
Atheroprotective effects of (poly)phenols: a focus on cell cholesterol metabolism
Ilaria
Zanotti
a,
Margherita
Dall'Asta
b,
Pedro
Mena
b,
Laura
Mele
b,
Renato
Bruni
b,
Sumantra
Ray†
c and
Daniele
Del Rio†
 *b
*b
aDepartment of Pharmacy, University of Parma, Viale delle Scienze 27/A, 43124 Parma, Italy
bThe Laboratory of Phytochemicals in Physiology, Department of Food Science, University of Parma, Medical School, Building C, Via Volturno 39, 43125 Parma, Italy. E-mail: daniele.delrio@unipr.it; Tel: +0039-0521-903830
cCambridge University Hospitals, School of Clinical Medicine, University of Cambridge, Cambridge, UK
First published on 15th October 2014
Abstract
Collated observations from several epidemiological studies have demonstrated that dietary intake of (poly)phenols from nuts, coffee, cocoa, grapes, and berries may protect against the development of atherosclerosis. Whereas this beneficial activity has previously been linked mainly to antioxidant or anti-inflammatory properties, recently emerging data suggest mechanisms by which (poly)phenolic substances can modulate cellular lipid metabolism, thereby mitigating atherosclerotic plaque formation. In this review, both experimental studies and clinical trials investigating the atheroprotective effects of the most relevant dietary (poly)phenols are critically discussed.
1. Introduction
The aim of this review is to provide an overview and critical analysis of the literature related to the atheroprotective activity of the principal dietary polyphenols. Whereas this class of compounds possesses well-documented, beneficial anti-inflammatory and antioxidant mechanisms, in the present paper we focus on those effects related to the modulation of lipid metabolism. From the large volume of data available on the atheroprotective impact of polyphenols, we critically selected the most consistent human studies, pointing out key questions, such as primary mechanisms of action and active compounds.It is worth noting that a significant bias characterizes most of the in vitro studies currently published, where human or animal cell lines have been exposed to polyphenols in their chemical form occurring in plant foods. However, following ingestion, these compounds are extensively modified by human and microbial enzymes and they appear at the cellular level as metabolites, often with relevant differences from a chemical standpoint. Therefore, in the attempt to identify the substances actually involved in the putative in vivo beneficial activities of this class of compounds and to describe their actual mode of action, these metabolites, and not their in planta precursors, should be tested in vitro, ideally at concentrations congruent with human physiology.
2. Dietary (poly)phenols and their metabolism
Polyphenols are one of the most copious and ubiquitous groups of secondary plant metabolites, occurring in a wide number of foods and beverages.1 Polyphenolic compounds are produced in planta by a very plastic system of biosynthetic pathways and are involved in a plethora of physiological and ecological roles, supporting diverse functions such as structure, pigmentation, pollination, allelopathy, pathogen and predator resistance, growth and development.2,3 Their roles and ecological significance warrant a wide array of biological activities, chemical diversity and abundance. After being labelled as mostly anti-nutritional food constituents, their role as potential healthy compounds, when introduced in the human diet in proper amounts, has been outlined only recently. In the last few decades, a growing number of clinical trials and epidemiological studies have attracted the combined attention of nutritionists and clinicians on the potential role of polyphenols in the prevention of several degenerative diseases. Furthermore, several in vitro investigations have recently tried to investigate the underlying mechanisms of action of these phytochemicals in several different cellular cultures.4From a chemical viewpoint, polyphenols constitute a rather heterogeneous class, characterized by the common presence of at least one aromatic ring in their structure, linked with other phenolic-, hydroxyl-, carbon- or other chemical groups showing an extensive variety of molecular structures. They encompass both low molecular weight and simple structures (e.g. phenolic acids) and high molecular weight and complex polymeric compounds (e.g. tannins), with obvious consequences in terms of solubility, membrane permeability, and putative bioactivities. Polyphenols are generally classified into flavonoids and non-flavonoids, depending on their chemical structure. The first group comprises a large family of compounds, further divided into several subclasses, of which six offer greater dietary relevance, i.e. flavonols, flavanones, flavones, isoflavones, anthocyanins, and flavan-3-ols,5 while non-flavonoids include mainly condensed and hydrolysable tannins, stilbenes, phenolic acids, and hydroxycinnamates. Polyphenols in planta, are principally linked together (to form both oligomers and polymers) or esterified with other chemical compounds, usually a wide array of sugar moieties or with organic acids, whereas they are only rarely available as isolated aglycones.5 According to their structure and role in a given plant tissue, these compounds may be dissolved in the aqueous content of the vacuole, or more or less tightly joined to the polysaccharidic constituents of the cell wall. Such positioning confers some differences in terms of their potential bioavailability, as some glycosides may be enzymatically hydrolysed in planta before being introduced through the diet (e.g., during leaf wilting or after grinding), and/or some polyphenols may bind to cellulose and pectin, or to a number of proteins during ingestion, de facto altering their fate during the gastrointestinal transit.6 The large chemodiversity of polyphenols available in nature is well described by the thousands of structures which have been reported, and given the evidence that many of them could still be discovered, this synthetic overview offers only a glimpse of the potential role of this class of natural compounds in food science, nutrition research, drug discovery, and ultimately in health promotion.
2.1. Metabolism of (poly)phenolic substances in humans
The positive modulation of human health attributed to polyphenols has steadily emerged over the years through different in vitro/ex vivo models, in in vivo experiments, and in clinical trials.4 Considerable attention has been paid to the investigation of the metabolism and bioavailability of polyphenols in the human organism, as an essential step in understanding their biological activity. The latter depends also on the capacity of specific compounds to actually reach tissues, organs and cells, before exerting their positive actions beyond the limited context of the gastrointestinal tract. To understand that, and to elucidate the potential of polyphenols not only in terms of lipidemic regulation but also as potential health enhancers, it must be clearly defined which transformations may occur to these dietary compounds before they may become systemic in the circulation or where some significant effects may occur.After ingestion, polyphenols are metabolised following the typical detoxification pathway common to xenobiotics and drugs, starting from the mouth (Fig. 1). Although the capability of the oral saliva and gastric secretion to modify the native polyphenolic structures is known, but rather weak,7–10 the first organ strongly involved in polyphenol modification and digestion is the small intestine. In this tract, a limited part of in planta conjugated forms unmodified in the oral cavity is absorbed, mainly following a specific pathway through which they can pass the enteric barrier and reach the portal circulation. This absorption step is mediated by the lactase phlorizin hydrolase, an enzyme present in the brush-border of the small intestine epithelial cells; its involvement in the modification of several glucoside flavonoids is well known.11 An alternative hydrolytic pathway for polyphenol intestinal absorption is catalysed by a cytosolic β-glucosidase,12 which seems able to operate after the intake of polyphenols into the cells due to the active sodium-dependent glucose transporter, SGLT-1.13
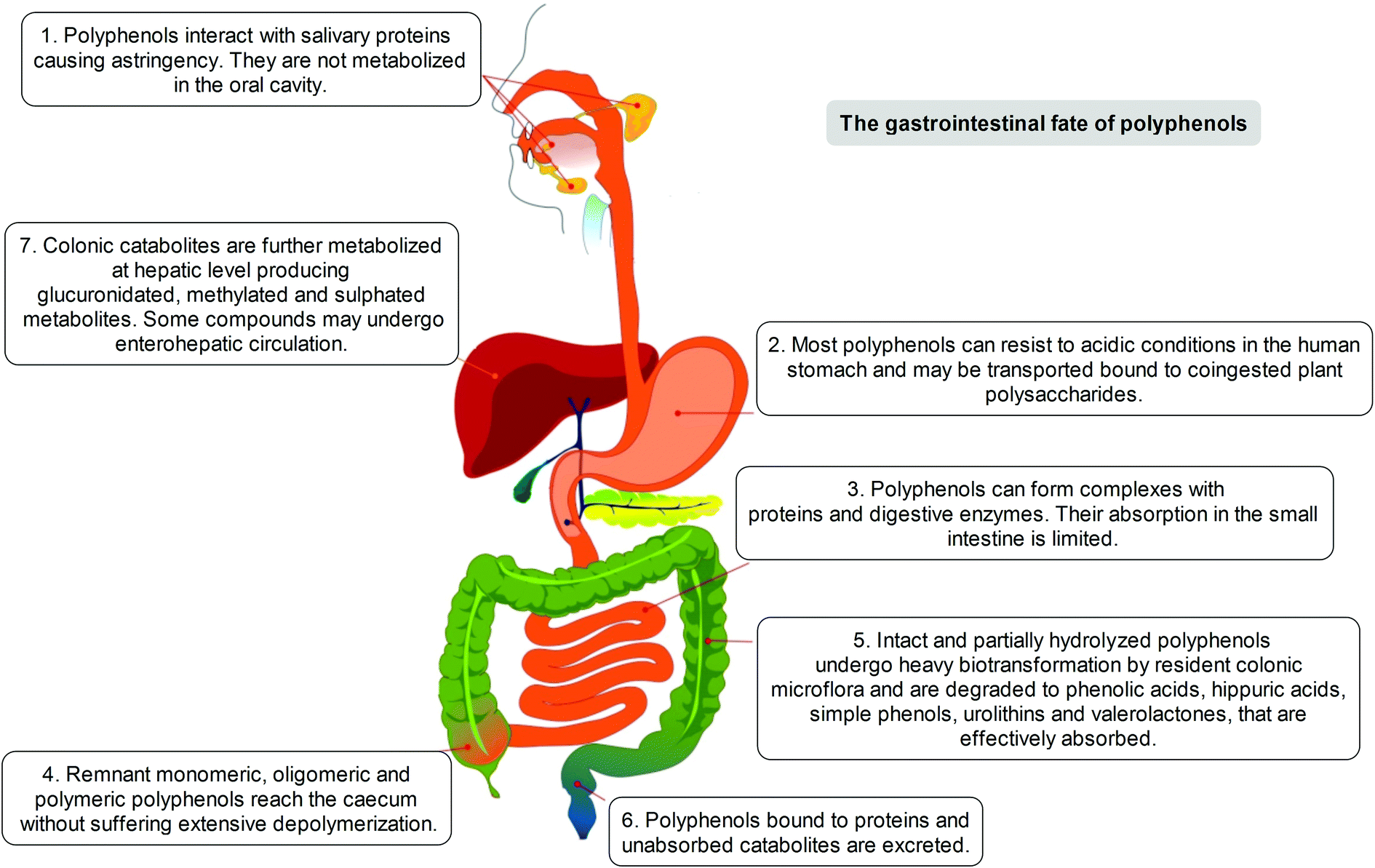 | ||
| Fig. 1 The fate of dietary polyphenols after ingestion. | ||
Before entering systemic circulation, polyphenols undergo phase II enzymatic detoxification through conjugation with chemical groups such as glucuronic acid, sulphate, and methyl groups operated by the action of uridine 5′-diphospho-glucuronosyltransferases, sulphotransferases, and catechol-O-methyltransferase, respectively.14 This conjugation step first occurs in the small intestine before entering the bloodstream, but subsequently takes place also in the liver, where the metabolites are further modified by the phase II enzymes before being transferred to the systemic circulation, or to enterohepatic re-circulation, and are finally excreted.15
However, it has been estimated that only a limited part of the ingested polyphenols (5–10%) is actually absorbed, independently of conjugation, in the small intestine. The remaining unmodified polyphenols (90–95%), together with conjugates excreted into the intestinal lumen through the bile, may proceed through the gastrointestinal tract and accumulate in the large intestine at high concentrations (up to the mM range), where they are exposed to the gut microbiota enzyme arsenal. Such events are increased and modulated by the presence of co-ingested plant polysaccharides, whose presence represents one of the relevant distinctions between the common dietary intake (e.g. fruits, vegetables) and purified or heavily processed materials and infusions (e.g. food supplements, teas, coffee).14,16–22
Indeed, the colon is a large ecosystem that works as a powerful bioreactor capable of structurally modifying polyphenolic compounds, leading to the production of metabolites with different physiological relevance. Actually, the gut microbiota is responsible for the breakdown of the original polyphenolic structures into a large number of low-molecular-weight compounds, which could be more absorbable than their original counterparts found in plant foods. This transformation occurs via multiple and intertwined steps of ester and glycoside hydrolysis, demethylation, dehydroxylation, and decarboxylation by different bacteria.18 Metabolites produced in the large intestine subsequently undergo further phase II metabolism, locally and/or in the liver level after absorption. They then enter the blood compartment, reach peripheral tissues, and are finally excreted in the urine in substantial amounts, largely exceeding the excretion of phenolic metabolites formed in the upper gastrointestinal tract.12 Ellagitannins are a striking example in this regard, as described in Fig. 2. As a result, given the longer transit time in the colon and their binding to co-ingested substances, the absorption of polyphenolic metabolites takes more time and leads to a prolonged permanence of these compounds in the bloodstream. In fact, while substances like quercetin are excreted after 2–3 hours, some phenolic metabolites can circulate for more than 3 days. The phenolic metabolites originating from microbial degradation plus phase II conjugation have been recovered in urine in extremely high amounts, with respect to their simple phase II counterparts,17,23–25 and may actually represent the true ‘actors’ in the framework of the health effects derived from polyphenol-rich food consumption.
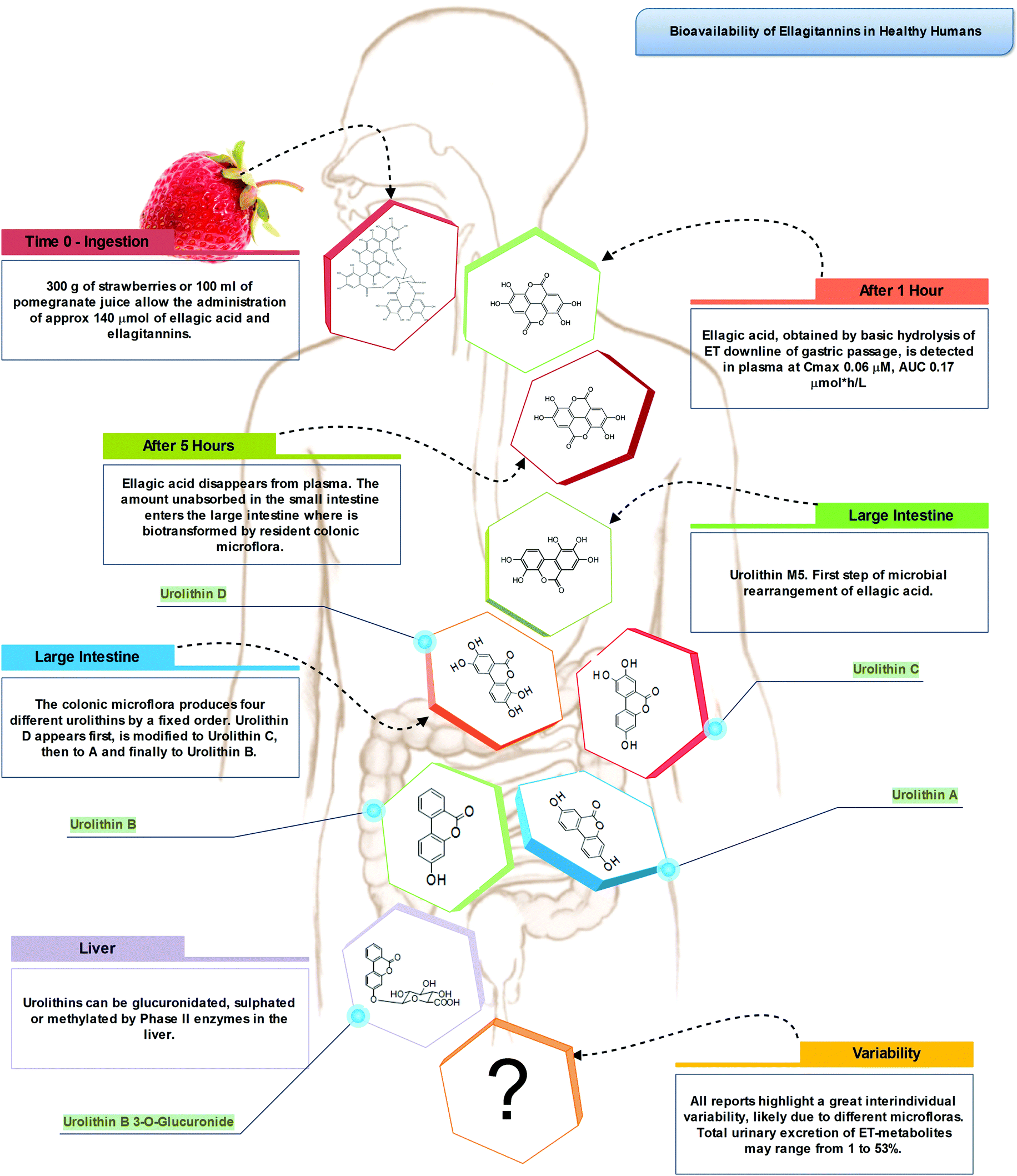 | ||
| Fig. 2 Bioavailability and metabolism of dietary ellagitannins. | ||
2.2. Phenolic acids
Phenolic acids (C6–C1) are the most abundant and widespread non-flavonoidic dietary phenolics, with a dietary intake of ∼600 mg per day. Among them, hydroxybenzoic and hydroxycinnamic acids are the most common representatives in plant foods and beverages (Fig. 3). Gallic acid is certainly the most relevant hydroxybenzoic acid. Red wine, tea, and some berries and nuts are its richest dietary sources and it is widely present in the form of complex sugar esters (gallotannins) as well as in non-sugar galloyl esters.5 Among hydroxycinnamic acids, the most representative compounds are caffeic, ferulic and p-coumaric acids. These substances occur mainly as conjugates named chlorogenic acids, where the phenolic skeleton is esterified with quinic acid to form structures known as caffeoylquinic acids, feruloylquinic acids, and p-coumaroylquinic acids. Coffee is one of the major dietary sources of chlorogenic acids (up to 1750 mg L−1), together with apples (600 mg kg−1), tea, berries (blueberries in particular, 2200 mg kg−1), plums, grapes, wine and many green vegetables (aubergines, up to 660 mg kg−1). Ferulic acid is the most abundant hydroxycinnamate found in whole cereal grains, which constitute its main food source.14 In human subjects fed coffee drinks rich in 5-caffeoylquinic acid, pharmacokinetic studies showed the appearance of free and sulphated dihydrocaffeic and dihydroferulic acids and feruloylglycine in blood. This result is explained by the action of the colonic microflora-mediated conversion of caffeoylquinic acids into caffeic acid and dihydrocaffeic acid, which is further metabolized to dihydro-isoferulic acid (Fig. 3). The microbial modification of the feruloylquinic acids induces their conversion to ferulic and dihydroferulic acid and feruloylglycine.26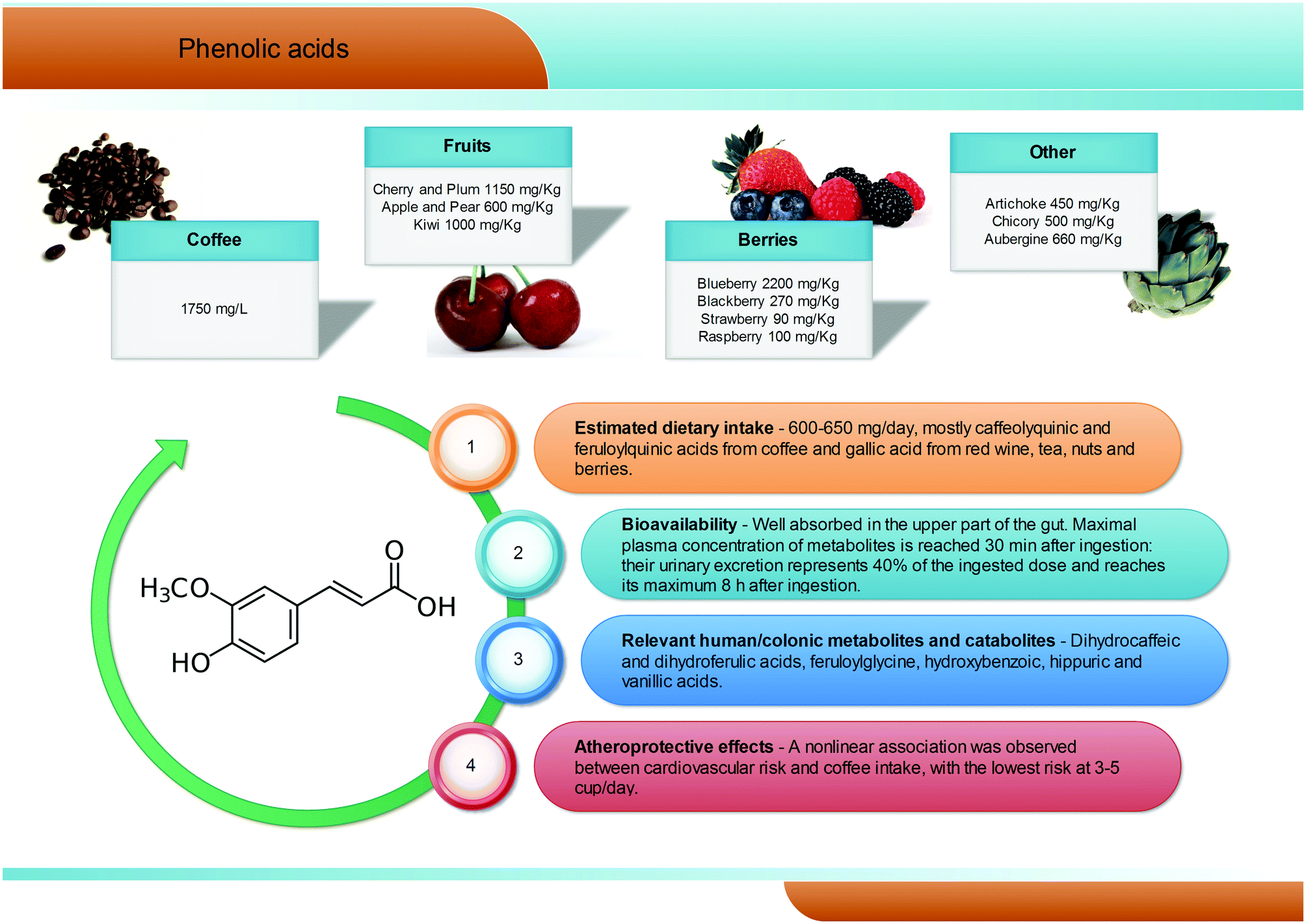 | ||
| Fig. 3 Dietary sources, bioavailability, metabolism and atheroprotective effects of phenolic acids. | ||
2.3. Stilbenes
Stilbenoids (C6–C2–C6 structure) are phytoalexins and constitute a group of non-flavonoids less widespread in plant foods compared to other classes of polyphenols. They are not of particular dietary relevance, since their daily intake has been estimated to be negligible.14 Resveratrol (3,5,4′-trihydroxystilbene) is the most famous and studied stilbene, occurring, as a trans-isomer, in foods and drinks such as red wine, grapes, peanuts, pistachios, and berries.27 Bioavailability studies suggested that the very low bioavailable fraction of trans-resveratrol consumed as well as its conjugated derivatives, including trans-resveratrol-3-O-glucoside (trans-piceid) are rapidly absorbed and metabolized in humans.28–32 Resveratrol is absorbed in the upper gastrointestinal tract and can be modified by both enterocytes and hepatocytes33,34 leading to the production of the glucuronide and sulphate forms, which are the major plasmatic and urinary metabolites, with the sulphates being predominant.31,35,36 It is known that intestinal bacteria are able to convert resveratrol into dihydro-resveratrol, which, at least partially, is absorbed and further metabolized to conjugated forms that can be excreted in urine.35,37–40 The main gut-derived metabolites after the consumption of trans-resveratrol were found to be dihydroresveratrol, 3,4′-dihydroxy-trans-stilbene and 3,4′-dihydroxybibenzyl (lunularin).41 The overall scenario regarding the actual properties of these substances is far from complete, and the scientific knowledge on the bioactivity of resveratrol catabolites and metabolites is limited, with contradicting results.2.4. Flavonols
Flavonols are the most common flavonoid form in the plant kingdom, with an average dietary intake estimated at 50 mg per day, with differences according to the specific diet (Fig. 4). Overall, flavonols are present in vegetables and fruits such as kale, onion, broccoli, tomato, and berries, with values ranging from 1200 mg kg−1 (onion) to 40 mg kg−1 (apples). Quercetin is certainly the most widespread dietary flavonol, and together with kaempferol, isorhamnetin, and myricetin is abundant in onions, apples, tea, broccoli, and red wine, and is typically present in the glycosidic form, with conjugation occurring at the 5, 7, 3′, 4′, and 5′ positions42 and with a clear prevalence of the rutinoside conjugate, named rutin. In the gastrointestinal tract, quercetin glycosides remain almost unaltered until the small intestine, where the aglyconic form is cleaved and subsequently converted into glucuronide, sulphate, and methylated metabolites both at the enterocytes and at the liver level. Like for the other phenolic compounds, the largest fraction of flavonols ingested reaches the colon,43 where the large amount and variety of microbial enzymes has been shown to break down the flavonol skeleton, inducing carbon cleavage and ring fissions that lead to the release of several polar metabolites with low molecular weight (Fig. 4). The main quercetin metabolites produced by the human microbiota are 3′,4′-dihydroxyphenylacetic acid and its dehydroxylated counterpart, 3′-hydroxyphenylacetic acid.3,44 Overall, a great degree of variability has been reported,3 highlighting a very subjective behaviour, likely related to the different composition of the gut microflora in different subjects.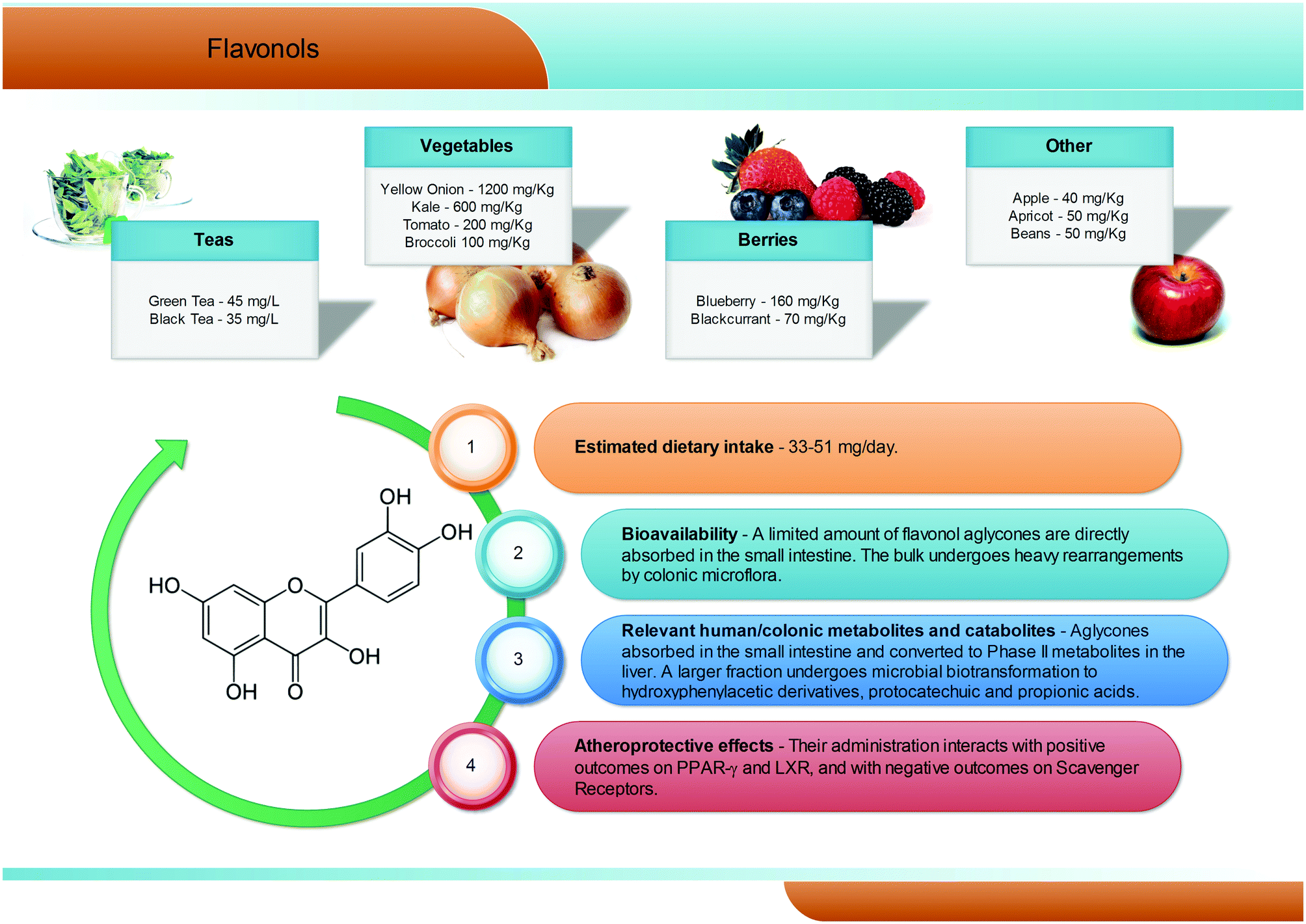 | ||
| Fig. 4 Dietary sources, bioavailability, metabolism and atheroprotective effects of flavonols. | ||
2.5. Anthocyanins
Anthocyanins are a group of natural pigments responsible for the attractive red-blue colour of flowers and many fruits; their estimated average dietary intake is about 60 mg per day (Fig. 5), but great variability is known according to specific dietary habits, and daily intakes in excess of 1 g are feasible.45 They are glycosides of polyhydroxy- and polymethoxy-derivatives of 2-phenylbenzopyrylium or flavilium salts. Six anthocyanidins, the aglyconic version of anthocyanins, are commonly found in plants: cyanidin, pelargonidin, peonidin, delphinidin, petunidin, and malvidin. The sugars most commonly bound to anthocyanidins are glucose, galactose, rhamnose, and arabidose. Some aliphatic or aromatic acids can also bind to the sugar residue.46,47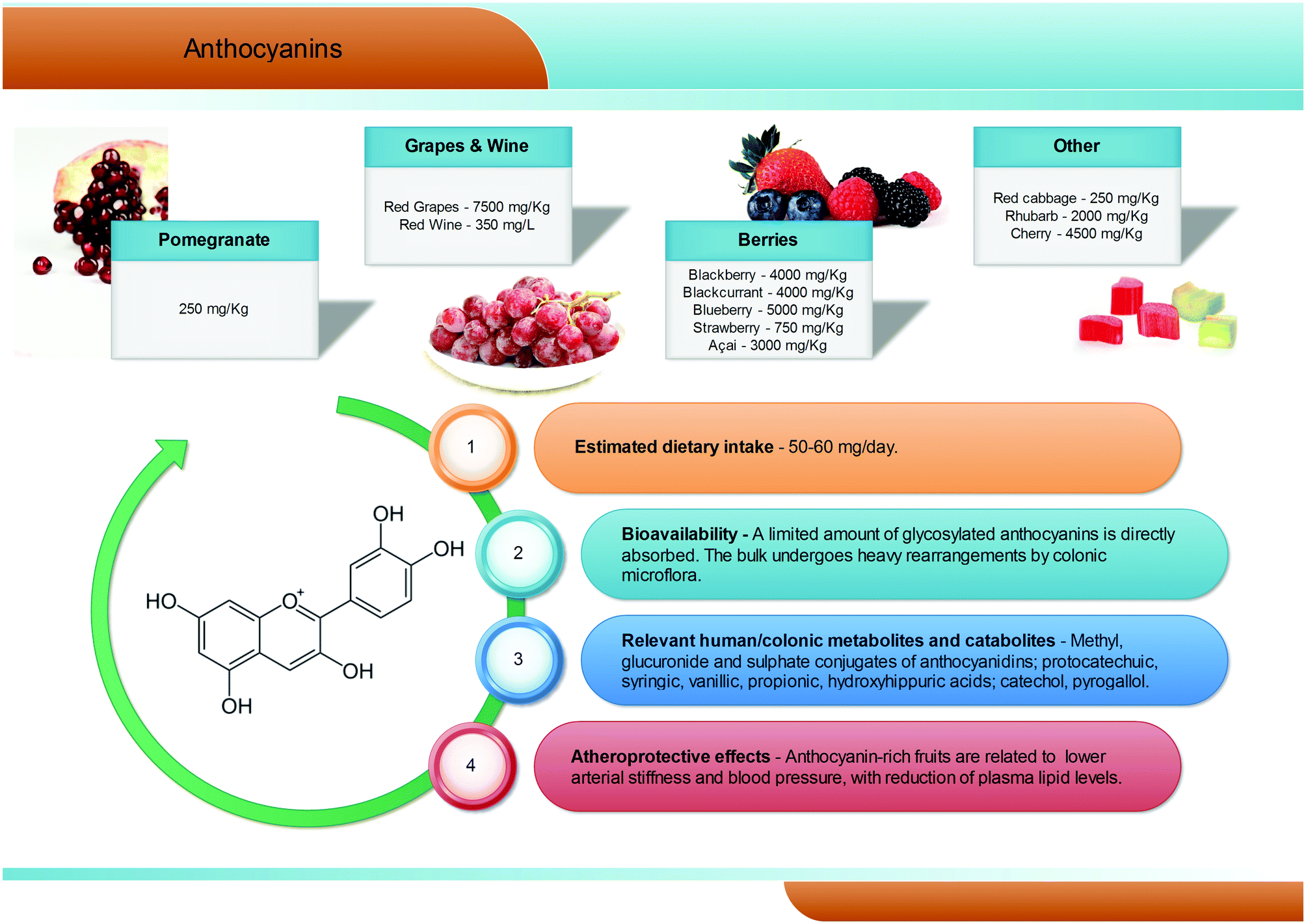 | ||
| Fig. 5 Dietary sources, bioavailability, metabolism and atheroprotective effects of anthocyanins. | ||
The main dietary sources of anthocyanins are berries (up to 5000 mg kg−1), red grapes (up to 7500 mg kg−1) and red wine (up to 350 mg L−1). Pomegranates, red cabbages, purple carrots, purple potatoes, and purple corn can also contribute to the dietary intake of anthocyanins but, despite the high anthocyanin content, their current contribution to the diet is still limited.48–50
Anthocyanins appear to have low bioavailability, with typical recoveries of <1% of the intake.51–53 Human feeding studies have showed that, after consumption of anthocyanin-rich foodstuffs, anthocyanins, but mainly their phase II conjugates (methyl, glucuronide, and sulphate), rapidly reach maximum plasma concentrations never overcoming the nanomolar range, typical of absorption in the upper gastrointestinal tract. However, the vast majority of the ingested anthocyanins reach the large intestine, where they are metabolised by the gut microbiota (Fig. 5).54–56 Consumption of raspberries by subjects with an ileostomy exhibited a >40% recovery of the ingested dose in the ileal fluid, although notable differences were recorded among volunteers.57 Once in the large intestine, anthocyanins are hydrolysed by the local microbiota through to β-glucosidase activity.58 The resulting aglycones are broken down by cleavage of the C3-ring, and further metabolised into a broad array of phenolic and aldehydic constituents, with a scheme similar to that of flavonols. Protocatechuic acid has been pointed out as the main catabolite of cyanidin-3-O-glucoside in humans.59 Gallic, syringic, and vanillic acids have been identified as the major degradation products of delphinidin-3-O-glucoside, malvidin-3-O-glucoside, and peonidin-3-O-glucoside, respectively.60,61
2.6. Flavanones
Flavanones are a sub-group of flavonoids mainly present in citrus fruits, particularly in the albedo. They can be also found in tomatoes. Hesperetin and its derivatives are characteristic flavanones of sweet orange, tangelo, lemon and lime, while naringenin and its derivatives are those of grapefruit and sour orange.62 The most common flavanone glycoside is hesperetin-7-O-rutinoside (hesperidin)63 and the daily mean intake of flavanones ranges from 7.6 to 93.7 mg per day.64The major studies on absorption and metabolism of flavanones have been performed with orange juice, the main source of flavanones in the diet, and have highlighted the importance of structure, food matrix, and absorption/excretion capacity on the bioavailability of flavanones.65–67 After consumption of 250 mL of orange juice containing 168 μmol of hesperetin-7-O-rutinoside and 12 μmol of naringenin-7-O-rutinoside by healthy volunteers, hesperetin-7-O-glucuronide and an unassigned hesperetin-O-glucuronide were detected in plasma. The combined Cmax for the hesperetin glucuronides was 922 nM at a Tmax of 4.4 h, which is indicative of absorption in the colon.66 Both glucuronide and sulphoglucuronide metabolites of hesperetin and naringenin, while absent in plasma, were recovered in urine, emphasizing substantial post-absorption phase II metabolism.66,68 The quantities of metabolites excreted in 0–24 h urine accounted for 6.5% of the ingested hesperetin-7-O-rutinoside, in contrast to the 17.3% recovered from the naringenin-7-O-rutinoside dose.66 The higher level of excretion of naringenin metabolites could be related to the impact on absorption of different substituents on the flavanone B ring66 rather than the amounts ingested.68 Similar relative levels of urinary excreted flavanone metabolites have been reported for differently treated orange juices.67 Tomás-Navarro and colleagues67 pointed out that the intake of similar amounts of soluble flavanones, irrespective of the juice treatment, leads to relatively similar levels of urinary metabolites. Urinary excretion of citrus flavanones may also vary depending on the flavanone source, as noted with the excretion of naringenin metabolites after the consumption of grapefruit and orange juices.65
2.7. Flavan-3-ols and proanthocyanidins
Flavan-3-ols are a complex subclass of polyphenolic substances lacking glycoside residues and ranging from simple monomers to oligomeric and polymeric proanthocyanidins. Monomeric flavan-3-ols include (+)-catechin, (−)-epicatechin, gallocatechin, epigallocatechin, their galloyl substituted derivatives (epicatechin-gallate and epigallocatechin-gallate), (+)-afzelechin and (−)-epiafzelechin. Proanthocyanidins, also known as condensed tannins, encompass a large span of polymerisation degrees of flavan-3-ol monomeric units and, due to limitations in analytical methods, their actual dietary intake has not been adequately evaluated, but is estimated to be around 300 mg per day (Fig. 6). These substances can be classified according to the structure of their monomers: those consisting entirely of epicatechin units are called procyanidins; if they contain epigallocatechin or epiafzelechin they are named prodelphinidins and propelargonidins, respectively. Proanthocyanidin dimers can also be classified according to the position of C–C or C–O intermolecular bonds: the most common B-type proanthocyanidins share a C–C bond in the 4 → 6 or 4 → 8 position, while the least frequent A-type proanthocyanidins are formed by an additional C–O linkage in the 2 → 7 or 2 → 5 position.69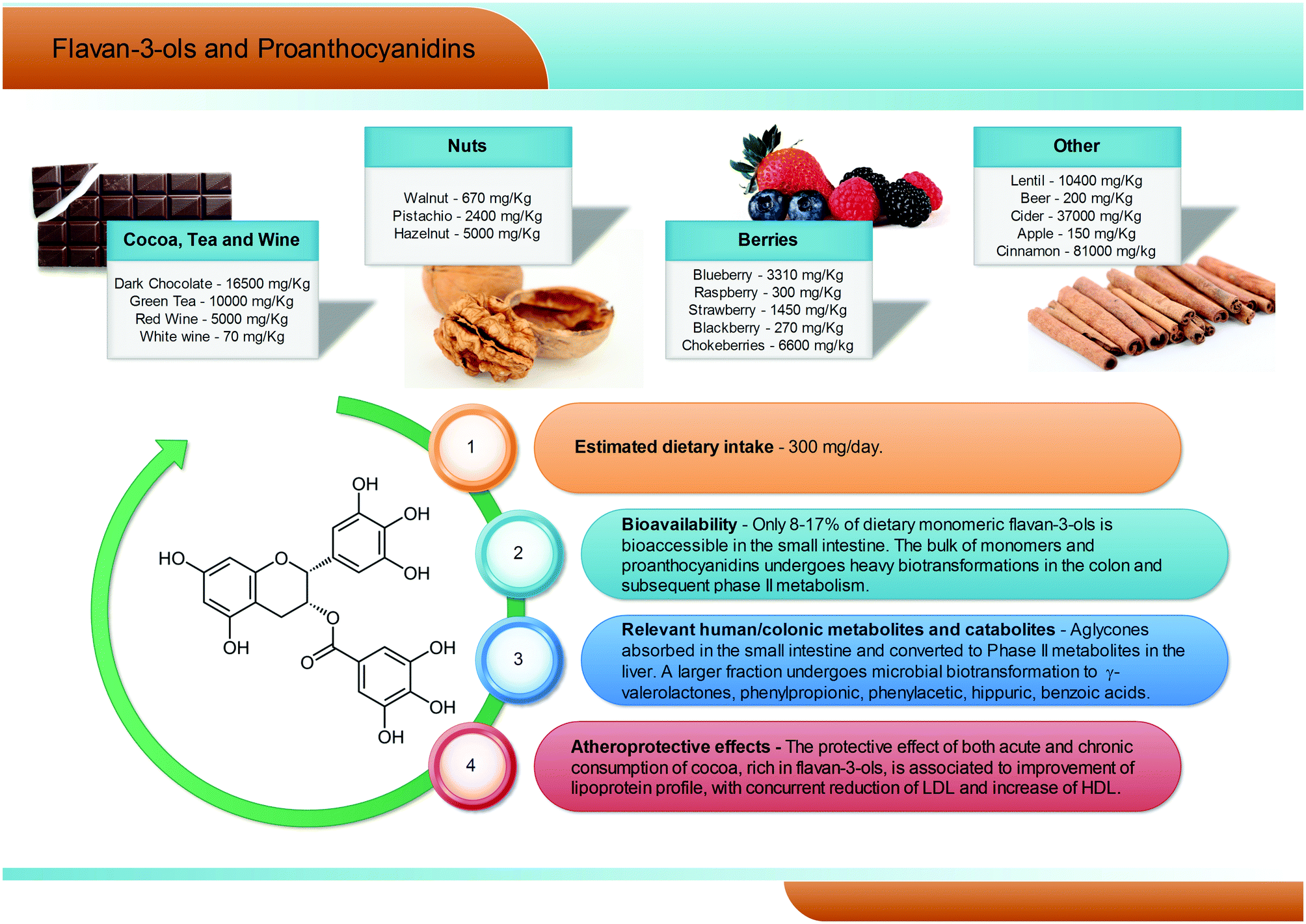 | ||
| Fig. 6 Dietary sources, bioavailability, metabolism and atheroprotective effects of flavan-3-ols and proanthocyanidins. | ||
Flavan-3-ol monomers and proanthocyanidins are widely spread in fruits and vegetables (mainly tea, cocoa and dark chocolate, apples, pears, grapes, berries, plums, nuts, and red wine, see Fig. 6), being the most largely consumed polyphenols in Western populations. Dark chocolate alone can contain up to 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 mg kg−1 of procyanidins.70–73 Characterized by a large span of molecular weights and structures, the solubility, absorbability, and bioavailability of these compounds may vary greatly, but some common trends may be outlined.
500 mg kg−1 of procyanidins.70–73 Characterized by a large span of molecular weights and structures, the solubility, absorbability, and bioavailability of these compounds may vary greatly, but some common trends may be outlined.
In humans, (−)-epicatechin-3′-O-glucuronide (reaching plasma levels ∼600 nM) has been pointed out as the main (−)-epicatechin metabolite after cocoa consumption, followed by (−)-epicatechin 3′-O-sulfate (∼300 nM).74 Regarding tea consumption, (−)-epigallocatechin-3-O-gallate is the only circulating unmetabolised compound, and the highest in absolute concentration with respect to (−)-epigallocatechin and (−)-epicatechin conjugates.75 However, it has been estimated that only about 8–17% of dietary flavan-3-ols are bioaccessible in the small intestine, while the remaining unabsorbed fraction of flavan-3-ol monomers and proanthocyanidins has been reported to reach the large intestine almost intact.76,77 Here, the colonic host microbiota is able to break down the flavonoidic skeleton, generating several low molecular weight metabolites, namely phenylpropionic, phenylacetic, hippuric, and benzoic acids with different hydroxylation patterns and, largely exceeding all the other metabolites, γ-valerolactones (Fig. 6).75,76,78–80
2.8. Hydrolysable tannins: gallotannins and ellagitannins
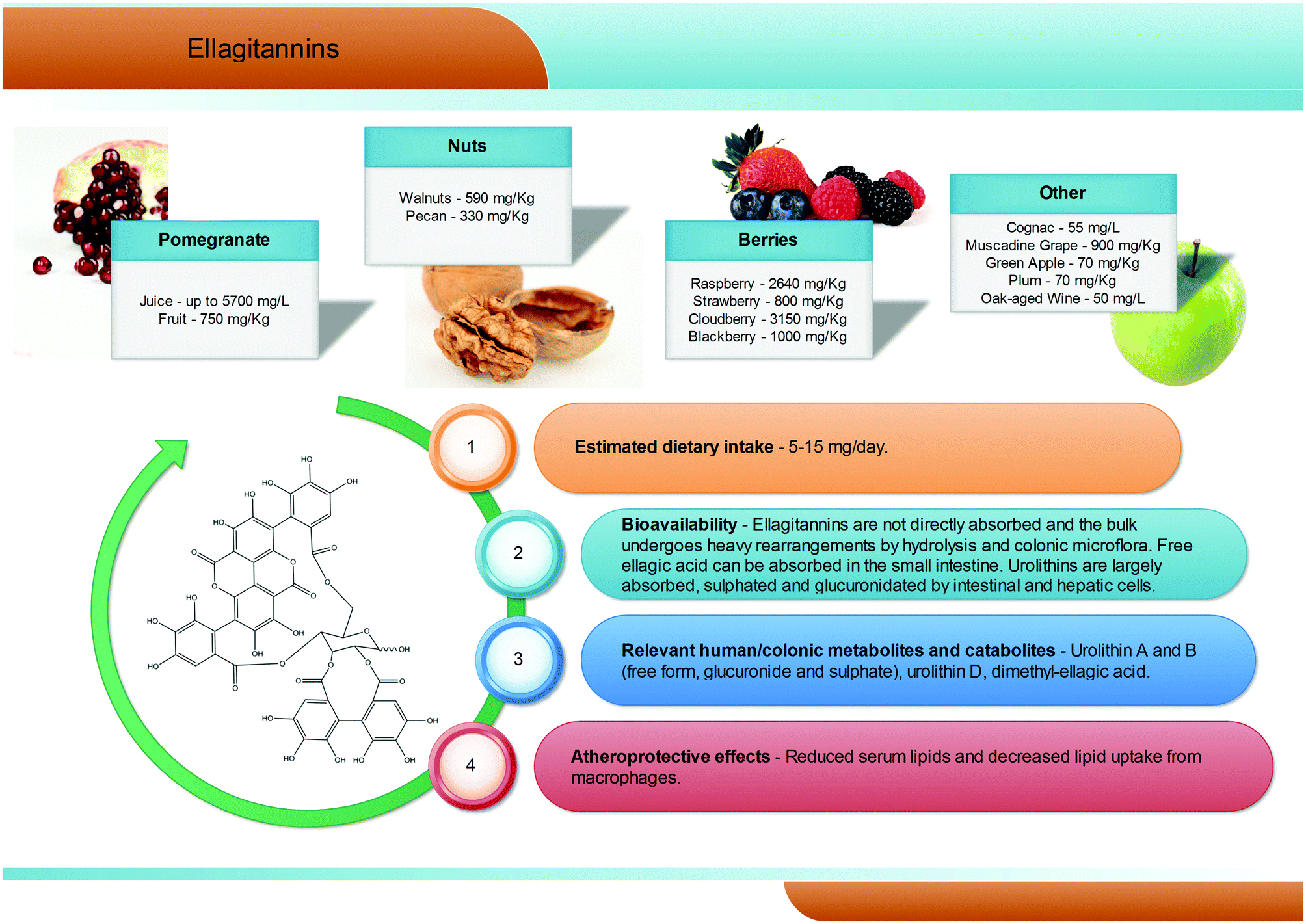
Hydrolysable tannins are the main group of plant tannins, with more than 500 structures hitherto identified. Hydrolysable tannins, polyesters of sugars (usually glucose) and phenolic acids, are divided into two subclasses according to their structural characteristics: gallotannins when the represented phenolic is gallic acid, and ellagitannins, characterised by the presence of at least one hexahydroxydiphenoyl group, that spontaneously rearranges into ellagic acid upon hydrolysis.81 Ellagic acid can also be present in its free form or as ellagic acid derivatives through methylation, methoxylation, and glycosylation.82Ellagitannins are typical constituents of many plant families, whilst the distribution of gallotannins in nature is rather limited (Fig. 7).83 However, despite their wide distribution, the occurrence of ellagitannins is restricted to a few fruits and nuts (Fig. 7) including raspberries (up to 2600 mg kg−1), strawberries, blackberries, blueberries, pomegranate (up to 5700 mg L−1 in juice), muscadine grapes, and persimmon, as well as walnuts (up to 590 mg kg−1), hazelnuts, and oak-aged wines (where tannins are released from the oak barrels, up to 50 mg L−1).84 Given their bitter and astringent taste, and as a consequence of their presence in plant tissues and organs with limited nutritional value (barks, wood), the presence of these substances in plant foods has been gradually reduced over centuries by means of agronomic selection and food processing. At present a normal western diet may provide 5–15 mg per day of ellagitannins.
 | ||
| Fig. 7 Dietary sources, bioavailability, metabolism and atheroprotective effects of ellagitannins. | ||
After ingestion of hydrolysable tannins, these polymeric structures are hydrolysed to yield gallic acid and/or ellagic acid for gallotannins, and ellagitannins, respectively. The released gallic acid may be metabolised into pyrogallol and pyrocatechol by the gut microbiota85 or may be absorbed and appear in the circulatory system free or methylated (3-O-methylgallic acid, 4-O-methylgallic acid, 3,4-di-O-methylgallic acid). In some cases, 2-O-sulfate-pyrogallol86,87 has also been described in human plasma after consumption of hydrolysable tannin sources. Free ellagic acid can be absorbed and undergoes extensive phase II metabolism; however, the bulk of the ellagic acid hydrolysed from ellagitannins (more than the 99%) is metabolised into urolithins by the gut microbiota (Fig. 2).57 Urolithins are microbial metabolites possessing a 6H-dibenzo[b,d]pyran-6-one structure with different phenolic hydroxylation patterns: urolithin D, tetrahydroxydibenzopyranone; urolithin C, trihydroxydibenzopyranone; urolithin A, dihydroxydibenzopyranone; and urolithin B, monohydroxydibenzopyranone (Fig. 7).88 It must be noticed that these substances differ largely from their starting counterparts and are not present in food sources. After absorption at the colonic region, urolithins appear in the circulatory system almost exclusively as glucuronide, sulphate and methylated metabolites in amounts that rarely exceed nM concentrations.61 Urolithin A glucuronide has been pointed out as the main metabolite excreted in urine after ellagitannin consumption, followed by urolithin B glucuronide and the free forms urolithin A and urolithin B (Fig. 2).57,61,89,90 However, considerable inter-individual variability in urolithin excretion levels has been observed, clearly associated with different colonic microbiota composition.
3. Cholesterol metabolism in atherosclerosis pathogenesis
3.1. Atherosclerosis
Cardiovascular disease (CVD) underpinned by atherosclerosis represents the leading cause of death in industrialized societies.91 Epidemiological studies have identified at least two classes of risk factors for developing atherosclerosis, behavioural (smoking, physical inactivity, unhealthy diet), and metabolic (high blood pressure, diabetes, high lipid levels, obesity). However, elevated levels of serum cholesterol have been identified as the key driving force for the development of atherosclerosis, in the absence of other known risk factors and therefore CVD has long been characterized as a lipid related disease due to the nature of its end stage pathology.92 In particular, low-density lipoprotein cholesterol (LDL-C) represents one of the major predictors of coronary heart diseases. Conversely, several epidemiologic studies have strongly supported the inverse correlation between high-density lipoproteins (HDL) levels and the risk of cardiovascular disease.93 In this context the central role of cholesterol metabolism in the cascade of events that leads to atherosclerosis is clear, the accumulation of LDL in the subendothelial matrix (intima of vessel) being a primary initiating factor. Accumulation is greater when levels of circulating LDL are raised, as it occurs in hypercholesterolemia. LDL in the subendothelial matrix become minimally oxidized, thus acquiring pro-inflammatory activity. However, they are not sufficiently modified to be recognized by macrophage scavenger receptors and are still uptaken by the LDL receptor (LDLR).94 Accumulation of minimally oxidized LDL stimulates further pro-inflammatory processes, including the differentiation of monocytes into macrophages, and their expression of scavenger receptors, such as SR-A and CD36. This latter event is responsible for modified LDL particle uptake by macrophages, leading to uncontrolled cholesterol accumulation and foam cell formation. With lesion progression, LDL becomes ‘highly oxidized’ and can be taken up more efficiently by foam cells. Cells can protect themselves from excessive cholesterol loading by effluxing cholesterol to HDL particles, which represents the first step of the reverse cholesterol transport (RCT).95 Nonetheless, cholesterol efflux is not sufficient to block disease progression, and the continuous accumulation of cholesterol leads to macrophage death and formation of a necrotic core that includes cholesteryl esters, cell debris, and macrophages.Although advanced atherosclerotic lesions can lead to ischemic symptoms as a result of progressive narrowing of vessels, pathological studies suggest that acute cardiovascular events, which result in myocardial infarction and stroke, generally are a consequence of plaque rupture and thrombosis.96
3.2. Cholesterol homeostasis, cholesterol efflux and reverse cholesterol transport
Another important source of cholesterol is represented by cholesterol absorbed in the intestinal lumen, which is derived from the diet as well as from the bile. Cholesterol absorption mostly occurs in the duodenum and in the jejunum. Net influx from intestinal lumen into the enterocytes results from the balance between absorption through the transporter Niemann-Pick C1-Like 1 (NPC1L1) and efflux through two half-transporters, named the ATP binding cassette transporter G5/G8 (ABCG5 and ABCG8). Intracellular cholesterol is esterified in the endoplasmatic reticulum by the enzyme Acyl-CoA cholesterol acyltransferase 2 (ACAT2). Once esterified, cholesterol is incorporated into chylomicrons together with triglycerides, phospholipids and apoB48 by microsomal triglyceride transfer protein (MTP). Chylomicrons are delivered to the lymph and they reach the blood circulation through the thoracic duct. Chylomicrons are mainly composed of triglycerides and few cholesteryl esters. They are remodelled while travelling through the systemic circulation by the action of lipoprotein lipase (LPL), which hydrolyses their triglycerides. These particles are normally cleared rapidly and efficiently from the plasma by the liver, through binding to LDLR.102
In the hepatocytes, lipids and proteins are assembled by MTP, forming the very low density lipoproteins (VLDL). VLDL are primarily composed of triglycerides (around 45–50% w/w), but also contain free and esterified cholesterol and ApoB100 protein.
LDL are primarily composed of free cholesterol and cholesteryl esters. They contain ApoB100, which allows LDL to be cleared from plasma by the LDLR pathway. LDL represent the main cholesterol transporter responsible for the delivery of cholesterol to peripheral tissues in humans, since 60–70% of the total serum cholesterol is carried by these lipoproteins.103 Therefore LDLR plays a crucial role in maintaining cholesterol homeostasis, and its expression is strictly regulated at both transcription and posttranscriptional levels.104,105
The first and limiting step of this pathway is the efflux of cholesterol from peripheral cells to HDL, that occurs through different transporters: ABCA1, ATP binding cassette G1 (ABCG1) and scavenger receptor B1 (SR-BI).107 ABCA1 and ABCG1 mediate unidirectional flux of cholesterol, thus leading to net removal of cell cholesterol, whereas SR-BI mediates a bidirectional flux of free cholesterol, whose net movement is determined by the concentration gradient.108 HDL capacity to mediate cell cholesterol removal has been recently associated with cardiovascular protection.109 Thus, the efflux potential of sera could be proposed as a novel biomarker characterizing the individual cardiovascular risk and the HDL function beyond their circulating levels, and could be considered a target for innovative atheroprotective strategies. Macrophage expression of ABCA1 and ABCG1 is transcriptionally up-regulated in response to elevated cellular cholesterol levels by the nuclear liver X receptors α and β (LXRα and LXRβ) which act as heterodimers with their partner, the retinoid X receptor. In vitro and in vivo studies revealed that synthetic LXR agonists may increase cholesterol efflux and macrophage RCT,110 inhibit atherosclerosis progression,111 and promote atherosclerosis regression.112 Moreover, it has been demonstrated that ABCA1 can also be up-regulated by peroxisome proliferator-activated receptor (PPAR)-α as well as PPAR-γ agonists,113,114 promoting macrophage cholesterol efflux in vitro and limiting macrophage foam cell formation and atherosclerosis progression in vivo.115 Conversely, miR-10b has been shown to directly repress ABCA1 and ABCG1, negatively regulating the cholesterol efflux from lipid-loaded macrophages.116
Along the RCT pathway, the cholesterol released from peripheral cells may be taken up by the liver through several distinct routes. The most direct involves the selective uptake of both unesterified and esterified cholesterol mediated by SR-BI.117 Alternatively, HDL cholesterol can be transferred to apoB-containing lipoproteins by lecithin-cholesterol acyltransferase and cholesteryl ester transfer protein (CETP) and taken up by the LDLR.117
To be excreted, HDL-derived cholesteryl esters need to be hydrolysed to generate free cholesterol. The latter can be secreted into bile, either directly, through the heterodimers ABCG5/G8, or after conversion into bile acids.118
4. Effect of (poly)phenolic compounds in the development of atherosclerotic cardiovascular disease
Several epidemiological observations have linked (poly)phenolic intake to atheroprotective effects in humans. In particular, the improvement of lipid metabolism causing the reduction of cardiovascular risk factors has been reported after diets enriched with nuts, coffee, cocoa, grapes, and berries. In this part of the review, pomegranate and olive oil have also been added, although the cardioprotective activities of these foods have been mainly associated with antioxidant and anti-inflammatory effects. However, emerging data, including those from our research groups, suggest that these compounds can positively modulate lipid metabolism. In the following narrative, the main relevance has been attributed to data from epidemiological and clinical trials in which the impact of polyphenol-containing food on the occurrence of cardiovascular disease was the principal end point. In vitro and in vivo studies were considered relevant and were included only when they offered suitable insights and elucidations of the putative underlying mechanisms of action and of the individual active substances. A visual synthesis of present knowledge is provided in Fig. 8 and 9.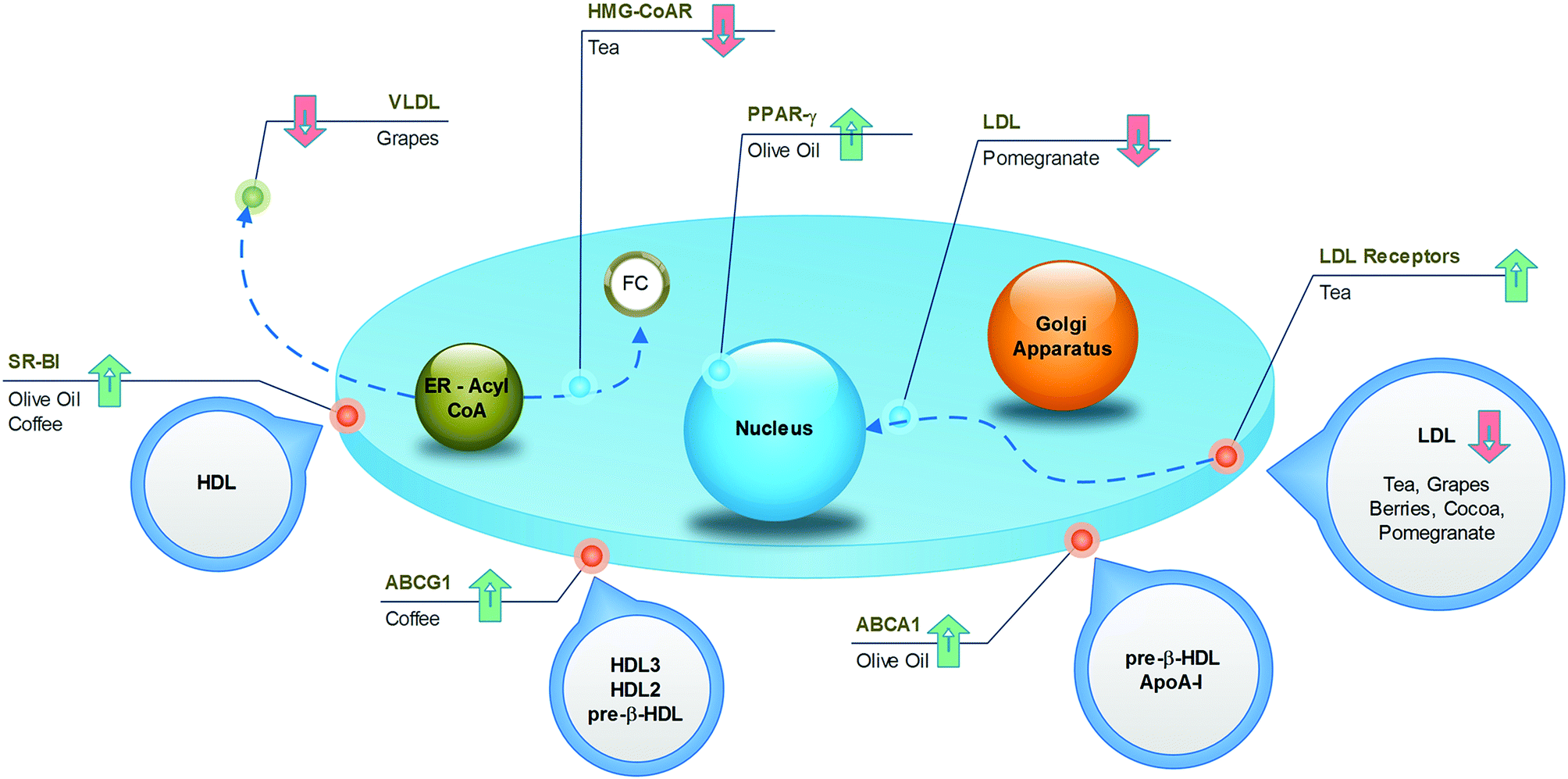 | ||
| Fig. 8 Cellular targets of anti-atherosclerotic effects of polyphenol-containing food. | ||
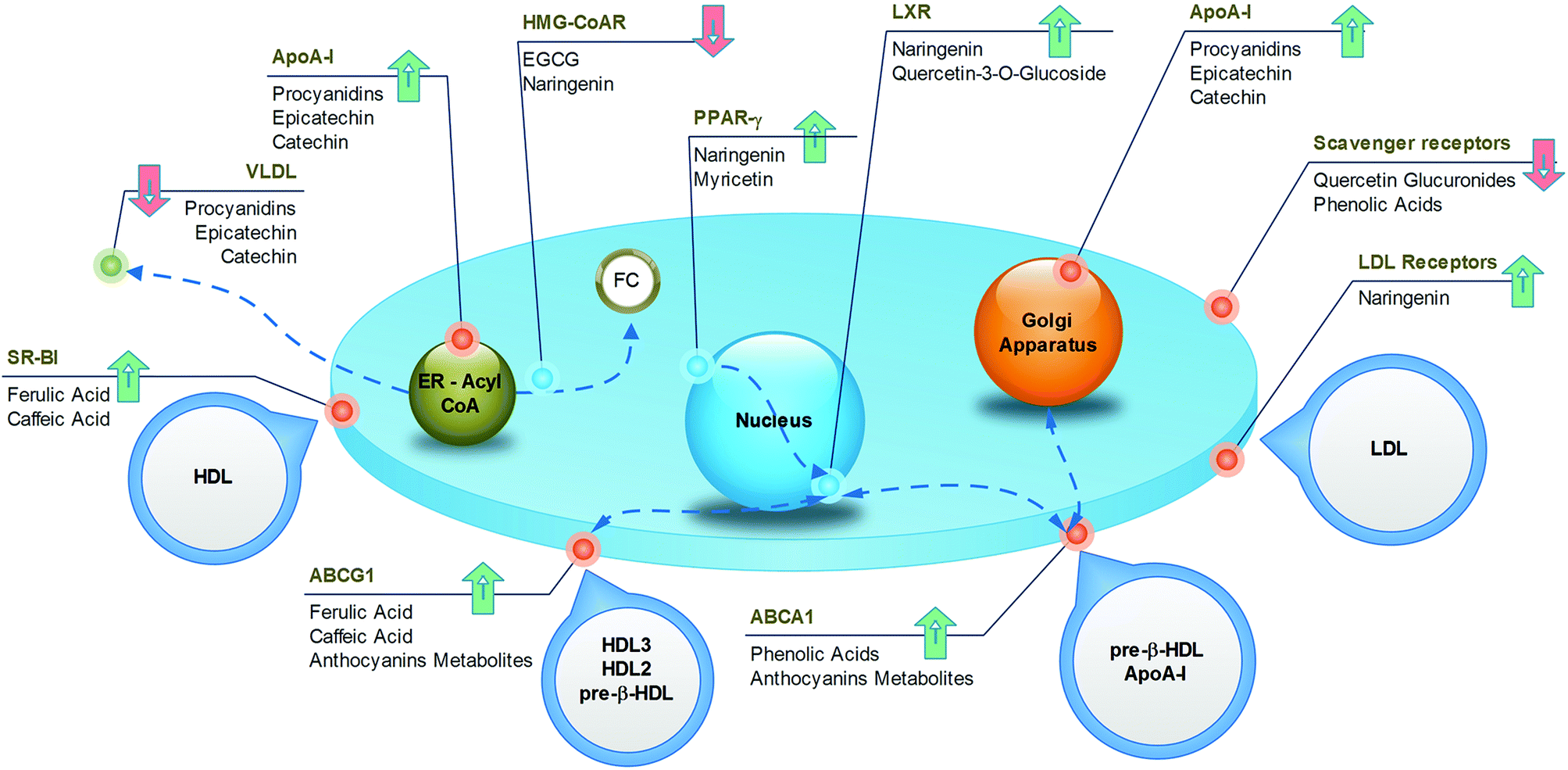 | ||
| Fig. 9 Cellular targets of anti-atherosclerotic effects of individual (poly)phenols. | ||
4.1. Nuts
Cardiovascular improvement following a high consumption of nuts has been known for a number of years,119,120 and was recently further documented by trials assessing the cardiovascular benefits of the Mediterranean diet.121 Review of several studies, including large prospective trials of primary prevention, allowed the establishment of the fact that daily ingestion of at least 30 g of nuts, or frequent weekly consumption (≥4 times), may reduce cardiovascular risk by 37%. A mean reduction of 8.3% for each incremental serving per week has been estimated.122 This effect is at least partly related to the reduction of total and LDL cholesterol, apoB and apoB/apoA ratio, and increase of HDL, as demonstrated in different intervention trials involving hypercholesterolemic men and women.123 In addition, a more detailed analysis evidenced the positive influence of nut consumption on lipoprotein remodelling in subjects at high cardiovascular risk fed a nut-enriched diet. Specifically, decreased LDL is accompanied by a shift towards increased particle size and a reduction in the most atherogenic small, dense LDL.121 Importantly, the effects of nut intake are dose-related and are likely to be independent of nut types; almonds, hazelnuts, pecans, pistachios, walnuts, peanuts are all effective, with a reduction in the total cholesterol by 5% and LDL concentration by 10%, as well as an increase in the HDL/LDL ratio by 8.3%.119,123 Several components of nuts may play a crucial role in these cardiovascular benefits, in addition to phenolic substances, such as γ-tocopherol, α-linolenic acid, linoleic acid, phytosterols, micronutrients and L-arginine.124 However, a study performed in atherosclerosis-susceptible mice suggested that the combined presence of polyunsaturated fatty acids and polyphenols is necessary for atheroprotective activity to occur. In fact, only the consumption of whole walnuts, rich in both components, and not walnut oil, containing only polyunsaturated fatty acids, caused the reduction of atherosclerotic plaques and decreased levels of circulating and hepatic lipids.125 Unfortunately, in this paper, the exact composition of the diet cannot be provided, but it is possible to postulate that whole walnuts contain a significant amount of phenolic acids, flavan-3-ols, proanthocyanidins and ellagitannins. The importance of associating polyphenols and polyunsaturated acids was also confirmed in humans, where a 3.3% increase in cholesterol efflux potential of serum was observed in moderate hypercholesterolemic subjects undergoing acute consumption of whole walnuts.126 This study indicates a novel mechanism of atheroprotection following nut ingestion.4.2. Coffee
As one of the most widely consumed beverages worldwide, the impact of coffee on cardiovascular health has been extensively studied for decades. More recent meta-analysis, including large prospective cohort studies, established a U-shaped association between cardiovascular risk and coffee intake, with the lowest risk at 3–5 cups per day.127,128 The reason for this non-linear trend could be found in the complex mixture of substances constituting coffee, in which both beneficial and detrimental compounds coexist. For example, it is well known that caffeine induces cardiac dysrhythmias and increases blood pressure, whereas the diterpenes (kahweol and cafestol) raise plasma cholesterol. However, the phenolic compounds may exert cardioprotective activities. From this point of view, even the brewing method is important, because it influences the final composition of the beverage. The current habit of drinking filtered coffee, where the diterpenes are absent, may result in more beneficial effects compared to the consumption of boiled, unfiltered coffee.The specific mechanisms accounting for the atheroprotective effects of phenolics from coffee have recently been elucidated in a translational study.129 Here, the coffee consumption by healthy subjects resulted in increased expression of SR-BI and ABCG1 in macrophages derived from circulating monocytes (Fig. 8), with a consequent 1.4-fold increase in cholesterol efflux. Concomitantly, sera collected after the intake of coffee was enriched with phenolic compounds, and promoted cell cholesterol efflux in cultured THP-1 macrophages more efficiently than pre-coffee sera. In vitro and in vivo investigations revealed that incubation of macrophages with ferulic or caffeic acid increased the cholesterol efflux to HDL (Fig. 9), whereas the administration of ferulic acid to mice caused the elimination of cholesterol via the RCT pathway.129 Despite interest in revealing the potential active compounds, the in vitro studies should focus more on the phenolic metabolites, in order to confirm the compounds that were actually present in plasma, which enabled definition of the brewing protocol that guarantees the best potential with regard to atheroprotection.
4.3. Tea
Tea is a very popular beverage worldwide, existing in six types (white, yellow, green, oolong, black, and post-fermented), depending on the processing method of Camellia sinensis leaves. All types of tea are rich in polyphenols, but the preparation method influences the qualitative/quantitative composition. Although oxidised black tea contains theaflavins and thearubigins, non-oxidised green tea is a source of catechins, especially epigallocatechin gallate. The potential health benefits of tea have been widely investigated and have received a large amount of attention in recent times. Data from prospective cohort studies revealed that the observed decrease in the risk of death from cardiovascular disease, by as much as 44%,130 could, at least partially, be associated with a reduction in total cholesterol (−7 mg dL−1) and LDL cholesterol (−2 mg dL−1).131 The impact of tea on HDL cholesterol is more controversial, as both increased132 and unchanged131 levels have been observed in subjects fed green tea. The underlying mechanisms have been investigated in different animal models, whose diets were supplemented with green tea. An increase in fecal lipid elimination was observed as well as a decrease in cholesterol synthesis and LDLR activity and expression (Fig. 8).133 From a molecular point of view, it is likely that the most active compound in green tea is epigallocatechin gallate, which is able to increase cholesterol fecal elimination in vivo by displacing cholesterol from intestinal micelles (Fig. 9)134 and increasing hepatic ABCG5/G8 expression.135 In addition, this compound potently inhibits HMG-CoA reductase in vitro.136 The flavonoid myricetin also plays a relevant role by reducing the content of hepatic and plasma lipids in rats through a PPAR-α-mediated mechanism (Fig. 9).137 Although most studies have focused on the beneficial activities of green tea, some reports have suggested that even black tea consumption may exert potential atheroprotective effects in humans by reducing plasma lipid levels. This could be related to the ability of theaflavins and their metabolites to reduce lipid uptake from the liver, as assessed in cultured hepatic cells,138 and to decrease the cholesterol solubility in micelles in vitro.139 These effects were confirmed in vivo, where the administration of theaflavins to rodents reduced intestinal cholesterol absorption.138,1394.4. Cocoa
Dark chocolate is a popular food in western countries, where it represents a dietary source of flavan-3-ols and proanthocyanidins. The protective effect of both the acute and chronic consumption of cocoa against atherosclerosis can be attributed to several mechanisms, including the improvement in the lipoprotein profile.140 Although systematic review and meta-analysis of randomized trials have associated the high intake of flavan-3-ols and phenolic acids, through habitual cocoa-derived product consumption, with the reduction of LDL (−15%)141 and the increase of HDL (by +24%) (Fig. 8),142,143 further studies are necessary to identify the active compounds and the mechanisms of action. Notably, the concentrations of all phytochemicals can vary largely between cocoa-based preparations, depending on the source of the beans and the processing conditions. Studies focusing on epicatechin, catechins and procyanidins have demonstrated that these cocoa polyphenols affect the apolipoprotein synthesis in cultured human hepatic and intestinal cells, two cell types that are mainly responsible for apoA1 and apoB production (Fig. 9).144 In addition, cocoa polyphenols such as (−)-epicatechin, (+)-catechin, procyanidins, increased LDLR expression in vitro.144 Although this evidence provides a consistent mechanism to account for the increased HDL and decreased LDL levels, these data should be confirmed with polyphenol metabolites at concentrations congruent with human physiology. Consistent with this concept, Guerrero and colleagues demonstrated that administration of a proanthocyanidin-rich extract of cocoa to rats produced metabolites that affected the hepatic de novo lipid synthesis in vitro. Therefore, this study provides the active substances and the hypolipidemic mechanism of cocoa in vitro under actual physiological conditions.1454.5. Grapes
Several dietary products originate from grapes, such as grape juice, de-alcoholised wine, grape-based extracts from grape seeds and grape pomace and, obviously, wine. It is well established that red wine, in particular, produces cardioprotective effects. However, wine was excluded in the present discussion, owing to the potentially confounding effects of alcohol. Many studies have demonstrated that all non-alcoholic grape products may produce atheroprotective effects.146 Grapes, especially the red varieties, contain significant amounts of anthocyanins, flavan-3-ol monomers, proanthocyanidins, phenolic acids (i.e., gallic acid), hydroxycinnamates, as well as trace levels of resveratrol and flavonols. The ingestion of grape products has been associated with a decrease in the amount of LDL (−17%), triglycerides (−15%) and apoB, in addition to a possible increase in the amount of HDL (+12%) and apoA1, in both healthy subjects and in patients with high cardiovascular risk, such as postmenopausal women, haemodialysis patients and diabetics (Fig. 8).147–149 Although the mechanisms, as well as the active compounds underlying these effects, are not completely elucidated, some hypotheses can be proposed. Some reports highlight the influence of the major quercetin metabolite, quercetin-3-O-glucuronide, on lipid metabolism in cultured macrophages; this compound is likely to enhance ABCA1 expression through a LXR-mediated mechanism (Fig. 9),150 with a potentially positive impact on the cell cholesterol efflux. It has also been indicated to inhibit CD36 expression,151 thus preventing foam-cell formation. Interestingly, quercetin glucuronides have been found to accumulate in macrophages of human atherosclerotic vessels.151 If the above described macrophage function effects were confirmed in vivo, they would represent a relevant mechanism of atherosclerotic plaque regression.It is likely that the triglyceride metabolism is a relevant target for different grape components. Resveratrol has been demonstrated to reduce apoB48 and apoB100 production in the liver and intestine of obese individuals,152 whereas animal studies have revealed a possible interference with lipoprotein lipase expression,153 which could be in agreement with the reported effects on triglycerides. Naringenin, a flavanone present at low concentrations in grapes, may affect VLDL assembly through different mechanisms described in vivo, including inhibition of ACAT2, apoB secretion and MTP activity. Moreover, naringenin may cause a reduction in the cholesterol level by inhibiting HMG-CoA reductase.154 Finally, naringenin is likely to give additional benefits, as demonstrated through in vitro studies, revealing the induction of LDLR expression as well as the regulation of PPARα, PPARγ and LXRα, leading to the induction of the apoA1 gene (Fig. 9).155 Interestingly, these effects have also been attributed to naringenin metabolites, thus increasing the physiological relevance of these data.156
Serum metabolites of rats that were fed a proanthocyanidin-rich extract of grape seeds demonstrated a remarkable decrease in intracellular cholesterol and triglycerides in hepatic cultured cells.145 Moreover, it has recently been demonstrated that chronic administration of proanthocyanidins to dyslipidemic rats can counteract the abnormal increase in the amount of miR-33 and miR-122, whose dysregulation has been linked to the development of metabolic diseases.157
The pivotal role of the colonic metabolism in producing bioactive compounds was recently demonstrated in a study where gut microbiota of mice that had been administered cyanidin-3-O-β-glucoside, or its metabolite protocatechuic acid, were manipulated. Wang and colleagues showed that only the latter, originating upon ingestion of cyanidin-3-O-β-glucoside in mice with intact gut microbiota, were able to exert multiple anti-atherogenic effects at physiologically reachable concentrations, both in vitro and in vivo. Among the described effects, promotion of the macrophage cholesterol efflux and RCT in vivo, as well as reduction of the extent of atherosclerosis, are the most prominent.116 These effects are mediated by the repression of miR-10b, leading to induction of ABCA1 and ABCG1 in macrophages.
4.6. Berries
It has long been recognised that a diet enriched in berries has atheroprotective effects. Large observational studies revealed that this is mostly attributed to the anthocyanin content of berries, whose consumption is associated with a protective effect against myocardial infarction,158 mainly due to its impact on arterial stiffness and blood pressure.159 As mentioned in section 2.5, anthocyanin-mediated effects on lipid metabolism have recently been documented in vivo.116Studies assessing the atheroprotective effects of berries have been performed using extracts from different types of berries (Fig. 8). In a study that used rats fed with blueberries, the impact of phenolic acid derivatives was demonstrated. In fact, the metabolites isolated from the serum of these animals were able to reduce foam-cell formation in vitro by inhibiting CD36 expression. Moreover, these compounds increased the expression of ABCA1, possibly contributing to the removal of excess cholesterol from macrophages (Fig. 9).160
In another study, human intestinal cells were incubated with a black chokeberry extract, whose consumption is associated with a hypolipidemic effect in hypercholesterolemic subjects.161 These compounds caused the inhibition of NPC1L1, SR-BI and ABCA1 expression and an increase in ABCG5, ABCG8 and LDLR expressions in vitro (Fig. 9). As a consequence, increased apical cholesterol efflux to the intestinal lumen and reduced lipid uptake was observed.162 Although these data are of interest for suggesting novel mechanisms for the reduction of plasma lipid levels, they should be confirmed in experiments using the metabolites of polyphenols that are naturally present in a chokeberry extract. As described above, in planta, a significant concentration of polyphenols will never be reached in plasma, whereas their combined human and colonic metabolites will. These latter molecules should, therefore, be the object of future investigations in this regard.
4.7. Olive oil
Evidence from several studies demonstrates that olive oil provides protection against different risk factors for coronary heart disease. It was also documented that olive-oil consumption positively affects lipid profiles.163 Many components can be responsible for this activity, including unsaturated fatty acids, simple phenols, triterpenic acids and phytosterols.The specific benefits associated with olive oil-derived polyphenols have been demonstrated in a randomised, crossover trial, in which the intake of olive oil containing a high content of polyphenols by subjects with moderate cardiovascular risk factors was associated with the up-regulation of genes involved in cholesterol efflux in blood cells. The increased expression of ABCA1, SR-BI and PPARs, by promoting macrophage cholesterol efflux, may represent an antiatherogenic mechanism (Fig. 8).164
4.8. Pomegranate
The cardiovascular benefits of pomegranates are mainly associated with their antioxidant and antinflammatory activities. However, some studies have also revealed a hypolipidemic effect on hypercholesterolemic subjects.165In animals fed with pomegranate juice, or extracts from different parts of the fruit, a reduction in atherosclerosis was observed (Fig. 8). This was associated with a reduced amount of serum lipids and decreased lipid uptake from macrophages,166 which was confirmed in macrophages from healthy subjects receiving pomegranate juice.167 In an attempt to identify the active compounds, the phenolic compounds in pomegranates were incubated with cultured macrophages exposed to oxLDL. Although this experiment revealed the inhibition of cholesterol uptake,166 the result must be confirmed using metabolites circulating after pomegranate intake and not simply with the structures present in the food matrix. Similarly, the demonstration that ellagic acid attenuates foam-cell formation in vitro by suppressing SR-BI expression and promoting cell cholesterol efflux through LXR-PPARγ and ABCA1168 should be confirmed with urolithins.
5. Conclusions
Cardiovascular disease (CVD), which refers to a complex group of related disorders with multifactorial aetiologies, unified mostly through the underpinning pathophysiology of atherosclerosis and its sequels, remains the topmost cause of global morbidity and mortality. Diet can occupy a key role in both primary as well as secondary prevention of atherosclerotic CVD. Epidemiological evidence suggests that diets rich in polyphenols may decrease the risk of cardiovascular disease by improving lipidemic profiles. Therefore, foods enriched with these compounds can be considered as positive contributors to a balanced and health-promoting diet. It is important to note that the beneficial effects of these foods largely depend on the final product consumed, because processing and cooking the raw material may alter the actual phytochemical composition. Thus, an accurate assessment of the active content is required to postulate the beneficial effects. However, it is not yet clear which substances are involved in such trends in vivo or what their actual mode of action is. Currently, in the vast majority of in vitro studies, the tested compounds are those that occur in foods or in raw plant material, at doses ranging from low micromolar to millimolar concentrations; although, following ingestion, these compounds are extensively modified by human and microbial enzymes, and their appearance in the internal compartments of the human body rarely exceeds nanomolar concentrations. Thus, the correct characterisation of the biological effects of polyphenols must be evaluated through the cellular activity of their principal metabolites, as assessed in bioavailability studies. In particular, despite growing evidence of their significant distribution in human plasma and tissues, human and colonic metabolites, such as urolithins from ellagitannins and γ-valerolactones from flavan-3-ols and proanthocyanidins, have never been properly investigated in terms of their effect on cholesterol homeostasis at the concentrations that are actually circulating. Finally, as the pattern of human and colonic metabolism may vary significantly among different individuals and populations, its possible consequences on the design of clinical trials and other intervention studies concerning dietary polyphenols and their atheroprotective effects should be carefully taken into account.Future studies may be conducted as multicentre randomised controlled trials in high-risk cardiovascular groups such as those who are overweight/obese, or with metabolic syndrome, Type-II diabetics or patients with early carotid plaques and other primary CV risk factors, including endothelial dysfunction as a precursor of atherosclerosis.
Acknowledgements
Pedro Mena was funded by a grant of the Postdoctoral Fellowship Programme from Fundación Séneca (Murcia Region, Spain).References
- J. Perez-Jimenez, V. Neveu, F. Vos and A. Scalbert, Eur. J. Clin. Nutr., 2010, 64(Suppl 3), S112–S120 CrossRef CAS PubMed.
- A. Crozier, T. Yokota, I. B. Jaganath, S. Marks, M. Saltmarsh and M. N. Clifford, Plant Secondary Metabolites: Occurrence, Structure and Role in the Human Diet, 2006, pp. 208–302 Search PubMed.
- I. B. Jaganath and A. Crozier, Phenolic Compounds of Plant Origin and Health: The Biochemistry behind their Nutritional and Pharmacological Value, 2009, pp. 11–48 Search PubMed.
- D. Del Rio, A. Rodriguez-Mateos, J. P. E. Spencer, M. Tognolini, G. Borges and A. Crozier, Antioxid. Redox Sign., 2013, 18, 1818–1892 CrossRef CAS PubMed.
- A. Crozier, I. B. Jaganath and M. N. Clifford, Nat. Prod. Rep., 2009, 26, 1001–1043 RSC.
- A. Padayachee, G. Netzel, M. Netzel, L. Day, D. Zabaras, D. Mikkelsen and M. J. Gidley, Food Chem., 2012, 135, 2287–2292 CrossRef CAS PubMed.
- T. Walle, A. M. Browning, L. L. Steed, S. G. Reed and U. K. Walle, J. Nutr., 2005, 135, 48–52 CAS.
- C. S. Yang, M. J. Lee and L. Chen, Cancer Epidemiol. Biomarkers Prev., 1999, 8, 83–89 CAS.
- M. J. Lee, J. D. Lambert, S. Prabhu, X. Meng, H. Lu, P. Maliakal, C. T. Ho and C. S. Yang, Cancer Epidemiol. Biomarkers Prev., 2004, 13, 132–137 CAS.
- S. Hirota, T. Nishioka, T. Shimoda, K. Miura, T. Ansai and U. Takahama, Food Sci. Technol. Res., 2011, 7, 239–245 CrossRef.
- A. J. Day, F. J. Canada, J. C. Diaz, P. A. Kroon, R. Mclauchlan, C. B. Faulds, G. W. Plumb, M. R. A. Morgan and G. Williamson, FEBS Lett., 2000, 468, 166–170 CrossRef CAS.
- A. Crozier, D. Del Rio and M. N. Clifford, Mol. Aspects Med., 2010, 31, 446–467 CrossRef CAS PubMed.
- J. M. Gee, M. S. DuPont, A. J. Day, G. W. Plumb, G. Williamson and I. T. Johnson, J. Nutr., 2000, 130, 2765–2771 CAS.
- C. Manach and J. L. Donovan, Free Radical Res., 2004, 38, 771–785 CrossRef CAS.
- M. C. Donovan JL, C. Manach, R. M. Faulks and P. A. Kroon, Plant Secondary Metabolites. Occurrence, Structure and Role in the Human Diet, 2006, pp. 303–351 Search PubMed.
- M. M. Appeldoorn, J. P. Vincken, A. M. Aura, P. C. Hollman and H. Gruppen, J. Agric. Food Chem., 2009, 57, 1084–1092 CrossRef CAS PubMed.
- L. Calani, D. Del Rio, M. Luisa Callegari, L. Morelli and F. Brighenti, Int. J. Food Sci. Nutr., 2012, 63, 513–521 CrossRef CAS PubMed.
- M. V. Selma, J. C. Espín and F. A. Tomás-Barberán, J. Agric. Food Chem., 2009, 57, 6485–6501 CrossRef CAS PubMed.
- M. Urpi-Sarda, I. Garrido, M. Monagas, C. Gomez-Cordoves, A. Medina-Remon, C. Andres-Lacueva and B. Bartolome, J. Agric. Food Chem., 2009, 57, 10134–10142 CrossRef CAS PubMed.
- S. Stoupi, G. Williamson, J. W. Drynan, D. Barron and M. N. Clifford, Mol. Nutr. Food Res., 2010, 54, 747–759 CAS.
- S. Deprez, C. Brezillon, S. Rabot, C. Philippe, I. Mila, C. Lapierre and A. Scalbert, J. Nutr., 2000, 130, 2733–2738 CAS.
- J. C. Espin, R. Gonzalez-Barrio, B. Cerda, C. Lopez-Bote, A. I. Rey and F. A. Tomas-Barberan, J. Agric. Food Chem., 2007, 55, 10476–10485 CrossRef CAS PubMed.
- S. Roowi, A. Stalmach, W. Mullen, M. E. Lean, C. A. Edwards and A. Crozier, J. Agric. Food Chem., 2010, 58, 1296–1304 CrossRef CAS PubMed.
- A. Stalmach, W. Mullen, H. Steiling, G. Williamson, M. E. Lean and A. Crozier, Mol. Nutr. Food Res., 2010, 54, 323–334 CAS.
- I. B. Jaganath, W. Mullen, C. A. Edwards and A. Crozier, Free Radical Res., 2006, 40, 1035–1046 CrossRef CAS PubMed.
- A. Stalmach, W. Mullen, D. Barron, K. Uchida, T. Yokota, C. Cavin, H. Steiling, G. Williamson and A. Crozier, Drug Metab. Dispos., 2009, 37, 1749–1758 CrossRef CAS PubMed.
- R. Zamora-Ros, C. Andres-Lacueva, R. M. Lamuela-Raventos, T. Berenguer, P. Jakszyn, C. Martinez, M. J. Sanchez, C. Navarro, M. D. Chirlaque, M. J. Tormo, J. R. Quiros, P. Amiano, M. Dorronsoro, N. Larranaga, A. Barricarte, E. Ardanaz and C. A. Gonzalez, Br. J. Nutr., 2008, 100, 188–196 CrossRef CAS PubMed.
- L. Almeida, M. Vaz-da-Silva, A. Falcao, E. Soares, R. Costa, A. I. Loureiro, C. Fernandes-Lopes, J. F. Rocha, T. Nunes, L. Wright and P. Soares-da-Silva, Mol. Nutr. Food Res., 2009, 53(Suppl 1), S7–15 Search PubMed.
- M. Vaz-da-Silva, A. I. Loureiro, A. Falcao, T. Nunes, J. F. Rocha, C. Fernandes-Lopes, E. Soares, L. Wright, L. Almeida and P. Soares-da-Silva, Int. J. Clin. Pharmacol. Ther., 2008, 46, 564–570 CrossRef CAS.
- P. Vitaglione, S. Sforza, G. Galaverna, C. Ghidini, N. Caporaso, P. P. Vescovi, V. Fogliano and R. Marchelli, Mol. Nutr. Food Res., 2005, 49, 495–504 CAS.
- D. J. Boocock, K. R. Patel, G. E. Faust, D. P. Normolle, T. H. Marczylo, J. A. Crowell, D. E. Brenner, T. D. Booth, A. Gescher and W. P. Steward, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 848, 182–187 CrossRef CAS PubMed.
- K. R. Patel, E. Scott, V. A. Brown, A. J. Gescher, W. P. Steward and K. Brown, Ann. N. Y. Acad. Sci., 2011, 1215, 161–169 CrossRef CAS PubMed.
- C. Henry-Vitrac, A. Desmouliere, D. Girard, J. M. Merillon and S. Krisa, Eur. J. Nutr., 2006, 45, 376–382 CrossRef CAS PubMed.
- C. Andres-Lacueva, M. Urpi-Sarda, R. Zamora-Ros and R. M. Lamuela-Raventos, Plant Phenolics and Human Health: Biochemistry, Nutrition, and Pharmacology, 2009 Search PubMed.
- A. Burkon and V. Somoza, Mol. Nutr. Food Res., 2008, 52, 549–557 CAS.
- M. Urpi-Sarda, R. Zamora-Ros, R. Lamuela-Raventos, A. Cherubini, O. Jauregui, R. de la Torre, M. I. Covas, R. Estruch, W. Jaeger and C. Andres-Lacueva, Clin. Chem., 2007, 53, 292–299 CAS.
- T. Walle, F. Hsieh, M. H. DeLegge, J. E. Oatis Jr. and U. K. Walle, Drug Metab. Dispos., 2004, 32, 1377–1382 CrossRef CAS PubMed.
- M. Azorin-Ortuno, M. J. Yanez-Gascon, F. Vallejo, F. J. Pallares, M. Larrosa, R. Lucas, J. C. Morales, F. A. Tomas-Barberan, M. T. Garcia-Conesa and J. C. Espin, Mol. Nutr. Food Res., 2011, 55, 1154–1168 CAS.
- C. M. Jung, T. M. Heinze, L. K. Schnackenberg, L. B. Mullis, S. A. Elkins, C. A. Elkins, R. S. Steele and J. B. Sutherland, FEMS Microbiol. Lett., 2009, 297, 266–273 CrossRef CAS PubMed.
- D. Wang, T. Hang, C. Wu and W. Liu, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2005, 829, 97–106 CrossRef CAS PubMed.
- L. M. Bode, D. Bunzel, M. Huch, G. S. Cho, D. Ruhland, M. Bunzel, A. Bub, C. M. Franz and S. E. Kulling, Am. J. Clin. Nutr., 2013, 97, 295–309 CrossRef CAS PubMed.
- C. Manach, G. Williamson, C. Morand, A. Scalbert and C. Remesy, Am. J. Clin. Nutr., 2005, 81, 230S–242S CAS.
- W. Mullen, C. A. Edwards and A. Crozier, Br. J. Nutr., 2006, 96, 107–116 CrossRef CAS.
- A. M. Aura, K. A. O'Leary, G. Williamson, M. Ojala, M. Bailey, R. Puupponen-Pimia, A. M. Nuutila, K. M. Oksman-Caldentey and K. Poutanen, J. Agric. Food Chem., 2002, 50, 1725–1730 CrossRef CAS PubMed.
- M. N. Clifford, J. Sci. Food Agric., 2000, 80, 1063–1072 CrossRef CAS.
- P. Bridle and C. F. Timberlake, Food Chem., 1997, 58, 103–109 CrossRef CAS.
- A. Castañeda-Ovando, M. d. L. Pacheco-Hernández, M. E. Páez-Hernández, J. A. Rodríguez and C. A. Galán-Vidal, Food Chem., 2009, 113, 859–871 CrossRef PubMed.
- S. de Pascual-Teresa, D. A. Moreno and C. García-Viguera, Int. J. Mol. Sci., 2010, 11, 1679–1703 CrossRef CAS PubMed.
- X. Wu, G. R. Beecher, J. M. Holden, D. B. Haytowitz, S. E. Gebhardt and R. L. Prior, J. Agric. Food Chem., 2006, 54, 4069–4075 CrossRef CAS PubMed.
- P. Mena, C. García-Viguera, J. Navarro-Rico, D. A. Moreno, J. Bartual, D. Saura and N. Martí, J. Sci. Food Agric., 2011, 91, 1893–1906 CrossRef CAS PubMed.
- C. D. Kay, G. Mazza and B. J. Holub, J. Nutr., 2005, 135, 2582–2588 CAS.
- T. K. McGhie and M. C. Walton, Mol. Nutr. Food Res., 2007, 51, 702–713 CAS.
- C. Manach, G. Williamson, C. Morand, A. Scalbert and C. Rémésy, Am. J. Clin. Nutr., 2005, 81, 230S–242S CAS.
- R. M. de Ferrars, C. Czank, Q. Zhang, N. P. Botting, P. A. Kroon, A. Cassidy and C. D. Kay, Br. J. Pharmacol., 2014, 171, 3268–3282 CrossRef CAS PubMed.
- R. González-Barrio, C. A. Edwards and A. Crozier, Drug Metab. Dispos., 2011, 39, 1680–1688 CrossRef PubMed.
- C. Czank, A. Cassidy, Q. Zhang, D. J. Morrison, T. Preston, P. A. Kroon, N. P. Botting and C. D. Kay, Am. J. Clin. Nutr., 2013, 97, 995–1003 CrossRef CAS PubMed.
- R. González-Barrio, G. Borges, W. Mullen and A. Crozier, J. Agric. Food Chem., 2010, 58, 3933–3939 CrossRef PubMed.
- M. Ávila, M. Hidalgo, C. Sánchez-Moreno, C. Pelaez, T. Requena and S. d. Pascual-Teresa, Food Res. Int., 2009, 42, 1453–1461 CrossRef PubMed.
- P. Vitaglione, G. Donnarumma, A. Napolitano, F. Galvano, A. Gallo, L. Scalfi and V. Fogliano, J. Nutr., 2007, 137, 2043–2048 CAS.
- R. González-Barrio, P. Truchado, H. Ito, J. C. Espín and F. A. Tomás-Barberán, J. Agric. Food Chem., 2011, 59, 1152–1162 CrossRef PubMed.
- C. García-Muñoz, L. Hernández, A. Pérez and F. Vaillant, Food Res. Int., 2014, 55, 161–169 CrossRef PubMed.
- M. K. Khan, E. H. Zill and O. Dangles, J. Food Compos. Anal., 2014, 33, 85–104 CrossRef CAS PubMed.
- D. Del Rio, A. Rodriguez-Mateos, J. P. E. Spencer, M. Tognolini, G. Borges and A. Crozier, Antioxid. Redox Signal., 2013, 18, 1818–1892 CrossRef CAS PubMed.
- P. J. Mink, C. G. Scrafford, L. M. Barraj, L. Harnack, C. P. Hong, J. A. Nettleton and D. R. Jacobs Jr., Am. J. Clin. Nutr., 2007, 85, 895–909 CAS.
- I. Erlund, E. Meririnne, G. Alfthan and A. Aro, J. Nutr., 2001, 131, 235–241 CAS.
- W. Mullen, M. A. Archeveque, C. A. Edwards, H. Matsumoto and A. Crozier, J. Agric. Food Chem., 2008, 56, 11157–11164 CrossRef CAS PubMed.
- M. Tomás-Navarro, F. Vallejo, E. Sentandreu, J. L. Navarro and F. A. Tomás-Barberán, J. Agric. Food Chem., 2014, 62, 24–27 CrossRef PubMed.
- C. Manach, C. Morand, A. Gil-Izquierdo, C. Bouteloup-Demange and C. Rémésy, Eur. J. Clin. Nutr., 2003, 57, 235–242 CrossRef CAS PubMed.
- C. Santos-Buelga and A. Scalbert, J. Sci. Food Agric., 2000, 80, 1094–1117 CrossRef CAS.
- R. Zamora-Ros, V. Knaze, L. Luján-Barroso, I. Romieu, A. Scalbert, N. Slimani, A. Hjartåker, D. Engeset, G. Skeie, K. Overvad, L. Bredsdorff, A. Tjønneland, J. Halkjær, T. J. Key, K. T. Khaw, A. A. Mulligan, A. Winkvist, I. Johansson, H. B. Bueno-de-Mesquita, P. H. M. Peeters, P. Wallström, U. Ericson, V. Pala, M. S. de Magistris, S. Polidoro, R. Tumino, A. Trichopoulou, V. Dilis, M. Katsoulis, J. M. Huerta, V. Martínez, M. J. Sánchez, E. Ardanaz, P. Amiano, B. Teucher, V. Grote, B. Bendinelli, H. Boeing, J. Förster, M. Touillaud, F. Perquier, G. Fagherazzi, V. Gallo, E. Riboli and C. A. González, Br. J. Nutr., 2013, 109, 1498–1507 CrossRef CAS PubMed.
- J. Pérez-Jiménez, L. Fezeu, M. Touvier, N. Arnault, C. Manach, S. Hercberg, P. Galan and A. Scalbert, Am. J. Clin. Nutr., 2011, 93, 1220–1228 CrossRef PubMed.
- A. Vogiatzoglou, A. A. Mulligan, R. N. Luben, M. A. H. Lentjes, C. Heiss, M. Kelm, M. W. Merx, J. P. E. Spencer, H. Schroeter and G. G. C. Kuhnle, Br. J. Nutr., 2014, 111, 1463–1473 CrossRef CAS PubMed.
- A. Tresserra-Rimbau, E. B. Rimm, A. Medina-Remón, M. A. Martínez-González, R. de la Torre, D. Corella, J. Salas-Salvadó, E. Gómez-Gracia, J. Lapetra, F. Arós, M. Fiol, E. Ros, L. Serra-Majem, X. Pintó, G. T. Saez, J. Basora, J. V. Sorlí, J. A. Martínez, E. Vinyoles, V. Ruiz-Gutiérrez, R. Estruch and R. M. Lamuela-Raventós, Nutr. Metab. Cardiovasc. Dis., 2014, 24, 639–647 CrossRef CAS PubMed.
- J. I. Ottaviani, T. Y. Momma, G. K. Kuhnle, C. L. Keen and H. Schroeter, Free Radical Biol. Med., 2012, 52, 1403–1412 CrossRef CAS PubMed.
- D. Del Rio, L. Calani, C. Cordero, S. Salvatore, N. Pellegrini and F. Brighenti, Nutrition, 2010, 26, 1110–1116 CrossRef CAS PubMed.
- A. Stalmach, S. Troufflard, M. Serafini and A. Crozier, Mol. Nutr. Food Res., 2009, 53, S44–S53 Search PubMed.
- W. Mullen, G. Borges, J. L. Donovan, C. A. Edwards, M. Serafini, M. E. J. Lean and A. Crozier, Am. J. Clin. Nutr., 2009, 89, 1784–1791 CrossRef CAS PubMed.
- S. Sang, M. J. Lee, I. Yang, B. Buckley and C. S. Yang, Rapid Commun. Mass Spectrom., 2008, 22, 1567–1578 CrossRef CAS PubMed.
- M. M. Appeldoorn, J. P. Vincken, A. M. Aura, P. C. H. Hollman and H. Gruppen, J. Agric. Food Chem., 2009, 57, 1084–1092 CrossRef CAS PubMed.
- S. Stoupi, G. Williamson, J. W. Drynan, D. Barron and M. N. Clifford, Mol. Nutr. Food Res., 2010, 54, 747–759 CAS.
- E. Bakkalbasi, O. Mentes and N. Artik, Crit. Rev. Food Sci. Nutr., 2009, 49, 283–298 CrossRef CAS PubMed.
- J. L. Maas, S. Y. Wang and G. J. Galletta, HortScience, 1991, 26, 66–68 Search PubMed.
- R. Niemetz and G. G. Gross, Phytochemistry, 2005, 66, 2001–2011 CrossRef CAS PubMed.
- J. M. Landete, Food Res. Int., 2011, 44, 1150–1160 CrossRef CAS PubMed.
- S. Roowi, A. Stalmach, W. Mullen, M. E. J. Lean, C. A. Edwards and A. Crozier, J. Agric. Food Chem., 2010, 58, 1296–1304 CrossRef CAS PubMed.
- C. A. Daykin, J. P. M. Van Duynhoven, A. Groenewegen, M. Dachtler, J. M. M. Van Amelsvoort and T. P. J. Mulder, J. Agric. Food Chem., 2005, 53, 1428–1434 CrossRef CAS PubMed.
- J. M. Hodgson, L. W. Morton, I. B. Puddey, L. J. Beilin and K. D. Croft, J. Agric. Food Chem., 2000, 48, 2276–2280 CrossRef CAS PubMed.
- J. C. Espín, R. González-Barrio, B. Cerdá, C. López-Bote, A. I. Rey and F. A. Tomás-Barberán, J. Agric. Food Chem., 2007, 55, 10476–10485 CrossRef PubMed.
- S. Tulipani, M. Urpi-Sarda, R. García-Villalba, M. Rabassa, P. López-Uriarte, M. Bulló, O. Jáuregui, F. Tomás-Barberán, J. Salas-Salvadó, J. C. Espín and C. Andrés-Lacueva, J. Agric. Food Chem., 2012, 60, 8930–8940 CrossRef CAS PubMed.
- P. Truchado, M. Larrosa, M. T. García-Conesa, B. Cerdá, M. L. Vidal-Guevara, F. A. Tomás-Barberán and J. C. Espín, J. Agric. Food Chem., 2012, 60, 5749–5754 CrossRef CAS PubMed.
- D. Lloyd-Jones, R. J. Adams, T. M. Brown, M. Carnethon, S. Dai, G. De Simone, T. B. Ferguson, E. Ford, K. Furie, C. Gillespie, A. Go, K. Greenlund, N. Haase, S. Hailpern, P. M. Ho, V. Howard, B. Kissela, S. Kittner, D. Lackland, L. Lisabeth, A. Marelli, M. M. McDermott, J. Meigs, D. Mozaffarian, M. Mussolino, G. Nichol, V. L. Roger, W. Rosamond, R. Sacco, P. Sorlie, R. Stafford, T. Thom, S. Wasserthiel-Smoller, N. D. Wong, J. Wylie-Rosett, C. American Heart Association Statistics and S. Stroke Statistics, Circulation, 2010, 121, 948–954 CrossRef PubMed.
- C. K. Glass and J. L. Witztum, Cell, 2001, 104, 503–516 CrossRef CAS.
- C. G. Santos-Gallego, C. Giannarelli and J. J. Badimon, Curr. Atheroscler. Rep., 2011, 13, 266–276 CrossRef CAS PubMed.
- M. Navab, J. A. Berliner, A. D. Watson, S. Y. Hama, M. C. Territo, A. J. Lusis, D. M. Shih, B. J. Van Lenten, J. S. Frank, L. L. Demer, P. A. Edwards and A. M. Fogelman, Arterioscler., Thromb., Vasc. Biol., 1996, 16, 831–842 CrossRef CAS.
- T. Y. Chang, C. C. Chang, N. Ohgami and Y. Yamauchi, Annu. Rev. Cell Dev. Biol., 2006, 22, 129–157 CrossRef CAS PubMed.
- R. T. Lee and P. Libby, Arterioscler., Thromb., Vasc. Biol., 1997, 17, 1859–1867 CrossRef CAS.
- J. M. Dietschy, S. D. Turley and D. K. Spady, J. Lipid Res., 1993, 34, 1637–1659 CAS.
- S. D. Turley, D. K. Spady and J. M. Dietschy, J. Lipid Res., 1995, 36, 67–79 CAS.
- J. L. Goldstein and M. S. Brown, Nature, 1990, 343, 425–430 CrossRef CAS PubMed.
- T. Rezen, D. Rozman, J. M. Pascussi and K. Monostory, Biochim. Biophys. Acta, 2011, 1814, 146–160 CrossRef CAS PubMed.
- C. Fernandez-Hernando, C. M. Ramirez, L. Goedeke and Y. Suarez, Arterioscler., Thromb., Vasc. Biol., 2013, 33, 178–185 CrossRef CAS PubMed.
- R. W. Mahley and Z. S. Ji, J. Lipid Res., 1999, 40, 1–16 CAS.
- E. National Cholesterol Education Program Expert Panel on Detection and A. Treatment of High Blood Cholesterol in,Circulation, 2002, 106, 3143–3421 Search PubMed.
- W. Annema and A. von Eckardstein, Circ. J., 2013, 77, 2432–2448 CrossRef CAS PubMed.
- C. Martel, W. Li, B. Fulp, A. M. Platt, E. L. Gautier, M. Westerterp, R. Bittman, A. R. Tall, S. H. Chen, M. J. Thomas, D. Kreisel, M. A. Swartz, M. G. Sorci-Thomas and G. J. Randolph, J. Clin. Invest., 2013, 123, 1571–1579 CAS.
- K. M. Wasan, D. R. Brocks, S. D. Lee, K. Sachs-Barrable and S. J. Thornton, Nat. Rev. Drug Discovery, 2008, 7, 84–99 CrossRef CAS PubMed.
- A. R. Tall, J. Intern. Med., 2008, 263, 256–273 CrossRef CAS PubMed.
- D. J. Rader, E. T. Alexander, G. L. Weibel, J. Billheimer and G. H. Rothblat, J. Lipid Res., 2009, 50(Suppl), S189–S194 Search PubMed.
- A. V. Khera, M. Cuchel, M. de la Llera-Moya, A. Rodrigues, M. F. Burke, K. Jafri, B. C. French, J. A. Phillips, M. L. Mucksavage, R. L. Wilensky, E. R. Mohler, G. H. Rothblat and D. J. Rader, N. Engl. J. Med., 2011, 364, 127–135 CrossRef CAS PubMed.
- S. U. Naik, X. Wang, J. S. Da Silva, M. Jaye, C. H. Macphee, M. P. Reilly, J. T. Billheimer, G. H. Rothblat and D. J. Rader, Circulation, 2006, 113, 90–97 CrossRef CAS PubMed.
- S. B. Joseph, E. McKilligin, L. Pei, M. A. Watson, A. R. Collins, B. A. Laffitte, M. Chen, G. Noh, J. Goodman, G. N. Hagger, J. Tran, T. K. Tippin, X. Wang, A. J. Lusis, W. A. Hsueh, R. E. Law, J. L. Collins, T. M. Willson and P. Tontonoz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7604–7609 CrossRef CAS PubMed.
- N. Levin, E. D. Bischoff, C. L. Daige, D. Thomas, C. T. Vu, R. A. Heyman, R. K. Tangirala and I. G. Schulman, Arterioscler., Thromb., Vasc. Biol., 2005, 25, 135–142 CrossRef CAS PubMed.
- A. Chawla, W. A. Boisvert, C. H. Lee, B. A. Laffitte, Y. Barak, S. B. Joseph, D. Liao, L. Nagy, P. A. Edwards, L. K. Curtiss, R. M. Evans and P. Tontonoz, Mol. Cell, 2001, 7, 161–171 CrossRef CAS.
- G. Chinetti, S. Lestavel, V. Bocher, A. T. Remaley, B. Neve, I. P. Torra, E. Teissier, A. Minnich, M. Jaye, N. Duverger, H. B. Brewer, J. C. Fruchart, V. Clavey and B. Staels, Nat. Med., 2001, 7, 53–58 CrossRef CAS PubMed.
- A. C. Li, C. J. Binder, A. Gutierrez, K. K. Brown, C. R. Plotkin, J. W. Pattison, A. F. Valledor, R. A. Davis, T. M. Willson, J. L. Witztum, W. Palinski and C. K. Glass, J. Clin. Invest., 2004, 114, 1564–1576 CrossRef CAS PubMed.
- D. Wang, M. Xia, X. Yan, D. Li, L. Wang, Y. Xu, T. Jin and W. Ling, Circ. Res., 2012, 111, 967–981 CrossRef CAS PubMed.
- R. S. Rosenson, H. B. Brewer Jr., W. S. Davidson, Z. A. Fayad, V. Fuster, J. Goldstein, M. Hellerstein, X. C. Jiang, M. C. Phillips, D. J. Rader, A. T. Remaley, G. H. Rothblat, A. R. Tall and L. Yvan-Charvet, Circulation, 2012, 125, 1905–1919 CrossRef PubMed.
- W. Annema and U. J. Tietge, Nutr. Metab., 2012, 9, 25 CAS.
- N. R. Damasceno, A. Perez-Heras, M. Serra, M. Cofan, A. Sala-Vila, J. Salas-Salvado and E. Ros, Nutr. Metab. Cardiovasc Dis., 2011, 21(Suppl 1), S14–S20 CrossRef CAS PubMed.
- D. Zhou, H. Yu, F. He, K. H. Reilly, J. Zhang, S. Li, T. Zhang, B. Wang, Y. Ding and B. Xi, Am. J. Clin. Nutr., 2014, 100, 270–277 CrossRef CAS PubMed.
- N. R. Damasceno, A. Sala-Vila, M. Cofan, A. M. Perez-Heras, M. Fito, V. Ruiz-Gutierrez, M. A. Martinez-Gonzalez, D. Corella, F. Aros, R. Estruch and E. Ros, Atherosclerosis, 2013, 230, 347–353 CrossRef CAS PubMed.
- J. H. Kelly Jr. and J. Sabate, Br. J. Nutr., 2006, 96(Suppl 2), S61–S67 CrossRef.
- J. Sabate, K. Oda and E. Ros, Arch. Intern. Med., 2010, 170, 821–827 CrossRef PubMed.
- P. M. Kris-Etherton, F. B. Hu, E. Ros and J. Sabate, J. Nutr., 2008, 138, 1746S–1751S CAS.
- R. Nergiz-Unal, M. J. Kuijpers, S. M. de Witt, S. Heeneman, M. A. Feijge, S. C. Garcia Caraballo, E. A. Biessen, G. R. Haenen, J. M. Cosemans and J. W. Heemskerk, Thromb. Res., 2013, 131, 411–417 CrossRef PubMed.
- C. E. Berryman, J. A. Grieger, S. G. West, C. Y. Chen, J. B. Blumberg, G. H. Rothblat, S. Sankaranarayanan and P. M. Kris-Etherton, J. Nutr., 2013, 143, 788–794 CrossRef CAS PubMed.
- N. D. Freedman, Y. Park, C. C. Abnet, A. R. Hollenbeck and R. Sinha, N. Engl. J. Med., 2012, 366, 1891–1904 CrossRef CAS PubMed.
- M. Ding, S. N. Bhupathiraju, A. Satija, R. M. van Dam and F. B. Hu, Circulation, 2014, 129, 643–659 CrossRef CAS PubMed.
- H. Uto-Kondo, M. Ayaori, M. Ogura, K. Nakaya, M. Ito, A. Suzuki, S. Takiguchi, E. Yakushiji, Y. Terao, H. Ozasa, T. Hisada, M. Sasaki, F. Ohsuzu and K. Ikewaki, Circ. Res., 2010, 106, 779–787 CrossRef CAS PubMed.
- N. Khan and H. Mukhtar, Life Sci., 2007, 81, 519–533 CrossRef CAS PubMed.
- X. X. Zheng, Y. L. Xu, S. H. Li, X. X. Liu, R. Hui and X. H. Huang, Am. J. Clin. Nutr., 2011, 94, 601–610 CrossRef CAS PubMed.
- D. J. Maron, G. P. Lu, N. S. Cai, Z. G. Wu, Y. H. Li, H. Chen, J. Q. Zhu, X. J. Jin, B. C. Wouters and J. Zhao, Arch. Intern. Med., 2003, 163, 1448–1453 CrossRef CAS PubMed.
- S. Sae-tan, K. A. Grove and J. D. Lambert, Pharmacol. Res., 2011, 64, 146–154 CrossRef CAS PubMed.
- M. Kobayashi, M. Nishizawa, N. Inoue, T. Hosoya, M. Yoshida, Y. Ukawa, Y. M. Sagesaka, T. Doi, T. Nakayama, S. Kumazawa and I. Ikeda, J. Agric. Food Chem., 2014, 62, 2881–2890 CrossRef CAS PubMed.
- P. Hirsova, G. Kolouchova, E. Dolezelova, J. Cermanova, R. Hyspler, Z. Kadova and S. Micuda, Eur. J. Pharmacol., 2012, 691, 38–45 CrossRef CAS PubMed.
- M. Cuccioloni, M. Mozzicafreddo, M. Spina, C. N. Tran, M. Falconi, A. M. Eleuteri and M. Angeletti, J. Lipid Res., 2011, 52, 897–907 CrossRef CAS PubMed.
- C. J. Chang, T. F. Tzeng, S. S. Liou, Y. S. Chang and I. M. Liu, Evidence-based Complementary Altern. Med., 2012, 2012, 787152 Search PubMed.
- C. L. Lin, H. C. Huang and J. K. Lin, J. Lipid Res., 2007, 48, 2334–2343 CrossRef CAS PubMed.
- I. Ikeda, T. Yamahira, M. Kato and A. Ishikawa, J. Agric. Food Chem., 2010, 58, 8591–8595 CrossRef CAS PubMed.
- L. Hooper, C. Kay, A. Abdelhamid, P. A. Kroon, J. S. Cohn, E. B. Rimm and A. Cassidy, Am. J. Clin. Nutr., 2012, 95, 740–751 CrossRef CAS PubMed.
- D. Grassi, S. Necozione, C. Lippi, G. Croce, L. Valeri, P. Pasqualetti, G. Desideri, J. B. Blumberg and C. Ferri, Hypertension, 2005, 46, 398–405 CrossRef CAS PubMed.
- J. Kim, J. Kim, J. Shim, C. Y. Lee, K. W. Lee and H. J. Lee, Crit. Rev. Food Sci. Nutr., 2014, 54, 1458–1472 CrossRef CAS PubMed.
- S. Baba, N. Osakabe, Y. Kato, M. Natsume, A. Yasuda, T. Kido, K. Fukuda, Y. Muto and K. Kondo, Am. J. Clin. Nutr., 2007, 85, 709–717 CAS.
- A. Yasuda, M. Natsume, N. Osakabe, K. Kawahata and J. Koga, J. Agric. Food Chem., 2011, 59, 1470–1476 CrossRef CAS PubMed.
- L. Guerrero, M. Margalef, Z. Pons, M. Quinones, L. Arola, A. Arola-Arnal and B. Muguerza, J. Nutr. Biochem., 2013, 24, 2092–2099 CrossRef CAS PubMed.
- M. M. Dohadwala and J. A. Vita, J. Nutr., 2009, 139, 1788S–1793S CrossRef CAS PubMed.
- P. Castilla, R. Echarri, A. Davalos, F. Cerrato, H. Ortega, J. L. Teruel, M. F. Lucas, D. Gomez-Coronado, J. Ortuno and M. A. Lasuncion, Am. J. Clin. Nutr., 2006, 84, 252–262 CAS.
- T. L. Zern, R. J. Wood, C. Greene, K. L. West, Y. Liu, D. Aggarwal, N. S. Shachter and M. L. Fernandez, J. Nutr., 2005, 135, 1911–1917 CAS.
- L. M. Vislocky and M. L. Fernandez, Nutr. Rev., 2010, 68, 656–670 CrossRef PubMed.
- K. Ohara, H. Wakabayashi, Y. Taniguchi, K. Shindo, H. Yajima and A. Yoshida, Biochem. Biophys. Res. Commun., 2013, 441, 929–934 CrossRef CAS PubMed.
- Y. Kawai, T. Nishikawa, Y. Shiba, S. Saito, K. Murota, N. Shibata, M. Kobayashi, M. Kanayama, K. Uchida and J. Terao, J. Biol. Chem., 2008, 283, 9424–9434 CrossRef CAS PubMed.
- S. Dash, C. Xiao, C. Morgantini, L. Szeto and G. F. Lewis, Arterioscler., Thromb., Vasc. Biol., 2013, 33, 2895–2901 CrossRef CAS PubMed.
- M. Azorin-Ortuno, M. J. Yanez-Gascon, A. Gonzalez-Sarrias, M. Larrosa, F. Vallejo, F. J. Pallares, R. Lucas, J. C. Morales, F. A. Tomas-Barberan, M. T. Garcia-Conesa and J. C. Espin, J. Nutr. Biochem., 2012, 23, 829–837 CrossRef CAS PubMed.
- U. J. Jung, M. K. Lee, Y. B. Park, M. A. Kang and M. S. Choi, Int. J. Biochem. Cell Biol., 2006, 38, 1134–1145 CrossRef CAS PubMed.
- J. Goldwasser, P. Y. Cohen, E. Yang, P. Balaguer, M. L. Yarmush and Y. Nahmias, PLoS One, 2010, 5, e12399 Search PubMed.
- S. M. Jeon, H. K. Kim, H. J. Kim, G. M. Do, T. S. Jeong, Y. B. Park and M. S. Choi, Transl. Res., 2007, 149, 15–21 CrossRef CAS PubMed.
- L. Baselga-Escudero, C. Blade, A. Ribas-Latre, E. Casanova, M. J. Salvado, L. Arola and A. Arola-Arnal, J. Nutr. Biochem., 2014, 25, 151–156 CrossRef CAS PubMed.
- A. Cassidy, K. J. Mukamal, L. Liu, M. Franz, A. H. Eliassen and E. B. Rimm, Circulation, 2013, 127, 188–196 CrossRef CAS PubMed.
- A. Jennings, A. A. Welch, S. J. Fairweather-Tait, C. Kay, A. M. Minihane, P. Chowienczyk, B. Jiang, M. Cecelja, T. Spector, A. Macgregor and A. Cassidy, Am. J. Clin. Nutr., 2012, 96, 781–788 CrossRef CAS PubMed.
- C. Xie, J. Kang, J. R. Chen, S. Nagarajan, T. M. Badger and X. Wu, J. Agric. Food Chem., 2011, 59, 10381–10387 CrossRef CAS PubMed.
- R. Poreba, A. Skoczynska, P. Gac, M. Poreba, I. Jedrychowska, A. Affelska-Jercha, B. Turczyn, A. Wojakowska, J. Oszmianski and R. Andrzejak, Ann. Agric. Environ. Med., 2009, 16, 305–308 CAS.
- B. Kim, Y. Park, C. J. Wegner, B. W. Bolling and J. Lee, J. Nutr. Biochem., 2013, 24, 1564–1570 CrossRef CAS PubMed.
- M. I. Covas, V. Konstantinidou and M. Fito, J. Cardiovasc. Pharmacol., 2009, 54, 477–482 CrossRef CAS PubMed.
- M. Farras, R. M. Valls, S. Fernandez-Castillejo, M. Giralt, R. Sola, I. Subirana, M. J. Motilva, V. Konstantinidou, M. I. Covas and M. Fito, J. Nutr. Biochem., 2013, 24, 1334–1339 CrossRef CAS PubMed.
- M. Aviram and M. Rosenblat, Evidence-based Complementary Altern. Med., 2012, 2012, 382763 Search PubMed.
- M. Aviram, N. Volkova, R. Coleman, M. Dreher, M. K. Reddy, D. Ferreira and M. Rosenblat, J. Agric. Food Chem., 2008, 56, 1148–1157 CrossRef CAS PubMed.
- M. Rosenblat, T. Hayek and M. Aviram, Atherosclerosis, 2006, 187, 363–371 CrossRef CAS PubMed.
- S. H. Park, J. L. Kim, E. S. Lee, S. Y. Han, J. H. Gong, M. K. Kang and Y. H. Kang, J. Nutr., 2011, 141, 1931–1937 CrossRef CAS PubMed.
Footnote |
| † c/o UK Medical Research Council (MRC) Human Nutrition Research Unit (HNR), Elsie Widdowson Laboratory, 120 Fulbourn Road, Peterhouse Technology Park, Cambridge CB1 9NL, UK. |
| This journal is © The Royal Society of Chemistry 2015 |