
Heterogeneously catalyzed strategies for the deconstruction of high density polyethylene: plastic waste valorisation to fuels
Anand S.
Burange
*ad,
Manoj B.
Gawande
*b,
Frank L. Y.
Lam
c,
Radha V.
Jayaram
d and
Rafael
Luque
ce
aCentre of Excellence on Surface Science, CSIR – National Chemical Laboratory, Dr Homi Bhabha Road, Pune 411 008, India. E-mail: a.burange@ncl.res.in
bRegional Centre of Advanced Technologies and Materials, Faculty of Science, Department of Physical Chemistry, Palacky University, Šlechtitelů 11, 783 71, Olomouc, Czech Republic. E-mail: manoj.gawande@upol.cz
cDepartment of Chemical and Biomolecular Engineering, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong
dDepartment of Chemistry, Institute of Chemical Technology (University under Section-3 of UGC ACT 1956), N. Parekh Marg, Matunga, Mumbai 400 019, India
eDepartamento de Quimica Organica, Universidad de Cordoba, Edificio Marie Curie (C-3), Ctra Nnal IV-A, Km 396, E14014, Cordoba, Spain
First published on 9th October 2014
Abstract
The plastic industry generates enormous quantities of plastics at projected rates (both production and consumption) which can significantly threaten our environment in terms of plastic waste generation. High density polyethylene (HDPE) is one of the main fractions of municipal solid waste which has a remarkable potential to be valorised into fuels (e.g. bio-oils). Catalytic degradation is an innovative alternative process to transform plastic waste into such value added products. This mini review was aimed to discuss the most relevant and recent catalysts developed for the catalytic degradation of HDPE including metal oxides, sulphated metal oxides, zeolites, nanostructured zeolites, molecular sieves, fluid catalytic cracking (FCC) catalysts, metal carbonates and mesoporous materials for the production of chemicals and fuels (e.g. diesel and gasolines). Activities and selectivities as well as important effects of additives, particle size, catalyst to polymer ratios and also recent approaches for waste management will be discussed.
 Anand S. Burange | Dr Anand Sudhakar Burange was born in Morshi, Amravati, Maharashtra, India in 1986. He received his master's degree in Chemistry from Sant Gadge Baba Amravati University, India. In 2008 he joined for Ph.D. at the Institute of Chemical Technology, Mumbai, India with Prof. R. V. Jayaram and received a UGC Green-tech fellowship. During his research tenure, he worked on the application of metal oxide based catalysts in organic transformations and in photocatalysis. Then, he joined Johnson Matthey Catalyst Company, Taloja, India where he worked on supported noble metal catalysis, deprotection of protected drugs and other high pressure reactions. Currently he is working, with Dr C. S. Gopinath's research group, as a Research Associate at the National Chemical Laboratory, Pune, India in the surface science and catalysis arena. His research interests focus on exploration of new materials as catalysts in value added organic transformations, surface science and photo-catalysis. |
 Manoj B. Gawande | Dr Manoj B. Gawande received his Ph.D. degree in Chemistry in 2008 from the Department of Chemistry, Institute of Chemical Technology (formerly UDCT), Matunga, Mumbai, India, with Prof. R.V. Jayaram. During his research tenure, he has been awarded several prestigious fellowships, including Brain Korea-21 (BK-21) Research Fellowship from the Ministry of Science, South Korea and FCT Research Fellowship from MCTES, Lisbon, Portugal. After several research stints in Germany, South Korea and Portugal, he worked as a Visiting Professor at CBC-SPMS, Nanyang Technological University, Singapore and a visiting scientist at the Department of Chemistry, Cambridge University, Cambridge, UK with Prof. Steve Ley. Presently, he works as a Senior Researcher and the Head of the Nanocatalysis and Organic Synthesis Research laboratories at the Regional Centre of Advanced Technologies and Materials, Faculty of Science, Palacky University, Olomouc, Czech Republic. His research interests include nanocatalysis, magnetically recyclable nanomaterials and their applications in organic transformations. He has published over 50 scientific publications, including reviews, patents and articles. He is currently supervising several doctoral and postdoctoral students. |
 Rafael Luque | Rafael Luque (Ph.D. in 2005 from Universidad de Cordoba, Spain) has significant experience in biomass and waste valorisation practices to materials, fuels and chemicals as well as nanoscale chemistry and microwave and flow chemistry. He has published over 220 research articles, filed 3 patent applications and edited 7 books, as well as numerous contributions to book chapters and invited, guest, keynote and plenary lectures in scientific events worldwide. He is also heavily involved in chemical education and in promoting science in developing countries. Among recent awards, Rafael has received the Marie Curie Prize from Instituto Andaluz de Quimica Fina in Spain (2011), the Green Talents award from the Federal Ministry of Education and Research in Germany (2011), the TR35 Spain from Technology Review and MIT (2012) and the RSC Environment, Sustainability and Energy Early Career Award (2013) from the Royal Society of Chemistry. Rafael has also been recently honored as 2013 Distinguished Engineering Fellow and Visiting Professor from CBME at Hong Kong University of Science and Technology in Hong Kong and currently the Chinese Academy of Sciences Visiting Professor at the State Key Laboratory of Electroanalytical Chemistry of the Changchun Institute of Applied Chemistry. Prof. Luque combines his academic duties with his activities as a young entrepreneur after co-founding Green Applied Solutions S.L.U. and more recently taking part in Posidonia Oceanica S.L. in Spain. |
1. Introduction
The major production of plastics in current society is based on petrol-derived feedstocks. Plastics production accounts for over 280 million tonnes (Mt) in 2011, a 4% increase with respect to 2010.1 From 2010 to 2016, global plastic consumption is expected to continuously grow in similar yearly numbers to those previously stated.1 The continuous production and use of plastics however leads to significant generation of plastic waste due to their inherent non-degradable nature, with over 24.9 Mt plastic waste produced only in EU-27, Norway and Switzerland in 2008.2 Packaging accounted for the most important contribution to plastic waste generation (>60%).2Current options to manage plastic waste are generally limited to recycling and incineration (with or without energy recovery) as well as landfill disposal. These basic waste management practices were able to drive a total recovery (energy recovery and recycling) to over 50% of plastic waste, with ca. 21% of recycling rate.2 What is more important, practices including incineration generate in some cases hazardous emissions of potentially carcinogenic compounds (e.g. polychlorinated dioxins and polychlorinated furans) as well as poisonous gases such as phosgene. Comparatively, plastic recycling based on pelletizing and moulding to low grade plastics has attracted the interest of many scientists worldwide, but the recycled plastics possess poor mechanical strength and color qualities and hence have low market value and restricted applications.
These figures and inherent issues of plastic recycling clearly evidence the need to seek alternative advanced sustainable valorisation practices able to provide technologies for the production of high added value products from plastic waste including chemicals and fuels. Plastic recycling which includes conversion of waste plastics into valuable chemicals and fuels has attracted significant interest worldwide.3–7
Plastic depolymerisation strategies to monomers or relatively higher complexity units could offer a promising alternative to plastic recycling.8 Nevertheless, complete plastic depolymerization into monomers is only possible in restricted cases and generally never exceed 95–97% (e.g. monomer recovery for polymethyl methacrylate (PMAA) is high, close to 97%, while that for polystyrene is just about 70%).9 Plastic waste valorisation can also be approached by thermal degradation methods. However, these protocols generally involve high temperatures (>500 °C) and generate a wide product distribution.10,11
In the light of these premises, it is clear that the conversion of plastic waste into fuels and valuable chemicals can only be approached in a sustainable way by means of catalytic degradation techniques. Suitable catalysts and reactor design can in principle be potentially able to control product yield, product distribution as well as to reduce the reaction temperature. As an example, vinyl polymers (e.g. polyethylene and polypropylene) are very difficult to decompose to their monomers because of random C–C bond fission of the polymer chain during the reaction mechanism which generates a broad range of products, when decomposed at high temperatures.12,13 Catalytic degradation of such plastics can yield a much narrower product distribution of carbon number and also reduce the reaction temperature, thus offering a more environmentally sound approach that can potentially lead to more cost effective processes for a range of valuable products.
There are various types of polymers discarded every year in large quantities from which high density polyethylene has a low tendency towards degradation. The decomposition properties of waste plastic follow the order depicted in Fig. 1.
 | ||
| Fig. 1 Decomposition of a series of waste plastics. | ||
HDPE is part of the polyolefins family and one of the main polymers in municipal solid waste,14,15 being very rich in carbon and hydrogen. Its H/C ratio is closer to that of petroleum.16 The HDPE volume in 2007 accounted for 23% (ca. 5 million tons) of Western Europe's total polyolefin production, which represents at the same time close to 40% of total plastic production in Europe.17 Obviously, these significant numbers illustrate the high volumes of discarded HDPE annually only in Europe.
This contribution is aimed to discuss strategies and possibilities in the deconstruction of HDPE to valuable fuels (e.g. diesel, bio-oils) using a series of heterogeneously catalyzed protocols.
2. Catalytic degradation of HDPE
2.1 Seminal studies and key processes
Conversion of waste plastics/coal mixtures into valuable hydrocarbon fuels requires the use of different catalysts.18–20 These waste plastics can be converted into bio-oils either via pyrolysis of plastics using acid catalysts or by direct liquefaction of plastics in the presence of acid catalysts.21–28 Higher average molecular weight and less branched HDPE is comparably more difficult to degrade with respect to other resins, while low density polyethylene (LDPE) is relatively easy to degrade to gaseous or liquid fuel products.29–40The detailed thermal depolymerization mechanism of linear HDPE was originally proposed by Holmstrom in 1976. The mechanism involves a radical chain mechanism, showing two possible termination reactions as depicted in Fig. 2.41
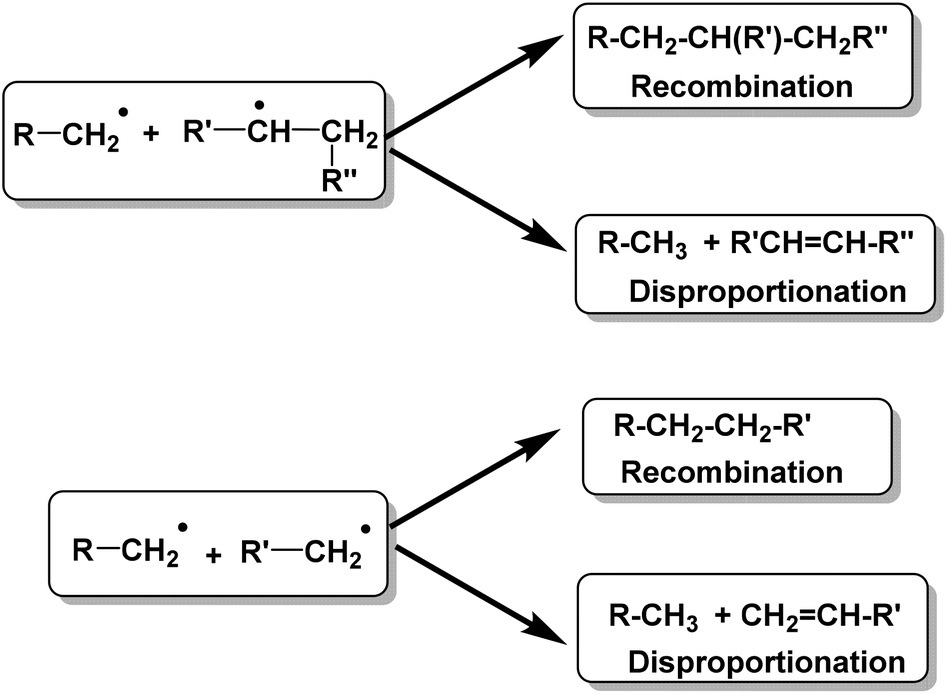 | ||
| Fig. 2 Holmstrom-proposed radical chain mechanism for thermal depolymerization of linear HDPE. | ||
2.2 Catalysts employed in HDPE catalytic degradation
Clinoptilolite is a silica-rich member of the heulandites family as compared to the ZSM-5 catalyst which contains pores of relatively small size (about 5 × 5 Å).42 HZSM-5 was initially reported to be useful for HDPE depolymerization using tetralin as a solvent,43 followed by a combination with TiCl3 in both tubing and autoclave reactors.44 Pyrolysis of high-density polyethylene (HDPE) using a specially developed laboratory fluidized-bed reactor operated isothermally at ambient pressure was proved to produce valuable hydrocarbons in the range of C3–C5 carbon numbers with a high olefinic content.44 The addition of TiCl3 or HZSM-5 was found to have a significant effect on solid yields (i.e. char) which remarkably decreased as compared to those of conventional thermal runs. As an example to illustrate such an important effect, solid yields were reduced to ca. 3% for catalytic runs with respect to a staggering 44.8% for non-catalytic runs at 430 °C.44 No significant non-catalytic reactions were found to take place below 430 °C. The chemistry behind the process encompassed HZSM-5 catalyzed cyclization and aromatization reactions as well as TiCl3 catalyzed recombination and disproportionation reactions. Optimum oil yields (88.7%) were obtained for HDPE catalysed depolymerisation using TiCl3, while maximum gas yields of 21.2% were observed, when HZSM-5 was employed as a catalyst in the autoclave reactor under the investigated reaction conditions.44
Further studies on the catalytic degradation of HDPE indicated that catalysts such as ultrastable zeolite-Y (USY) had a profound effect in reducing the catalytic degradation temperatures in semi-batch reactors as compared to purely thermal non-catalytic degradation.45 USY is a microporous zeolite containing supercages (12 Å) tetrahedrally connected by 12-membered ring windows of 7.4 Å diameter. The particle size of the reported catalyst was 1 μm. Gel permeation chromatographic studies in fact confirmed that solid state reactions occurred in the presence of the microporous catalyst. As the system reached its final temperature, the reaction rate for the formation of volatile products decreased rapidly. C3–C15 range products were observed in the liquid fraction, isobutene and isopentane being the main gaseous products (Table 1).45
| Mass fraction of liquid (%) | ||||
|---|---|---|---|---|
| Conditions | Alkanes | Alkenes | Cycloproducts | Aromatics |
| 100 K min−1 to 649 K | 73 | 7 | 4 | 16 |
| 100 K min−1 to 570 K | 75 | 5 | 3 | 17 |
| 100 K min−1 to 529 K | 83 | 2 | 4 | 11 |
| 2 K min−1 to 559 K | 86 | 3 | 3 | 8 |
Alkene-rich liquid products were also obtained which underwent a series of bimolecular transfer hydrogenation reactions to give iso-paraffins as major products. These compounds have associated high octane numbers which illustrate their potential for catalytic degradation in terms of commercial technology for plastic waste valorisation. The observed selectivity to the liquid fraction (ca. 50%) was interestingly found to be insensitive to temperature.
In a separate study, USY zeolite was also employed as a catalyst in the catalytic decomposition of both LDPE and HDPE under dynamic conditions in a batch reactor.46 The particle size of the investigated catalyst was 1 μm. Interestingly, no appreciable differences could be observed in terms of product distribution among the gaseous products formed by both LDPE and HDPE. Aromatic compounds were obtained from 440 °C to the end of the catalytic decomposition process.46
The catalytic activities of the various aforementioned zeolites including FCC, HZSM-5 and clinoptilolite were compared by Miskolczi and co-workers for the catalytic degradation of plastic waste (HDPE and polystyrene) in batch reactors.47 HZSM-5 was proved to significantly increase the yield of gaseous products due to its higher microporous surface area as compared to clinoptilolite, in good agreement with previous reports.43,44 In any case, both FCC and natural clinoptilolite catalysts exhibited good catalytic activities for the production of light hydrocarbon liquids comprising C5–C28 compounds of various types including linear non-branched structures (mostly n-paraffins and alkenes) with some aromatics (e.g. ethyl-benzene, styrene, toluene, and benzene). Product variability was dependent upon cracking parameters and an interesting difference in yield to products was observed between crushed and uncrushed catalysts, particularly for FCC and ZSM-5 catalysts (Fig. 3).47
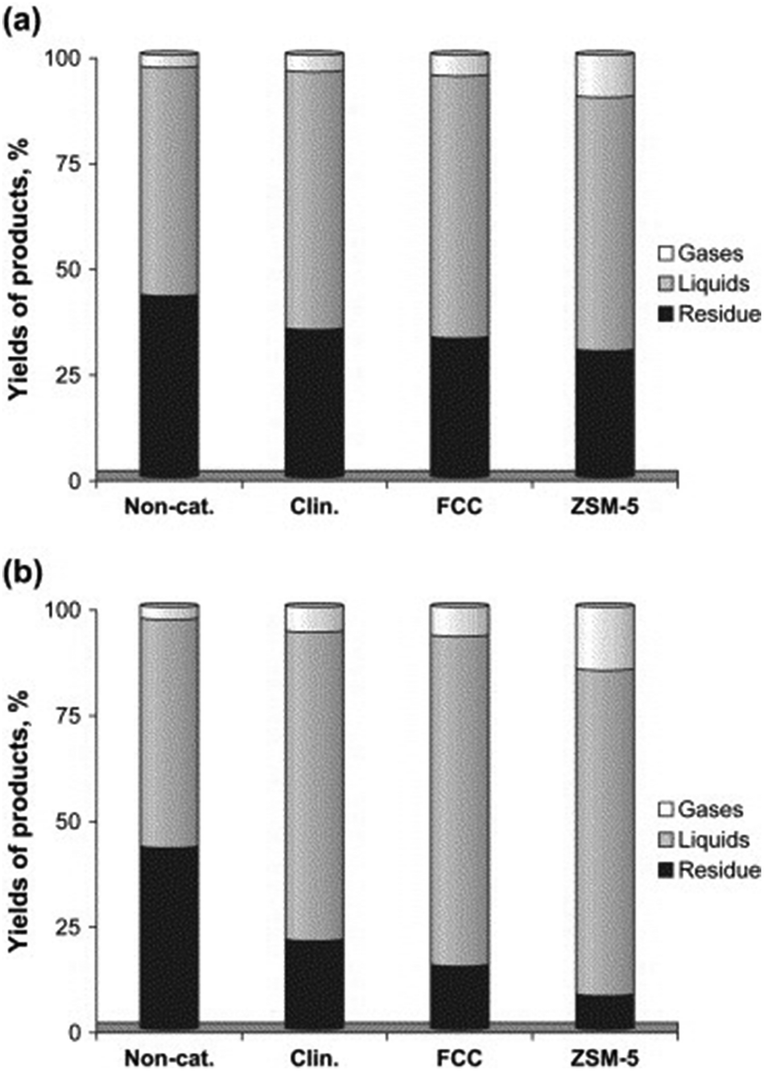 | ||
| Fig. 3 Yields of products (a) over uncrushed and (b) crushed catalysts at 430 °C. Reprinted with permission from ref. 47. Copyright 2013 Elsevier Ltd. | ||
Nano-sized crystals of HZSM-5 can be more active for external cracking processes to generate molecules small enough to pass through the pores to further undergo overcracking to increase the gas yields. In some cases, poor contact between the catalyst and the plastic residue reduces the gas production.48–50
HDPE was thus catalytically degraded under these reaction conditions over nanocrystalline H-ZSM-5 (crystal size ∼100 nm) to produce improved gas yields between 350 and 550 °C. Of note was the higher yield of butenes (25%) in the gas fractions under the investigated conditions. Waxes obtained in the catalytic degradation comprised linear and branched paraffins, in the C10 to C20 range. Either an increase in the polymer to catalyst ratio or keeping operation temperatures below 450 °C had an associated detrimental effect on gas generation in the systems.51 Interestingly, catalytic degradation at temperatures over 500 °C pointed to significant changes in gas and liquid compositions due to the thermal degradation of HDPE, increasing the olefin concentration in the waxes. A plausible mechanism was suggested in which polyethylene was cracked to waxes on external surface acid sites of the zeolite which then further diffused into the internal active surface sites and overcracked to produce lighter compounds.51 Intensive and strong external acid sites of nanocrystalline H-ZSM-5 lead to the remarkable advantages to promote higher yields of gaseous fractions and to provide a higher selectivity to desired products.49,51
Zeolite acidity is one of the key influencing factors in catalytic degradation studies. Coelho and co-workers studied the influence of ZSM-5 zeolite acidity on catalytic degradation of HDPE using simultaneous DSC/TG analysis.52 For degradation of HDPE, both pure HDPE and a mixture of HDPE with catalysts were placed in an alumina TG pan and experiments were conducted to find out the temperature for maximum HDPE degradation rates (Table 3).52
Notably, (80), (63) and (50) are three different NaHZSM-5 zeolites obtained by a single exchange with a 0.5 M NaNO3 solution, with a single exchange with 1.0 M NaNO3 solution and by two successive exchanges with 1.0 M solutions, respectively.
Most of the zeolite catalysts showed nearly good catalytic activity at elevated temperature while the increase in acidity (Si/Al ratio in NaHZSM-5 = 50) could reduce degradation temperatures from 465.1 to 405.7 °C, similar to those of H-ZSM-5 (Table 2, last entry).
| Sample | T degrad. (°C) |
|---|---|
| HDPE | 483.2 |
| HDPE + NaHZSM-5 (80) | 465.1 |
| HDPE + NaHZSM-5 (63) | 413.1 |
| HDPE + NaHZSM-5 (50) | 405.7 |
| HDPE + HZSM-5 | 402.2 |
Comparably, nanocrystalline beta zeolites were developed as an efficient solid acid catalyst for the degradation of HDPE in the liquid phase.53 As previously mentioned, zeolites with smaller crystallite sizes have been regarded as better catalysts for plastic waste degradation providing the large external surface areas and high acidity required for the initial cracking step. For this purpose, six different beta zeolite catalysts having different Si/Al ratios or crystallite sizes were synthesized53 and used for HDPE degradation in a round bottom flask, purged with nitrogen. The reaction temperature was set at 380 °C to avoid thermal degradation of HDPE favoured at temperatures above 400 °C. Nanocrystalline H-beta zeolite with crystallite size around 10 nm and a low Si/Al ratio (highly acidic) was found to be an efficient catalyst for the degradation of HDPE in the liquid phase. Liquid products were collected in a burette through a condenser maintained at −4 °C and the amount of gaseous products was determined by measuring continuously the product flow using a Ritter TG05 gas flow meter. Liquid product yields were found to be ca. 80%, with high selectivities to C7–C12 hydrocarbons.53
Fluidized bed systems are capable of a better isothermal control and mass transfer. Powdered catalysts, namely silica-alumina (SA) and silica-alumina-Na2O (denoted as F9), have been employed by Luo et al. for the HDPE in a fluidized bed system.56
In terms of textural properties, both (F9 and SA) catalysts were similar except for a higher specific surface area with a smaller pore volume of the F9 catalyst. The authors found that SA exhibited a better catalytic activity, producing higher yields of liquid fuels and more valuable gas products in a temperature range between 400 and 550 °C.53
Various zeolites including HZSM-5, H-Y, H-beta, SAPO-37, AlMCM-41 and Al-TUD-1 have been utilised for the degradation of polyethylene (PE and/or HDPE) in recent years.57–77 SAPO-37, belonging to the class of silicoaluminophosphates, was prepared using a hydrothermal approach by Fernandes and co-workers65 for HDPE degradation. Silicoaluminophosphate (SAPO) molecular sieves represent an important class of materials generated by the introduction of silicon into its aluminophosphate framework.55 According to thermogravimetric analysis and Vyazovkin model–free kinetic methods, the activation energy required for HDPE degradation in the absence of a catalyst decreased from 290 to 220 kJ mol−1 for SAPO-37/HDPE. It was also found to be selective for the decomposition of HDPE into light hydrocarbons (C2–C12).56
Pyrolysis of HDPE resulted in higher yields of gaseous products (up to 95%)77 at temperatures between 500 and 800 °C with respect to a combined gas and oil yield of 52 wt% achieved via catalytic degradation of HDPE over a silica-alumina catalyst (catalyst/sample ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 320 °C, within 60 min).78 A significant temperature reduction in the catalytic valorisation of HDPE could provide superior yields to products. Moreover, an 80 wt% liquid yield could be obtained using sulphated zirconia at temperatures ranging between 420 and 450 °C with high initial H2 pressures (500–2000 psi) for 0.5–3.0 h reaction time.79 Other catalysts with additional metal elements supported on sulphated zirconia, including Ni/ZrO2/SO42− and Pt/ZrO2/SO42−, could even maximize bio-oil conversion to 99 wt% with 19–30 wt% gas yield at 375 °C.80 The addition of a large volume of catalyst provided large yields of gaseous products.
:1, 320 °C, within 60 min).78 A significant temperature reduction in the catalytic valorisation of HDPE could provide superior yields to products. Moreover, an 80 wt% liquid yield could be obtained using sulphated zirconia at temperatures ranging between 420 and 450 °C with high initial H2 pressures (500–2000 psi) for 0.5–3.0 h reaction time.79 Other catalysts with additional metal elements supported on sulphated zirconia, including Ni/ZrO2/SO42− and Pt/ZrO2/SO42−, could even maximize bio-oil conversion to 99 wt% with 19–30 wt% gas yield at 375 °C.80 The addition of a large volume of catalyst provided large yields of gaseous products.
Another important parameter influencing the thermal degradation of HDPE is the effect of additives.81,82 In principle, additives, including Al(OH)3·xH2O, AlCl3, CaCO3, Al2O3, CuO, Fe2O3 and ZrO2, are thermally stable below 600 °C except Al(OH)3·xH2O and AlCl3. Findings pointed out that large quantities of gaseous products were obtained in the presence of zirconia at 460 °C, while oil production (up to 80%) was maximized under analogous conditions using calcium carbonate.83 Several interesting results were disclosed related to the advantages of catalytic HDPE degradation as compared to non-catalytic degradation that generally does not take place in relevant quantities into products at temperatures under 490 °C.
| Catalysta | Distribution of hydrocarbon range products (%) | ||||
|---|---|---|---|---|---|
| C6–C12 | C13–C16 | C17–C20 | C20–C30 | ≥C30 | |
| a Catalyst loading: 1%. | |||||
| No catalyst | 32.56 | 30.80 | 14.19 | 12.35 | 10.95 |
| BaTiO3 | 15.45 | 33.83 | 16.71 | 24.38 | 8.05 |
| Pb/BaTiO3 | 17.91 | 27.45 | 19.64 | 31.49 | 3.49 |
| Co/BaTiO3 | 24.34 | 35.45 | 22.42 | 13.05 | 4.4 |
| Pb–Co/BaTiO3 | 18.55 | 31.34 | 25.71 | 16.02 | 7.88 |
Products C6 to C16 comprised over 62% of total selectivity under purely thermal conditions. Comparatively, the addition of catalysts generally provided a shift towards longer C chain products C13 to C30 (Table 3), mostly alkanes as indicated in Table 4. Hydrocarbon distribution also showed reduced naphthalene and olefin yields after doping with Pb and/or Co which points to a preferential catalytic activity towards hydrogen transfer reactions.86
The use of FCC spent catalysts has also been considered as a candidate to plastic waste degradation, particularly for the liquid-phase catalytic degradation of waste HDPE and polystyrene (PS) mixtures in a semi-batch reactor at 400 °C.87 Different mixing proportions of reactants had an important effect on the final characteristics of the obtained liquid products. Increasing the PS content from 0 to 100 wt% produced larger quantities of gasoline fraction (from ca. 85 to ca. 98 wt%). Additionally, PS addition was also found to accelerate the production of aromatic components, showing the highest selectivity (>60 wt%) for ethyl benzene and styrene.87
Pyrolysis of HDPE has also been similarly reported using laboratory fluidized-bed reactors isothermally at ambient pressure over various catalysts using HZSM-5 as compared to non-zeolitic catalysts including MCM-41 and SiO2–Al2O3 (SAHA) and the mixtures zeolite/MCM and zeolite/SAHA. Degradation over HZSM-5 resulted in much larger amounts of volatile hydrocarbons as compared to those observed using non-zeolitic catalysts. SAHA and MCM-41 materials facilitate bulky reactions to occur, leading to the generation of coke and subsequent deactivation of the catalyst. Yields of volatile hydrocarbons using such catalysts followed the order (HZSM-5 > MCM-41/HZSM-5 > SAHA/HZSM-5). The highest product yield (nearly 94 wt%) was obtained for HZSM-5. A higher BTX formation was also observed using HZSM-5.90
Microporous and mesoporous materials, namely ZSM-11 and a modified MCM-41 as a mesoporous material, were also compared in terms of efficiency and product distribution in the degradation of HDPE.57 Zn, Mo and H-ZSM-11 materials were found to be selective for liquid hydrocarbon (LHC) production, namely benzene, toluene and xylenes (BTX). Zn-zeolite exhibited the highest levels of LHC (>57 wt%), with a BTX selectivity of 47 wt%. Comparatively, H-zeolite produced a higher fraction of gaseous hydrocarbons (C1–C4) than those observed for Mo- and Zn-zeolites. In contrast, Zn-MCM-41 produced mainly C5–C16 products. Complete conversion of HDPE could be achieved in the temperature range of 410–500 °C. LPG (C3–C4) levels were also interesting for all materials, following the order:

3. Latest trends
3.1 Continuous pyrolysis coupled with catalytic cracking
Conical spouted bed reactors (CSBR) have been reported to provide a good performance in continuous pyrolysis of HDPE for the production of waxes. A two-step system combining thermal and catalytic cracking of HDPE (Fig. 4) involves CSBR pyrolysis followed by catalytic cracking using zeolites in a fixed bed reactor. This combination was found to be useful for the conversion of HDPE to waxes (first step) and waxes to olefins (second step) as depicted in Fig. 4.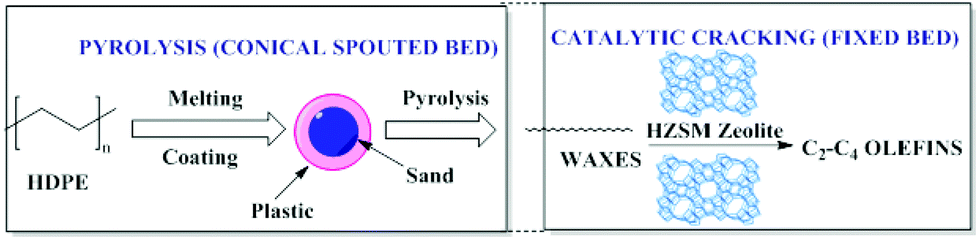 | ||
| Fig. 4 Two-step thermal and catalytic processes for HDPE cracking. Adapted from ref. 97. | ||
HDPE was converted into olefins using the proposed two-step methodology using HZSM-5 zeolites with different acidities. In this investigation, three different HZSM-5 catalysts were prepared with various SiO2/Al2O3 ratios (Table 5). HZSM-5 with a SiO2/Al2O3 ratio of 30 provided a high yield (58 wt%) of light olefins and propylene (32 wt%), which in any case were rather similar to those obtained in a single stage HDPE catalytic cracking process at the same temperature but at significantly higher catalyst loadings (ca. almost 4 times, Table 5).98
| Process | SiO2/Al2O3 ratio | Light olefin yield (wt%) | Propylene yield (wt%) |
|---|---|---|---|
| a Operating conditions: 500 °C, 30 gcat min gHDPE−1. b 500 °C + 500 °C, 8 gcat min gHDPE−1. | |||
| Catalytic process (single-step)a | 30 | 57.0 | 28.0 |
| 80 | 59.8 | 29.0 | |
| Pyrolysis + catalytic cracking (two-step)b | 30 | 58.0 | 32.0 |
| 80 | 43.6 | 24.1 | |
| 280 | 35.5 | 18.8 | |
Catalytic co-processing studies of binary mixtures of plastic and biomass waste using thermogravimetric analysis have also been recently reported.99 A comparison study of catalytic and non-catalytic co-processing of rubber seed shell (RSS) and HDPE mixtures using thermogravimetric analysis indicated that activation energies (EA) and pre-exponential factor (A) values for binary mixtures are comparatively lower than those of pure HDPE but comparable to pure RSS.99 These results may pave the way for future utilisation of plastic waste/biomass mixtures for catalytic valorisation of HDPE.
Similar to HDPE, plastic grocery bags are responsible for relevant environmental concerns which include the ever increasing concentration of plastic waste in the Pacific,100–102 lethal to animal health and society.103–105
Sharma et al. recently studied the thermochemical conversion of plastic grocery bags (HDPE) under batch conditions (Fig. 5).106a In a typical process, pyrolysis of HDPE waste grocery bags was followed by distillation resulting into a hydrocarbon mixture. Upon analysis of the obtained hydrocarbons, the mixture was found to contain primarily saturated aliphatic paraffinic compounds with a similar boiling point range to that of conventional petroleum diesel fuel.
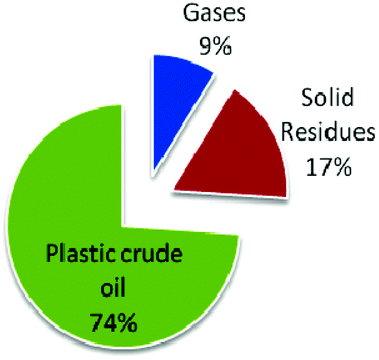 | ||
| Fig. 5 Product yields from plastic grocery bags. Adapted from ref. 106. | ||
Corn derived plastic bags have also been pyrolyzed to bio-oils under microwave-assisted irradiation.106b Bio-oil upgrading can subsequently lead to high quality hydrocarbon fuels with important advantages in terms of storage, transportation and implementation in current fuel infrastructure.107,108
4. Incineration energy recovery
Waste-to-energy technologies were recently discovered and re-invented, emerging as potentially useful protocols to create renewable energy from waste matter, including municipal solid waste (MSW), industrial waste, agricultural waste, and waste byproducts.109 Valorisation of plastic waste is prioritised as compared to agricultural waste due of their inherent previously discussed environmental issues and non-disposable nature. Another promising and important area for plastic waste valorisation relates to energy recovery from incineration of municipal solid waste (MSW). This has been accomplished in many developed countries since several decades ago in an effort to approach sustainable development.110–113 Incineration-based technologies yield a high volume of recoverable energy which is comparable (if not superior) to those obtained with other technologies, i.e. anaerobic digestion.Fig. 6 shows a general process flow of a typical refuse-derived fuel (RDF) plant aimed to convert mixed municipal waste into energy. Four main processing steps associated with the RDF process include physical separation of incombustible materials, drying of MSW, size reduction and pelletizing.
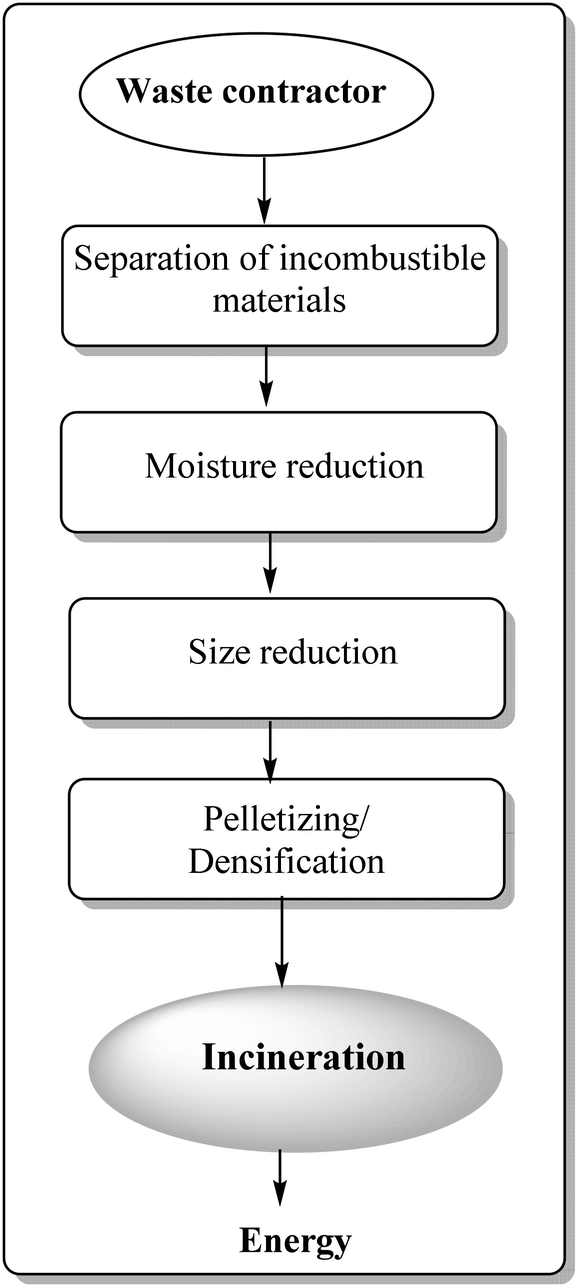 | ||
| Fig. 6 Process flow of a conventional RDF plant. | ||
In recent years, life cycle assessment,114 3R (reduce, reuse and recycle) methodologies,115 tertiary recycling116 and plasma pyrolysis and pyrolysis technology117 are protocols for HDPE conversion to energy and are among the most promising alternatives for future plastic waste management from MSW. Energetic recovery from polymers currently has promising future prospects to tackle plastic-related environmental problems.116
5. Conclusions and future prospects
This contribution is aimed to highlight some of the key developments in the design and preparation of heterogeneous catalysts for waste plastics conversion to fuel oil. A number of catalytic systems have been reported in this regard which have been investigated to better understand their effects on fuels production and product/chemical distribution. Some of these effects (e.g. for zeolite-based catalysts, solid basis, mesoporous materials, etc.) have been discussed and illustrated in plastic degradation studies in terms of conversion and selectivity. Our reported findings addressed (1) the use of fluidized bed systems which provide enhanced mass transfer and thermal homogeneity for catalysts and reactants mixtures, (2) certain structures in nano-sized zeolite crystals which possessed unique high conversion and selectivity to produce fuel oil from plastic waste, (3) the effects of additives on plastic waste conversion and (4) the effects of porous systems (micropores vs. mesopores) in the framework of the proposed “waste to oil” concept.At present, ideal “waste to oil” transformation processes are a great challenge because they require high reaction temperatures and pose difficulties to control conversion as well as selectivity to the different products, particularly for plastic waste. Lowering the reaction temperature is always a long-term goal in catalytic processes which can in principle be made possible by the appropriate design of advanced catalytic materials. Catalyst design, with a particular focus on active phases (benign by design nanomaterials) and porous networks, can undoubtedly pave the way for increasingly efficient protocols for plastic waste conversion. Future efforts should be geared towards enhanced and more controllable low-temperature processes using alternative technologies (e.g. microwave irradiation, continuous flow systems) able to provide an energy-saving, scalable and tunable process to maximise plastic waste conversion as well as target selectivities (e.g. diesel or gasoline fractions).
The authors of this contribution sincerely hope that the future can bring more sustainable advances in the field for an environmentally sound processing of waste plastics to valuable products including fuels and chemicals.
Acknowledgements
The authors gratefully acknowledge the support by the Operational Program Research and Development for Innovations – European Regional Development Fund (project CZ.1.05/2.1.00/03.0058) and by the Operational Program Education for Competitiveness – European Social Fund (project CZ.1.07/2.3.00/30.0041) of the Ministry of Education, Youth and Sports of the Czech Republic.References
- http://www.plasticseurope.org/plastics-industry/market-and-economics.aspx .
- http://ec.europa.eu/environment/waste/studies/pdf/plastics.pdf .
- M. Uddin, K. Koizumi, K. Murata and Y. Sakata, Polym. Degrad. Stab., 1997, 56, 37–44 CrossRef CAS
.
- P. N. Sharratt, Y. H. Lin, A. A. Garforth and J. Dwyer, Ind. Eng. Chem. Res., 1997, 36, 5118–5124 CrossRef CAS
- D. W. Park, E. Y. Hwang, J. R. Kim, J. K. Choi, Y. A. Kim and H. C. Woo, Polym. Degrad. Stab., 1999, 65, 193–198 CrossRef CAS
- T. Masuda, Y. Miwa, K. Hashimoto and Y. Ikeda, Polym. Degrad. Stab., 1998, 61, 217–224 CrossRef CAS
- M. J. Mclntosh, G. G. Arzoumanidis and F. E. Brockmeier, Environ. Prog., 1998, 17, 19–23 CrossRef
- S. C. Cardona and A. Corma, Appl. Catal., B, 2000, 25, 151–162 CrossRef CAS
- Y. Sakata, M. Azharuddin and A. Muto, Chem. Lett., 1998, 245–246 Search PubMed
- A. R. Songip, T. Masuda, H. Kuwahara and K. Hashimoto, Appl. Catal., B, 1993, 2, 153–164 CrossRef CAS
-
(a) A. R. Songip, T. Masuda, H. Kuwahara and K. Hashimoto, Energy Fuels, 1994, 8, 131–135 CrossRef CAS
- F. J. Mastral, E. Esperanza, P. Garcia and M. Juste, J. Anal. Appl. Pyrolysis, 2002, 63, 1–15 CrossRef CAS
- H. Bockhorn, J. Hentschel, A. Hornung and U. Hornung, Chem. Eng. Sci., 1999, 54, 3043–3051 CrossRef CAS
- F. Pinto, P. Costa, I. Gulyurtlu and I. Cabrita, J. Anal. Appl. Pyrolysis, 1999, 51, 57–71 CrossRef CAS
-
(a) P. T. Williams and E. A. Williams, J. Inst. Energy, 1998, 71(487), 81–93 CAS
- M. M. Ibrahim and M. S. Seehra, Energy Fuels, 1997, 11, 926–930 CrossRef CAS
- http://www.plasticseurope.org/Documents/Document/20100312112214-FINAL_HDPE_280409-20081215-017-EN-v1.pdf .
-
(a) A. M. Mastral, R. Murillo, J. M. Palacios, M. C. Mayoral and M. Callén, Energy Fuels, 1997, 11, 813–818 CrossRef CAS
-
(a) A. T. Townsend and J. Abbot, Energy Fuels, 1994, 8, 690–699 CrossRef CAS
- A. Schielffer, ACS Prep. Div. Fuel Chem., 1994, 39, 1060–1064 Search PubMed
- https://shareok.org/handle/11244/5475 .
- A. R. Songip, T. Masuda, H. Kuwahara and K. Hashimoto, Energy Fuels, 1994, 8, 136–140 CrossRef CAS
- D. E. Boone, Pet. Petrochem. Int., 1972, 12, 36–38 Search PubMed
- J. Aguado, D. P. Serrano, G. Vicente and N. Sánchez, Ind. Eng. Chem. Res., 2007, 46, 3497–3504 CrossRef CAS
- P. L. Beltrame, P. Camiti, G. Audisio and F. Bertini, Polym. Degrad. Stab., 1989, 26, 209–220 CrossRef CAS
- R. Mordi, J. Dwyer and R. Fields, Polym. Degrad. Stab., 1994, 46, 57–62 CrossRef CAS
- G. Audisio, A. Silvani, P. L. Beltrame and P. Carniti, J. Anal. Appl. Pyrolysis, 1984, 7, 83–90 CrossRef CAS
- Y. Kashiwaya, Y. Tsukiadate, M. Ayame and H. Kanoh, Bull. Chem. Soc. Jpn., 1983, 56, 2768–2773 CrossRef
- A. C. Albertsson and S. Karlsson, ACS Symp. Ser., 1990, 60–64 CrossRef CAS PubMed
- H. Ohkita, R. Nishiyama, Y. Tochihara, T. Mizushima, N. Kakuta, Y. Morioka, A. Ueno, Y. Namiki, S. Tanifuji, H. Katoh, H. Sunazuka, R. Nakayama and T. Kuroyanagi, Ind. Eng. Chem. Res., 1993, 32, 3112–3116 CrossRef CAS
- R. C. Mordi, R. Fields and J. Dwyer, J. Anal. Appl. Pyrolysis, 1994, 29, 45–55 CrossRef CAS
- Y. Uemichi, Y. Kashiwaya, A. Ayame and H. Kanoh, Chem. Lett., 1984, 41–44 CrossRef CAS
- D. S. Scott, S. R. Czemik, J. Piskorz and D. St. A. G. Radlein, Energy Fuels, 1990, 4, 407–411 CrossRef CAS
- Y. Uemichi, Y. Makino and T. Kanazuka, J. Anal. Appl. Pyrolysis, 1989, 14, 331–344 CrossRef CAS
- M. Sarkar, M. M. Rashid, Md. S. Rahman and M. Molla, Br. J. Environ. Clim. Change, 2012, 2, 1–11 CrossRef PubMed
- D. A. Anderson and E. S. Freeman, J. Polym. Sci., 1961, 54, 253–260 CrossRef CAS
- S. L. Madorsky, J. Polym. Sci., 1954, 9, 133–156 CrossRef
- H. H. G. Jellinek, J. Polym. Sci., 1949, 4, 13–16 CrossRef CAS
- L. A. Wall and S. Straus, J. Polym. Sci., 1960, 44, 313–323 CrossRef CAS
- R. D. Sedgwick, Y. Y. Al-Sultan and A. M. Abushihada, J. Polymer Sci., Polym. Chem. Ed., 1981, 19, 2007–2015 CrossRef CAS
-
(a) A. Holmstrom and E. M. Sorvik, Chromatographia, 1970, 53, 95–108 CrossRef CAS
- H. C. Woo, K. H. Lee and J. S. Lee, Appl. Catal., A, 1996, 134, 147–158 CrossRef CAS
- M. M. Taghiei, Z. Feng, F. E. Huggins and G. P. Huffman, Energy Fuels, 1994, 8, 1228–1232 CrossRef CAS
- W. Ding, J. Liang and L. L. Anderson, Fuel Process. Technol., 1997, 51, 47–62 CrossRef CAS
- G. Manos, A. Garforth and J. Dwyer, Ind. Eng. Chem. Res., 2000, 39, 1203–1208 CrossRef CAS
- A. Marcilla, M. I. Beltran and R. Navarro, Ind. Eng. Chem. Res., 2008, 47, 6896–6903 CrossRef CAS
- N. Miskolczi, L. Bartha and G. Deak, Polym. Degrad. Stab., 2006, 91, 517–526 CrossRef CAS PubMed
- V. R. Grieken, D. P. Serrano, J. Aguado, R. Garcia and C. Rojo, J. Anal. Appl. Pyrolysis, 2001, 127, 58–59 Search PubMed
- D. P. Serrano, J. Aguado, J. M. Escola, E. Garagorri, J. M. Rodriguez and L. Morselli, Appl. Catal., B, 2004, 49, 257–265 CrossRef CAS PubMed
- K. Takuma, Y. Uemichi and A. Ayame, Appl. Catal., A, 2000, 192, 273–280 CrossRef CAS
- J. F. Mastral, C. Berrueco, M. Gea and J. Ceamanos, Polym. Degrad. Stab., 2006, 91, 3330–3338 CrossRef CAS PubMed
- A. Coelho, L. Costa, M. M. Marques, I. M. Fonseca, M. A. N. D. A. Lemos and F. Lemos, Appl. Catal., A, 2012, 413, 183–191 CrossRef PubMed
- Y. J. Lee, J. Kim, S. H. Kim, S. B. Hong and G. Seo, Appl. Catal., B, 2008, 83, 160–167 CrossRef CAS PubMed
- K. Takuma, Y. Uemichi and A. Ayame, Appl. Catal., A, 2000, 192, 273–280 CrossRef CAS
- K. Takuma, Y. Uemichi, M. Sugioka and A. Ayame, Ind. Eng. Chem. Res., 2001, 40, 1076–1082 CrossRef CAS
- G. Luo, T. Suto, S. Yasu and K. Kato, Polym. Degrad. Stab., 2000, 70, 97–102 CrossRef CAS
- L. B. Pierella, S. Renzini and O. A. Anunziata, Microporous Mesoporous Mater., 2005, 81, 155–159 CrossRef CAS PubMed
- J. Aguado, D. P. Serrano, G. S. Miguel, J. M. Escola and J. M. Rodriguez, J. Anal. Appl. Pyrolysis, 2007, 78, 153–162 CrossRef CAS PubMed
- D. P. Serrano, J. Aguado, J. M. Rodriguez and A. Peral, J. Anal. Appl. Pyrolysis, 2007, 79, 456 CrossRef CAS PubMed
- J. F. Mastral, C. Berrueco, M. Gea and J. Ceamanos, Polym. Degrad. Stab., 2006, 91, 3330–3338 CrossRef CAS PubMed
- D. P. Serrano, J. Aguado, J. M. Escola and J. M. Rodriguez, J. Anal. Appl. Pyrolysis, 2005, 74, 353–360 CrossRef CAS PubMed
- Y. H. Lin and M. H. Yang, Appl. Catal., A, 2007, 328, 132–139 CrossRef CAS PubMed
- J. R. Kim, J. H. Van, D. W. Park and M. H. Lee, React. Kinet. Catal. Lett., 2004, 81, 73–81 CrossRef CAS
- A. Marcilla, A. G. Siurana and F. J. Valdes, Microporous Mesoporous Mater., 2008, 109, 420–428 CrossRef CAS PubMed
- G. J. T. Fernandes, V. J. Fernandes and A. S. Araujo Jr., Catal. Today, 2002, 75, 233–238 CrossRef CAS
- K. Gobin and G. Manos, Polym. Degrad. Stab., 2004, 83, 267–279 CrossRef CAS
- Y. S. You, J. S. Shim, J. H. Kim and G. Seo, Catal. Lett., 1999, 59, 221–227 CrossRef CAS
- J. Agullo, N. Kumar, D. Berenguer, D. Kubicka, A. Marcilla, A. Gomez, T. Salmi and D. Y. Murzin, Kinet. Catal., 2007, 48, 535–540 CrossRef CAS
- A. Marcilla, M. I. Beltran, A. G. Siurana, R. Navarro and F. Valdes, Appl. Catal., A, 2007, 328, 124–131 CrossRef CAS PubMed
- A. Marcilla, A. G. Siurana and D. Berenguer, Appl. Catal., A, 2006, 301, 222–231 CrossRef CAS PubMed
- A. Marcilla, A. G. Siurana and F. Valdes, J. Anal. Appl. Pyrolysis, 2007, 79, 433–442 CrossRef CAS PubMed
- A. Marcilla, A. G. Siurana and F. Valdes, Polym. Degrad. Stab., 2007, 92, 197 CrossRef CAS PubMed
- J. W. Park, J. H. Kim and G. Seo, Polym. Degrad. Stab., 2002, 76, 495–501 CrossRef CAS
- R. A. Garcia, D. P. Serrano and D. Otero, J. Anal. Appl. Pyrolysis, 2005, 74, 379–386 CrossRef CAS PubMed
- I. C. Neves, G. Botelho, A. V. Machado, P. Rebelo, S. Ramoa, M. F. R. Pereira, A. Ramanathan and P. Pescarmona, Polym. Degrad. Stab., 2007, 92, 1513–1519 CrossRef CAS PubMed
- M. R. Hernandez, A. N. Garcia and A. Marcilla, J. Anal. Appl. Pyrolysis, 2007, 78, 272–281 CrossRef PubMed
- J. A. Conesa, R. Font, A. Marcilla and A. N. Garcia, Energy Fuels, 1994, 8, 1238–1246 CrossRef CAS
- Y. Ishihara, H. Nanbu, T. Ikemura and T. Takesue, Fuel, 1990, 69, 978–984 CrossRef CAS
- X. Xiao, W. Zmierczak and J. Shabtai, Prepr. – Am. Chem. Soc., Div. Fuel Chem., 1995, 40, 4–8 CAS
- K. R. Venkatesh, J. Hu, J. W. Tiemey and I. Wender, Prepr. – Am. Chem. Soc., Div. Fuel Chem., 1995, 40, 788–792 CAS
-
J.-M. Charrier, Polymeric Materials and Processing: Plastics, Elastomers and Composites, Hanser, New York, 77th edn, 1989 Search PubMed
-
(a)
http://cdn.intechopen.com/pdfs/32828.pdf
;
(b) O. P. Korobeinichev, A. A. Paletsky, L. V. Kuibida, M. B. Gonchikzhapov and I. K. Shundrina, Proc. Combust. Inst., 2013, 34, 2699–2706 CrossRef CAS PubMed
- M. V. S. Murty, E. A. Grulke and D. Bhattacharyya, Polym. Degrad. Stab., 1998, 61, 421–430 CrossRef CAS
- M. R. Jan, J. Shah and H. Gulab, Fuel, 2010, 89, 474–480 CrossRef PubMed
- M. R. Jan, J. Shah and H. Gulab, Fuel Process. Technol., 2010, 91, 1428–1437 CrossRef CAS PubMed
- I. Ahmad, M. I. Khan, M. Ishaq, H. Khan, K. Gul and W. Ahmad, Polym. Degrad. Stab., 2013, 98, 2512–2519 CrossRef CAS PubMed
- K. H. Lee, D. H. Shin and Y. H. Seo, Polym. Degrad. Stab., 2004, 84, 123–127 CrossRef CAS PubMed
-
(a) D. Verboekend and J. Perez-Ramirez, Catal. Sci. Technol., 2011, 1, 879–890 RSC
- A. Garforth, S. Fiddy, Y. H. Lin, A. G. Siakhali, R. N. Sharratt and J. Dwyer, Thermochim. Acta, 1997, 294, 65–69 CrossRef CAS
- Y. H. Lina, M. H. Yang, T. F. Yeh and M. D. Ger, Polym. Degrad. Stab., 2004, 86, 121–128 CrossRef PubMed
- J. Y. Lee, S. M. Park, S. K. Saha, S. J. Cho and G. Seo, Appl. Catal., B, 2011, 108–109, 61–71 CAS
- A. N. C. Van Laak, L. Zhang, A. N. Parvulescu, P. C. A. Bruijincx, B. M. Weckhuysen, K. P. de Jong and P. E. de Jongh, Catal. Today, 2011, 168, 48–56 CrossRef CAS PubMed
- Y. Tao, H. Kanoh and K. Kaneko, Adsorption, 2006, 12, 309–316 CrossRef CAS
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896–910 CrossRef CAS PubMed
- A. Marcilla, M. I. Beltran and R. Navarro, Appl. Catal., A, 2007, 333, 57–66 CrossRef CAS PubMed
- E. F. B. Silva, M. P. Ribeiro, L. P. F. C. Galvão, V. J. Fernandes and A. S. Araujo, J. Therm. Anal. Calorim., 2011, 103, 465–469 CrossRef CAS
-
(a) A. Coelho, L. Costa, M. M. Marques, I. M. Fonseca, M. A. N. D. A. Lemos and F. Lemos, Appl. Catal., A, 2012, 413–414, 183–191 CrossRef CAS PubMed
- M. Artetxe, G. Lopez, M. Amutio, G. Elordi, J. Bilbao and M. Olazar, Ind. Eng. Chem. Res., 2013, 52, 10637–10645 CrossRef CAS
- B. L. F. Chin, S. Yusup, A. Shoaibi, P. Kannan, C. Shrinivasakannan and S. A. Sulaiman, J. Clean. Prod., 2014, 70, 303–314 CrossRef CAS PubMed
- E. A. Howell, S. J. Bograd, C. Morishige, M. P. Seki and J. J. Polovina, Mar. Pollut. Bull., 2012, 65, 16–22 CrossRef CAS PubMed
- J. G. B. Derraik, Mar. Pollut. Bull., 2002, 44, 842–852 CrossRef CAS
- D. K. A. Barnes, A. Walters and L. Goncalves, Mar. Environ. Res., 2010, 70, 250–252 CrossRef CAS PubMed
- B. Lazar and R. Gracan, Mar. Pollut. Bull., 2011, 62, 43–47 CrossRef CAS PubMed
- B. Page, J. McKenzie, R. McIntosh, A. Baylis, M. Morrissey and N. Calvert, Mar. Pollut. Bull., 2004, 49, 33–42 CrossRef CAS PubMed
- C. Muller, K. Townsend and J. Matschullat, Sci. Total Environ., 2012, 416, 464–467 CrossRef PubMed
-
(a) B. K. Sharma, B. R. Moser, K. E. Vermillion, K. M. Doll and N. Rajgopalan, Fuel Process. Technol., 2014, 122, 79–90 CrossRef CAS PubMed
- P. Lan, Q. Xu, M. Zhou, L. Lan, S. Zhang and Y. Yan, Chem. Eng. Technol., 2010, 33, 2021–2028 CrossRef CAS
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell III, J. Wang, L. Moens and J. E. Hensley, Green Chem., 2014, 16, 454–490 RSC
- Waste to Energy, The Cleantech Report, 2007, pp. 187–193.
- F. J. C. Pirotta, E. C. Ferreira and C. A. Bernardo, Energy, 2013, 49, 1–11 CrossRef PubMed
- N. Juul, M. Münster, H. Ravn and M. Ljunggren Söderman, Waste Manage., 2013, 33, 1918–1925 CrossRef CAS PubMed
- R. E. Hester, R. M. Harrison, J. L. Gascoigne and S. M. Oglivie, Waste Treat. Disposal, 1995, 3, 91–114 Search PubMed
- C. L. Simoes, L. M. C. Pinto and C. A. Bernardo, Waste Manage. Res., 2014, 32(5), 414–422 CrossRef CAS PubMed
- P. Usapein and O. Chavalparit, Waste Manage. Res., 2014, 1–9, DOI:10.1177/0734242×14533604
- S. Kumar, A. K. Panda and R. K. Singh, Resour. Energy Econ., 2011, 55, 893–910 Search PubMed
- B. Baytekin, H. T. Baytekin and B. A. Grzybowski, Energy Environ. Sci., 2013, 6(12), 3467–3482 CAS
-
(a) M. Punčochář, B. Ruj and P. K. Chatterj, Procedia Eng., 2012, 42, 420–430 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2015 |