
A novel D-glucosamine-derived pyridyl-triazole@palladium catalyst for solvent-free Mizoroki–Heck reactions and its application in the synthesis of Axitinib†
Chao
Shen
ab,
Hongyun
Shen
b,
Ming
Yang
b,
Chengcai
Xia
b and
Pengfei
Zhang
*b
aCollege of Biology and Environmental Engineering, Zhejiang Shuren University, Hangzhou 310015, China
bCollege of Material, Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou 310036, China. E-mail: zpf100@163.com; Fax: +86-571-28862867; Tel: +86-571-28862867
First published on 13th October 2014
Abstract
A green method for the synthesis of a D-glucosamine-derived triazole@palladium catalyst is described. The synthesized catalyst containing a 2-pyridyl-1,2,3-triazole ligand was prepared via a click route in high yields and was explored in Heck cross-coupling reactions between different aryl halides and olefins under solvent-free conditions. The catalyst can be separated from the reaction mixture and reused at least six times with superior activity. In addition, using this protocol, the marketed drug Axitinib (antitumor) could be synthesized easily.
Introduction
The palladium-catalyzed Mizoroki–Heck coupling reaction is one of the most powerful synthetic methods for the formation of C–C bonds between alkenes and aryl or alkyl halides.1 The Mizoroki–Heck coupling products find good applications as intermediates in the preparation of materials, natural products, and bioactive compounds.2 However, drawbacks like the high cost of palladium catalysts, harsh reaction conditions and low yields greatly hamper their large-scale practical application, especially in the pharmaceutical industry.3 Efforts to overcome these drawbacks include attempted syntheses of more efficient catalysts,4 performing coupling reactions in ionic liquids or water,5 utilization of solvent-free reaction conditions and the application of non-classical energy sources such as microwave irradiation, ultrasonication or high pressure and mechanochemical techniques.6 Recently, interest in the exploration of catalytic reactions under solvent-free conditions has been increasing dramatically as the solvent-free organic syntheses are generally faster, selective, higher yielding with cleaner products, environmentally benign and involve a simple operational procedure as compared to the classical reaction.7 Hence in the past few years, much effort has been made to design novel ligands/catalysts to promote transformations under solvent-free conditions,8 though how to retain the activity of a catalyst in solvent-free catalytic reactions is still a real challenge.To date, numerous attempts have been made to prepare new heterogeneous metal catalysts for the coupling reactions, because of their reusability compared to homogeneous catalysts.9 The advantages of heterogeneous processes, the ease of separation of the product, the reusability of the catalyst and better steric control of the reaction intermediate prompted researchers to immobilize a homogeneous catalytic site on various supports such as silica,10 zeolites,11 magnetic materials,12 and polymers,13 and from the published literature, we also know that most of these catalysts are of nano-size. Among these heterogeneous metal catalysts, biopolymers (such as starch, cellulose, chitosan or pectin) offer a unique set of environmentally benign properties such as biodegradability to harmless products, biocompatibility, stability in air and moisture, and cheapness.14 However, extensive progress in designing more sustainable chemical processes is achieved when biopolymers are used without any post-modification. On the other hand, one of the major reasons for poor recyclability is the aggregation and agglomeration of metal nanoparticles into less active large particles during the reaction due to the high surface energy of nanoparticles. Even for the systems using molecular catalysts, the aggregation and agglomeration still occurred because metal nanoclusters were formed as the reaction proceeded and all of these factors led to the drastic decrease in the activity of the reused catalysts. In this context, the development of highly active and easily recoverable catalysts which can be used not only in the academic field but also in industrial applications is full of significance.
Following our interest in carbohydrates as a source of highly efficient catalysts,15 we have recently reviewed D-glucosamine as a cheap and readily available chiral scaffold for the synthesis of a series of novel ligands and organocatalysts,16 and our previous investigation and other groups’ work revealed that the support structure and functional groups grafted on the support played an important role in preventing the aggregation of metal nanoparticles into the less active large particles.14a,17 The hydrogen bonding interaction and the special structure of the sugar ring may inhibit the motion of metal nanoparticles, thereby preventing the undesired aggregation and agglomeration.
Herein, we report an efficient method for the synthesis of a D-glucosamine-derived triazole@palladium catalyst. The synthesized catalyst containing a 2-pyridyl-1,2,3-triazole ligand was prepared via a click route and was explored in Heck cross-coupling reactions between different aryl halides and olefins under solvent-free conditions. The catalyst can be separated from the reaction mixture and reused at least six times with superior activity. In addition, using this protocol, the marketed drug Axitinib (antitumor) could be synthesized easily.
Results and discussion
Catalyst synthesis and characterization
The synthetic pathway for chelating palladium complexes is depicted in Scheme 1. Acetylation of the free D-glucosamine using AcONa/(AcO)2O afforded sugar substrate (2) and the azide group was introduced into 2 through a simple procedure and this was verified by the appearance of the N3 band at 2118 cm−1 in FT-IR spectra (Fig. S1, ESI†). Then azide (3) underwent a click process with commercially available 2-ethynylpyridine in the presence of copper sulfate and sodium ascorbate in tert-butanol. The resulting D-glucosamine-derived triazole (4) was further treated with Pd(OAc)2 in toluene to provide the desired solid catalyst (5). | ||
| Scheme 1 Synthesis of D-glucosamine-derived triazole@Pd catalyst 5. Reaction conditions: (a) AcONa/(AcO)2O, 12 h; (b) TMSN3, SnCl4, CH2Cl2, rt; (c) 2-ethynylpyridine, CuSO4/NaASc, tert-butanol, rt, 12 h; (d) Pd(OAc)2, toluene, rt, 6 h. | ||
Complex 5 is stable in air and moisture. It can be stored in air for a long period of time. It was characterized by 1H, 13C NMR, and 2D ROESY NMR spectroscopy, infra-red analyses (FT-IR), thermogravimetric analysis (TG) and inductively coupled plasma-atomic emission spectrometry (ICP-AES). It was clear that the proton signals on pyridine and triazole rings of Pd(OAc)2 in general were shifted downfield (Fig. 1), which indicated that the palladium ion was coordinated with both units of the ligand and the triazole unit worked as a part of the bidentate chelator. Meanwhile, because the triazole ligand could bind with the palladium metal through either the N2 or N3 atom, the spectra of PdL(OAc)2 showed two sets of proton signals, the ratio of which was around 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. Hence six-membered chelate rings formed by binding with the N3 atom and the nitrogen atom of pyridine are considered to be the predominant structures. Then a two-dimensional ROESY NMR experiment was conducted, with the aim of examining the potential spatial proximities among the different protons of catalyst 5 (Fig. S4, ESI†). From the spectrum, it turned out that no contacts were observed among the aromatic protons of triazole-pyridine and the sugar ring protons, implying that the functional group was not embraced in the hydrophobic cavity of the D-glucosamine.
:5. Hence six-membered chelate rings formed by binding with the N3 atom and the nitrogen atom of pyridine are considered to be the predominant structures. Then a two-dimensional ROESY NMR experiment was conducted, with the aim of examining the potential spatial proximities among the different protons of catalyst 5 (Fig. S4, ESI†). From the spectrum, it turned out that no contacts were observed among the aromatic protons of triazole-pyridine and the sugar ring protons, implying that the functional group was not embraced in the hydrophobic cavity of the D-glucosamine.
 | ||
| Fig. 1 Partial 1H NMR spectra (400 MHz, DMSO-d6, 298 K) of (a) the ligand D-glucosamine-derived triazoles 4, and (b) the palladium complex Pd@triazoles 5. | ||
The surface morphology of the synthesized catalyst 5 was also characterized by TEM. TEM analysis showed that the average diameter of the catalyst was about 3–7 nm and the dispersion of the Pd particles was very good (Fig. 2a). The TEM images also suggested that no palladium clusters were formed during the preparation.
 | ||
| Fig. 2 TEM images of (a) catalyst 5, and (b) recovered catalyst 5 after the sixth run. | ||
The catalyst was also characterized by TG to study its thermal behavior and stability at elevated temperatures and the TG shows that the catalyst 5 is stable up to 230.9 °C (Fig. 3), suggesting that its high thermal stability allows it to be compatible with most organic reactions. The palladium amount measured by inductively coupled plasma atomic emission spectrometry (ICP-MS) was 1.41 mmol g−1.
 | ||
| Fig. 3 Thermogravimetric plots of D-glucosamine-derived triazole@Pd catalyst 5. | ||
Catalytic studies
To evaluate the catalytic performance of the catalyst, the model coupling reaction between iodobenzene 6a and styrene 7a in the presence of catalyst 5 was conducted to screen the optimal reaction conditions, including solvents, bases, temperature and catalyst loadings. Initially, considering that the solvent always plays important roles in the Heck coupling reaction, we studied the solvent effect, and the results are shown in Table 1. After much experimentation on optimizing the solvent, it was found that the use of highly polar solvents like DMF and DMSO afforded product 3a in high yields (Table 1, entries 1 and 2). In stark contrast, the coupling reaction proceeded less efficiently in nonpolar solvents such as dioxane and toluene (Table 1, entries 3 and 4). With H2O as the solvent, no target product was obtained (Table 1, entry 5). Excellent yield was achieved under solvent-free conditions (Table 1, entry 6). In order to find a suitable base that would effect the desired reaction, we also screened several bases such as NaOH, KOAc, K2CO3, and K3PO4. All the reactions examined led to some conversion, albeit with quite different efficiencies (Table 1, entries 7–11), with Et3N being the most reactive, allowing the reaction to be complete in 6 h. However the coupling reactions did not proceed in the absence of a base (Table 1, entry 12). Lowering the reaction temperature to 60 °C decreased the yield to 77% even with a prolonged reaction time (Table 1, entry 13). When the amount of the catalyst was reduced to 0.05 mol%, the yields decreased obviously (Table 1, entry 14). Thus, we obtained the optimal conditions: 1.0 mmol iodobenzene, 2.0 mmol styrene, 0.1 mol% catalyst 5, 3.0 mmol Et3N, at 80 °C under solvent-free conditions.|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Catalyst (mol %) | Time (h) | Yieldb (%) |
| a Reaction conditions: iodobenzene (1 mmol), styrene (2 mmol), 0.1 mol% of 5, base (3 mmol), solvent (3 mL) at 80 °C under air. b Determined by GC-MS. c Isolated yield in parentheses. d At 60 °C. | |||||
| 1 | DMSO | Et3N | 0.1 | 6 | 97 |
| 2 | DMF | Et3N | 0.1 | 6 | 80 |
| 3 | Toluene | Et3N | 0.1 | 6 | 55 |
| 4 | Dioxane | Et3N | 0.1 | 6 | 61 |
| 5 | H2O | Et3N | 0.1 | 6 | Trace |
| 6 | Neat | Et3N | 0.1 | 6 | 99(95) |
| 7 | Neat | NaOH | 0.1 | 12 | 35 |
| 8 | Neat | KOAc | 0.1 | 12 | 35 |
| 9 | Neat | K2CO3 | 0.1 | 12 | 45 |
| 10 | Neat | Cs2CO3 | 0.1 | 12 | 61 |
| 11 | Neat | K3PO4 | 0.1 | 12 | 23 |
| 12 | Neat | — | 0.1 | 6 | 0 |
| 13 | Neat | Et3N | 0.1 | 12 | 77d |
| 14 | Neat | Et3N | 0.05 | 6 | 81 |
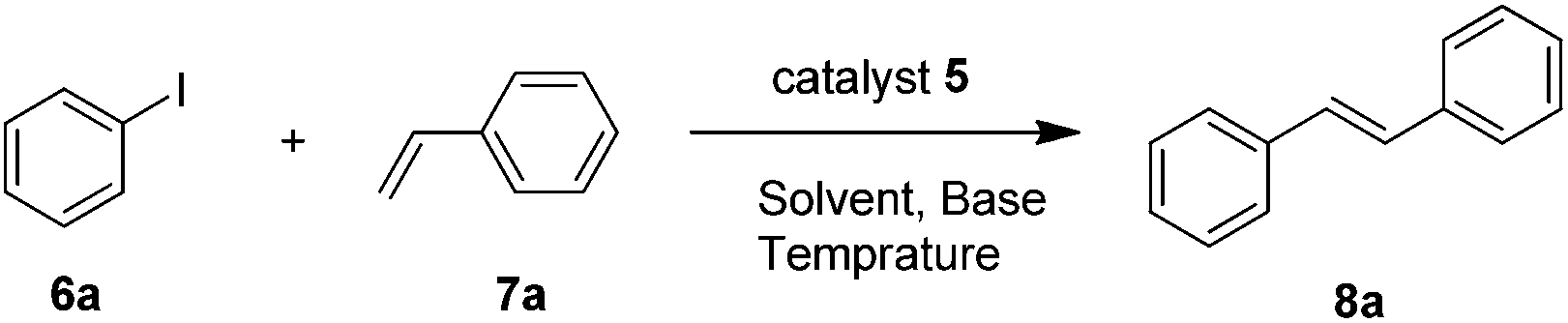
With optimal conditions determined, the scope of Heck coupling reactions of iodobenzene, bromobenzene and chlorobenzene with various olefins was investigated and the results are summarized in Table 2. Many valuable functional groups such as hydroxyl, chloro, acetyl and trifluoromethyl groups were well tolerated. We found that substrates with an electron-withdrawing group on the benzene ring performed better than those with an electron-donating group. Furthermore, substituents at meta-, or ortho-positions of the benzene ring do not affect the efficiency of this transformation (Table 2, entries 10–12). This catalytic system was also applied to bromobenzene and chlorobenzene. It is clear from Table 2 that aryl bromides containing electron donating, electron withdrawing, or electron neutral substituents are coupled in good yields (Table 2, entries 13–18). However, aryl chloride coupled with olefins in moderate yields within 24 h reaction time and also a much higher temperature was needed (Table 2, entries 19–21).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | X | R2 | Time (h) | Yieldb (%) |
| a Unless otherwise stated, the reactions were carried out using 1.0 mmol aryl halide, 2 mmol olefin, 3.0 mmol Et3N, 0.1 mol% of 5, at 80 °C under air. b Isolated yield. c At 120 °C. | |||||
| 1 | H | I | Ph | 6 | 95 (8a) |
| 2 | 4-CH3O | I | Ph | 6 | 83 (8b) |
| 3 | 4-CH3 | I | Ph | 6 | 86 (8c) |
| 4 | 4-NO2 | I | Ph | 6 | 95 (8d) |
| 5 | 4-CF3 | I | Ph | 6 | 94 (8e) |
| 6 | 4-Cl | I | Ph | 6 | 82 (8f) |
| 7 | 4-OH | I | Ph | 12 | 71c (8g) |
| 8 | 4-CH3CO | I | Ph | 6 | 84 (8h) |
| 9 | 4-Ph | I | Ph | 6 | 85 (8i) |
| 10 | 3-NO2 | I | Ph | 6 | 96 (8j) |
| 11 | 3-NO2 | I | Pyridine | 6 | 92 (8k) |
| 12 | 2-CH3 | I | Ph | 12 | 81 (8l) |
| 13 | H | Br | Ph | 12 | 82 (8a) |
| 14 | 4-CH3O | Br | Ph | 12 | 80 (8b) |
| 15 | 4-CH3 | Br | Ph | 12 | 83 (8c) |
| 16 | 4-NO2 | Br | Ph | 12 | 92 (8d) |
| 17 | 4-CF3 | Br | Ph | 12 | 90 (8e) |
| 18 | 4-Ph | Br | Ph | 12 | 80 (8i) |
| 19 | H | Cl | Ph | 24 | 70c (8a) |
| 20 | 4-CH3O | Cl | Ph | 24 | 62c (8b) |
| 21 | 4-NO2 | Cl | Ph | 24 | 73c (8k) |
| 22 | H | I | CO2Me | 6 | 93 (8m) |
| 23 | H | Br | CO2Me | 6 | 85 (8n) |
| 24 | H | I | CO2t-Bu | 6 | 95 (8m) |
| 25 | H | Br | CO2t-Bu | 6 | 83 (8n) |

The optimized conditions were also applied to Heck reactions of methyl acrylate and t-butyl acrylate with iodobenzene and bromobenzene; the two olefins gave the products in excellent yield and the t-butyl acrylate substrate was the best (Table 2, entries 22–25).
Further experiments were performed to verify the catalyst recyclability using the reaction of iodobenzene with styrene as a model system. After the first use, the catalyst was recovered by simple filtration and reused in the next run after a simple workup. We were pleased to find that the recovered catalyst was successfully reused in the subsequent six cycles with a consistent catalytic activity, giving the products in excellent yields (84–94%). No Pd metal was detected in the solution by ICP analysis. Furthermore, the TEM image of the catalyst obtained after the sixth cycle of the reaction does not show any significant change in the morphology and the size of the catalyst (5–9 nm) (Fig. 2b), which indicates the retention of the catalytic activity after recycling (Table 3).
With this methodology in hand, we turned our attention to the synthesis of Axitinib, an important anticancer agent targeting the vascular endothelial growth factor (VEGF) (Scheme 2).18 Axitinib exhibits a tendency to retain palladium, a fact that likely is due to the indazole ring that may enable formation of a relatively stable complex with the metal. As a result, the separation of the Axitinib API from the residual palladium has been a challenging task.19,20 So we planned to enable the palladium removal process using our system to reduce the residual Pd content. Firstly, a Pd2(dba)3-catalyzed Migita coupling reaction between commercially available 6-iodoindazole 9 and 2-mercapto-N-methylbenzamide 10 and iodination reactions afforded the iodoindazole intermediate 11 in 90% yield. Then the intermediate 11 was treated with 2-vinylpyridine 7e (2 mmol) in the presence of 0.1 mol% of catalyst 5 and Et3N (3.0 mmol) at 100 °C for 24 h, and Axitinib was isolated in 70% yield.
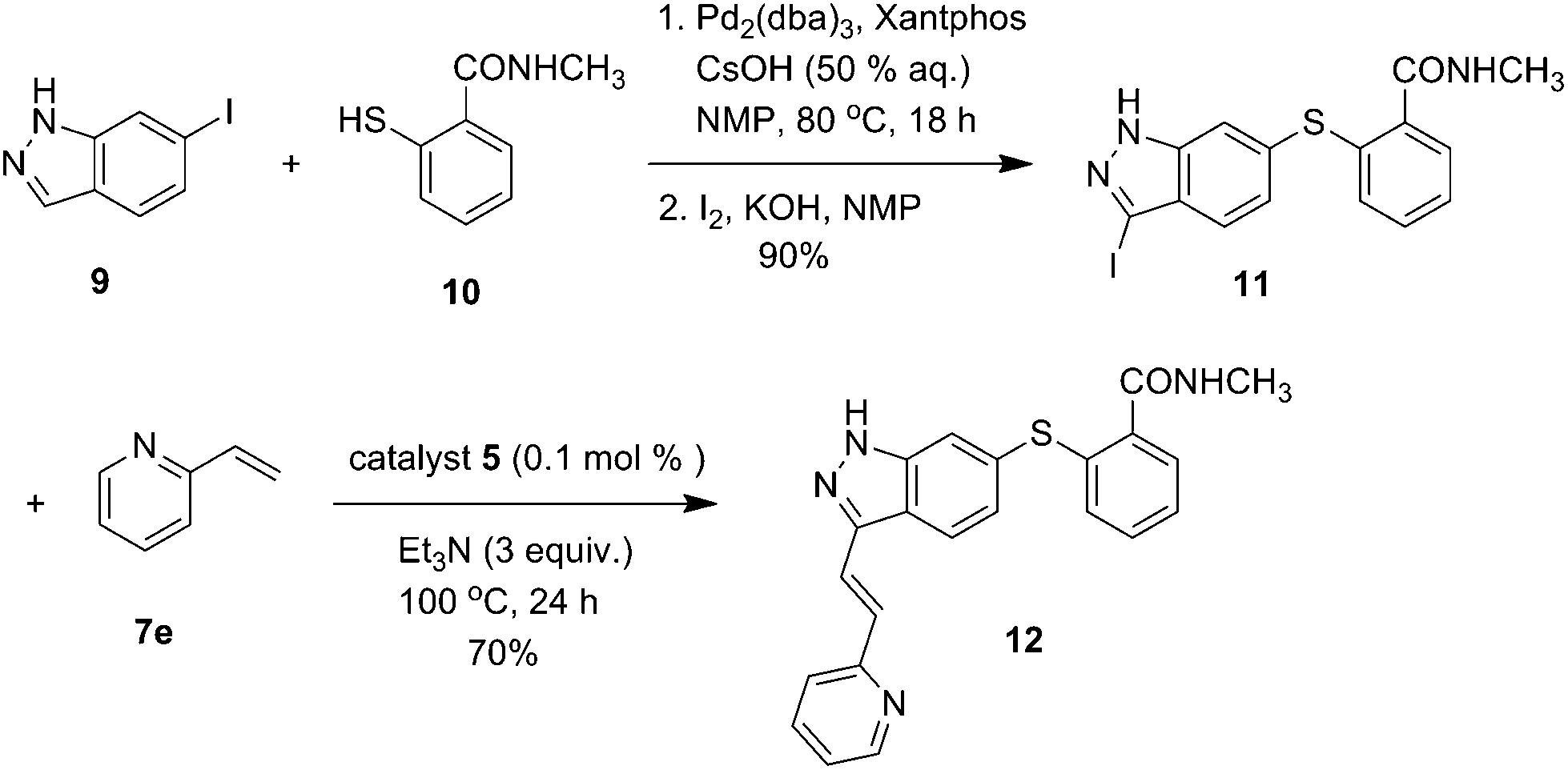 | ||
| Scheme 2 Application of the catalyst 5 in the synthesis of Axitinib. | ||
Such a heterogeneous catalyst not only gave the product in satisfying yield but also provided an efficient method for removal of the residual palladium from the Axitinib drug substance material. The residual Pd content in the system was determined to be not more than 20 ppm by atomic absorption spectroscopy. Next the method was also successfully applied to the synthesis of novel fluoroquinolone derivatives (Scheme S1, ESI†).
Conclusions
In conclusion, we prepared a new D-glucosamine-derived triazole@palladium catalyst via a “click” route and this was explored in Heck cross-coupling reactions between different aryl halides and olefins under solvent-free conditions. The catalyst was very stable and could be easily separated from the products and reused at least six times with superior activity. The significantly enhanced recyclability may be attributed to the catalyst which could efficiently prevent the aggregation and agglomeration of Pd particles formed during the catalytic reaction into the less active large particles, as evidenced by TEM. In addition, using this protocol, the marketed drug Axitinib (antitumor) could be synthesized easily and such an efficient process for removal of the residual palladium from the Axitinib drug is demonstrated. The applicability of the D-glucosamine-derived triazole@palladium catalyst in other fields of organic transformation is underway in our laboratory.Experimental section
The starting materials were commercially available and were used without further purification except for solvents. The products were isolated by column chromatography on silica gel (200–300 mesh) using petroleum ether (60–90 °C) and ethyl acetate. Melting points were determined on X-5 Data microscopic melting point apparatus. 1H NMR and 13C NMR spectra were recorded on a Bruker Advance 400 spectrometer at ambient temperature with CDCl3 or DMSO-d6 as a solvent unless otherwise noted and tetramethylsilane (TMS) as the internal standard. 1H NMR data are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = double-doublet, m = multiplet and br = broad), coupling constant (J values, Hz). Mass spectra (EI-MS) were acquired on an Agilent 5975 spectrometer. Analytical thin layer chromatography (TLC) was performed on Merk precoated TLC (silica gel 60 F254) plates. The sugar substrates were prepared according to our previous reports.15 All compounds were characterized by 1H NMR and mass spectroscopy, and the results are consistent with those reported in the literature.1–3General procedure for solvent-free Mizoroki–Heck reactions
To a flask, a mixture of D-glucosamine-derived triazole@palladium catalyst 5 (0.1 mol%), aryl halide (1 mmol), olefins (2 mmol) and Et3N (3 mmol) was added and heated at 80 °C under solvent-free conditions. After completion of the reaction, ethylacetate (10 mL) was added to the flask. The catalyst was separated by simple filtration. Water (3 × 15 mL) was added to the ethylacetate phase and decanted. The organic layer was dried over anhydrous Na2SO4. After evaporation of the solvent, the resulting crude products were purified by column chromatography (hexane–ethylacetate) giving the pure products in excellent yields.General procedure for recycling of the catalyst used in solvent-free Mizoroki–Heck reactions
After completion of the reaction at the first run, the reaction mixture was cooled down to room temperature and ethylacetate (5 mL) was added to the reaction mixture to extract organics. The ethylacetate phase was sucked from the vial using a syringe and the catalyst was dried under vacuum. After complete drying, the catalyst was reused for a similar reaction. This process was repeated for six runs.General procedure for synthesis of Axitinib under solvent-free conditions
To a flask, a mixture of intermediate 11 (1 mmol), 2-vinylpyridine 7e (2 mmol), Et3N (3 mmol) and D-glucosamine-derived triazole@palladium catalyst 5 (0.1 mol%) was added and heated at 100 °C under solvent-free conditions. The reaction progress was monitored by TLC. After completion of the reaction, the mixture was allowed to cool to room temperature, and ethylacetate (10 mL) was added to the flask. The catalyst was separated by simple filtration and the aqueous phase was extracted with CH2Cl2 3 times (3 × 2 mL). Then the combined organic layers were dried over anhydrous Na2SO4, concentrated under vacuum and purified by column chromatography (hexane–ethyl acetate 10:1) to afford the desired product. The residual Pd content in the solvent was determined to be not more than 20 ppm by atomic absorption spectroscopy.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21376058, 21302171), the Zhejiang Provincial Natural Science Foundation of China (no. LZ13B020001) and the Science and Technology Plan of Zhejiang Province (no. 2014C31153).Notes and references
-
(a) J. Ruan and J. Xiao, Acc. Chem. Res., 2011, 44, 614 CrossRef CAS PubMed
; (b) A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973 RSC
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440 CrossRef CAS PubMed
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177 CrossRef CAS PubMed
- T. Nishikata, Y. Noda, R. Fujimoto and T. Sakashita, J. Am. Chem. Soc., 2013, 135, 16372 CrossRef CAS PubMed
-
(a) H. Yang, X. Han, G. Li and Y. Wang, Green Chem., 2009, 11, 1184 RSC
- W. Susanto, C. Y. Chu, W. J. Ang, T. C. Chou, L. C. Lo and Y. Lam, J. Org. Chem., 2012, 77, 2729 CrossRef CAS PubMed
-
(a) Y. N. Zhao, J. Li, C. J. Li, K. Yin, D. Y. Ye and X. S. Jia, Green Chem., 2010, 12, 1370 RSC
-
(a) M. Tamura, S. M. A. H. Siddiki and K. Shimizu, Green Chem., 2013, 15, 1641 RSC
- L. Yin and J. Leibescher, Chem. Rev., 2007, 107, 133 CrossRef CAS PubMed
- B. Li, M. Li, C. H. Yao, Y. F. Shi, D. R. Ye, J. Wu and D. Y. Zhao, J. Mater. Chem., 2013, 1, 6742 RSC
-
(a) L. Gu, D. Ma, S. Yao, C. Wang, W. Shen and X. Bao, Chem. Commun., 2010, 46, 1733 RSC
-
(a) V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J. M. Basset, Chem. Rev., 2011, 111, 3036 CrossRef CAS PubMed
- J. Zhang, D. Han, H. Zhang, M. Chaker, Y. Zhao and D. Ma, Chem. Commun., 2012, 48, 11510 RSC
-
(a) C. Shen, J. Xu, W. B. Yu and P. F. Zhang, Green Chem., 2014, 16, 3007 RSC
-
(a) C. Shen, P. F. Zhang and X. Z. Chen, Helv. Chim. Acta, 2010, 93, 2433 CrossRef CAS
- C. Shen and P. F. Zhang, Curr. Org. Chem., 2013, 17, 1507 CrossRef CAS
- J. E. Camp, J. J. Dunsford, E. P. Cannons, W. J. Restorick, A. Gadzhieva, M. W. Fay and R. J. Smith, ACS Sustainable Chem. Eng., 2014, 2, 500 CrossRef CAS
- E. J. Flahive, B. L. Ewanicki, N. W. Sach, S. A. O'Neill-Slawecki, N. S. Stankovic, S. Yu, S. M. Guinness and J. Dunn, Org. Process Res. Dev., 2008, 12, 637 CrossRef CAS
- Y. Xiang, P. Y. Caron, P. Y. Lillie and R. Vaidyanathan, Org. Process Res. Dev., 2008, 12, 116 CrossRef CAS
- B. P. Chekal, S. M. Guinness, B. M. Lillie, R. W. McLaughlin, C. W. Palmer, R. J. Post, J. E. Sieser, R. A. Singer, G. W. Sluggett, R. Vaidyanathan and G. J. Withbroe, Org. Process Res. Dev., 2014, 18, 266 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, the 13C NMR spectrum, GC/MS profile, and HRMS profile. See DOI: 10.1039/c4gc01606h |
| This journal is © The Royal Society of Chemistry 2015 |