
Direct synthesis of aryl substituted pyrroles from calcium carbide: an underestimated chemical feedstock†
Narongpol
Kaewchangwat
a,
Rangsarit
Sukato
a,
Viwat
Vchirawongkwin
b,
Tirayut
Vilaivan
b,
Mongkol
Sukwattanasinitt
c and
Sumrit
Wacharasindhu
*c
aProgram of Petrochemistry and Polymer Science, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand
bDepartment of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand
cNanotec-CU Center of Excellence on Food and Agriculture, Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand. E-mail: sumrit.w@chula.ac.th
First published on 18th September 2014
Abstract
In this work, a novel synthetic methodology for the preparation of aryl pyrroles directly from the reaction of calcium carbide with oxime is reported. Various pyrrole derivatives are generated from the corresponding oximes in satisfactory yields (49–88%) under the optimized conditions. The one-pot synthesis of aryl pyrrole from widely available ketone is also successfully developed. A new near-infrared fluorescent BODIPY dye containing a phenyl substitution at the C-3 position is expediently prepared from the aryl pyrrole derived from this methodology. The key benefit of this methodology is the use of an inexpensive and less hazardous primary chemical feedstock, calcium carbide, in a wet solvent without any metal catalysts. This process offers a novel cost-efficient route for the synthesis of functionalized pyrrole.
Introduction
One of the key conversion routes along the value added chain in the petrochemical industry is the formation of calcium carbide from an electric arc furnace of coke and lime. The hydrolysis of calcium carbide produces acetylene gas as one of the major primary chemical feedstocks for downstream products in the chemical industry such as acetic acid, ethanol, aromatics, vinyl acetate, etc. (Scheme 1).1 Although acetylene gas is widely available and inexpensive, its highly flammable gaseous nature poses a serious disadvantage in requiring an extra level of operation control, and a higher cost to prevent leakage and explosion. To achieve an industrial safety improvement without additional cost, a less hazardous and more economical starting material is highly desirable.2 As a main source of acetylene gas, solid calcium carbide is a promising candidate for a safer and cheaper alternative to acetylene gas, for the production of acetylene-derived chemicals.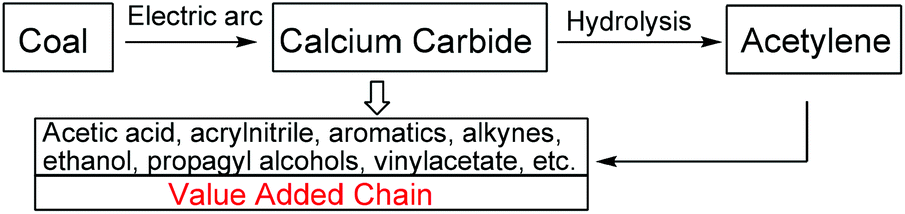 | ||
| Scheme 1 Conversion of coal to commodity chemicals. | ||
In 2006, Zhang et al. reported the use of calcium carbide as a C–C triple bond moiety for the synthesis of diarylethynes via a Sonogarshira coupling reaction.3 Following this work, our group and others recently developed methodologies for the direct use of calcium carbide in the synthesis of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-containing compounds, such as diarylethynes,4 poly(p-phenyleneethynylene)s (PPEs),5 polyenynes,6 propagylamine,7 enaminones8 and acetylenic alcohols.9 All reported processes rely on the slow release of acetylene gas from calcium carbide which then turns into the corresponding anion (C22− anion). Subsequently, the reactive intermediate acts as either a coupling partner for a metal-catalyzed reaction or as a nucleophile for a carbonyl addition.
C-containing compounds, such as diarylethynes,4 poly(p-phenyleneethynylene)s (PPEs),5 polyenynes,6 propagylamine,7 enaminones8 and acetylenic alcohols.9 All reported processes rely on the slow release of acetylene gas from calcium carbide which then turns into the corresponding anion (C22− anion). Subsequently, the reactive intermediate acts as either a coupling partner for a metal-catalyzed reaction or as a nucleophile for a carbonyl addition.
One of the highest value compounds that can be obtained from acetylene is 2-aryl pyrrole. It is a basic building block for pharmaceuticals10 and fluorescent dyes (BODIPY).11 Current methods for the preparation of 2-aryl pyrrole involve using either unsubstituted pyrrole or ketoxime (Scheme 2). The first method involves a metal-catalyzed reaction to introduce the aryl group to C2-halogenated pyrroles.12 In addition, direct C–H activation of pyrrole has recently been developed.13 The drawbacks of these methods are the requirement of a precious metal catalyst and additional protection/deprotection steps in some cases. Moreover, direct arylation of N-substituted pyrroles with aryl iodides in the presence of lithium tert-butoxide, without use of a transition metal catalyst, has also been reported.14
 | ||
| Scheme 2 Synthetic approaches for 2-aryl pyrrole. | ||
On the other hand, the readily and inexpensively available ketoxime can be converted to 2-aryl pyrrole in one step by a reaction with substituted alkynylcarbonyl compounds or acetylene gas. The former uses a reagent metal catalyst such as Au and Ni to activate the triple bond.15 The latter (the so-called Trofimov reaction) uses acetylene gas, bubbled into the reactor in the presence of a strong base.16 The Trofimov reaction is the preferred method for 2-aryl pyrrole synthesis, because metal is not required in the transformation and the product is directly obtained without an additional deprotection step.
In this study, we propose a novel process for the direct transformation of calcium carbide into 2-aryl pyrroles (Scheme 2). This is not only an unprecedented application of calcium carbide as an electrophile precursor in a vinylation reaction to produce vinylic derivatives, but also its first utilization in heterocycle synthesis. Our methods successfully overcome the challenges of the poor solubility of calcium carbide in organic solvent and the unpolarized nature of the acetylenic triple bond, to achieve excellent product yields. The developed method uses economical starting materials (ketoximes) and avoids the handling of gaseous acetylene. The use of heavy metal is eliminated, and overall process is significantly shortened in terms of the number of steps. Therefore it can be considered a convenient, safe, and more environmentally conscious, green and sustainable process. Moreover, our process can be easily applied in large scale manufacturing in the chemical industries as well as for routine laboratory synthesis.
Results and discussion
To design the reaction conditions for the calcium carbide we considered the main problems; these are the low solubility of calcium carbide, and the poor electrophilic nature of acetylene gas, which limits its application in organic synthesis. However, previous work by Trofimov et al. demonstrated that the use of a superbase can promote the O-alkylation of oximes by acetylene gas.16 Moreover, our work has shown that the use of a wet polar aprotic solvent can slowly hydrolyze the calcium carbide into acetylene in situ.4 Therefore, we began our optimization study by reacting acetophenone oxime (1) with calcium carbide in the presence of a base to provide NH-2-aryl pyrrole (1a) along with N-vinyl pyrrole (1b); the results are depicted in Table 1. Among several bases studied, including KOH, NaOH and CsOH (Table 1, entries 1, 3–4), the best results were obtained with KOH, providing the desired pyrrole 1a in 58% yield along with the vinylation product 1b in 3% yield. Switching from DMSO to DMF resulted in no reaction occurring (Table 1, entry 2). In order to drive the reaction forward, the temperature was raised from 100 to 120 °C for 24 hours; however, the yield of the pyrrole 1a decreased along with an increase in the vinylation product 1b (Table 1, entry 5). Therefore, controlling the temperature and reaction time are vital for the reaction conditions. To increase the base strength, we therefore added a catalytic amount of 18-crown-6 as a catalyst (3% mol) to the reaction mixture. The yields of both pyrroles (1a and 1b) increased to 65% and 8%, respectively (Table 1, entry 6). It should be noted that under these conditions, the crude NMR indicated that the oxime was completely consumed, and no hydrolysis product (acetophenone) was detected in the reaction mixture. The slightly low yield of pyrrole 1a is perhaps due to its sensitivity to air, meaning that some decomposition may have occurred during chromatographic purification.|
|
|||||
|---|---|---|---|---|---|
| Entrya | Additive | Base [equiv.] | Solvent | Yieldb1a [%] | Yieldb1b [%] |
| a Acetophenone oxime (1 equiv.), CaC2 (6 equiv.), 18-crown-6 (3 mol%) and solvent (0.074 M) were heated at 100 °C in a sealed tube for 15 h. b Isolated yields after purified by column chromatography on neutral alumina. c Starting material was recovered in 50% yield. d The reaction was heated to 120 °C. e Large excess (10 equiv.) of CaC2 was added. | |||||
| 1 | — | KOH [1.5] | DMSO | 58 | 3 |
| 2c | — | KOH [1.5] | DMF | 0 | 0 |
| 3 | — | NaOH [1.5] | DMSO | 44 | 0 |
| 4 | — | CsOH [1.5] | DMSO | 48 | 0 |
| 5d | — | KOH [1.5] | DMSO | 44 | 8 |
| 6 | 18-crown-6 | KOH [1.5] | DMSO | 65 | 8 |
| 7e | 18-crown-6 | KOH [3.0] | DMSO | 32 | 12 |
| 8 | — | KOH [1.5] | 2% H2O–DMSO | 60 | 0 |
| 9 | — | KOH [1.5] | 10% H2O–DMSO | 32 | 0 |
| 10 | 18-crown-6 | KOH [1.5] | 2% H2O–DMSO | 73 | 0 |

All reactions mentioned above were performed in the undried solvent. In order to increase the liberation of acetylene, we added a small amount of water to the reaction (Table 1, entries 8–10). The presence of water at 2% w/w with 18-crown-6 gave the best yield of the product 1a (73%) and this has been used as the optimized conditions for further studies. The deliberate addition of water can suppress the formation of vinyl pyrrole, and only trace amounts were detected in the crude 1H NMR spectra. In the presence of water, N-vinylation is sluggish due to the reduction of the basic strength of the KOH in DMSO by the so-called “leveling effect”. However, too much added water (10%) gave a significantly lower yield of 1a, perhaps due to the hydrolysis of the calcium carbide occurring too fast, and making acetylene escape from the system faster than it could react with the oxime; it could also be due to the leveling effect lowering the KOH–DMSO basic strength. Importantly, in comparison with the original Trofimov reaction, our reaction based on calcium carbide gave a comparable efficiency, offering an alternative synthesis of 2-aryl pyrroles that is both convenient and safer to perform. The proposed mechanism of the reaction between calcium carbide and oxime is presented in Scheme 3. The added water initially hydrolyzed the calcium carbide to liberate the acetylene gas. The reaction then proceeded as in the standard Trofimov reaction, which involves addition to acetylene followed by tautomerization to generate an enamine.1c Next, the enamine underwent a sigmatropic rearrangement to form an aldehyde–imine intermediate. The desired aryl pyrrole 1a was finally obtained following a tautomerization-addition-dehydration sequence of the aldehyde–imine intermediate.
 | ||
| Scheme 3 Proposed mechanism for the synthesis of pyrrole 1a from oxime 1. | ||
With the optimized conditions in hand, we next expanded the scope of this reaction by subjecting a variety of oximes (2–11) to the reaction conditions (Table 1). These oxime substrates were easily prepared from commercially available ketones using hydroxylamine hydrochloride, and were purified by recrystallization. A panel of acetophenone oximes (2–6) bearing methyl, chloro, methoxy, butoxy and dimethylamino were successfully transformed into aryl pyrroles (2a–6a) in moderate yields, along with small amounts of vinylation products (2b–6b) of less than 6% yields. The oxime-containing bicyclic structures such as 7–9 were also tested in the optimized conditions and generated highly conjugated pyrroles 7a–9a in fair-to-good yields. The scope of the reaction also extends to preparation of highly substituted pyrroles. Propiophenone oxime 10 was reacted with calcium carbide under the described conditions to afford the 2,3-disubstituted pyrrole 10a in 57% yield along with the corresponding vinylpyrrole 10b in 2% yield (Table 2).
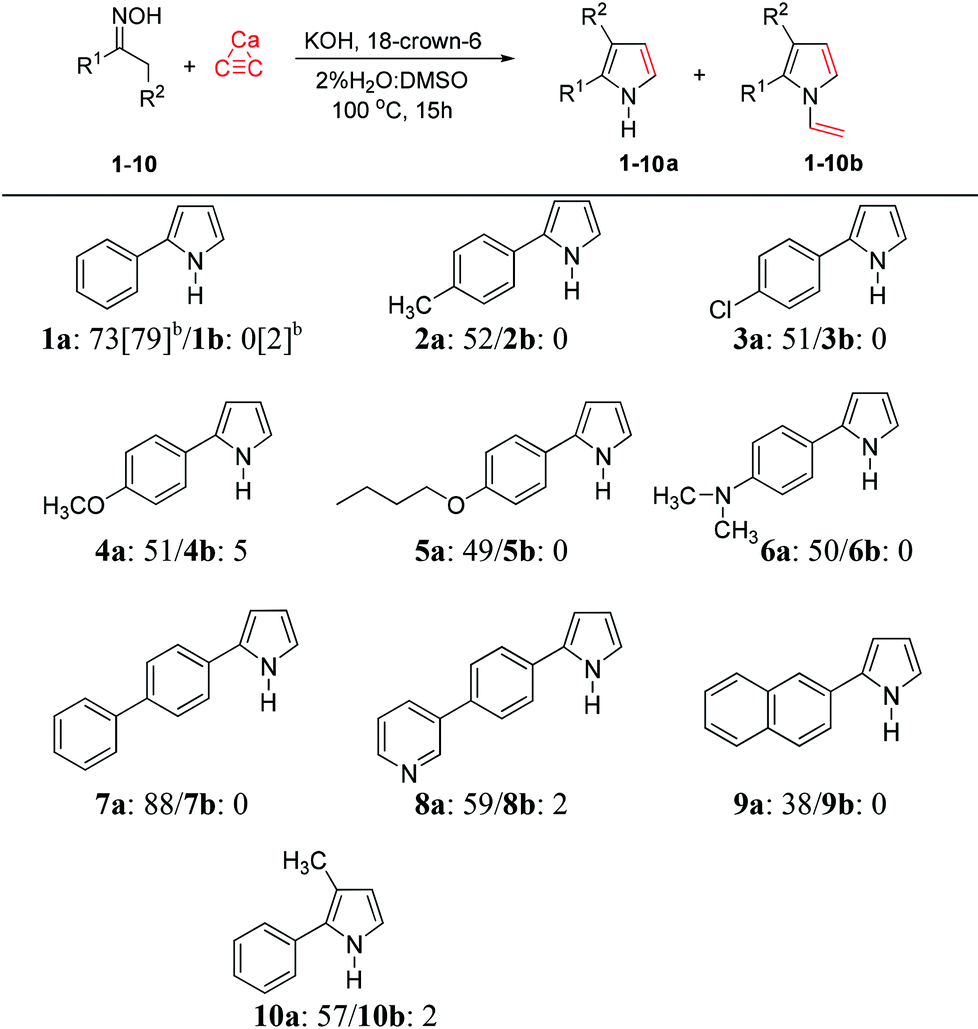
Although oxime can be prepared easily, an additional oxime preparation step is still required. On the other hand, the use of ketone (the precursor of oxime) as the starting material is preferred because it allows the convenient one-pot preparation of 2-arylpyrroles. Here, we successfully developed such one-pot pyrrole synthesis directly from ketone, which combines oxime formation with the Trofimov reaction, as shown in Scheme 4. The ketone 12 was reacted with hydroxylamine hydrochloride in DMSO at 60 °C in the presence of a base (NaHCO3). Addition of calcium carbide and catalytic amounts of 18-crown-6 to the reaction mixture led to the formation of aryl pyrrole (1a) in 56% yield as the sole product. This one-pot process is comparable to the two-step transformation in terms of isolated yield, but is shorter and more convenient to perform.
 | ||
| Scheme 4 One-pot synthesis of 2-phenyl pyrrole 1a from acetophenone 12. | ||
To demonstrate an application of our methodology, we next synthesized 3-substituted BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) using the aryl pyrrole prepared by our methods. Fluorescent BODIPY dyes are widely used as sensors, for optoelectronic materials and in imaging applications.11b,17 In recent years, near-infrared (NIR) dyes have attracted much interest for in vivo imaging applications18 because they allow bioimaging with low interference from the tissues. The high penetration power and the low energy of the light also causes less cell damage. To make NIR BODIPY, post-structural modification of the BODIPY scaffold by an extension of the π-conjugation, such as an alkenyl or aryl substitution at the 3-position, is generally employed.11b The 3-alkenyl substituted BODIPY is obtained from the Knoevenagel reaction of 3-methyl BODIPY.11a The preparation of 3-aryl substituted BODIPY usually involves a metal-catalyzed cross-coupling reaction of the halo-substituted BODIPY, which not only requires additional steps but also causes the partial decomposition of the BODIPY during the coupling reaction.19
With the readily accessible 2-aryl pyrroles in hand, we have the opportunity to perform the direct synthesis of a BODIPY fluorophore containing an aromatic substitutent at the C-3 position. Using the standard method,20 the synthesis was accomplished via a two-step procedure presented in Scheme 5. Initially, the condensation of biphenyl-substituted pyrrole 7a with 4-bromobenzaldehyde led to the formation of the dipyrrane 13 in 71% yield (Scheme 5). The oxidation of 13 using DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone), followed by complexation with BF3·OEt2, gave the desired BODIPY 14 with a biphenyl substitution at the C-3 position in 50% yield. The peripheral bromobenzene moiety at the C-5 position of 14 provides an opportunity for further probe attachment for fluorescence sensing applications. The absorption and emission spectra of the BODIPY 14 are shown in Fig. 1. The maximum absorption and emission wavelengths are at 582 and 632 nm, respectively. It is apparent that the conjugation extension of the BODIPY scaffold by installation of additional phenyl groups can shift both absorption and emission maxima into the far red region.
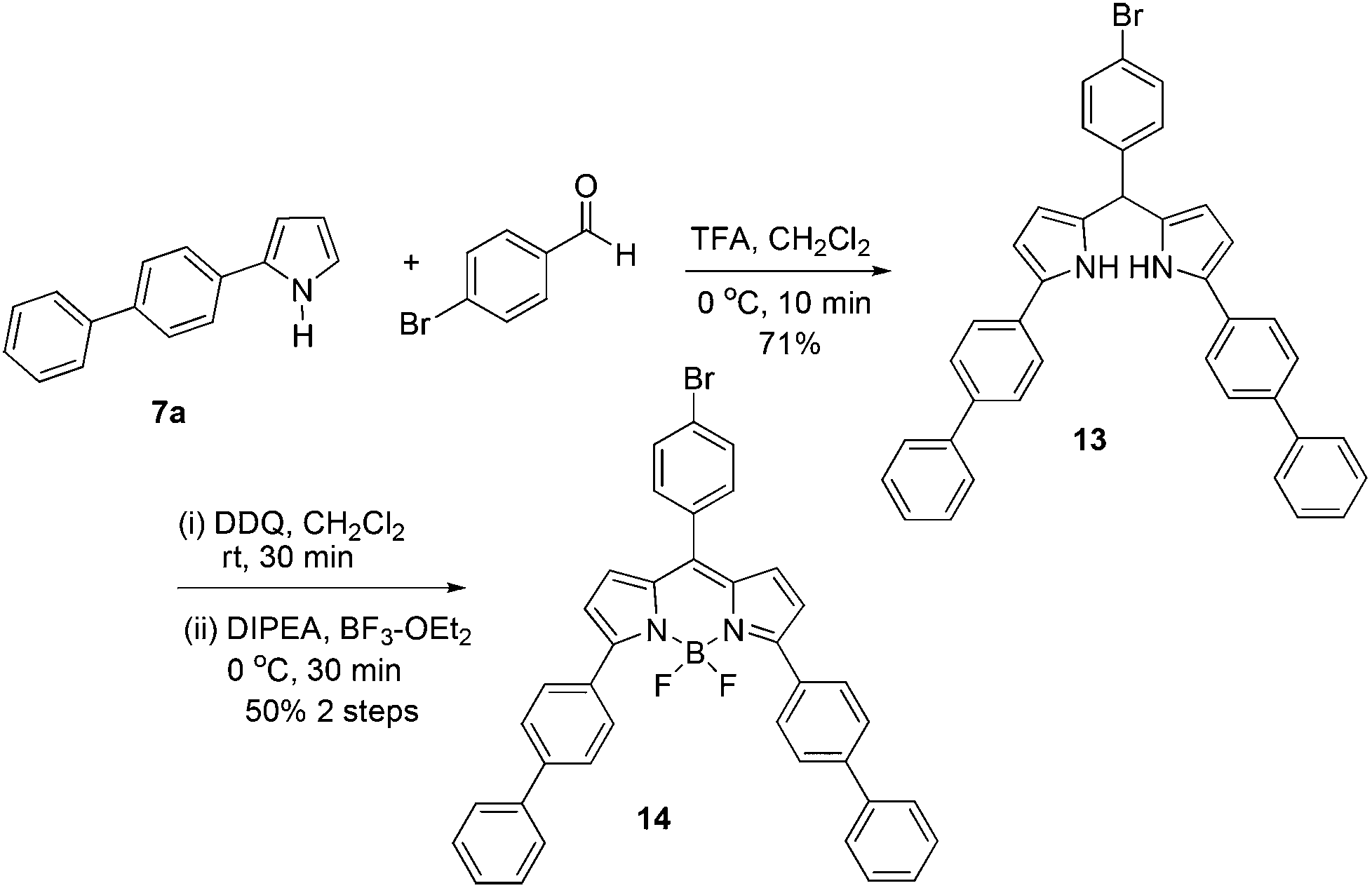 | ||
| Scheme 5 The synthesis of BODIPY 14 from pyrrole 7a. | ||
 | ||
| Fig. 1 UV-vis absorption and fluorescence emission spectra of BODIPY 14 in THF. λex = 582 nm. | ||
Materials and methods
All chemicals were obtained from commercial suppliers (Sigma Aldrich), and were used without further purification. All solvents were used directly without drying, except for dimethyl sulfoxide (DMSO), which was dried over 4 Å molecular sieves. Calcium carbide was ground before use. Analytical thin-layer chromatography (TLC) was performed on Kieselgel F254 pre-coated plastic TLC plates from EM Science. Visualization was performed with a 254 nm ultraviolet lamp. Column chromatography was carried out with aluminium oxide (90 active neutral, 70–230 mesh) from Merck and silica gel (60, 230–400 mesh) from ICN Silitech. The 1H and 13C NMR spectra were recorded on a Varian Mercury 400 or Bruker Avance 400 for 1H (400 MHz) and Bruker Avance 400 for 13C (100 MHz) in CDCl3 or (CD3)2SO solution. Mass spectrometry was performed with a Micromass Quattro micro TM API and triple quadrupole GC/MS from Agilent technologies. The UV-visible absorption spectra were obtained with a Varian Cary 50 UV-Vis spectrometer and the fluorescence emission spectra were recorded on a Varian Cary Eclipse spectrofluorometer.General procedure for synthesis of 2-arylpyrroles
A mixture of calcium carbide (6.0 equiv.), ketone oxime (1.0 equiv.), potassium hydroxide (1.5 equiv.) and 18-crown-6 (3.0 mol%) was suspended in 10 mL of (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) DMSO–H2O in a sealed tube. The reaction mixture was stirred at 100 °C overnight. The reaction was cooled to room temperature and diluted by the dropwise addition of H2O (10 mL). The reaction mixture was filtered and the solution was extracted with ether (5 × 30 mL). The combined extracts were washed with brine (2 × 30 mL), dried over MgSO4 and evaporated under reduced pressure to give the crude product, which was further purified by column chromatography on alumina (eluted with ethyl acetate–hexanes = 1:3) to afford the desired compound.
:1) DMSO–H2O in a sealed tube. The reaction mixture was stirred at 100 °C overnight. The reaction was cooled to room temperature and diluted by the dropwise addition of H2O (10 mL). The reaction mixture was filtered and the solution was extracted with ether (5 × 30 mL). The combined extracts were washed with brine (2 × 30 mL), dried over MgSO4 and evaporated under reduced pressure to give the crude product, which was further purified by column chromatography on alumina (eluted with ethyl acetate–hexanes = 1:3) to afford the desired compound.
General procedure for one-pot synthesis of 2-phenylpyrrole (1a)
Hydroxylamine hydrochloride (0.83 mmol, 57 mg) was dissolved in DMSO (10 mL) in a sealed tube with a magnetic stirrer bar, and then NaHCO3 (0.83 mmol, 93 mg) and acetophenone (0.83 mmol, 100 mg) were added. The reaction mixture was stirred at 60 °C for 4 h. After completion of the reaction, calcium carbide (4.98 mmol, 319 mg), potassium hydroxide (1.25 mmol, 70 mg) and 18-crown-6 were added and the mixture was heated to 100 °C overnight. The reaction was cooled to room temperature and diluted by the dropwise addition of H2O (10 mL). The reaction mixture was filtered and the solution was extracted with ether (5 × 30 mL). The combined extracts were washed with brine (2 × 30 mL), dried over MgSO4 and evaporated under reduced pressure to give the crude product, which was further purified by column chromatography on alumina (eluted with ethyl acetate–hexanes = 1:3) to afford the corresponding 2-phenylpyrrole as a purple solid (66 mg, 56% yield).
Synthesis of BODIPY 14
4-Bromobenzaldehyde (0.28 mmol, 52 mg) and 2-(biphenyl-4-yl)pyrrole (7a) (0.558 mmol, 122 mg) were dissolved in dichloromethane (30 mL) in a round bottomed flask with a magnetic stirrer bar. The reaction mixture was stirred at room temperature for 5 minutes, then a few drops of trifluoroacetic acid were added and the reaction was stirred for another 5 minutes. The reaction was washed with water (3 × 30 mL), dried over MgSO4, and the solvent was removed under reduced pressure. Purification by column chromatography on silica gel (dichloromethane–hexanes = 1:2) gave compound 13 as a dark red solid (240 mg, 71% yield). The intermediate 13 (0.196 mmol, 119 mg) and DDQ (0.196 mmol, 45 mg) were dissolved in dichloromethane (10 mL) in a round bottomed flask with a magnetic stirrer bar under a nitrogen atmosphere. The reaction mixture was stirred at room temperature for 30 minutes. Then N,N-diisopropylethylamine (1.97 mmol, 0.34 mL) and boron trifluoride diethyl etherate (2.36 mmol, 0.30 mL) were added at 0 °C and the mixture was stirred for 30 minutes. The reaction was washed with saturated NaHCO3 (3 × 30 mL) and brine (3 × 30 mL), dried over MgSO4 and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel (dichloromethane–hexanes = 1:2) to give the product as a red solid (129 mg, 50% yield). 1H NMR (400 MHz, CDCl3): δ ppm 8.00 (d, J = 8.4 Hz, 4H, Ar–H), 7.73–7.61 (m, 10H, Ar–H), 7.52–7.42 (m, 6H, Ar–H), 7.37 (d, J = 7.4 Hz, 2H, Ar–H), 6.89 (d, J = 4.4 Hz, 2H, CH), 6.72 (d, J = 4.2 Hz, 2H, CH); 13C NMR (100 MHz, CDCl3): δ ppm 142.4, 140.5, 136.4, 133.3, 132.0, 131.7, 131.4, 130.5, 130.0, 130.0, 129.9, 128.8, 127.6, 127.2, 127.0, 124.7, 121.2. HRMS (ESI) calculated for [M + Na]+: 673.1238, found at 673.1232.
Conclusions
In summary, a practical synthesis of 2-arylpyrroles from calcium carbide and oximes was successfully developed. The reaction was carried out in wet solvent without the use of toxic metals. Moreover, the process was extended to preparing 2-arylpyrroles in a one-pot manner starting from acetophenone and hydroxylamine. The resulting aryl pyrroles prove to be excellent building blocks for NIR-BODIPY fluorophores. These novel synthetic methods provide an opportunity to establish calcium carbide as a sustainable and cost effective carbon source in modern chemical industries.Acknowledgements
This study is financially supported by the Thailand Research Fund (TRF-RSA5780055) and National Nanotechnology Center (NANOTEC), NSTDA, Ministry of Science and Technology, Thailand, through its program of the Center of Excellence Network. This work is part of the Project for Establishment of Comprehensive Center for Innovative Food, Health Products and Agriculture supported by the Thai Government Stimulus Package 2 (TKK2555, SP2), the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission (AM1006A-56) and the Ratchadaphiseksomphot Endowment Fund of Chulalongkorn University (RES560530126-AM). The student scholarship was received from 90th Anniversary of Chulalongkorn University fund (Ratchadaphiseksomphot Endowment Fund).Notes and references
- (a) H. Schobert, Chem. Rev., 2014, 114, 1743–1760 CrossRef CAS PubMed; (b) I. T. Trotuş, T. Zimmermann and F. Schüth, Chem. Rev., 2014, 114, 1761–1782 CrossRef PubMed; (c) B. A. Trofimov, Curr. Org. Chem., 2002, 6, 1121–1162 CrossRef CAS.
- R. Diercks, J. D. Arndt, S. Freyer, R. Geier, O. Machhammer, J. Schwartze and M. Volland, Chem. Eng. Technol., 2008, 31, 631–637 CrossRef CAS.
- W. Zhang, H. Wu, Z. Liu, P. Zhong, L. Zhang, X. Huang and J. Cheng, Chem. Commun., 2006, 4826–4828 RSC.
- P. Chuentragool, K. Vongnam, P. Rashatasakhon, M. Sukwattanasinitt and S. Wacharasindhu, Tetrahedron, 2011, 67, 8177–8182 CrossRef CAS PubMed.
- N. Thavornsin, M. Sukwattanasinitt and S. Wacharasindhu, Polym. Chem., 2014, 5, 48–52 RSC.
- F. Cataldo, Tetrahedron Lett., 2005, 46, 3665–3667 CrossRef CAS PubMed.
- Z. Lin, D. Yu, Y. N. Sum and Y. Zhang, ChemSusChem, 2012, 5, 625–628 CrossRef CAS PubMed.
- D. Yu, Y. N. Sum, A. C. C. Ean, M. P. Chin and Y. Zhang, Angew. Chem., Int. Ed., 2013, 52, 5125–5128 CrossRef CAS PubMed.
- Y. N. Sum, D. Yu and Y. Zhang, Green Chem., 2013, 15, 2718–2721 RSC.
- (a) V. Onnis, A. De Logu, M. T. Cocco, R. Fadda, R. Meleddu and C. Congiu, Eur. J. Med. Chem., 2009, 44, 1288–1295 CrossRef CAS PubMed; (b) G. A. Pinna, G. Loriga, G. Murineddu, G. Grella, M. Mura, L. Vargiu, C. Murgioni and P. La Colla, Chem. Pharm. Bull., 2001, 49, 1406–1411 CrossRef CAS; (c) Y. Zhao, C. Mao, Y. Li, P. Zhang, Z. Huang, F. Bi, R. Huang and Q. Wang, J. Agric. Food Chem., 2008, 56, 7326–7332 CrossRef CAS PubMed.
- (a) Y. Ni, L. Zeng, N. Y. Kang, K. W. Huang, L. Wang, Z. Zeng, Y. T. Chang and J. Wu, Chem. – Eur. J., 2014, 20, 2301–2310 CrossRef CAS PubMed; (b) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- (a) A. Burghart, H. Kim, M. B. Welch, L. H. Thoresen, J. Reibenspies, K. Burgess, F. Bergström and L. B. Å. Johansson, J. Org. Chem., 1999, 64, 7813–7819 CrossRef CAS; (b) L. Li, B. Nguyen and K. Burgess, Bioorg. Med. Chem. Lett., 2008, 18, 3112–3116 CrossRef CAS PubMed; (c) H. L. Phil, Bull. Korean Chem. Soc., 2008, 29, 261–264 CrossRef; (d) R. D. Rieth, N. P. Mankad, E. Calimano and J. P. Sadighi, Org. Lett., 2004, 6, 3981–3983 CrossRef CAS PubMed; (e) J. Wen, S. Qin, L. F. Ma, L. Dong, J. Zhang, S. S. Liu, Y. S. Duan, S. Y. Chen, C. W. Hu and X. Q. Yu, Org. Lett., 2010, 12, 2694–2697 CrossRef CAS PubMed.
- (a) P. Ehlers, A. Petrosyan, J. Baumgard, S. Jopp, N. Steinfeld, T. V. Ghochikyan, A. S. Saghyan, C. Fischer and P. Langer, ChemCatChem, 2013, 5, 2504–2511 CrossRef CAS; (b) B. Li, J. Ma, W. Xie, H. Song, S. Xu and B. Wang, Chem. – Eur. J., 2013, 19, 11863–11868 CrossRef CAS PubMed; (c) Y. Y. Qian, K. L. Wong, M. W. Zhang, T. Y. Kwok, C. T. To and K. S. Chan, Tetrahedron Lett., 2012, 53, 1571–1575 CrossRef CAS PubMed; (d) L. Chen, C. Bruneau, P. H. Dixneuf and H. Doucet, ChemCatChem, 2013, 5, 1956–1963 CrossRef CAS.
- O. Vakuliuk, B. Koszarna and D. T. Gryko, Adv. Synth. Catal., 2011, 353, 925–930 CrossRef CAS.
- (a) S. Ngwerume and J. E. Camp, J. Org. Chem., 2010, 75, 6271–6274 CrossRef CAS PubMed; (b) S. Ngwerume and J. E. Camp, Chem. Commun., 2011, 47, 1857–1859 RSC; (c) H. Y. Wang, D. S. Mueller, R. M. Sachwani, H. N. Londino and L. L. Anderson, Org. Lett., 2010, 12, 2290–2293 CrossRef CAS PubMed.
- (a) A. I. Mikhaleva, B. A. Trofimov and A. N. Vasil'ev, Zh. Org. Khim., 1979, 15, 602 CAS; (b) B. A. Trofimov and A. I. Mikhaleva, Heterocycles, 1994, 37, 1193–1232 CrossRef CAS; (c) S. E. Korostova, A. I. Mikhaleva, A. M. Vasil'tsov and B. A. Trofimov, Russ. J. Org. Chem., 1998, 34, 911–948 CAS; (d) E. Y. Schmidt, A. I. Mikhaleva, A. M. Vasil'tsov, A. B. Zaitsev and N. V. Zorina, ARKIVOC, 2005, 11–17 CrossRef CAS; (e) A. I. Mikhaleva, O. V. Petrava and L. N. Sobenina, Chem. Heterocycl. Compd., 2012, 47, 1367–1371 CrossRef CAS.
- (a) A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC; (b) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed; (c) B. C. Popere, A. M. Della Pelle and S. Thayumanavan, Macromolecules, 2011, 44, 4767–4776 CrossRef CAS.
- (a) E. I. Altinoǧlu and J. H. Adair, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 461–477 CrossRef PubMed; (b) D. R. Leff, O. J. Warren, L. C. Enfield, A. Gibson, T. Athanasiou, D. K. Patten, J. Hebden, G. Z. Yang and A. Darzi, Breast Cancer Res. Treat., 2008, 108, 9–22 CrossRef PubMed; (c) K. H. Song, C. Kim, C. M. Cobley, Y. Xia and L. V. Wang, Nano Lett., 2009, 9, 183–188 CrossRef CAS PubMed; (d) M. Vendrell, D. Zhai, J. C. Er and Y. T. Chang, Chem. Rev., 2012, 112, 4391–4420 CrossRef CAS PubMed; (e) L. Yuan, W. Lin, S. Zhao, W. Gao, B. Chen, L. He and S. Zhu, J. Am. Chem. Soc., 2012, 134, 13510–13523 CrossRef CAS PubMed.
- (a) J. Han, O. Gonzalez, A. Aguilar-Aguilar, E. Peña-Cabrera and K. Burgess, Org. Biomol. Chem., 2009, 7, 34–36 RSC; (b) T. Rohand, W. Qin, N. Boens and W. Dehaen, Eur. J. Org. Chem., 2006, 4658–4663 CrossRef CAS.
- E. Y. Schmidt, N. V. Zorida, M. Y. Dvorko, N. I. Protsuk, K. V. Beleaeva, G. Clavier, R. Méallet-Renault, T. T. Vu, A. I. Mikhaleva and B. A. Trofimov, Chem. – Eur. J., 2011, 17, 3069–3073 CrossRef CAS PubMed.
- A. V. Afonin, I. A. Ushakov, D. E. Simonenko, E. Yu. Shmidt, N. V. Zorina, A. I. Mikhaleva and B. A. Trofimov, Russ. J. Org. Chem., 2005, 41, 1515–1521 CrossRef PubMed.
- D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18327–18339 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01615g |
| This journal is © The Royal Society of Chemistry 2015 |