxide, protons and electrons, which is often represented by the oxidation of acetate:
| CH3COO− + 4H2O → 2HCO3− + 9H+ + 8e− | (5) |
As the electrode can act as an electron acceptor, the produced electrons are transferred by the microorganisms to the anode, which is connected over an external circuit to the cathode where a reduction reaction takes place.
Depending on the cathode reaction, BESs can be divided in two types: galvanic and electrolytic cells. In galvanic cells, electricity is produced by coupling the anodic oxidation to the reduction of a suitable electron acceptor (i.e. O2, Fe3+, and Cu2+).24–26 These systems are called microbial fuel cells (MFCs). In electrolytic cells, electricity is needed to drive the reduction reaction at the cathode. In a so-called microbial electrolysis cell (MEC), the anodic oxidation is coupled to reduction of protons to hydrogen gas.8,27–29
The cathodic reactions of an MFC (oxygen reduction reaction, ORR) and an MEC (hydrogen evolution reaction, HER) under neutral to alkaline pH conditions are given in reactions (6) and (7), respectively.
| 2H2O + O2 + 4e− → 4OH− | (6) |
| 2H2O + 2e− → H2 + 2OH− | (7) |
BESs are seen as a potential sustainable solution to treat wastewaters while at the same time producing energy and/or chemicals. Anode and cathode chambers are often separated by an ion exchange membrane to prevent mixing of the oxidation and reduction products.8,9 The ion exchange membrane allows for anions and/or cations to be transported to maintain electron neutrality of the electrochemical system.
BESs allow for energetically and chemically efficient ammonium recovery from wastewater. The organic matter in wastewater is oxidized at the anode by bacteria, while ammonium (present in the wastewater) is transported over a cation ion exchange membrane (CEM) to the cathode chamber, where the high catholyte pH allows for recovery in the form of ammonia. Therefore, no addition of caustics is required compared to other ammonia recovery technologies. In this way, BESs can create a shortcut in the nitrogen cycle by removing NH3 from the wastewater, not by reduction to N2 but by direct recovery in the form of NH3 (Fig. 1).
Ammonium recovery using an electrochemical cell (EC) was investigated by Desloover et al. (2012).30 Ammonium recovery from anaerobic digestate was investigated using an electrolysis cell, in which ammonium transported over a CEM was stripped from the catholyte by the produced hydrogen. The current and thus the rate of ammonium transport in such an abiotic system are not limited by the biological catalyzed anode reaction. Furthermore, the common limitation of high internal resistance in MFCs by the cathodic reaction (ORR), which is the result of the prevalent conditions found in MFCs (temperature, pH and oxygen concentration), is not found in such an EC. As a result, the reported ammonium transport rates (120 gN m−2 d−1) are significantly higher than those reported in MFCs, whereas the ammonium transport accounted for approximately 40% of the overall charge transport over the membrane, similar to results found in BESs.13,14 Desloover et al. (2012) reported an energy demand of 5 kW h kgN−1 (or 18 kJ gN−1) for ammonium recovery by an EC,30 which is lower than the reported energy demand of 9 kW h kgN−1 (or 32.4 kJ gN−1) for conventional ammonia stripping.31 However, they did not take into account the energy used for stripping and absorption. Finally, similar to the BES approach, no caustic is necessary to increase the pH at the cathode.
3 Bioelectrochemical systems for ammonium removal and recovery
As an alternative to conventional nitrogen removal processes, bioelectrochemical systems offer new advantages. Several nitrogen-based reactions in BESs, both at the anode and at the cathode, have been reported so far. In this section, we will give an overview of the reported ammonia removal and recovery mechanisms in BESs (Fig. 2). The first mechanism of ammonium removal/recovery is based on active and passive transport through the ion exchange membrane, combined with acid/base equilibrium (1). NH4+ concentrations in wastewater are considerable: in urine >4 g L−1 (ref. 32–34) and in domestic wastewater 0.04 g L−1 (ref. 18 and 35). Consequently, especially in highly concentrated wastewaters, NH4+ is the main ion transported through the membrane, either passively in its non-charged NH3 form (1a) or actively in its charged NH4+ form (1b). In the catholyte, it can leave the system in the form of ammonia as the chemical equilibrium shifts from ammonium to ammonia because of the elevated pH in the cathode (1c). The second mechanism is reduction (denitrification) of nitrate to inert N2 gas by microorganisms at the cathode (2a). In this case, nitrate needs to be formed first, for example, via biological oxidation of ammonium by aeration (2b). Alternatively, ammonium can be converted into N2via Anammox, which is, in principle, independent of the cathode. The third mechanism that has been suggested occurs at the anode, where ammonium is directly converted into nitrogen gas by microorganisms (3), but clear evidence is still lacking. The fourth mechanism is incorporation of ammonium in biomass during microbial growth in the anode (4) or in the cathode compartment. | ||
| Fig. 2 Overview of the ammonia removal mechanisms in an MFC. This figure is representative of BES in general. Ammonium can be transported through the membrane (1) either passively via diffusion of ammonia (1a) or actively via migration in the form of ammonium (1b). Ammonia can leave the system by evaporating into its gaseous form as a result of the elevated pH in the cathode (1c). At the cathode chamber, it can be (biologically) oxidized by oxygen and denitrified by microorganisms at the cathode (2a) or in solution (2b) – the dashed lines show processes that are independent of the electrodes. At the anode, it has been suggested that ammonia can be directly nitrified/denitrified to nitrogen gas by microorganisms; however, solid proof is still lacking (3). Finally, ammonium can be incorporated in biomass for growth (4), either in the anode or in the cathode chamber. | ||
Concerning the direct oxidation of ammonium to nitrogen gas at the anode (3), a study reports that ammonium is involved in electricity generation either directly as the anodic fuel or indirectly as substrates for nitrifiers to produce organic compounds for heterotrophs.36 On the contrary, it was reported in the study of Kim et al. (2008) that ammonia removal, instead of being biologically oxidized at the anode to nitrogen gas, was mainly due to physicochemical factors, such as diffusion through the membrane and volatilization of ammonia due to pH increase at the cathode.37 Zang et al. (2012) also reported that direct ammonia oxidation in the anode was not a source of electricity.38 Cyclic voltammetry tests did not detect redox couples, nitrite and nitrate were not found and nitrifying or Anammox bacteria were not identified in the anode compartment.38 In conclusion, the occurrence of direct oxidation of ammonium to nitrogen gas so far lacks solid proof.
Table 1 gives an overview of the reported mechanisms for the removal and recovery of ammonium via routes 1 and 2 and their performance.
| Reactor type | Type of wastewater | Removal mechanism | N concentration influent (mgN L−1) | Removal efficiency/rate | Current density (A m−2) | Ref. |
|---|---|---|---|---|---|---|
| a Based on cathodic volume. b NCC: net cathodic compartment. c TN: total nitrogen. d TCV: total compartment volume. Both cathode and anode have the same volume. e Insufficient data to determine current density. f Based on anodic volume. | ||||||
| Single-chamber MFC | Swine wastewater | Ammonia removal | 188 ± 6 | 60% | 0.5 mAe | 37 |
| Dual-chamber MFC | Swine wastewater | Ammonia removal | 219 ± 2 | 68% | 0.5 mAe | 37 |
| Dual-chamber MFC | Swine wastewater | Ammonia removal | 219 ± 2 | 89% | 0.3 mAe | 37 |
| Dual-chamber MFC | Urine | Ammonia removal | 4050 | 3.3 gN m−2 d−1 | 0.5 | 13 |
| Dual-chamber MFC | Urine | Ammonia removal | 4050 | 9.7 gN m−2 d−1 | 2.7 | 33 |
| Dual-chamber MEC | Urine | Ammonia removal | 700 | 162.18 gN m−2 d−1 | 14.7 | 39 |
| Dual-chamber MFC | Synthetic | Nitrate reduction | 2615 | 146 gN m−3 d−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
35 A m−3a |
40 |
| Dual-chamber MFC | Synthetic | Nitrification coupled to nitrate reduction | 88 | 67.4% and 410 gN m−3 NCC d−1b |
133 A m−3b |
41 |
| Dual-chamber MFC | Synthetic | Simultaneous nitrification and denitrification | 41 | 94.1% TNc and 104 gN m−3 TCV d−1d |
39.7 A m−3d |
42 |
| Dual-cathode MFC | Synthetic | Nitrification and denitrification | 30–90 | 67–90% TNc and 140 gTN m−3 d−1c |
43 A m−3f |
43 |
| Single-chamber MFC | Synthetic | Simultaneous nitrification and denitrification | 100 | 96.8% | 3.6 | 44 |
| Dual-chamber MFC/MEC | Synthetic | Ammonia removal | 3000 | 2.94 gN m−2 d−1 | 3.6 | 45 |
| Dual-chamber MFC/MEC | Synthetic | Ammonia removal | 3000 | 8.5 gN m−2 d−1 | 3.6 | 45 |
| Dual-chamber MFC/MEC | Synthetic | Urea removal | 4000 | 37.8 gN m−2 d−1 | 5 | 45 |
3.1 Diffusion and migration of NH3 and NH4+
In a BES equipped with a CEM, ammonium (and other cations) can be transported from the anode to the cathode chamber by two processes: migration and diffusion. Transport by migration is induced by the electric field across the ion exchange membrane. Diffusional transport is induced by a concentration gradient across the ion exchange membrane. As a result of the production of hydroxyl ions in the ORR and HER, and the insufficient H+ and OH− transport over the CEM, the pH increases in the cathode chamber.46–48 Therefore, ionic ammonium is transformed into volatile ammonia, which can be removed from the cathode compartment by NH3 stripping with a suitable gas stream. Hence, one of the advantages of using BES for nitrogen recovery is that no additional caustics need to be added to increase the pH of the solution.13Kim et al. (2008) studied ammonia loss from swine wastewater in both single- and two-chamber MFCs.37 An air cathode was used in the single-chamber MFC, while both aerated phosphate buffer and ferricyanide were tested as catholyte in the two-chamber MFC. An ammonia removal of 60% was reached in 5 days in the single-chamber MFC (cathode exposed to air), while 68% was accomplished in 13 days when using ferricyanide as a catholyte in the two-chamber MFC. The authors concluded that the main mechanism for this ammonia removal was ammonium transport (migration and diffusion) to the cathode with subsequent ammonia volatilization.
Exploiting this ammonia loss mechanism, future work of research groups focused on ammonia as a suitable proton shuttle for pH control in BESs and ammonium recovery by NH3 stripping from the cathode.13,14,49 Cord-Ruwisch et al. (2011) investigated the feasibility of more sustainable pH control in MFCs by proton shuttling from anode to cathode via ammonia addition to the anode chamber.49 Their results showed that ammonium accounts for 90% of the ionic flux in the BES and that ammonia recovered from the cathode could be recycled to the anode. Further investigation by Cheng et al. (2013) demonstrated the feasibility of such ammonium recycle for anolyte pH control in an MEC also demonstrating a possible pathway for ammonia recovery from wastewater.50
In domestic wastewater, most of the nitrogen (75%) originates from urine.51 This nitrogen is excreted by humans in the form of urea ((NH2)2CO),32 which is hydrolyzed to ammonia and carbamate by the enzyme urease.52,53
 | (8) |
Subsequently, carbamate decomposition leads to the formation of ammonia and carbon dioxide:
| NH2COOH + H2O → NH3 + H2CO3 | (9) |
Ammonium recovery from urine in an MFC was first reported by Kuntke et al. (2011), where the authors investigated the feasibility of ammonium recovery in a two-chamber MFC with a CEM using a sacrificial potassium ferricyanide cathode system.14 The authors showed that ammonium concentrations up to 4 gN L−1 did not affect the performance of the MFC and that total ammonium transport from anode to cathode accounted for up to 50% of the charge transport. Following up on this proof of principle, Kuntke et al. (2012) demonstrated and evaluated the feasibility of using a two-chamber MFC with a gas diffusion cathode.13 Here, the volatile ammonia was removed from the cathode chamber by the gas stream used for aeration and subsequently collected in a gas wash bottle containing an acid (boric acid). They reported an ammonium recovery rate of 3.3 gN m−2 d−1 from urine with an energy yield of −3.46 kJ gN−1 at a current density of 0.5 A m−2. Later results presented by Kuntke et al. (2013) showed an ammonium recovery rate of 9.7 gN m−2 d−1 from urine with an energy yield of −10 kJ gN−1 at a current density of 2.6 A m−2 (ref. 33). Following the studies of Cheng et al. (2013)50 and Desloover et al. (2012),30 Kuntke et al. (2014) investigated simultaneous ammonium recovery and hydrogen production from urine in an MEC.39 They achieved a stable ammonium removal rate of 162.2 gN m−2 d−1 at a current density of 14.7 A m−2, while a maximum removal rate of 173.3 gN m−2 d−1 at a current density of 23.1 A m−2 was reported.39
The consumed COD also represents a certain amount of energy. In this respect, it is interesting to compare the COD consumption for different nitrogen removal and recovery processes. Using an MFC or an MEC, for each kg of N recovered, 0.57 kg of COD is required, assuming that each mole of COD corresponds to 4 moles of electrons, that the COD is converted into electricity at 100% coulombic efficiency, that all cation transport occurs through NH4+, and that all NH4+ is recovered at the cathode. For nitrification/denitrification, theoretically, 2.86 kg COD is required for removal of 1 kg of N.54 On the other hand, nitrification and Anammox require no COD at all.
3.2 Nitrification/denitrification at the cathode
The second part of Table 1 summarizes the performance of systems that studied denitrification at the biocathode. Clauwaert et al. (2007) showed that complete denitrification could be performed by microorganisms in the cathode of an MFC.40 Acetate was used as the electron donor in the biological anode, while nitrate was used as the electron acceptor in the biological cathode. Electricity production was thus coupled to the reduction of nitrate to nitrogen gas by denitrifying microorganisms. Nitrate was removed at a rate of 146 gN m−3 d−1.Virdis et al. (2008) coupled the anodic oxidation of organic matter and cathodic nitrate reduction in an MFC to an external nitrification reactor enabling the removal of ammonium from wastewater by nitrification and denitrification while producing electricity. During their experiments, organics present in the anode influent containing ammonium were oxidized to produce electricity. The anode effluent containing ammonia was fed to an external biofilm reactor for nitrification followed by denitrification at the cathode.41 The authors reported a removal rate of 0.41 kgN per m3 of net cathode compartment per day and a removal efficiency of 67.4%.
Virdis et al. (2010) also used nitrate as an electron acceptor at the cathode of an MFC.42 The system accomplished simultaneous nitrification and denitrification by optimising the oxygen supply in the aerated cathode chamber. The anode effluent was directed to the cathode through a loop connection to mitigate the pH increase in the cathode. Low levels of nitrate and ammonium in the effluent were accomplished (1 and 2 mgN L−1, respectively). The highest nitrogen removal efficiency obtained in this system was 94%.
Zhang and He (2012) also achieved high ammonia and total nitrogen removal efficiencies (96% and between 67% and 90%, respectively) by testing a tubular dual-cathode MFC, in which both anion and cation exchange membranes were used.43 The design allowed for an anoxic inner cathode in which bioelectrochemical denitrification could take place and an aerobic outer cathode for the nitrification process, which shared the same anode. A final nitrate concentration of 3 mgN L−1 was achieved in this system.
Finally, higher ammonia removal efficiencies compared to the previous system were reached without the extra energy input of aeration by Yan et al. (2012).44 The MFC consisted of a single chamber with an air cathode which was pre-enriched with a nitrifying biofilm. The cathode was especially prepared with partial positive charges to promote the nitrifying biofilm formation by using a diethylamine-functionalized polymer (DEA) as a catalyst binder instead of the conventional Nafion-type binder and went through an enrichment period of 75 days. The MFC with the DEA binder had an ammonia removal efficiency of up to 97%, while that with the Nafion binder (which also went through the pre-enrichment) had an ammonia removal efficiency of 91%. The maximum total nitrogen efficiencies were between 75% and 95%, but nitrate concentrations of up to 30 mg L−1 were detected in the MFC effluent. Furthermore, the enrichment process improved the maximum power densities of the MFCs regardless of the catalyst binder, while without the enrichment, power production was about 25% lower.
4 Ammonia recovery from urine in an MFC and an MEC
Kuntke et al. (2013) proposed a full treatment concept for urine, in which ammonium is recovered using a BES33 (Fig. 3). | ||
| Fig. 3 Proposed full treatment concept for urine, in which ammonium is recovered using a BES. | ||
The treatment concept includes phosphate recovery via struvite (also known as MAP, MgNH4PO4·6H2O) precipitation as a pre-treatment step followed by a BES to recover energy and ammonia simultaneously. Notably, the recovered ammonia can be used either as a fertilizer or as an energy source, for example, in a fuel cell.55 In the proposed scheme, caustics can be obtained as a by-product. The BES could be either an MFC for electricity production or an MEC, in which case hydrogen would be produced instead of electricity. The advantage of using an MEC is that a voltage is applied, which helps overcome the internal resistances of the system, and higher current densities can be achieved compared to an MFC. On the other hand, the advantage of an MFC is its electricity production, as applying a voltage in an MEC means an additional energy input.
In this section, we present an analysis of the potential energy recovery from urine, in the form of electricity or hydrogen, and ammonia, using a BES under different conditions. Urine, collected by separation toilets and water-free urinals, has a COD and an ammonia concentration of 10–1.6 gCOD L−1 and 8.1–0.4 gN L−1, respectively.34 In this case, to resemble the treatment concept shown in Fig. 3, a urine supernatant after struvite precipitation with COD and NH4-N concentrations of 4 g L−1 was used as a model, corresponding to earlier reported concentrations.13 The typical pH difference observed in previous experiments performed in two-chamber BESs with urine is 6 (pH 7 in the anode and 13 in the cathode). This pH gradient means that a higher applied voltage would be required in an MEC and a higher energy loss would be experienced in an MFC.16 The effect of this gradient, which can be considered as an additional internal resistance, was included in the calculations. Furthermore, in this calculation, it was assumed that there is no energy input for the MFC, and the energy outputs come from electricity production and ammonia recovery. For MECs, the energy input consists of electricity or external power supply, and the energy outputs are hydrogen production and ammonia recovery. To calculate the theoretical energy output, the lower heating values (LHV) for both hydrogen and ammonia were used: −10817 J L−1 for hydrogen56 and −18577 J g−1 for ammonia (adapted from Hacker et al. (2003) (ref. 57)). For comparison, the values for both MFC and MEC are presented in units of kJ per gram of nitrogen recovered. The potential NH4-N removal depends on both the coulombic efficiency of the system and the ammonium transport efficiency across the membrane. Therefore, the amount of removed ammonium from the anode compartment can be calculated based on assumptions for coulombic efficiency, loading rate and transport efficiency as will be described below.
Fig. 4A shows the theoretical current density (contour lines) as a function of the coulombic efficiency and the COD loading rate. The coulombic efficiency represents the amount of degraded COD that ends up in electricity, where one mole of COD corresponds with 4 moles of electrons, while the loading rate is the amount of COD that is added to the system per time unit. This figure shows that current density increases with increasing loading rate and increasing coulombic efficiency. In addition, the same current density can be achieved by combining a high loading rate and a low coulombic efficiency or vice versa.
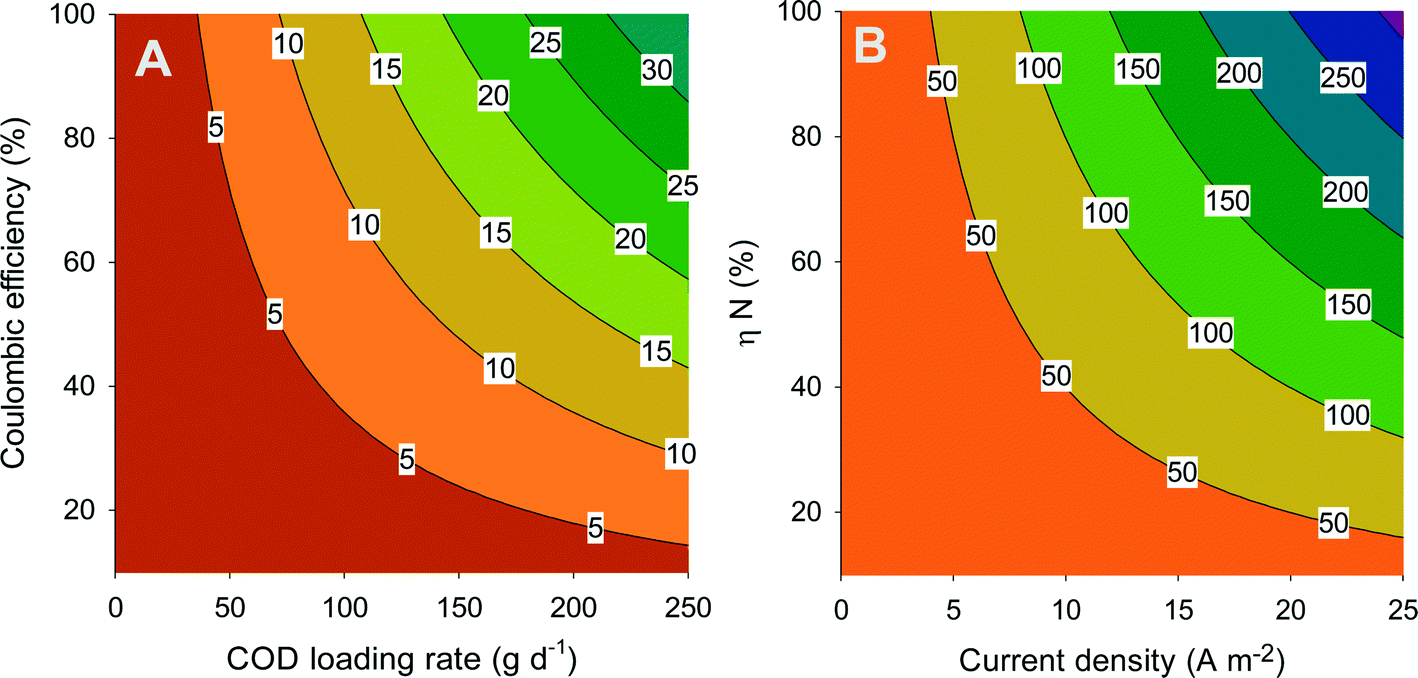 | ||
| Fig. 4 (A) Current density (A m−2) as a function of coulombic efficiency (%) and loading rate (g d−1) and (B) ammonium flux from anode to cathode (g d−1) as a function of transport efficiency (%) and current density (A m−2). | ||
In BESs, the produced charge (related to current) is equal to the amount of ions that are transported through the membrane. The transport efficiency (ηN) represents the percentage of ammonium that is transported through the membrane compared to the total amount of ions transported. Thus, at a transport efficiency of 100%, all the transported charge through the system is in the form of ammonium. The overall ammonium flux increases with increasing current density and increasing ammonium transport efficiency. From the combination of Fig. 4A and B, the total amount of ammonium that is transported from the anode compartment to the cathode compartment can be related to ammonium transport efficiency, coulombic efficiency, and COD loading rate. Here, it is assumed that 100% of the ammonia which is removed is also recovered. Clearly, to maximize the ammonium recovery, the BES needs to be operated at high coulombic efficiency and high loading rate (to achieve a high current density), in combination with high ammonium transport efficiency. These aspects will be further discussed in section 6.
Reported values (Table 2) for MFCs and MECs for urine treatment are well in line with Fig. 4A. In MFC mode, the COD loading rate was 286 gCOD m−2 d−1, but not all COD was removed, while in the calculation for Fig. 4, it is assumed that all COD is removed. Therefore, we compare the performance of the BES treating urine (Table 2) with Fig. 4 based on COD removal rate. At 181 gCOD m−2 d−1 and 10% coulombic efficiency, indeed the expected current from Fig. 4A is below 5 A m−2 (measured value of 2.6 A m−2). In MEC mode, at a COD removal rate of 171 gCOD m−2 d−1 and 96% coulombic efficiency, the expected current is between 20 and 25 A m−2, matching well with the measured value. Now, the current can be linked with the N removal rate (Table 2) to determine the N transport efficiency. For the MFC, the N transport efficiency derived from Fig. 4 is 30%, a value also reported in this study; for the MEC, it is 60%.
| Microbial fuel cell | Microbial electrolysis cell | |
|---|---|---|
| Current density (A m−2) | 2.6 ± 0.1 | 23.1 ± 1.15 |
| COD loading rate (gCOD m−2 d−1) | 285.7 ± 7.1 | 466.6 ± 14.0 |
| COD removal rate (gCOD m−2 d−1) | 180.9 ± 5.8 | 171.0 ± 16.9 |
| Coulombic efficiency (%) | 10.4 ± 0.5 | 95.6 ± 3.1 |
| N removal rate (gN m−2 d−1) | 10 | 173.34 ± 18.07 |
| Internal resistance (mΩ m2) | 95 | 43 ± 2.2 |
| References | 33 | 39 |
Fig. 5A shows the energy yield for a scaled-up MEC treating urine for ammonia recovery, using the concept proposed by Kuntke et al. (2013).33 All calculations were performed for current densities from 0.5 to 25 A m−2 (normalized by the membrane surface area) and for high (200 mΩ m2) and low (25 mΩ m2) internal resistances. Negative values for energy recovery represent actual energy recovery. Fig. 5A shows that systems with low internal resistance (25 mΩ m2) can be used in the full current range to recover energy from urine. At high internal resistance though (200 mΩ m2), operating in the low current density range (up to 13 A m−2) is required to recover energy from the system. The reason for this is that at high internal resistance, the power input required to overcome the internal resistance to achieve a certain current density increases much faster than the recovery of energy in the form of hydrogen and ammonium. This can be seen in more detail in Fig. 5B. Here, the effect of the different contributions to the total energy balance for a system with 200 mΩ m2 internal resistance is shown. The input of electrical energy increases linearly with increasing internal resistance, as more energy is required to overcome the internal resistance at higher current density. The output energy (kJ gN−1 recovered) in the form of H2 and NH3 in Fig. 5B is independent of current density because it is normalized by the nitrogen recovered. The nitrogen recovered is directly related to the current density through the transport efficiency of ammonium. Overall, energy can only be recovered at high internal resistance when the electrical energy input is limited, and thus current density is lower than 13 A m−2. These results show that achieving a low internal resistance in these systems is crucial to actually recover energy from urine, because low internal resistance leads to both high current density and low power consumption.
 | ||
| Fig. 5 (A) Energy recovery in an MEC as a function of current density for high (200 mΩ m2) and low (25 mΩ m2) internal resistances. The energy recovery for an MFC is not shown since it is comparable to the MEC. (B) A more detailed overview of the energy input (electrical) and output (hydrogen and ammonia) for the MEC with an internal resistance of 200 mΩ m2 as a function of current density. | ||
The results of the analysis for an MFC are not shown here but are comparable. The main difference is that the current range that can be achieved in the MFC is much smaller (3.5 A m−2) compared to the MEC. In an MFC, the cell voltage is limited by the internal resistance. This internal resistance causes the cell voltage to decrease to short circuit conditions (0 V, reached at 3.5 A m−2 in our analysis). Currents higher than short circuit cannot be achieved in MFC mode, which means that to achieve higher current densities, the MFC would operate as an MEC. Here, the applied voltage acts as a driving force for the reactions to proceed at a high rate, and thus, current densities higher than 3.5 A m−2 can be achieved. Although MFCs do not rely on the input of electrical energy to drive the reactions (applied voltage), this additional applied energy in MECs is recovered in the form of hydrogen. The recovered energy in the form of nitrogen is the same for both systems as it is directly correlated to the produced current. As a result, the energy balance for both MFC and MEC, at low current densities (<3.5 A m−2), is similar.
In the comparison shown above, the energy required for pumping and stripping is not taken into account, because at this early stage of development no realistic estimations can be made in terms of flow rates required in scaled-up systems. Of course, these processes will negatively affect the energy recovery from urine.
5 Economic analysis for ammonia recovery from urine in an MFC and an MEC
Sleutels et al. (2012) have shown an approach to determine the maximum internal resistance at which point it is still economically feasible to apply MFCs and MECs for the removal of COD from wastewater.16 In this analysis, the costs (both operational and capital) and benefits of both systems for the treatment of COD in wastewaters were estimated. Here, we extend the analysis for the case that ammonium is recovered in an MFC or an MEC. The economic value of the treatment of urine, products and by-products is presented in Table 3. Similar to the example of COD removal, the value for ammonia removal mainly originates from the fact that no additional treatment is required.| Parameter | Unit | References | |
|---|---|---|---|
| Electricity | 0.06 | € kW h−1 | 58 |
| Hydrogen | 0.35 | € m−3 | 59 |
| Capital and operational costs | 0.05 | € per kg COD removed | Calculated from ref. 60 |
| COD removal | 0.35 | € per kg COD removed | 61 |
| Ammonia removal efficiency | 30 | % | Based on ref. 13 |
| Ammonia removal | 1.63 | € per kg of N removed | 62 |
| pH difference between anode and cathode | 6 | — | Estimated from ref. 63 |
| Applied voltage | 1 | V | — |
| Interest rate | 6 | % | — |
As an indication for feasibility of simultaneous ammonium recovery and wastewater treatment in MFCs and MECs, costs and benefits have been calculated for current densities in the range of 0 to 50 A m−2. The results for the economic comparison for the removal of ammonia are shown in Fig. 6A for MFCs and in Fig. 6B for MECs. To study the effect of internal resistance, the analysis was done at both 25 and 200 mΩ m2.
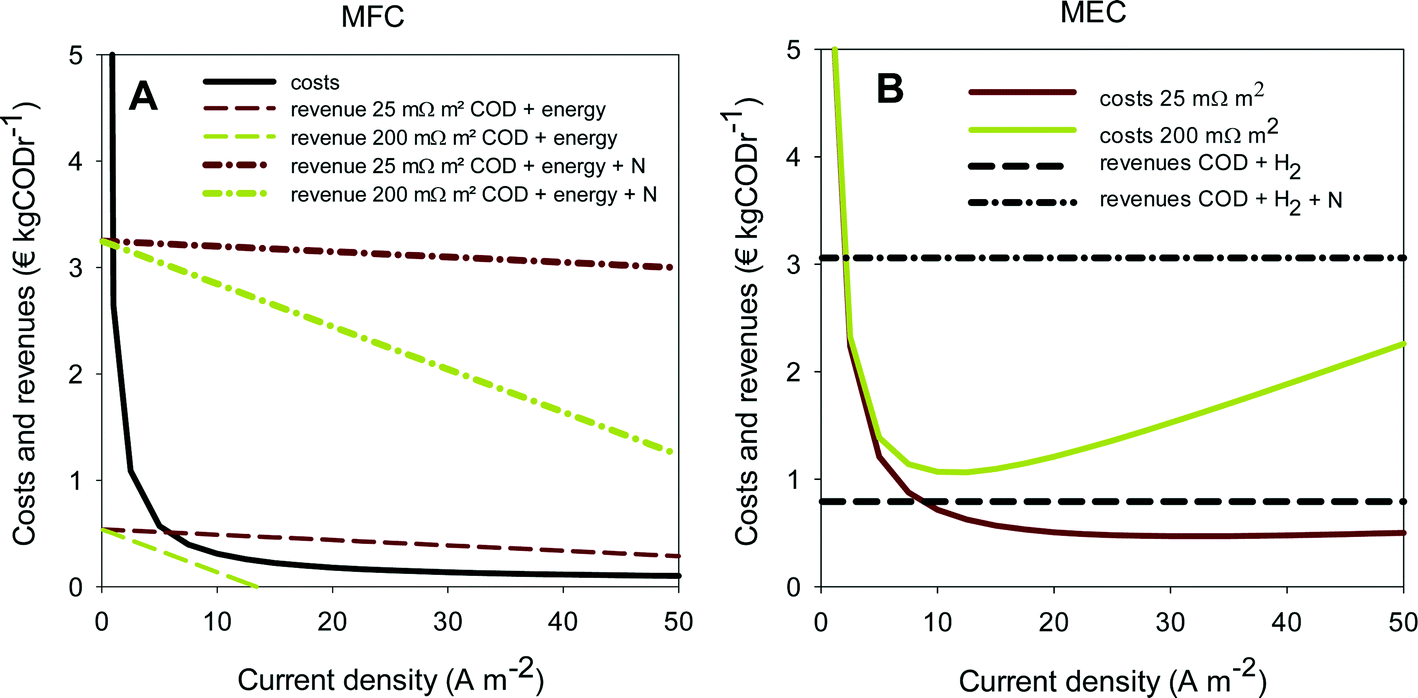 | ||
| Fig. 6 Comparison of the costs and revenues of ammonia recovery in an MFC (A) and in an MEC (B). The costs and revenues are shown at different internal resistances of the system and with and without the recovery of ammonia. | ||
The solid lines in Fig. 6 represent the costs for the MFC and MEC consisting of both capital and operational costs. The dotted lines represent the revenues at high and low internal resistances and with and without N recovery. When revenues are higher than costs, the system becomes profitable. Clearly, when ammonium recovery is included in the economic analysis in addition to COD removal, the revenues increase for both MFC and MEC at both internal resistances (Fig. 6A and B) compared to the situation without ammonium recovery. Both systems become profitable at almost all current densities, even at higher internal resistance.
The capital costs for both MFCs and MECs are similar, since these systems have the same configuration and are constructed from the same materials. However, the capital costs decrease with increasing current density when expressed in € kgCODr−1, because the rate of COD removal increases at higher current densities. In contrast with the capital costs, the operational costs are different for MECs and MFCs. For MFCs, the operational costs are independent of current densities, while for MECs, the electrical energy input and thus the operational costs need to be increased to achieve a higher rate of hydrogen production. Therefore, the total costs for MECs have a minimum value, determined by the current density and the internal resistance, and the total costs for MFCs decrease with increasing current density. For the MFC, low internal resistances are required to make production of electricity from wastewater economically attractive. Only at 25 mΩ m2, the revenues become higher than the costs at current densities that have been achieved in MFCs so far (5–10 A m−2). Like Sleutels et al. (2012) reported before, hardly any MFCs so far have reached such a low internal resistance.16
When the recovery of ammonia is taken into account, much higher internal resistances can be allowed for both systems at which the process is still economic, mainly because of the savings for wastewater treatment that are reflected in the value of ammonia per kg of N removed. This means that, for example, distances between electrodes can be allowed to be higher to still achieve higher revenue than cost, making scale-up of the systems easier. When ammonium recovery is taken into account, the costs become relatively low compared to the revenue for both the MFC and the MEC. It should be noted however that, when locally produced, the production of electricity compared to hydrogen is probably more attractive.
These calculations show that the removal of ammonia in BESs gives a significant economic value in addition to the production of electricity in an MFC or hydrogen in an MEC, which leads to less stringent design criteria (e.g. in terms of internal resistance), and an easier road towards practical application. Kuntke et al. (2014) reported an internal resistance of 43 mΩ m2 (Table 2) for urine treatment using an MEC with platinum-coated titanium electrodes. The challenge now is to reach similar internal resistances with less expensive materials.
6 Challenges in bioelectrochemical systems for nitrogen recovery
In the previous sections, we have shown and compared the different processes for nitrogen removal and recovery in BESs. Although our analysis shows a promising prospect for application of ammonium recovery via membrane transport in an MFC or an MEC, there are still several challenges to overcome before the technology can be applied in practice.Most importantly, we need to optimize the ammonium recovery and therefore the ammonium transport across the membrane. At present, the reported values for the ammonium removal efficiency from urine, e.g. the fraction of ammonium that is removed from urine, are around 30%. The removal of ammonium from the anode waste stream is dependent on several factors, most importantly (i) the concentration of ammonium in the wastewater, (ii) the removal of ammonium from the catholyte/cathode head space by stripping, (iii) the current density, (iv) the catholyte pH, (v) the type of membrane, and (vi) the equilibria of other ions at the anode and cathode. Many of these factors are interrelated, e.g. the current density determines the catholyte pH63 and is in turn dependent on the type of membrane and the ions present in solution.48 These interactions should be studied in more detail to clarify how the removal efficiency and ammonium recovery can be improved. It has been shown that removing the ammonium/ammonia batch-wise from the catholyte by replenishing the catholyte results in higher ammonium removal rates.39 In a continuous setup, removal of ammonium can be achieved by stripping, where the soluble ammonia evaporates to gaseous ammonia by contact with a N2 or H2 gas stream and subsequently absorbed in sulphuric acid, as has been done in the electrochemical ammonium recovery process.30 Up until now, such a system has not been tested and the performance of a BES connected to a stripper, and the maximum achievable ammonia recovery, needs to be determined. Another approach to understand the processes that determine ammonium transport is modelling. Dykstra et al. (2014) derived a one-dimensional steady-state model of the urine MEC used by Kuntke et al. (2014).64 The model includes the transport of ions through the membrane and allowed us to identify one of the main limiting factors of the system, which is the inert gas flow along the cathode. It was concluded that a higher N2 flow or a decrease in pressure in the cathode chamber was needed to increase ammonia recovery. Modelling the system gave valuable insight and understanding of its performance. In urine, COD concentrations are approximately 10 g L−1, and ammonium concentrations are approximately 9 g L−1. Because ideally 0.57 kg of COD is required for the removal of 1 kg N, urine contains sufficient COD to recover all the ammonium, and no additional carbon source is required. In this ideal case, we assume that all the COD is converted into electrons and that ammonium is the single transported cation. Further experiments are needed to study which COD removal efficiency, coulombic efficiency, and nitrogen recovery can be achieved in a BES.
While all current nitrogen removal technologies require energy for the removal of nitrogen gas mainly through aeration, BESs, though not applied in practice yet, are a promising technology for energy-efficient ammonia recovery. The main reason for the low-energy requirement for MFCs and MECs, or even energy recovery, is the fact that no oxygen is required to oxidize the ammonium to nitrate or nitrite. Instead, the movement of electrons from anode to cathode drives the transport and recovery of ammonium at the cathode.
The next step is to bring BESs in general, and also specifically for the application of nitrogen recovery, to a larger scale. A recent review article summarizes all the work performed until now on scaling-up of MFCs.65 Their major observations are that in terms of design, both tubular and flat-plate systems still suffer from too high internal resistance. The main causes for these high internal resistances are that the electrode spacing is too high, electrode materials have limited conductivity, and contact between electrodes and current collectors is often not optimal. They point out that separators, like membranes, are crucial to obtain high treatment efficiency, although separators also increase the total internal resistance. Janicek et al. (2014) stress that waste streams with high organic concentrations are likely to generate higher performance,65 although the drawback is that COD removal is generally lower, and as a result, treatment efficiencies are limited, when a single system is used. They conclude that pilot studies are essential to demonstrate the practical feasibility of BESs.
In 2013, Wetsus has set up a pilot BES at the water board of Fryslân located in Leeuwarden, The Netherlands in cooperation with partners from industry. The objective of this pilot experiment is to purify urine by removing the COD and ammonium and recover energy in the form of hydrogen, in addition to ammonia gas. This installation consists of 4 flat-plate cells that are stacked in parallel with a total surface area of 0.16 m2. With this pilot, we will demonstrate the feasibility of on-site treatment of urine and conversion into ammonium and hydrogen gas.
Acknowledgements
This work was performed in the cooperation framework of Wetsus, Centre of Excellence for Sustainable Water Technology (www.wetsus.nl). Wetsus is co-funded by the Dutch Ministry of Economic Affairs and Ministry of Infrastructure and Environment, the European Union Regional Development Fund, the Province of Fryslân, and the Northern Netherlands Provinces. The authors would like to thank the participants of the research theme “Resource Recovery” for the fruitful discussions and their financial support. This research has received funding from the European Union's Seventh Programme for research, technological development and demonstration under grant agreement no. 308535.References
- US Bureau Census, World Population, http://www.census.gov/population/international/data/worldpop/table_population.php (accessed May 2014)
.
- UN, World population projected to reach 9.6 billion by 2050 with most growth in developing regions, especially Africa – says UN, http://esa.un.org/unpd/wpp/Documentation/pdf/WPP2012_Press_Release.pdf (accessed May 2014)
- J. Rockstrom, W. Steffen, K. Noone, A. Persson, F. S. Chapin, E. F. Lambin, T. M. Lenton, M. Scheffer, C. Folke, H. J. Schellnhuber, B. Nykvist, C. A. de Wit, T. Hughes, S. van der Leeuw, H. Rodhe, S. Sorlin, P. K. Snyder, R. Costanza, U. Svedin, M. Falkenmark, L. Karlberg, R. W. Corell, V. J. Fabry, J. Hansen, B. Walker, D. Liverman, K. Richardson, P. Crutzen and J. A. Foley, Nature, 2009, 461, 472–475 CrossRef PubMed
-
M. T. Madigan, J. M. Martinko and J. Parker, Brock biology of microorganisms, Prentice Hall, Upper Saddle River, NJ, 9th edn, 2000 Search PubMed
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed
- A. M. Leach, J. N. Galloway, A. Bleeker, J. Willem Erisman, R. Kohn and J. Kitzes, Environ. Dev., 2012, 1, 40–66 CrossRef
- S. Cheng, D. Xing, D. F. Call and B. E. Logan, Environ. Sci. Technol., 2009, 43, 3953–3958 CrossRef CAS PubMed
- B. E. Logan, D. Call, S. Cheng, H. V. M. Hamelers, T. H. J. A. Sleutels, A. W. Jeremiasse and R. A. Rozendal, Environ. Sci. Technol., 2008, 42, 8630–8640 CrossRef CAS PubMed
- B. E. Logan, B. Hamelers, R. A. Rozendal, U. Schröder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Environ. Sci. Technol., 2006, 40, 5181–5192 CrossRef CAS PubMed
- R. A. Rozendal, E. Leone, J. Keller and K. Rabaey, Electrochem. Commun., 2009, 11, 1752–1755 CrossRef CAS
- K. J. J. Steinbusch, H. V. M. Hamelers, J. D. Schaap, C. Kampman and C. J. N. Buisman, Environ. Sci. Technol., 2009, 44, 513–517 CrossRef PubMed
- M. C. A. A. Van Eerten-Jansen, A. Ter Heijne, T. I. M. Grootscholten, K. J. J. Steinbusch, T. H. J. A. Sleutels, H. V. M. Hamelers and C. J. N. Buisman, ACS Sustainable Chem. Eng., 2013, 1, 1069–1069 CrossRef CAS
- P. Kuntke, K. M. Śmiech, H. Bruning, G. Zeeman, M. Saakes, T. H. J. A. Sleutels, H. V. M. Hamelers and C. J. N. Buisman, Water Res., 2012, 46, 2627–2636 CrossRef CAS PubMed
- P. Kuntke, M. Geleji, H. Bruning, G. Zeeman, H. V. M. Hamelers and C. J. N. Buisman, Bioresour. Technol., 2011, 102, 4376–4382 CrossRef CAS PubMed
- P. T. Kelly and Z. He, Bioresour. Technol., 2014, 153, 351–360 CrossRef CAS PubMed
- T. H. J. A. Sleutels, A. Ter Heijne, C. J. N. Buisman and H. V. M. Hamelers, ChemSusChem, 2012, 5, 1012–1019 CrossRef CAS PubMed
- J. L. Barnard, Elimination of eutrophication through resource recovery, in Proceedings of International Conference on Nutrient Recovery from Wastewater Streams, ed. D. Mavinic, K. Ashley and F. Koch, IWA publishing, London, 2009, pp. 1–22.
-
G. Tchobanoglous, F. L. Burton and H. D. Stensel, Wastewater engineering: treatment and reuse, McGraw-Hill, New York, 4th edn, International Edition, 2004 Search PubMed
- M. S. M. Jetten, I. Cirpus, B. Kartal, L. van Niftrik, K. T. van de Pas-Schoonen, O. Sliekers, S. Haaijer, W. van der Star, M. Schmid, J. van de Vossenberg, I. Schmidt, H. Harhangi, M. van Loosdrecht, J. G. Kuenen, H. O. den Camp and M. Strous, Biochem. Soc. Trans., 2005, 33, 119–123 CrossRef CAS PubMed
-
L. G. J. M. Dongen, M. S. M. Jetten, M. C. M. van Loosdrecht and K. Kujawa-Roeleveld, The combined Sharon/Anammox process: a sustainable method for N-removal from sludge water, IWA Publishing, London, 2001 Search PubMed
- U. van Dongen, M. S. M. Jetten and M. C. M. van Loosdrecht, Water Sci. Technol., 2001, 44, 153–160 CAS
- M. S. M. Jetten, M. Wagner, J. Fuerst, M. van Loosdrecht, G. Kuenen and M. Strous, Curr. Opin. Biotechnol., 2001, 12, 283–288 CrossRef CAS PubMed
- Y. D. Scherson, G. F. Wells, S.-G. Woo, J. Lee, J. Park, B. J. Cantwell and C. S. Criddle, Energy Environ. Sci., 2013, 6, 241–248 CAS
- A. ter Heijne, H. V. M. Hamelers, V. de Wilde, R. A. Rozendal and C. J. N. Buisman, Environ. Sci. Technol., 2006, 40, 5200–5205 CrossRef CAS PubMed
- A. Ter Heijne, F. Liu, R. van der Weijden, J. Weijma, C. J. N. Buisman and H. V. M. Hamelers, Environ. Sci. Technol., 2010, 44, 4376–4381 CrossRef CAS PubMed
- F. Zhao, F. Harnisch, U. Schröder, F. Scholz, P. Bogdanoff and I. Herrmann, Environ. Sci. Technol., 2006, 40, 5193–5199 CrossRef CAS PubMed
- H. V. M. Hamelers, A. Ter Heijne, T. H. J. A. Sleutels, A. W. Jeremiasse, D. P. B. T. B. Strik and C. J. N. Buisman, Appl. Microbiol. Biotechnol., 2010, 85, 1673–1685 CrossRef CAS PubMed
- H. Liu, S. Grot and B. E. Logan, Environ. Sci. Technol., 2005, 39, 4317–4320 CrossRef CAS PubMed
- R. A. Rozendal, H. V. M. Hamelers, G. J. W. Euverink, S. J. Metz and C. J. N. Buisman, Int. J. Hydrogen Energy, 2006, 31, 1632–1640 CrossRef CAS
- J. Desloover, A. Abate Woldeyohannis, W. Verstraete, N. Boon and K. Rabaey, Environ. Sci. Technol., 2012, 46, 12209–12216 CrossRef CAS PubMed
- M. Maurer, P. Schwegler and T. A. Larsen, Water Sci. Technol., 2003, 48, 37–46 CAS
-
K. Diem and C. Lentner, Scientific tables, Ciba-Geigy Limited, Basle, 7th edn, 1970 Search PubMed
-
P. Kuntke, Ph.D. Thesis, Wageningen University, 2013 Search PubMed
- M. Maurer, W. Pronk and T. A. Larsen, Water Res., 2006, 40, 3151–3166 CrossRef CAS PubMed
-
J. Wilsenach and M. van Loosdrecht, Separate urine collection and treatment; option for sustainable wastewater systems and mineral recovery, 2002 Search PubMed
- Z. He, J. Kan, Y. Wang, Y. Huang, F. Mansfeld and K. H. Nealson, Environ. Sci. Technol., 2009, 43, 3391–3397 CrossRef CAS PubMed
- J. R. Kim, Y. Zuo, J. M. Regan and B. E. Logan, Biotechnol. Bioeng., 2008, 99, 1120–1127 CrossRef CAS PubMed
- G.-L. Zang, G.-P. Sheng, W.-W. Li, Z.-H. Tong, R. J. Zeng, C. Shi and H.-Q. Yu, Phys. Chem. Chem. Phys., 2012, 14, 1978 RSC
- P. Kuntke, T. H. J. A. Sleutels, M. Saakes and C. J. N. Buisman, Int. J. Hydrogen Energy, 2014, 39, 4771–4778 CrossRef CAS
- P. Clauwaert, K. Rabaey, P. Aelterman, L. De Schamphelaire, T. H. Pham, P. Boeckx, N. Boon and W. Verstraete, Environ. Sci. Technol., 2007, 41, 3354–3360 CrossRef CAS PubMed
- B. Virdis, K. L. Rabaey, Z. Yuan and J. Keller, Water Res., 2008, 42, 3013–3024 CrossRef CAS PubMed
- B. Virdis, K. Rabaey, R. A. Rozendal, Z. Yuan and J. Keller, Water Res., 2010, 44, 2970–2980 CrossRef CAS PubMed
- F. Zhang and Z. He, Process Biochem., 2012, 47, 2146–2151 CrossRef CAS
- H. Yan, T. Saito and J. M. Regan, Water Res., 2012, 46, 2215–2224 CrossRef CAS PubMed
- S. Haddadi, E. Elbeshbishy and H.-S. Lee, Bioresour. Technol., 2013, 142, 562–569 CrossRef CAS PubMed
- R. A. Rozendal, H. V. M. Hamelers and C. J. N. Buisman, Environ. Sci. Technol., 2006, 40, 5206–5211 CrossRef CAS PubMed
- R. A. Rozendal, T. H. J. A. Sleutels, H. V. M. Hamelers and C. J. N. Buisman, Water Sci. Technol., 2008, 57, 1757–1762 CrossRef CAS PubMed
- T. H. J. A. Sleutels, H. V. M. Hamelers, R. A. Rozendal and C. J. N. Buisman, Int. J. Hydrogen Energy, 2009, 34, 3612–3620 CrossRef CAS
- R. Cord-Ruwisch, Y. Law and K. Y. Cheng, Bioresour. Technol., 2011, 102, 9691–9696 CrossRef CAS PubMed
- K. Y. Cheng, A. H. Kaksonen and R. Cord-Ruwisch, Bioresour. Technol., 2013, 143, 25–31 CrossRef CAS PubMed
- T. A. Larsen and W. Gujer, Water Sci. Technol., 1996, 34, 87–94 CrossRef CAS
- H. L. T. Mobley and R. P. Hausinger, Microbiol. Rev., 1989, 53, 85–108 CAS
- K. M. Udert, T. A. Larsen, M. Biebow and W. Gujer, Water Res., 2003, 37, 2571–2582 CrossRef CAS PubMed
- E. Volcke, K. Villez, S. Van Hulle, M. van Loosdrecht and P. Vanrolleghem, Afvalwaterwetenschap, 2004, 3, 297–318 Search PubMed
- A. Fuerte, R. X. Valenzuela, M. J. Escudero and L. Daza, J. Power Sources, 2009, 192, 170–174 CrossRef CAS
- U.S. Department of Energy, Hydrogen Analysis Resource Center, http://hydrogen.pnl.gov/cocoon/morf/hydrogen/site_specific/fuel_heating_calculator?canprint=false (accessed May 2014)
-
V. Hacker and K. Kordesch, in Handbook of fuel cells-Fundamentals, technology and applications, ed. W. Vielstich, A. Lamm and H. A. Gasteiger, John Wiley & Sons, Chichester, 2003, vol. 3, pp. 121–127 Search PubMed
- Centraal Bureau voor de Statistiek, Aardgas en elektriciteit, gemiddelde prijzen van eindverbruikers, http://statline.cbs.nl/StatWeb/publication/?VW=T&DM=SLNL&PA=81309NED&D1=1,3,5,8,11,15&D2=0&D3=l&D4=0-3,5-8,10-13,15-18,20-23,l&HD=120813-1059&HDR=T&STB=G2,G1,G3 (accessed May 2014)
- International Energy Agency, Hydrogen Production & Distribution, http://www.iea.org/techno/essentials5.pdf (accessed June 2014)
-
J. W. Post, Ph.D. Thesis, Wageningen University, 2009 Search PubMed
- S. Aiyuk, I. Forrez, D. K. Lieven, A. van Haandel and W. Verstraete, Bioresour. Technol., 2006, 97, 2225–2241 CrossRef CAS PubMed
-
S. J. Kang, K. Olmstead, K. Takacs and J. Collins, Municipal Nutrient Removal Technologies Reference Document, U.S. Environmental Protection Agency, 2008 Search PubMed
- T. H. J. A. Sleutels, A. Ter Heijne, C. J. N. Buisman and H. V. M. Hamelers, Int. J. Hydrogen Energy, 2013, 38, 7201–7208 CrossRef CAS
- J. E. Dykstra, P. M. Biesheuvel, H. Bruning and A. Ter Heijne, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 90 Search PubMed
- A. Janicek, Y. Fan and H. Liu, Biofuels, 2014, 5, 79–92 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2015 |