DOI:
10.1039/C4GC01169D
(Paper)
Green Chem., 2015,
17, 271-278
An atom transfer radical polymerization system: catalyzed by an iron catalyst in PEG-400†
Received
23rd June 2014
, Accepted 12th August 2014
First published on 13th August 2014
Abstract
A green and highly efficient AGET ATRP (activators generated by electron transfer for atom transfer radical polymerization) system was constructed in the absence of any additional ligands, using FeCl3·6H2O as a catalyst, and methyl methacrylate as a model monomer in polyethylene glycol 400 (PEG-400). The effects of various factors, such as the type of ATRP initiator, the molecular weight of PEG and the reducing agent type, polymerization temperature as well as solvent, on the polymerization were investigated. Polymerization kinetics demonstrated that the polymerization was a controlled/“living” process with molecular weight increasing linearly with conversion while maintaining a low molecular weight distribution. The living feature was further confirmed by chain extension experiments.
1. Introduction
Atom transfer radical polymerization (ATRP) is a robust and versatile reversible deactivation radical polymerization (RDRP)1 method, which has been used to obtain well-defined polymers and materials with predetermined molecular weight, narrow molecular weight distribution, a variety of architectures, and useful end-functionalities.2 ATRP3 is a catalytic process where an alkyl halide is activated by a transition metal catalyst in a lower oxidation state, to generate the corresponding alkyl radical and the transition metal complex in its higher oxidation state. The intermittent and repeated activation/deactivation cycles ensure that the majority of the polymer chains grow at the same rate, thus making it a controlled/“living” process. In ATRP, a lower oxidation state metal was utilized as a catalyst to mediate the propagation of the polymerization. However, the lower state transition metal is difficult to handle or preserve and is very sensitive to air or other oxidants. In order to overcome these drawbacks, several improved ATRP methods with higher state transition metals as the catalysts, such as AGET (Activators Generated by Electron Transfer) ATRP,4 ICAR (Initiators for Continuous Activators Regeneration) ATRP,5 photo-induced ATRP,6 electron chemically mediated ATRP (e-ATRP),7 as well as SARA (supplemental activators and reducing agents) ATRP,8 have been developed. It should be noted that AGET ATRP utilizes reducing agents such as ascorbic acid (AsAc), tinII 2-ethylhexanoate (Sn(EH)2) or glucose to react with the higher oxidation state catalyst and generate the lower oxidation state catalyst in situ. More importantly, with careful and delicate selection of polymerization conditions and components, the catalyst concentration can be reduced to the ppm level9 without sacrificing controllability. Thus, AGET ATRP has all the benefits of normal ATRP, but remains tolerant to air during operation.
Among various transition meal catalysts, iron catalysts have recently attracted extensive attention owing to their abundance, low toxicity, and biocompatibility,10 although iron complexes were generally considered to be inferior to copper complexes for the control of polymerization. A variety of ligands11 can be applied for iron catalysts, mainly phosphines and their derivates with a few amines. FeBr2-catalyzed ATRP in polar solvents (e.g., N,N-dimethylformamide, DMF)12 without any additional ligand or in the presence of a non-polar solvent and weakly coordinating anions13 was reported by Matyjaszewski et al.; thereafter, certain ionic liquids14 can also serve as ligands of iron catalysts. Unfortunately, these ligands are relatively either expensive or toxic, which is unbeneficial to the environment and biomedical applications. Solvents like DMF, however, are hazardous, toxic and difficult to remove; this partly counteracts the advantage of using iron as a “green” catalytic center. Recently, low molecular weight polyethylene glycols (PEGs) have attracted increasing interest as novel solvents due to their good biocompatibility, low cost, low toxicity to human beings and the environment, non-volatile, and easily degradable, and many have already been approved for internal consumption by the US FDA. PEGs as a new kind of environmentally friendly and less expensive solvent have been applied in organic synthesis,15 biphasic catalysis and phase-transfer catalysis.16
PEG has the ability to coordinate with many metal centers to form stable complexes and its coordination ability varies with its molecular weight and chain end groups. The use of low molecular weight PEGs as solvents for ATRP was first reported by Perrier and co-workers,17 using low oxidation state Cu(I) as the catalyst in PEG-400. The polymerization rate of methyl methacrylate (MMA) was found to be higher than that in toluene, but the rate was slower for styrene than that in xylene. They18 earlier used online 1H NMR spectroscopy to investigate whether the presence of oxyethylene groups within a solvent, monomer, or initiator led to an enhanced rate. Afterwards, PEGs as solvents in ATRP have been reported by several groups;19 the polymerization rate is significantly enhanced while maintaining control over the polymerization; furthermore, the transition metal residue is easily removable compared with other traditional volatile organic compounds (VOCs). PEG as both the solvent and the ligand in iron-catalyzed ATRP was reported only by Fabio di Lena et al.;20 ATRP of MMA using iron(II) chloride and various polyethers as catalytic complexes formed homogeneous mixtures in the absence of any additional ligands or solvents, but the controllability was moderate with Mw/Mn > 1.30 and the chain extension experiment was not so satisfactory. They also investigated the effects of polyether structure, molecular weight, and chain end functionality on the polymerization kinetics.
Inspired by the above mentioned results and the advantages of AGET ATRP, given the environmentally benign and biocompatibility feature of iron-PEG complexes, herein we report for the first time a facile and highly efficient AGET ATRP of methyl methacrylate in polyethylene glycol 400 using ethyl α-bromophenylacetate (EBrPA) as an initiator, FeCl3·6H2O as the catalyst without any additional ligand.
2. Experimental section
2.1. Materials
Methyl methacrylate (MMA) (>99%) was purchased from Shanghai Chemical Reagents Co. Ltd (Shanghai, China); the inhibitor was removed by passing through a short neutral alumina column. Ethyl α-bromophenylacetate (EBrPA) (>97%) was purchased from Alfa Aesar. Polyethylene glycol 400 (PEG-400) was purchased from TCI. Azobis(isobutyronitrile) (AIBN) and 1,1′-azobis(cyclohexanecarbonitrile) (ACHN) were obtained from Sigma-Aldrich and purified by recrystallizing twice from methanol. Iron(III) chloride hexahydrate (FeCl3·6H2O) (>99%) and sodium ascorbate (AsAc-Na) (≥99.0%) were purchased from Shanghai Chemical Reagents Co. Ltd (Shanghai, China) and used as-received. All the other chemicals were obtained from Shanghai Chemical Reagents Co. Ltd and were used as received unless mentioned.
2.2. Characterization
The number average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) values of the resulting polymers were determined using a TOSOH-HLC-8320 gel permeation chromatograph (GPC) equipped with a refractive index detector (Waters 2414), using TSK gel Super AWM-H columns (4.6 mm I.D. × 15 cm × 2) with measurable molecular weights ranging from 103 to 10 × 105 g mol−1. THF was used as the eluent at a flow rate of 0.35 mL min−1 and 40 °C. GPC samples were injected using a TOSOH plus autosampler and calibrated with poly(methyl methacrylate) or polystyrene standards purchased from TOSOH. The 1H NMR spectrum of the obtained polymer was recorded on a Bruker 300 MHz nuclear magnetic resonance instrument using CDCl3 or DMSO-d6 as the solvent and tetramethylsilane (TMS) as an internal standard.
2.3. General procedure for AGET ATRP of MMA
A typical polymerization procedure with the molar ratio of [MMA]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[EBrPA]0:[FeCl3·6H2O]0:[AsAC-Na]0 = 200:1:1:1 in the absence of air is as follows: a mixture was obtained by adding FeCl3·6H2O (12.7 mg, 0.047 mmol), AsAc-Na (9.4 mg, 0.047 mol), EBPA (8.25 μL, 0.047 mmol), MMA (1.0 mL, 9.44 mmol) and PEG-400 (desired volume) to a dried ampule. The reaction mixture was degassed by at least three freeze–pump–thaw cycles to eliminate the dissolved oxygen, and then the ampule was flame-sealed and transferred into an oil bath held at the desired temperature (90 °C) by a thermostat to polymerize under stirring. After the desired polymerization time, the ampule was cooled by immersing in iced water. Afterwards, it was opened and the contents were dissolved in THF (2 mL) and then precipitated into a large amount of methanol (250 mL). The polymer obtained by filtration was dried under vacuum at 35 °C until a constant weight. The monomer conversion was determined gravimetrically.
:[EBrPA]0:[FeCl3·6H2O]0:[AsAC-Na]0 = 200:1:1:1 in the absence of air is as follows: a mixture was obtained by adding FeCl3·6H2O (12.7 mg, 0.047 mmol), AsAc-Na (9.4 mg, 0.047 mol), EBPA (8.25 μL, 0.047 mmol), MMA (1.0 mL, 9.44 mmol) and PEG-400 (desired volume) to a dried ampule. The reaction mixture was degassed by at least three freeze–pump–thaw cycles to eliminate the dissolved oxygen, and then the ampule was flame-sealed and transferred into an oil bath held at the desired temperature (90 °C) by a thermostat to polymerize under stirring. After the desired polymerization time, the ampule was cooled by immersing in iced water. Afterwards, it was opened and the contents were dissolved in THF (2 mL) and then precipitated into a large amount of methanol (250 mL). The polymer obtained by filtration was dried under vacuum at 35 °C until a constant weight. The monomer conversion was determined gravimetrically.
2.4 Chain extension using PMMA as a macroinitiator
A predetermined quantity of PMMA (obtained by AGET ATRP of MMA) with a molar ratio of [MMA]0:[PMMA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 500:1:1:1 was added to a dried ampule, and then a predetermined quantity of MMA, FeCl3·6H2O, AsAc-Na, and PEG-400 was added. The rest of the procedure was the same as that described above. The chain-extension polymerization was carried out under stirring at 90 °C.
3. Results and discussion
3.1. Effect of reducing agents on the AGET ATRP of MMA in PEG-400 without any additional ligand
In an AGET ATRP process, reducing agents play a vital important role, since the reducing agents allow starting an ATRP with lower oxidation state Cu(I) or Fe(II) catalyst, which is in situ generated by reduction of oxidatively stable Cu(II) or Fe(III) species, reducing/reactivating cycles can also be employed to eliminate oxygen or other radical traps in the reaction system; and appropriate reducing agents can even reduce the transition metal catalyst amounts or allow a polymerization to be conducted at ambient temperature.11f,g,21 Therefore, we investigated the influence of various reducing agents on the polymerization. As shown in Table 1, when Sn(EH)2 or AsAc-Na was utilized, a controlled polymerization can be obtained. By the way, we also tried thermal initiators (AIBN or ACHN) as the reducing agent to conduct an ICAR ATRP, and only trace polymerization occurred. Taking the polymerization rate and controllability into consideration, AsAc-Na was selected as the reducing agent for our next polymerization. That is ascribed to the fact that base additives (e.g., AsAc-Na) can increase the basicity of polymerization system and lower the redox potential of the catalytic complexes, and therefore increase the polymerization rate as reported earlier by our group.22
Table 1 Effect of reducing agent (RA) types on the AGET ATRP of MMAa
| Entry |
Reducing agent |
Conv. (%) |
M
n,theo (g mol−1) |
M
n,GPC (g mol−1) |
M
w/Mn |
|
Polymerization conditions: [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[RA]0 = 200:1:1:1, VMMA = VPEG-400 = 1.0 mL, T = 90 °C; t = 12 h, except for entry 6, t = 3 h.
|
| 1 |
No |
NA |
NA |
NA |
NA |
| 2 |
AsAc |
NA |
NA |
NA |
NA |
| 3 |
Glucose |
NA |
NA |
NA |
NA |
| 4 |
Triamine |
NA |
NA |
NA |
NA |
| 5 |
Sn(EH)2 |
16.4 |
3300 |
12000 |
1.24 |
| 6 |
AsAc-Na |
82.0 |
16400 |
17800 |
1.18 |
The mechanism of AGET ATRP indicates that increasing the amount of AsAc-Na resulted in a higher concentration of FeII species produced by the in situ reduction of the FeIII complex, and then increased the concentration of the propagating radicals in the polymerization system, causing an increase in polymerization rate. As demonstrated in Table 2, when the molar ratio of [FeCl3·6H2O]0/[AsAc-Na]0 increased from 1/0.5 to 1/2, the polymerization rate increased significantly, which is demonstrated by an increasing conversion (0 to 97%) (entries 1–5) within a shorter polymerization time for a higher conversion (4.6 h for entry 5). This trend was also confirmed by reducing the amount of catalyst but maintaining a higher amount of the reducing agent AsAc-Na (entries 6–7).
Table 2 Effect of RA concentration on the AGET ATRP of MMAa
| Entry |
x:y |
Conv. (%) |
M
n,theo (g mol−1) |
M
n,GPC (g mol−1) |
M
w/Mn |
|
Polymerization conditions: [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 200:1:x : y, VMMA = VPEG-400 = 1.0 mL, T = 90 °C; for entries 1 and 2, t = 12 h, very trace polymer was obtained; for entry 3, t = 2.5 h; for entry 4, t = 5.2 h; for entries 5–7, t = 4.6 h.
|
| 1 |
1/0.5 |
NA |
NA |
NA |
NA |
| 2 |
1/0.6 |
5.3 |
1000 |
NA |
NA |
| 3 |
1/0.8 |
59.2 |
11800 |
16200 |
1.16 |
| 4 |
1/1 |
90.9 |
18200 |
21200 |
1.18 |
| 5 |
1/2 |
97.0 |
19400 |
15600 |
1.15 |
| 6 |
0.5/0.5 |
34.1 |
6800 |
11500 |
1.21 |
| 7 |
0.5/1 |
85.7 |
17100 |
16900 |
1.26 |
3.2. Effect of initiator type on the AGET ATRP of MMA in PEG-400 without any additional ligand
Different ATRP initiators were used to investigate the effect on polymerization of MMA, as presented in Table 3. It can be seen from Table 3 that all the listed initiators are suitable for the AGET ATRP of MMA. In order to facilitate the end chain characterization of PMMA, mono-functional EBrPA with a phenyl group was used as the initiator for further investigation. We also conducted AGET ATRP using BPN as the initiator as a comparison. The detailed information (polymerization kinetics and GPC traces) using BPN as the initiator for example is shown in Fig. S1 and S2 (ESI†).
Table 3 Effect of types of initiator on the AGET ATRP of MMAa
| Entry |
Initiator |
Conv. (%) |
M
n,theo (g mol−1) |
M
n,GPC (g mol−1) |
M
w/Mn |
|
Polymerization conditions: [MMA]0:[initiator]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 200:1:1:1, VMMA = VPEG-400 = 1.0 mL, T = 90 °C; t = 4.6 h, except for entry 1, t = 4 h; EBiB: ethyl α-bromoisobutyrate, BPN: 2-bromopropionitrile, BMPB-2: 1,4-(2-bromo-2-methylpropionato)benzene.
|
| 1 |
EBrPA |
85.6 |
17100 |
20000 |
1.18 |
| 2 |
EBiB |
95.2 |
19000 |
21000 |
1.14 |
| 3 |
BPN |
89.6 |
17900 |
19700 |
1.14 |
| 4 |
BMPB-2 |
75.7 |
15100 |
17100 |
1.13 |
3.3. Effect of PEG type on the AGET ATRP of MMA
It has been reported15,20,23 that PEGs with different molecular weights and chain end groups have different coordination ability, microviscosity, reactivity and solubility with metal catalyst centers. Generally, hydroxy-terminated PEGs coordinated with metal complexes are slightly more stable than those with ether end-groups. The stability constant of PEG/metal complexes increases, up to a plateau, with increasing molecular weight of PEG. More stable metal–ligand complexes means high KATRP (= kact/kdeact)24 and thus high catalytic activity. The effect of PEG type on the AGET ATRP of MMA is listed in Table 4. It can be seen that narrow molecular weight distributions of the resulting polymers could be obtained in all cases although Mn,GPC values deviated from the Mn,theo ones. The detailed information (polymerization kinetics and GPC traces) using MPEG-750 as the solvent for comparison was also investigated, which is shown in Fig. S3 and S4 (ESI†).
Table 4 Effect of PEG type and amounts on the AGET ATRP of MMAa
| Entry |
PEG type |
Conv. (%) |
M
n,theo (g mol−1) |
M
n,GPC (g mol−1) |
M
w/Mn |
|
Polymerization conditions: [MMA]0:[EBrPA]0:[FeCl3·6H2O]0: [AsAc-Na]0 = 200:1:1:1, t = 2 h, VMMA = VPEG = 1.0 mL, T = 90 °C.
|
| 1 |
PEG-200 |
51.8 |
10300 |
17800 |
1.20 |
| 2 |
PEG-600 |
84.0 |
16800 |
24000 |
1.21 |
| 3 |
MPEG-750 |
73.6 |
14700 |
18800 |
1.14 |
| 4 |
PEG-1000 |
75.8 |
15100 |
19500 |
1.16 |
We further investigated the effect of the amounts of PEG-400 on the polymerization. From Table 5, entries 1–8, it can be seen that with an increase in VPEG-400, the polymerization rate increases to a plateau and then goes down due to the dual function of PEG as both a ligand and a solvent. This trend was a comprehensive result of the stronger complexing ability between FeCl3·3H2O and PEG-400 as a ligand, better solubility and decreasing radical concentrations as a solvent. To our surprise, even catalytic amounts of PEG-400 could still maintain control over the polymerization, while when without PEG-400, almost no polymer was found (entry 1 in Table 5).
Table 5 Effect of the amounts of VPEG-400 on the AGET ATRP of MMAa
| Entry |
V
PEG-400 (mL) |
Conv. (%) |
M
n,theo (g mol−1) |
M
n,GPC (g mol−1) |
M
w/Mn |
|
Polymerization conditions: [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 200:1:1:1, T = 90 °C, t = 2 h, VMMA = 1.0 mL.
Entry 1 use toluene or 1,4-dioxane instead of PEG-400, t = 42 h, no polymer was detected. Except for entries 2, 3, 4 and 5, t = 18 h, 18 h, 5 h and 3 h, respectively.
|
| 1b |
0 |
NA |
NA |
NA |
NA |
| 2 |
0.02 |
NA |
NA |
NA |
NA |
| 3 |
0.03 |
18.5 |
3700 |
10000 |
1.17 |
| 4 |
0.05 |
49.9 |
10000 |
14500 |
1.15 |
| 5 |
0.1 |
40.8 |
8100 |
13900 |
1.14 |
| 6 |
0.15 |
41.5 |
8300 |
14400 |
1.14 |
| 7 |
0.2 |
51.3 |
10300 |
11000 |
1.12 |
| 8 |
0.4 |
75.4 |
15000 |
18400 |
1.14 |
| 9 |
0.8 |
61.3 |
12300 |
15700 |
1.16 |
3.4. Effect of temperature on the AGET ATRP of MMA in PEG-400 without any additional ligand
For an AGET ATRP process, catalyst regeneration depends on the redox reaction between higher oxidation state catalyst and reducing agent. Temperature can accelerate this process and shorten the induction period, so reaction temperature controls the concentration of propagating radicals. As presented in Table 6, it can be seen that as temperature decreased from 90 °C to 60 °C, the polymerization rate slowed down greatly, and the initiator efficiency (Ieff = Mn,theo/Mn,GPC × 100%) went from 76.3% to 49.3%.
Table 6 Effect of temperature on the AGET ATRP of MMAa
| Entry |
T (°C) |
Conv. (%) |
M
n,theo (g mol−1) |
M
n,GPC (g mol−1) |
M
w/Mn |
|
Polymerization conditions: [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 200:1:1:1, VMMA = VPEG-400 = 1.0 mL; for entries 1 and 2, t = 2 h; for entries 3 and 4, t = 4 h.
|
| 1 |
90 |
64.4 |
12900 |
16900 |
1.19 |
| 2 |
80 |
37.7 |
7500 |
13300 |
1.21 |
| 3 |
70 |
48.6 |
9700 |
16000 |
1.20 |
| 4 |
60 |
36.3 |
7500 |
16200 |
1.21 |
3.5. In contrast with ATRP in conventional polar solvents without any additional ligand
It has been reported by Matyjaszewski et al.12,25 that polar solvents or certain ionic liquids can be used as ligands for iron-catalyzed ATRP. In this work, some other solvents were used in the absence of any additional ligand. From Table 7, it is found that with polar solvents such as DMF, MeCN or DMSO as both the solvent and the ligand, Mn,theo deviated greatly with Mn,GPC, which means a low initiator efficiency, and the polymerization rate was rather slow. At the same time, there is a small ultra-high Mn,GPC shoulder in GPC curves, indicating poor controllability in the presence of these polar solvents, possibly caused by some side reactions.
Table 7 Conventional polar solvents as both ligand and solvent for AGET ATRP of MMAa
| Entry |
Solvent |
Conv. (%) |
M
n,theo (g mol−1) |
M
n,GPC (g mol−1) |
M
w/Mn |
|
Polymerization conditions: [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 200:1:1:1, VMMA = Vsolvent = 1.0 mL, T = 90 °C, t = 16 h.
|
| 1 |
DMF |
43.6 |
8700 |
15900 |
1.24 |
| 2 |
MeCN |
8.6 |
1700 |
8200 |
1.16 |
| 3 |
DMSO |
28.2 |
5600 |
15900 |
1.37 |
3.6. Polymerization kinetics of AGET ATRP of MMA in PEG-400 without any additional ligand
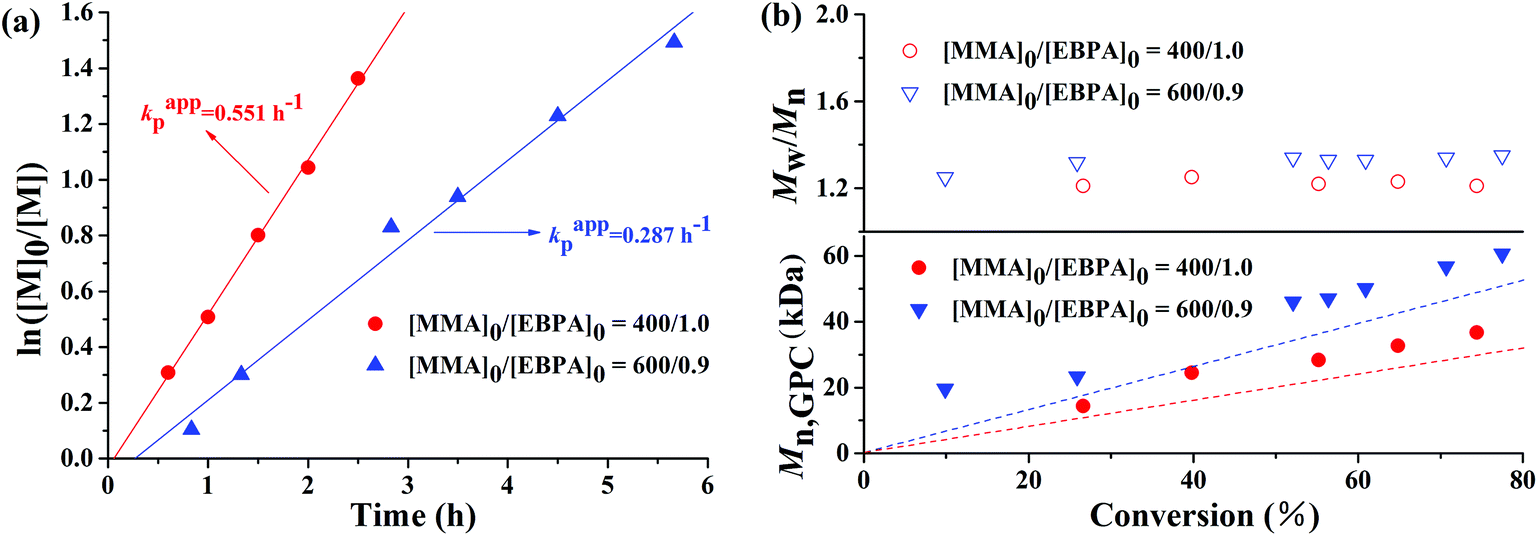
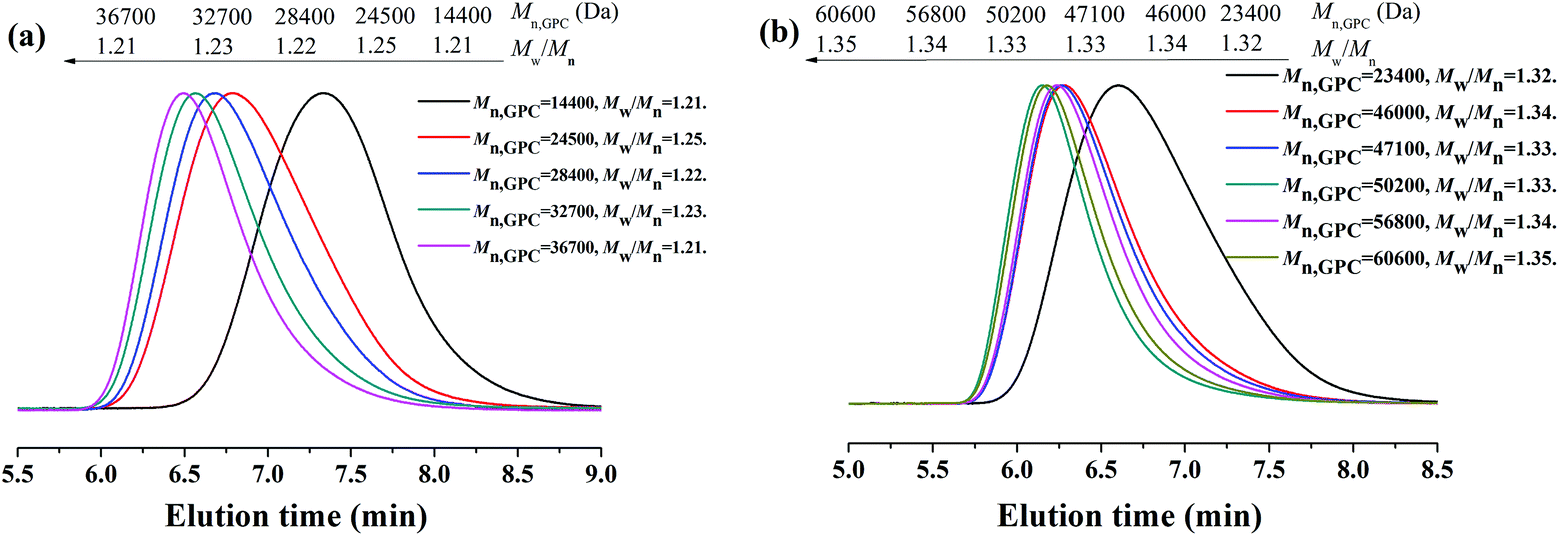
The polymerization kinetic plots for the AGET ATRP of MMA with a molar ratio of [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = x:y:1:1 (x:y = 400:1, or x:y = 600:0.9) at 90 °C are depicted in Fig. 1. It can be seen from Fig. 1(a) that polymerizations proceeded with approximately first-order kinetics in both cases, indicating a constant concentration of growing radicals during polymerizations. A short induction period was detected and the apparent rate constants were kappp = 0.551 h−1 in the case of 400/1 and 0.287 h−1 in the case of 600/0.9, respectively. Evolution of number average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) versus conversion (Fig. 1(b)) shows that during AGET ATRP of MMA with PEG-400 as both the solvent and the ligand, Mn,GPC values of the resulting polymers increased linearly with monomer conversion while keeping relatively low polydispersity (Mw/Mn < 1.35). It is noted that Mn,GPC values slightly deviated from the corresponding theoretical ones. The appearance of an apparent nonzero intercept of Mn,GPC values may be attributed to a low radical concentration at the beginning of polymerization and it needs time to establish a dynamic equilibrium between the Fe(II) and Fe(III) species, which results in a low initiator efficiency during low conversion. In addition, we further investigated AGET ATRP of MMA in the presence of limited amounts of air; however, within 24 hours, only trace PMMA (conversion = 9.2%) was obtained with Mn,GPC = 12800, Mw/Mn = 1.15. That was because oxygen may have a significant inhibition26or retardation effect on the polymerization. Fig. 2 shows GPC curves of the resulting PMMAs under the two different molar ratios mentioned above. The GPC peaks are both mono-modal, but not so symmetrical with a little tailing in the low molecular weight region.
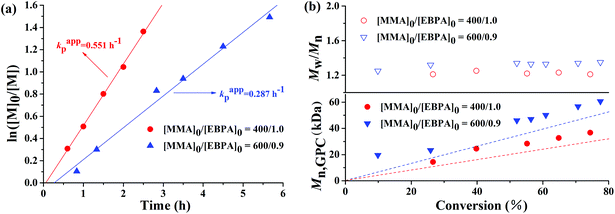 |
| | Fig. 1 ln([M]0/[M]) as a function of time (a) and evolution of number-average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) versus conversion (b) for AGET ATRP of MMA with various molar ratios of [MMA]0: [EBrPA]0 in PEG-400 without any additional ligand. Polymerization conditions: [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 400:1:1:1 and [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 600:0.9:1:1, VMMA = VPEG-400 = 1.0 mL, T = 90 °C. | |
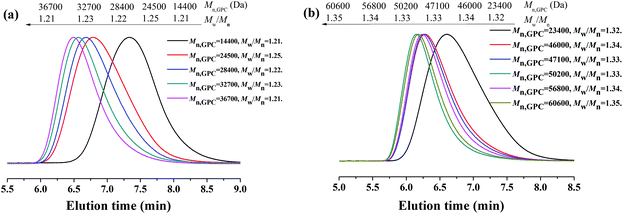 |
| | Fig. 2 GPC curves for AGET ATRP of MMA with various molar ratios of [MMA]0:[EBrPA]0, 400:1 (a) and 600:0.9 (b), in PEG-400 without any additional ligand. Polymerization conditions: [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 400:1:1:1 and [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 600:0.9:1:1, T = 90 °C, VMMA = VPEG-400 = 1.0 mL. | |
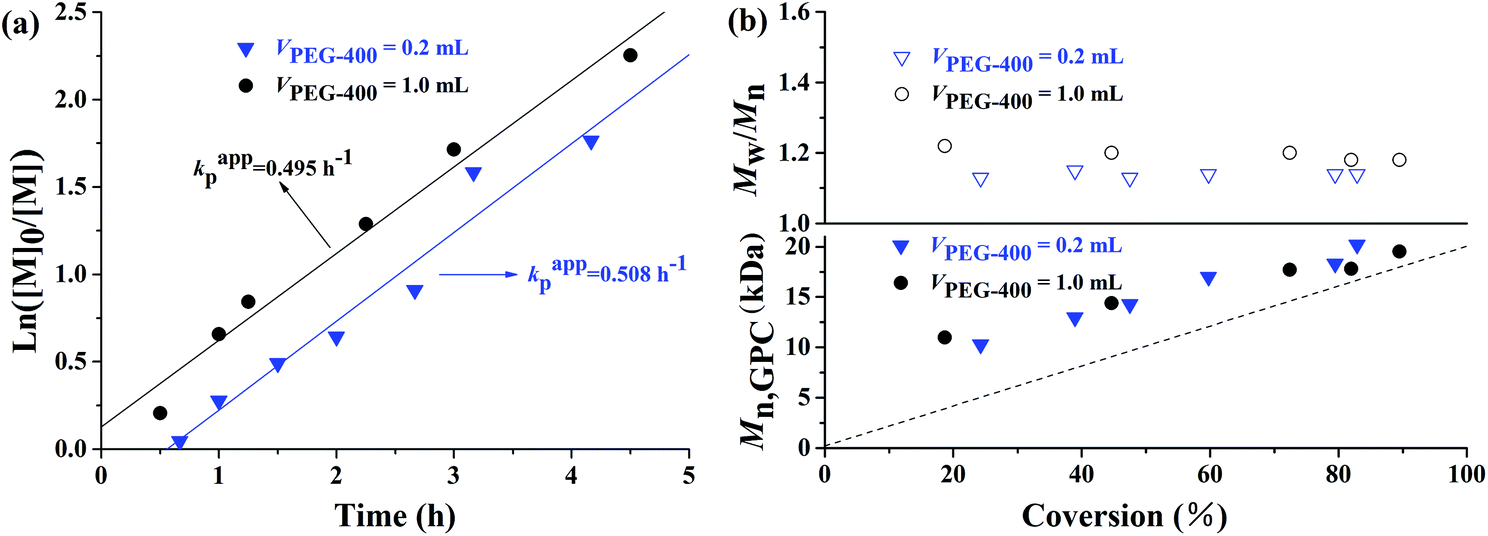
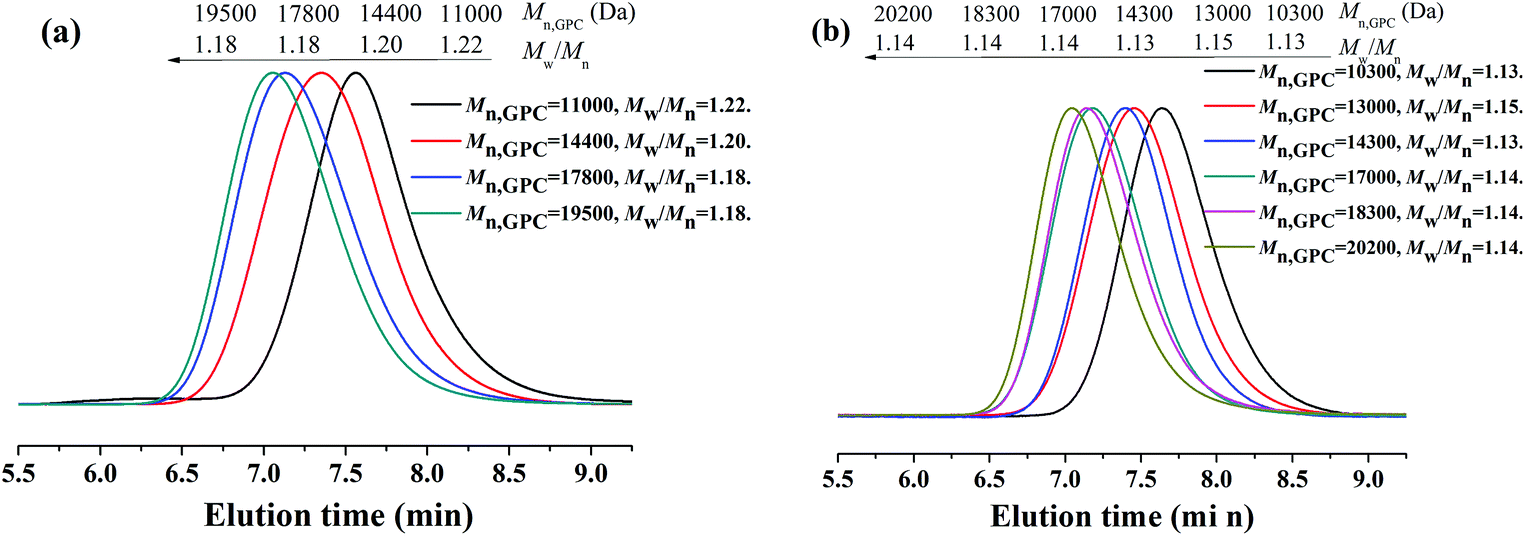
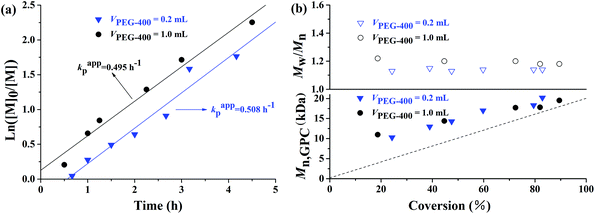
Because PEG-400 also serves as the ligand in the polymerization process, the effects of the amount of PEG-400 on the polymerization kinetics were investigated with the original molar ratio: [MMA]0:[EBPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 200:1:1:1 at 90 °C. It can be seen from Fig. 3 that both the polymerization kinetics showed linear plots, which indicates that the polymerizations were approximately first order with respect to the monomer concentration and the number of active species remained nearly constant during the polymerization process. It is also observed that the polymerization rate for the AGET ATRP using two different amounts of PEG-400 as the ligand was almost the same, while the induction period of the polymerization with VPEG-400 = 1.0 mL was only slightly shorter than that with VPEG-400 = 0.2 mL. Both Mn,GPC values of the resulting polymers with two different volumes of PEG-400 increased linearly with monomer conversion while keeping polymerization controllable (Mw/Mn < 1.25) and the Mn,GPC values slightly deviated from the corresponding theoretical ones. Furthermore, the GPC curves from Fig. 4 are obviously more symmetrical than those of Fig. 2.
 |
| | Fig. 3 ln([M]0/[M]) as a function of time (a) and evolution of number-average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) versus conversion (b) for AGET ATRP of MMA in the presence of various amounts of PEG-400 without any additional ligand. Polymerization conditions: [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 200:1:1:1, T = 90 °C. VMMA = 1.0 mL, VPEG-400 = 0.2 mL or VPEG-400 = 1.0 mL. | |
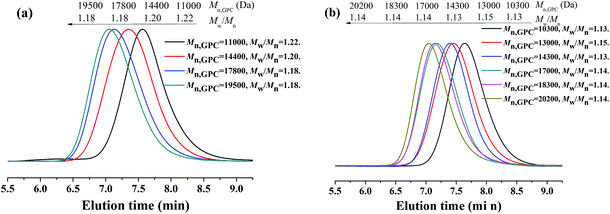 |
| | Fig. 4 GPC curves for AGET ATRP of MMA in the presence of various amounts of PEG-400, VPEG-400 = 1.0 mL (a) and VPEG-400 = 0.2 mL (b), without any additional ligand. Polymerization conditions: [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 200:1:1:1, T = 90 °C. VMMA = 1.0 mL. | |
All these results from the first order kinetics and the linear increase of Mn,GPC with conversion in Fig. 1 and 3 indicated the controlled/“living” features of the polymerizations under various molar ratios and amounts of PEG-400 discussed above.
3.7. Chain end analysis and chain extension:
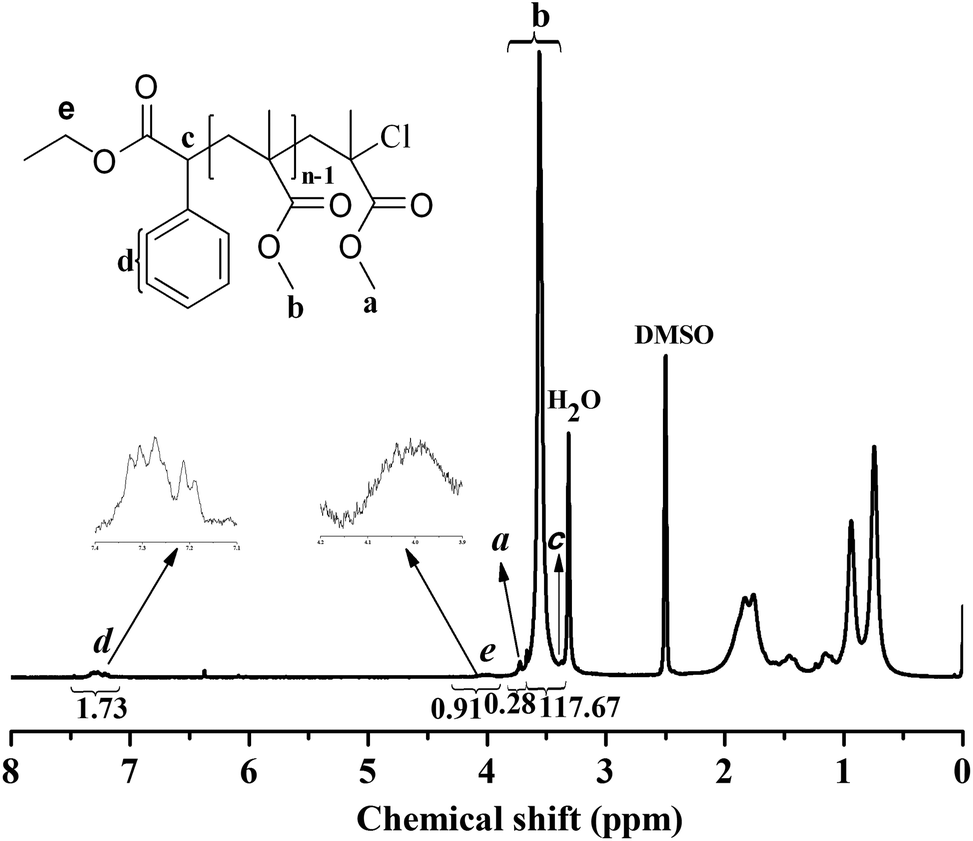
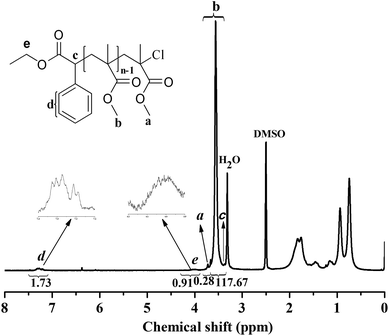
The chain end of the PMMA (Mn,GPC = 11000 g mol−1, Mw/Mn = 1.12) using EBrPA as the initiator and FeCl3·6H2O as the catalyst without any additional ligand was analyzed by 1H NMR spectroscopy, as shown in Fig. 5. The signal of a (3.8 ppm) was attributed to the methyl ester group at the chain end, as mentioned by Sawamoto et al.,11a which deviated from the chemical shift (3.60 ppm, b in Fig. 5) of other methyl ester groups in PMMA because of the electron-attracting function of the ω-Cl atom. The signals of c (3.4 ppm), d (7.1–7.4 ppm) and e (3.9–4.2 ppm) corresponded to the protons derived from EBrPA with methane (a little overlapped with H2O signal), phenyl, and methylene, respectively. The molecular weight (Mn,NMR) of the PMMA can be calculated from the integrals in the 1H NMR spectrum, according to eqn (1):| | | Mn,NMR (g mol−1) = (Ia,b/3) × 100.12/(Id/5) + 163.2 + 35.5 | (1) |
 |
| | Fig. 5
1H NMR spectrum of PMMA (Mn,GPC = 11000 g mol−1, Mw/Mn = 1.12) with DMSO-d6 as the solvent and tetramethylsilane (TMS) as the internal standard. Polymerization conditions: [MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 200:1:1:1, t = 2 h, T = 90 °C, conversion = 51.3%, VMMA = 1.0 mL, VPEG-400 = 0.2 mL. | |
The molecular weight calculated from the 1H NMR spectrum (Mn,NMR) was 11500 g mol−1, which is in good agreement with Mn,gpc, indicating that the PMMA obtained was end-capped by the EBrPA moieties with high fidelity. In addition, as calculated from the signal intensity ratio of the α-end methylene (e) to the ω-end methoxy (a) capped by chloride species, the percentage of chain-end functionality for the initiator fragment was 93.8%. These data revealed that although the initiator efficiency was less than 100%, most of the resulting polymers were active with the “living” polymer chain ends.
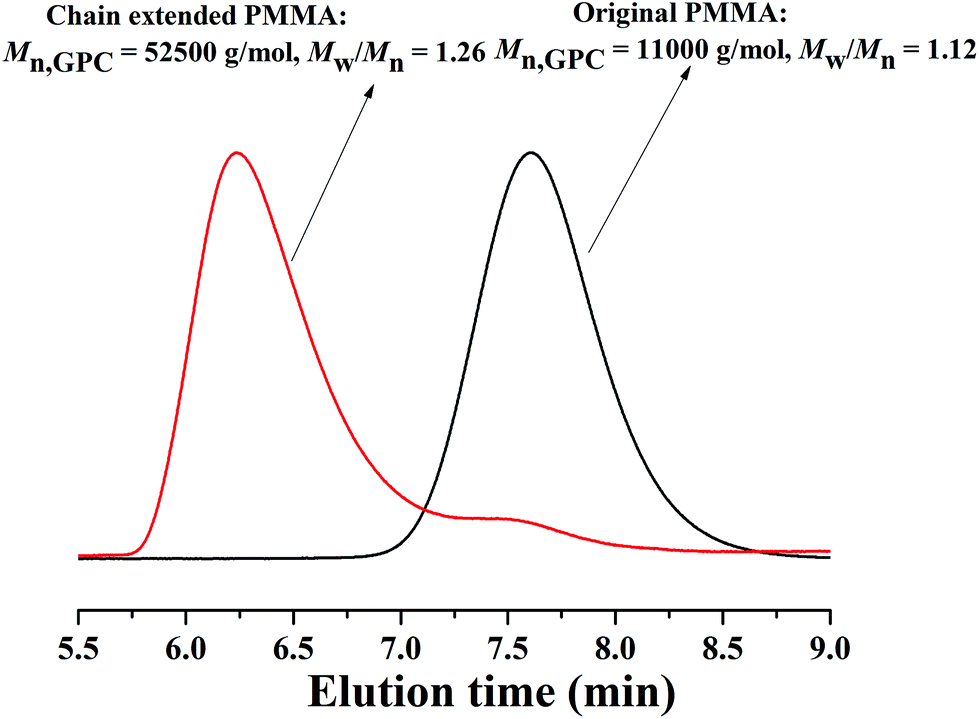
In addition, according to the mechanism of ATRP, the resulting polymers can be used as macro-initiators to conduct chain-extension reaction. Therefore, the obtained PMMA (Mn,GPC = 11000 g mol−1, Mw/Mn = 1.12) was used as a macro-initiator in the chain extension experiment. From Fig. 6, there is a peak shift from the macro-initiator to the chain extended PMMA with Mn,GPC = 52500 g mol−1 and Mw/Mn = 1.26. There is a small shoulder peak in the GPC curves of chain-extended PMMA, which resulted in an increase of Mw/Mn with respect to the macro-initiator. This may be due to a little dead polymer existing in the macro-initiator. The successful chain extension reaction further confirms the features of controlled/“living” radical polymerization of MMA in PEG-400 without any additional ligand.
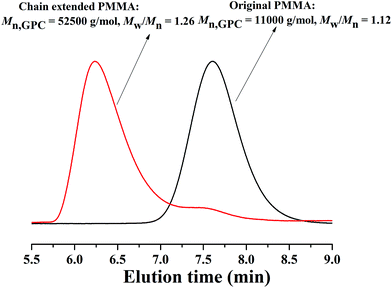 |
| | Fig. 6 GPC curves before and after chain extension using PMMA as the macro-initiator. Original PMMA:[MMA]0:[EBrPA]0:[FeCl3·6H2O]0:[AsAc-Na]0 = 200:1:1:1, T = 90 °C, VMMA = 1.0 mL, VPEG-400 = 0.2 mL, t = 2 h, conversion = 51.3%, Mn,theo = 10300 g mol−1. Chain extended PMMA:[MMA]0:[PMMA]0:[FeCl3·6H2O]0: [AsAc-Na]0 = 500:1:1:1, T = 90 °C, VMMA = 1.0 mL, VPEG-400 = 0.5 mL, t = 2.5 h, conversion = 59.1%, Mn,theo = 40500 g mol−1. | |
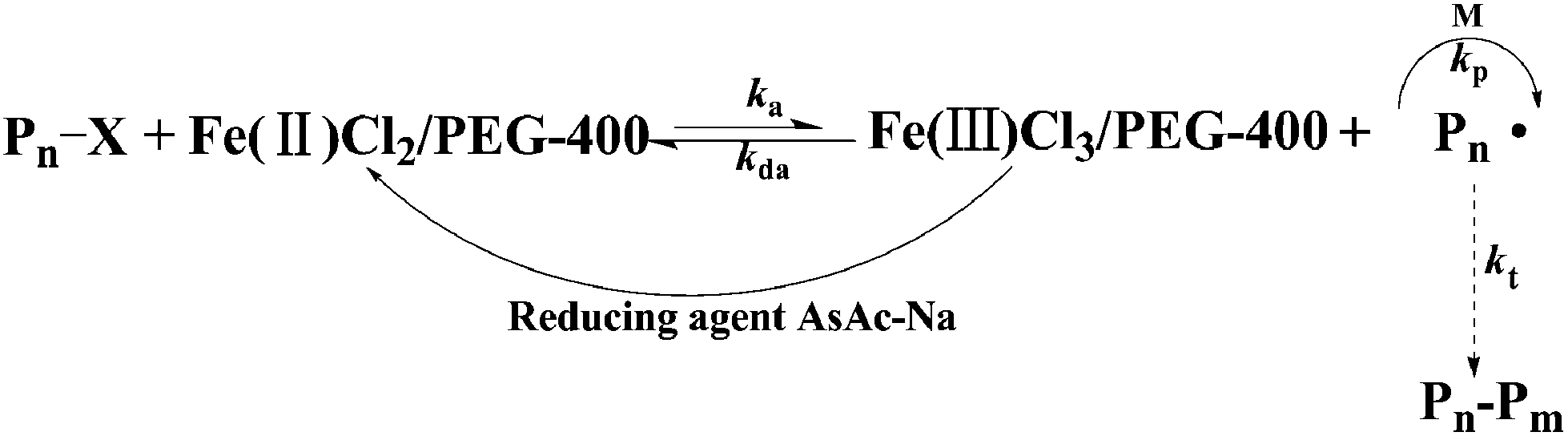
3.8. Mechanism of iron-catalyzed AGET ATRP in PEG-400 without any additional ligand
A possible mechanism for iron-catalyzed AGET ATRP using PEG-400 as both the solvent and the ligand is shown in Scheme 1. Lower oxidation state catalyst Fe(II) was generated by in situ reduction of higher oxidation state catalyst Fe(III) with the help of the reducing agent AsAc-Na, and then the process was proceeded as normal ATRP catalyzed by FeCl2. As a comparison, a direct Fe(II) catalyst (FeCl2) was used to conduct the polymerization of MMA successfully (Table S1, ESI†), which further confirmed the proposed mechanism as shown in Scheme 1.
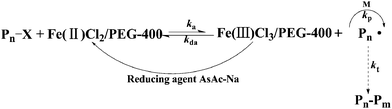 |
| | Scheme 1 A proposed mechanism for green AGET ATRP in PEG-400 without any additional ligand. | |
4. Conclusions
A facile and highly efficient iron-catalyzed AGET ATRP of MMA in the absence of any additional ligands was developed using FeCl3·6H2O as the catalyst and PEG-400 as both the solvent and the ligand. In comparison with most conventional iron-catalyzed ATRP systems reported, this system is much more environmentally benign and relatively cheap and the iron complex residue is easily removable. In addition, even when VPEG-400 is reduced to a catalytic amount, a controllable polymerization can also be established. Therefore, this novel iron-catalyzed AGET ATRP system may have potential applications in the synthesis of biomedical or functional polymers. More details such as extending the applicable scope of monomers or polymerization at ambient temperature with reduced iron catalyst are under way in our laboratory.
Acknowledgements
The financial support from the National Natural Science Foundation of China (no. 21174096, 21274100 and 21234005), the Project of Science and Technology Development Planning of Suzhou (no. ZXG201413 and SYG201430), and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) is gratefully acknowledged.
Notes and references
- A. D. Jenkins, G. J. Richard and G. Moad, Pure Appl. Chem., 2010, 82, 483 CAS.
-
(a) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed;
(b) M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS PubMed;
(c) Z. P. Cheng, X. L. Zhu, J. Y. Chen and J. M. Lu, J. Macromol. Sci., Part A: Pure Appl. Chem., 2003, 40, 1157 CrossRef PubMed;
(d) Z. P. Cheng, X. L. Zhu, L. F. Zhang, N. C. Zhou and X. R. Xue, Polym. Bull., 2003, 49, 363 CrossRef CAS PubMed;
(e) N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270 CrossRef CAS PubMed;
(f) M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS PubMed;
(g) F. J. Xu, K. G. Neoh and E. T. Kang, Prog. Polym. Sci., 2009, 34, 719 CrossRef CAS PubMed;
(h) T. Guo, L. F. Zhang, H. J. Jiang, Z. B. Zhang, J. Zhu, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2011, 2, 2385 RSC;
(i) X. Y. Du, J. L. Pan, M. T. Chen, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Chem. Commun., 2014, 50, 9266 RSC;
(j) J. Cao, L. F. Zhang, X. N. Zhao, Q. Ke, X. Q. Pan, Z. P. Cheng and X. L. Zhu, Macromol. Rapid Commun., 2013, 34, 1747 CrossRef CAS PubMed;
(k) F. J. Zhang and X. H. Liu, Chin. J. Polym. Sci., 2013, 31, 1613 CrossRef CAS;
(l) W. W. He, H. J. Jiang, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2013, 4, 2919 RSC;
(m) G. L. Lu, S. Zhang, Y. J. Li and X. Y. Huang, Chin. J. Polym. Sci., 2011, 29, 2791 CAS;
(n) X. Y. Jiang, Y. J. Li, G. L. Lu and X. Y. Huang, Polym. Chem., 2013, 4, 1402 RSC;
(o) Y. P. Liu, K. J. Yao, X. M. Chen, J. F. Wang, Z. K. Wang, H. J. Ploehn, C. P. Wang, F. X. Chu and C. B. Tang, Polym. Chem., 2014, 5, 3170 RSC;
(p) J. L. Pan, Z. Li, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Chin. J. Polym. Sci., 2014, 32, 1010 CrossRef CAS PubMed;
(q) Z. P. Cheng, X. L. Zhu, J. Zhu, J. M. Lu and J. H. Yu, J. Macromol. Sci., Part A: Pure Appl. Chem., 2003, 42, 341 CrossRef PubMed;
(r) G. H. Zhu, L. F. Zhang, X. Q. Pan, W. Zhang, Z. P. Cheng and X. L. Zhu, Macromol. Rapid Commun., 2012, 33, 2121 CrossRef CAS PubMed.
-
(a) M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS;
(b) J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS.
-
(a) W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482 CrossRef CAS PubMed;
(b) W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 39 CrossRef CAS;
(c) L. J. Bai, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2012, 3, 2685 RSC;
(d) W. W. He, L. F. Zhang, J. Miao, Z. P. Cheng and X. L. Zhu, Macromol. Rapid Commun., 2012, 33, 1067 CrossRef CAS PubMed;
(e) W. W. He, L. Cheng, L. F. Zhang, Z. Liu, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2014, 5, 638 RSC;
(f) J. L. Pan, L. F. Zhang, L. J. Bai, Z. B. Zhang, H. Chen, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2013, 4, 2876 RSC;
(g) L. J. Bai, L. F. Zhang, J. L. Pan, J. Zhu, Z. P. Cheng and X. L. Zhu, Macromolecules, 2013, 46, 2060 CrossRef CAS.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309 CrossRef CAS PubMed.
-
(a) M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58 CrossRef CAS PubMed;
(b) J. Mosnáček and M. Ilčíková, Macromolecules, 2012, 45, 5859 CrossRef.
-
(a) A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81 CrossRef CAS PubMed;
(b) N. Bortolamei, A. A. Isse, A. J. D. Magenau, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 11391 CrossRef CAS PubMed.
-
(a) C. M. R. Abreu, P. V. Mendonça, A. C. Serra, A. V. Popov, K. Matyjaszewski, T. Guliashvili and J. F. J. Coelho, ACS Macro Lett., 2012, 1, 1308 CrossRef CAS;
(b) V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156 CrossRef CAS PubMed.
-
(a) K. Yungwan, A. J. D. Magenau and K. Matyjaszewski, Macromolecules, 2011, 44, 811 CrossRef;
(b) Y. Wang, Y. Z. Zhang, B. Parker and K. Matyjaszewski, Macromolecules, 2011, 44, 4022 CrossRef CAS;
(c) X. X. Chen, M. Y. Khan and S. K. Noh, Polym. Chem., 2012, 3, 1971 RSC;
(d) T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087 RSC.
-
(a) C. Bolm, J. Legros, J. L. Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS PubMed;
(b) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317 CrossRef CAS PubMed.
-
(a) T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 1997, 30, 4507 CrossRef CAS;
(b) K. Matyjaszewski, M. Wei, J. Xia and N. E. McDermot, Macromolecules, 1997, 30, 8161 CrossRef CAS;
(c) G. H. Zhu, L. F. Zhang, Z. B. Zhang, J. Zhu, Y. F. Tu, Z. P. Cheng and X. L. Zhu, Macromolecules, 2011, 44, 3233 CrossRef CAS;
(d) Z. G. Xue, N. T. Ba Linh, S. K. Noh and W. S. Lyoo, Angew. Chem., Int. Ed., 2008, 47, 6426 CrossRef CAS PubMed;
(e) H. Aoshima, K. Satoh, T. Umemura and M. Kamigaito, Polym. Chem., 2013, 4, 3554 RSC;
(f) L. F. Zhang, Z. P. Cheng, S. P. Shi, Q. H. Li and X. L. Zhu, Polymer, 2008, 49, 3054 CrossRef CAS PubMed;
(g) L. F. Zhang, Z. P. Cheng, Y. T. Lu and X. L. Zhu, Macromol. Rapid Commun., 2009, 30, 543 CrossRef CAS PubMed;
(h) M. Teodorescu, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 2000, 33, 2335 CrossRef CAS;
(i) K. Mukumoto, Y. Wang and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 599 CrossRef CAS.
- Y. Wang and K. Matyjaszewski, Macromolecules, 2010, 43, 4003 CrossRef CAS.
- Y. Wang and K. Matyjaszewski, Macromolecules, 2011, 44, 1226 CrossRef CAS.
-
(a) T. Sarbu and K. Matyjaszewski, Macromol. Chem. Phys., 2001, 202, 3379 CrossRef CAS;
(b) Z. J. Deng, J. N. Guo, L. H. Qiu, Y. X. Zhou, L. Xia and F. Yan, Polym. Chem., 2012, 3, 2436 RSC;
(c) G. Q. Lai, F. M. Ma, Z. Q. Hu, H. Y. Qiu, J. X. Jiang, J. R. Wu, L. M. Chen and L. B. Wu, Chin. Chem. Lett., 2007, 18, 601 CrossRef CAS PubMed.
-
(a) J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64 RSC;
(b) S. Chandrasekhar, C. Narsihmulu, S. S. Sultana and N. R. Reddy, Org. Lett., 2002, 4, 4399 CrossRef CAS PubMed.
-
(a) J. H. David and G. J. Philip, J. Am. Chem. Soc., 2003, 125, 5600 CrossRef PubMed;
(b) V. J. Sangeeta and M. D. Raj, Catal. Today, 2008, 131, 353 CrossRef PubMed.
- S. Perrier, H. Gemici and S. Li, Chem. Commun., 2004, 604 RSC.
- D. M. Haddleton, S. Perrier and S. A. F. Bon, Macromolecules, 2000, 33, 8246 CrossRef CAS.
-
(a) Z. Q. Hu, X. R. Shen, H. Y. Qiu, G. Q. Lai, J. R. Wu and W. Q. Li, Eur. Polym. J., 2009, 45, 2313 CrossRef CAS PubMed;
(b) M. K. Solomon and C. M. Stephen, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1588 CrossRef PubMed.
- Y. H. Ng, F. D. Lena and C. L. L. Chai, Macromol. Res., 2012, 20, 552 CrossRef CAS PubMed.
-
(a) C. M. R. Abreu, A. C. Serra, A. V. Popov, K. Matyjaszewski, T. Guliashvili and J. F. J. Coelho, Polym. Chem., 2013, 4, 5629 RSC;
(b) R. A. Cordeiro, N. Rocha, J. P. Mendes, K. Matyjaszewski, T. Guliashvili, A. C. Serrad and J. F. J. Coelho, Polym. Chem., 2013, 4, 3088 RSC;
(c) J. R. Góis, N. Rocha, A. V. Popov, T. Guliashvili, K. Matyjaszewski, A. C. Serra and J. F. J. Coelho, Polym. Chem., 2014, 5, 3919 RSC;
(d) L. J. Bai, L. F. Zhang, J. Zhu, Z. P. Cheng and X. L. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2002 CrossRef CAS PubMed;
(e) L. J. Bai, L. F. Zhang, Z. B. Zhang, J. Zhu, N. C. Zhou, Z. P. Cheng and X. L. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3970 CrossRef CAS PubMed;
(f) L. J. Bai, L. F. Zhang, Z. B. Zhang, J. Zhu, N. C. Zhou, Z. P. Cheng and X. L. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3980 CrossRef CAS PubMed;
(g) H. J. Jiang, L. F. Zhang, J. L. Pan, X. W. Jiang, Z. P. Cheng and X. L. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2244 CrossRef CAS PubMed.
-
(a) M. X. Tao, L. F. Zhang, H. J. Jiang, Z. B. Zhang, J. Zhu, Z. P. Cheng and X. L. Zhu, Macromol. Chem. Phys., 2011, 212, 1481 CrossRef CAS PubMed;
(b) L. J. Bai, L. F. Zhang, Z. B. Zhang, Y. F. Tu, N. C. Zhou, Z. P. Cheng and X. L. Zhu, Macromolecules, 2010, 43, 9283 CrossRef CAS.
- A. G. West, C. B. Kowollik and S. Perrier, Polymer, 2010, 51, 3836 CrossRef CAS PubMed.
-
(a) W. Tang, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 1598 CrossRef CAS PubMed;
(b) T. Pintauer, P. Zhou and K. Matyjaszewski, J. Am. Chem. Soc., 2002, 124, 8196 CrossRef CAS PubMed;
(c) F. di Lena and C. L. L. Chai, Polym. Chem., 2010, 1, 922 RSC;
(d) W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702 CrossRef CAS PubMed.
- D. F. Yang, D. He, Y. G. Liao, Z. G. Xue, X. P. Zhou and X. L. Xie, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1020 CrossRef CAS PubMed.
- H. Zhang, D. M. Xu and K. D. Zhang, Chin. J. Chem., 2005, 23, 913 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01169d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.