
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-heme iron catalysts for epoxidation and aziridination reactions of challenging terminal alkenes: towards sustainability†
Anja
Fingerhut
,
Olga V.
Serdyuk
and
Svetlana B.
Tsogoeva
*
Department of Chemistry and Pharmacy, Organic Chemistry Chair I and Interdisciplinary Center for Molecular Materials (ICMM), University of Erlangen-Nürnberg, Henkestrasse 42, 91054 Erlangen, Germany. E-mail: svetlana.tsogoeva@fau.de; Fax: (+49)-(0)9131-85-26865; Tel: (+49)-(0)9131-85-22541
First published on 19th March 2015
Abstract
Through catalytic epoxidation and aziridination of olefins versatile synthetic intermediates and subunits of biologically active compounds and pharmaceuticals –epoxides and aziridines– can be obtained. The focus of this review is on recent advances in the epoxidation and aziridination of challenging terminal alkenes with respect to safety of solvents or reagents used and atom economy aspects, as well as scope and limitations of developed non-heme iron catalysts. The still large research potential for sustainable and environmentally attractive iron-based catalysts in enantioselective epoxide and aziridine synthesis is demonstrated.
 Anja Fingerhut | Anja Fingerhut was born in 1987 in Nuremberg, Germany. In 2007, she started with her academic studies in molecular science at the Friedrich-Alexander University (FAU) of Erlangen-Nuremberg. She carried out her Master Thesis in the working group of Professor S. B. Tsogoeva, before she graduated in 2012 with M.Sc. from the FAU. In the same year she started with her PhD studies in the Tsogoeva group focusing on the development of new non-heme iron epoxidation catalysts and visible light absorbing systems. |
 Olga V. Serdyuk | Olga V. Serdyuk was born in 1980 in Anapa, Russia. She graduated from Rostov State University in 2002. In 2005 she received PhD degree under the supervision of Professor A.V. Gulevskaya and Professor A.F. Pozharskii. After postdoctoral studies (2007–2008) in Leibniz Institute of Catalysis (Rostock, Germany), she moved to Southern Federal University (Rostov-on-Don, Russia). Since 2011, she has been a research associate in the group of Professor S.B. Tsogoeva the Friedrich-Alexander-University of Erlangen-Nuremberg. Her research interests include synthesis and modification of heterocyclic compounds, fluoroorganic chemistry, homogeneous catalysis, organocatalysis. |
 Svetlana B. Tsogoeva | Svetlana B. Tsogoeva was born in 1973, studied chemistry at St. Petersburg State University, where she completed her doctoral thesis in 1998. Then, she moved to the Johann Wolfgang Goethe-University, Frankfurt am Main, for a postdoctoral research. In July 2000 she joined the Degussa AG Fine Chemicals Division as a research scientist. In January 2002 she was appointed a first junior professor in Germany at the Georg-August-University of Göttingen. Since February 2007, she is professor of organic chemistry at the Friedrich-Alexander-University of Erlangen-Nuremberg. Her research is currently focused on asymmetric organocatalysis, asymmetric oxidations with non-heme iron complexes and synthesis of natural product hybrids for medicinal chemistry. |
Introduction
High on the list of existing challenges in organic chemistry is the development of ecologically and economically attractive and sustainable new catalytic synthetic approaches towards epoxides and their nitrogen equivalents aziridines, especially chiral representatives, which were found to be highly useful intermediates and building blocks widely used for the synthesis of fine chemicals and pharmaceuticals.1–3 These strained three-membered heterocycles offer versatile synthetic possibilities for modifications and generation of bioactive compounds and chiral drugs via ring-opening,4 ring-expansion,5 and intermolecular rearrangement reactions.6 The ring opening can take place with a broad range of nucleophiles with high or complete stereo- and regioselectivity (Fig. 1).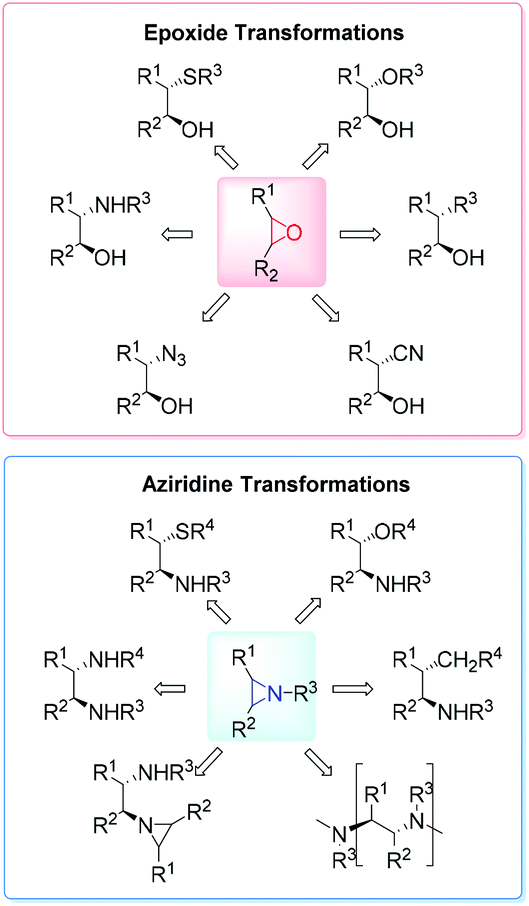 | ||
| Fig. 1 Broad range of nucleophilic ring-opening reactions of epoxides and aziridines. | ||
The 1,2-difunctional product itself represents common motifs in many important organic compounds. The general pharmaceutical relevance of epoxides and aziridines is proven by the fact that they found numerous application as subunits in biologically active compounds like antitumor agents,7,8 antibiotics,9 and enzyme inhibitors.10 Even naturally occurring aziridines and epoxides were found to be active against bacteria or tumor cells (Fig. 2).10,11 Hence, it is common sense that atom efficient and sustainable synthetic strategies towards epoxides and aziridines are very desirable.
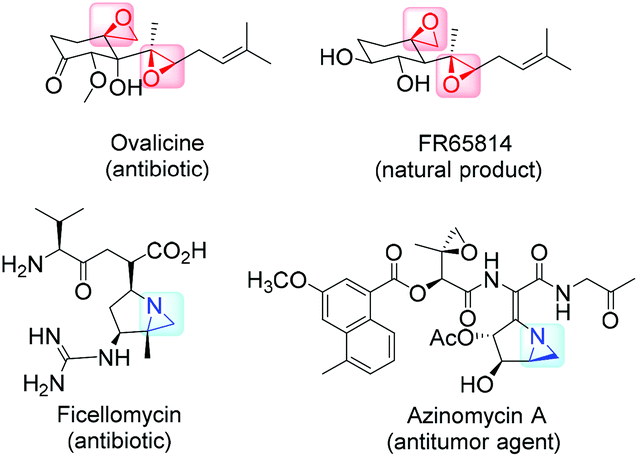 | ||
| Fig. 2 Epoxides and aziridines as part of natural products and bioactive compounds. | ||
The most venerable approaches in epoxide and aziridin synthesis use olefins as precursors, which are oxidized during oxygen or nitrogen transfer. Compared to aziridines, epoxides (both racemic and enantiopure) have been handled as desirable target compound for years. Especially concerning enantioselective catalysis, there are fewer methods for aziridination of alkenes comparing with epoxidation.12,13
Several efficient methods towards epoxides have been developed, including enantioselective Sharpless epoxidation of allylic alcohols with titanium-tartrate complexes,14,15 the Katsuki–Jacobsen epoxidation of unfunctionalized alkenes by chiral manganese-salen complexes16,17 and the enantioselective organocatalytic Shi epoxidation.18 But also aziridines attract increasing attention during the last decade due to their use in preparation of amino acids,19,20 β-lactams,21,22 polymers,23 and α-amido ketones.24 The easiest access to aziridines is a direct preparation via nitrogen atom transfer to olefins catalyzed by transition metal complexes.10 Already established catalytic systems employ manganese, copper, ruthenium, silver, gold, rhodium, and cobalt complexes.25,26
The main driver for extensively continued research activities and efforts to develop new more sustainable and economically more attractive and green processes is the high versatility of epoxides and aziridines for the synthesis of drugs, natural products and their intermediates (Fig. 1 and 2). In this context, iron-based catalysts receive increasing attention in enantioselective and non-enantioselective epoxidation and aziridination because they are inexpensive, readily available from earth-abundant element “Fe” and less toxic compared to other well established metal catalysts.
The iron-catalyzed procedures can be divided into biomimetic ones (orientated close to nature) and those with no enzymatic models. Analogously to natural models, iron containing enzyme-mimetic catalysts can be classified into heme and non-heme iron complexes. In the last three decades, biomimetic approaches to oxidation reactions mostly have concentrated on synthetic heme (porphyrin) iron complexes and their interactions with oxygen donors such as iodosobenzene, peracids and hydroperoxides.27,28
However, the active iron-porphyrin catalyst systems have not reached the level to achieve high enantioselectivities. While in particular moderately enantioselective heme iron containing oxidation catalysts are already well investigated, relatively less work has yet been devoted to developing non-heme iron complexes as oxidation and/or aziridination catalysts. This might be explained by the fact that non-heme iron active sites are more difficult to study because they do not exhibit the intense spectral features characteristic of the porphyrin ligand. However, non-heme iron complexes containing ligands with fine tunable and easily variable structural and electronic properties (in contrast to porphyrin-based systems) possess of particular appeal for the development of new highly active and enantioselective catalysts.
Application of non-heme iron catalysts might, therefore, provide an economical and environmentally friendly solution to epoxidation/aziridination and is of particular interest for future large scale industrial applications.29–31 Literature provides a range of efficient examples for both target compounds. Nonetheless, a generally applicable non-heme iron catalytic system for all classes of olefins with special attention to challenging substrates like terminal, electron deficient and highly substituted olefins has not yet been developed.29,32,33
In this review the considerable current interest in iron for the development of a generally applicable, highly efficient epoxidation and aziridination catalytic systems is pointed out first. Subsequently, the recent advances and remaining challenges (with regard to sustainability of processes, safety of solvents or reagents used and atom economy aspects) in non-heme iron catalysis towards epoxides and aziridines starting from olefins with a special focus on terminal alkenes are discussed.
Iron – a promising all-rounder
In the last several years considerable efforts have been made to enrich the chemistry of synthesis with more economical and environmentally friendly pathways and taking into account the principles of “Green Chemistry”.34 In order to reach a high level in sustainability, efficiency and selectivity, catalysts are applied in about 80% of all syntheses of chemicals and pharmaceutical compounds in industry.35 Organometallic compounds, especially containing palladium, rhodium, iridium and ruthenium are well established catalysts for a broad spectrum of reactions. Unfortunately, the named precious metals bear several disadvantages like toxicity and rareness leading to high costs, which are great problems especially for large scale applications in industry.36 In order to avoid these problems, the chemical community started to look around for new promising catalysts for example first row transition metal complexes which incorporate iron, copper, zinc or manganese. Among these, iron is the most beneficial one due to the fact that it is the second most abundant metal in the earth crust (4.7 wt%). Furthermore, as a most inexpensive and relatively nontoxic metal, iron meets the economically and environmentally requirements the community requests.35,37 The easy accessibility of iron is also demonstrated by the fact that many iron salts and complexes are either commercially available38,39 or the synthesis are described in literature.40 Noteworthy is the fact, that iron is involved and thus indispensable in many biological systems, like metalloproteins. These natural and highly efficient catalysts, for example metabolistic iron systems for activating oxygen, can be used as blueprint for designing new iron complexes. In order to get mechanistic information of these natural catalysts for the design of ‘bioinspired’ or ‘biomimetic’ complexes, studies like X-ray crystallography and spectroscopic characterizations of the active site are carried out.41 With its strong Lewis acidity and a facile change of the oxidation state, iron can be applied to a broad range of enantioselective and non-enantioselective catalytic reactions summarized in several excellent reviews: additions, substitutions, cycloadditions, hydrogenation, reductions, oxidations, coupling reactions, isomerizations, rearrangements and polymerizations.29,32,35,36,42 The reactivity and selectivity of the iron complex is influenced by the surrounding ligand, which implies clarification of relationships between structure and action and leads to the rational design of catalysts. The combination of iron center and sterically and electronically tuned chiral ligands might therefore result in development of efficient and low cost catalysts for enantioselective synthesis.43,44However, iron based homogeneous catalysis, especially enantioselective processes, still requires further advancement leading to a break-through. Further efforts in tuning iron catalysts concerning handling, stability, activity and selectivity have to be made to bring the catalytic systems closer to large scale industrial application. Herein we discuss numerous non-heme iron catalytic systems for epoxidation and aziridination and their attempt to meet the existing challenges with a special focus on enantioselective and non-enantioselective transformation of terminal alkenes as challenging substrates.
Prior to this, the main breakthroughs in epoxidation and aziridination reactions with enantioselective non-heme iron based catalysts are highlighted to get insight into the great potential and the ability to keep up with other highly efficient but less economically and ecologically benign catalytic systems.
Highlights in enantioselective non-heme iron catalyzed epoxidation and aziridination
One of the early approaches towards enantioselective epoxidation was presented by Jacobsen and co-workers in 1999.45 They demonstrated the epoxidation of trans-β-methylstyrene to its corresponding oxide as a sole product using polymer-supported non-heme iron complexes as catalysts and hydrogen peroxide as a simple, inexpensive and environmentally benign oxidizing agent (Scheme 1).46,47 Notably, the only side-product when using H2O2 as oxidant is a water molecule. While the oxidizing agent applied for this reaction is very attractive and desirable, the solvent used (dichloromethane) is, unfortunately, a suspected human carcinogen.48 | ||
| Scheme 1 | ||
The three most active complexes were identified from a library of 5760 compounds which are combinations from 192 different chiral peptide-like ligands anchored on polystyrene and a variety of metal ion sources. The most promising ligand-iron(II) combination led to 20% enantiomeric excess and a high conversion of 78%, which could not be reached up to that time (Scheme 1).
One of the main breakthroughs concerning enantioselective iron catalyzed epoxidation was presented by the Beller group in 2007.49 The simple reaction system works efficient with hydrogen peroxide and enables the in situ generation of a complex containing a chiral 1,2-diphenylethylenediamine derivative as ligand and pyridine-2,6-dicarboxylic acid (H2Pydic) as co-ligand. The most active in situ formed complex was applicable to a broad substrate scope containing several aromatic non-terminal olefins and gave yields up to 94% and ee-values up to 91% (Scheme 2). Yield and enantioselectivity showed high variation, strongly depending on substrate symmetry and bulkiness.
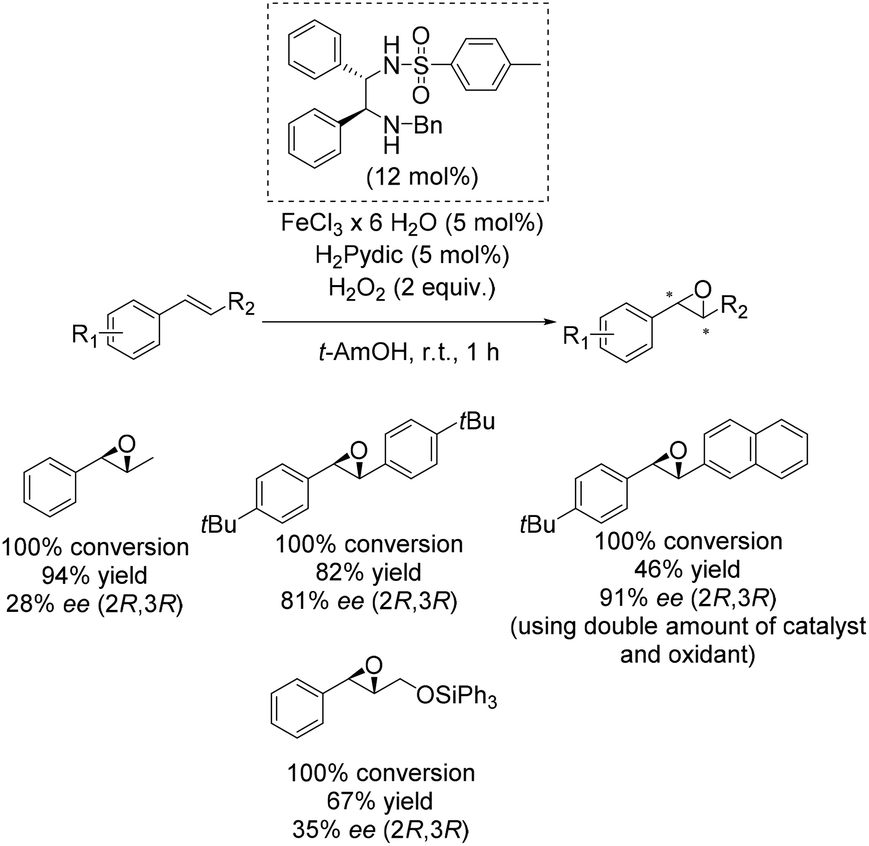 | ||
| Scheme 2 | ||
An excellent and also the first enantioselective catalytic system for epoxidation of β,β-substituted enones was reported in 2011 by Yamamoto and co-workers.50 They presented an in situ generated iron complex derived from Fe(OTf)2 and two equivalents of an axially chiral biaryl moiety linked to phenanthroline. A screening of several ligands which differ in substituents of the phenanthrolin moiety found the ligand depicted in Scheme 3 to form the most efficient catalyst. A broad range of substrates could be tested successfully with yields up to 88% and ee-values up to 92% (Scheme 3). The complex which is also applicable to epoxidation of nonactivated substrates like trans-α-methylstilbene (50% yield, 87% ee) requires peracetic acid as an oxidizing agent. Competitive experiments elucidated the electrophilic nature of the active oxidant species.
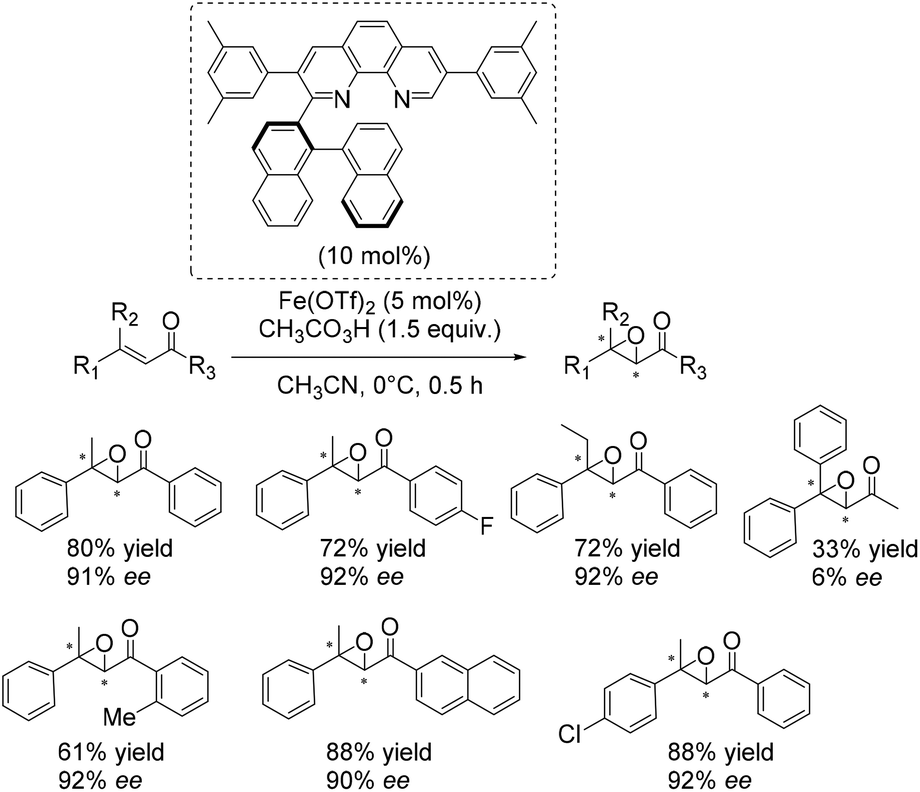 | ||
| Scheme 3 | ||
In the same year, the group of Sun suggested bioinspired chiral iron(II) complexes with N4 ligands for the enantioselective epoxidation of α,β-unsaturated ketones, employing hydrogen peroxide as an oxidant and acetic acid as additive, which assists in the O–O cleavage step.51 The ligands were based on a chiral ethylenediamine backbone combined with two pyridine moieties. Besides, they proposed that the intermediate LFe(V)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is the main catalytic active species formed after a reversible water binding step. Also a bridged complex [L2Fe(III)2(μ-O)(μ-CH3CO2)]3+ could be identified as intermediate during the reaction. Although the results concerning enantioselectivity (up to 87% ee) and yield (up to 90%) were promising, the application of the catalytic system was limited to α,β-unsaturated ketones. The enantioselective epoxidation of terminal olefins, e.g. styrene, was not possible. Further investigations were carried out and one year later Sun and co-workers introduced new chiral iron(II) complexes, containing ligands derived from proline and benzimidazole which a more rigid compared to their parent compounds.52 Using hydrogen peroxide as desirable oxidant, these complexes were found to be active in epoxidation of a broad range of multisubstituted enones. ee-values up to 98% in combination with good to very good yields could be reached (Scheme 4: best results for most promising iron(II)-complex).
O is the main catalytic active species formed after a reversible water binding step. Also a bridged complex [L2Fe(III)2(μ-O)(μ-CH3CO2)]3+ could be identified as intermediate during the reaction. Although the results concerning enantioselectivity (up to 87% ee) and yield (up to 90%) were promising, the application of the catalytic system was limited to α,β-unsaturated ketones. The enantioselective epoxidation of terminal olefins, e.g. styrene, was not possible. Further investigations were carried out and one year later Sun and co-workers introduced new chiral iron(II) complexes, containing ligands derived from proline and benzimidazole which a more rigid compared to their parent compounds.52 Using hydrogen peroxide as desirable oxidant, these complexes were found to be active in epoxidation of a broad range of multisubstituted enones. ee-values up to 98% in combination with good to very good yields could be reached (Scheme 4: best results for most promising iron(II)-complex).
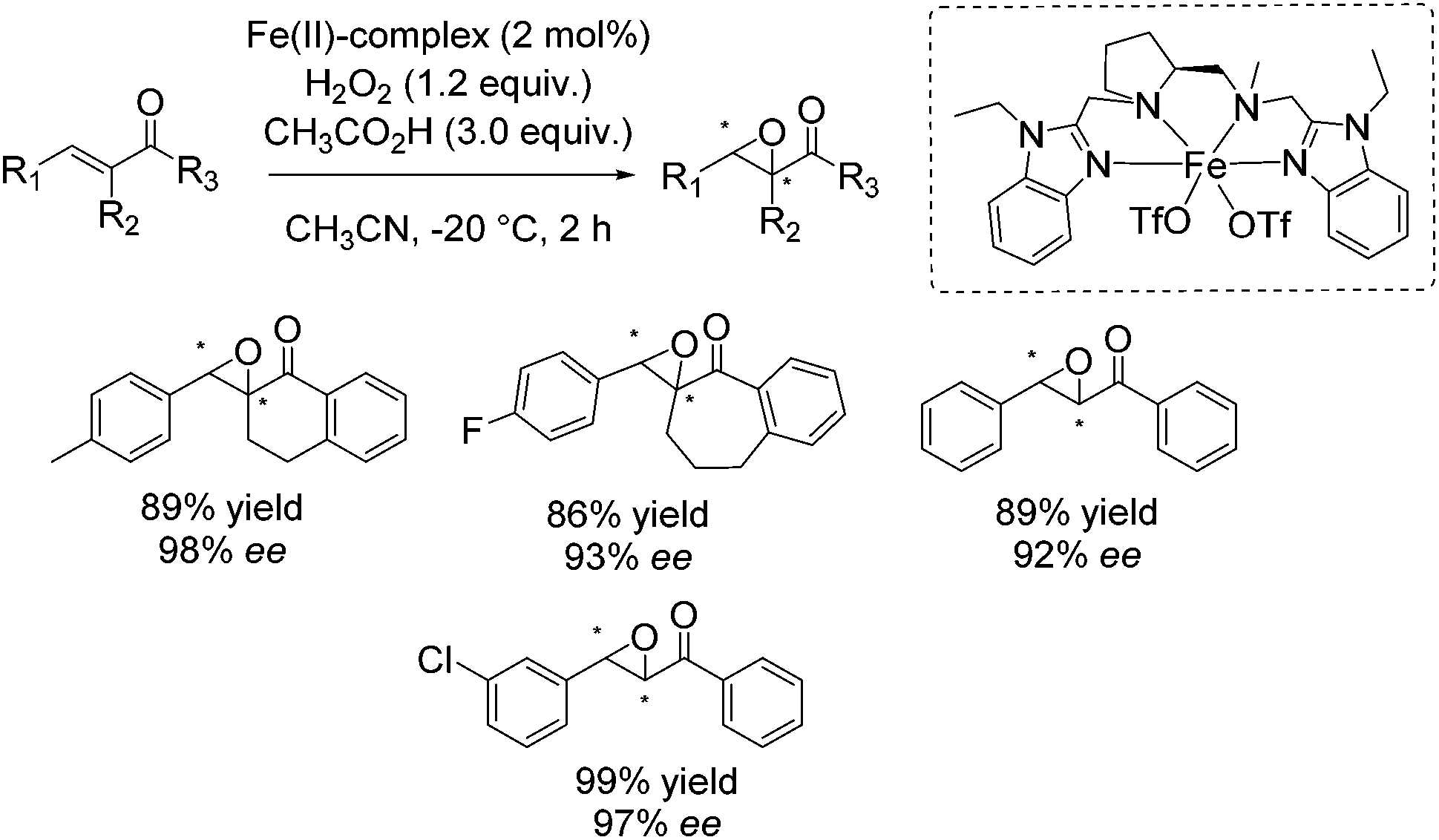 | ||
| Scheme 4 | ||
In 2013 Costas and co-workers presented several iron(II) complexes containing aminopyridine ligands for epoxidation reactions.53 Several electron donating or electron withdrawing groups were attached to the pyridyl moiety of the ligand and the influence on enantioselectivity and yield was studied. The best catalyst was found to be iron(II) catalyst depicted in Scheme 5, which was active with hydrogen peroxide as terminal oxidant for a broad substrate spectrum observing excellent enantioselectivities and very good yields. Carboxylic acid as for example (S)-ibuprofen or 2-ethylhexanoic acid (2-eha) was added in catalytic amount to the reaction mixture in order to assist in O–O cleavage favoring the formation of the electrophilic oxygen delivering iron species.
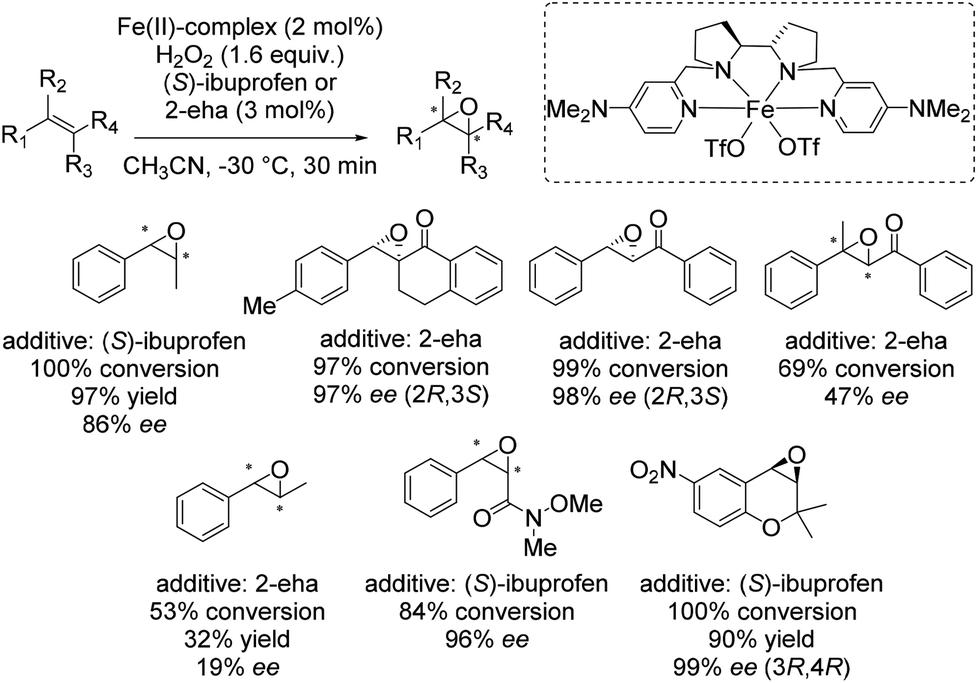 | ||
| Scheme 5 | ||
Literature provides several examples also for non-enantioselective non-heme iron catalyzed aziridination of aliphatic and aromatic, terminal and non-terminal alkenes. However, the examples for enantioselective non-heme iron catalyzed aziridinations remain rare. To the best of our knowledge, for several years there was just one approach, wherein enantioselective aziridination of styrene is demonstrated, which is presented in this review later.54
Notably, in 2013 Xu and co-workers reported their interesting studies concerning iron(II)-catalyzed intramolecular aminohydroxylation of olefins with functionalized hydroxylamines and presented herein their findings of intramolecular stereoselective aziridination as parallel reaction.55 On their way to explore enantioselective induction in hydroxyl oxazolidinone synthesis with chiral in situ formed Fe(II) bisoxazoline complexes as catalyst they obtained a chiral aziridine in 46% yield and an enantiomeric excess of 65% (Scheme 6). The sense of enantioinduction for both products is the same and they cannot interconvert. The aziridination is a competing pathway from the same intermediate. They postulated a mechanism for both reactions which involves an iron-nitrenoid species.
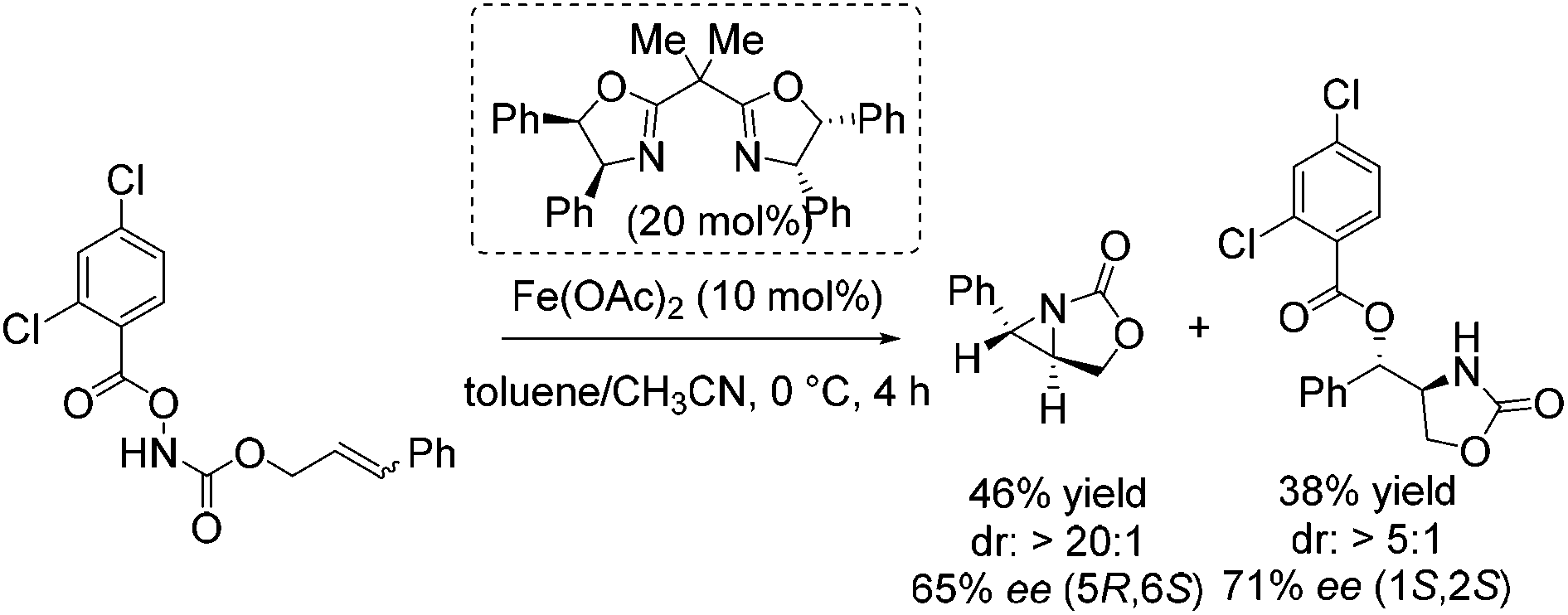 | ||
| Scheme 6 | ||
Non-heme iron-catalyzed epoxidation of terminal alkenes
During the last decades several approaches to epoxides via olefin oxidation with the use of non-heme iron complexes have been developed. However, the application of the reported methods to terminal aromatic and aliphatic alkenes provided predominantly moderate yields and selectivities and brought out racemic mixtures or products with low enantiomeric excesses. | ||
| Scheme 7 | ||
During their study on new synthetic analogues of the methane monooxygenase enzyme and hemerythrin, Que and co-workers developed in 1986 a synthetic binuclear iron complex activating the O–O bond for catalytic oxidation.57 The confacial bioctahedral complex (Me4N)[Fe(III)2(μ-L)(μ-CH3CO2)2] shows catalytic activity at only 0.1 mol% loading towards alkene epoxidation with hydrogen peroxide as oxidizing agent, but using DMF as a solvent. In general, DMF is known as a reproductive toxin48 and its replacement through less toxic to handle, more environmentally friendly and safe solvents in chemical processes is desirable.
The authors evaluated the capability of their new synthetic binuclear iron complex to catalyze epoxidation of alkenes. The catalyst was found to be active in styrene epoxidation under the selected reaction conditions with the turnover number of 3.2 (Scheme 8). Mechanistic investigations proposed that by the addition of hydrogen peroxide one bridging acetate is replaced and an active Fe(III)-peroxide complex is formed, while the C2 symmetry of the parent complex is destroyed.
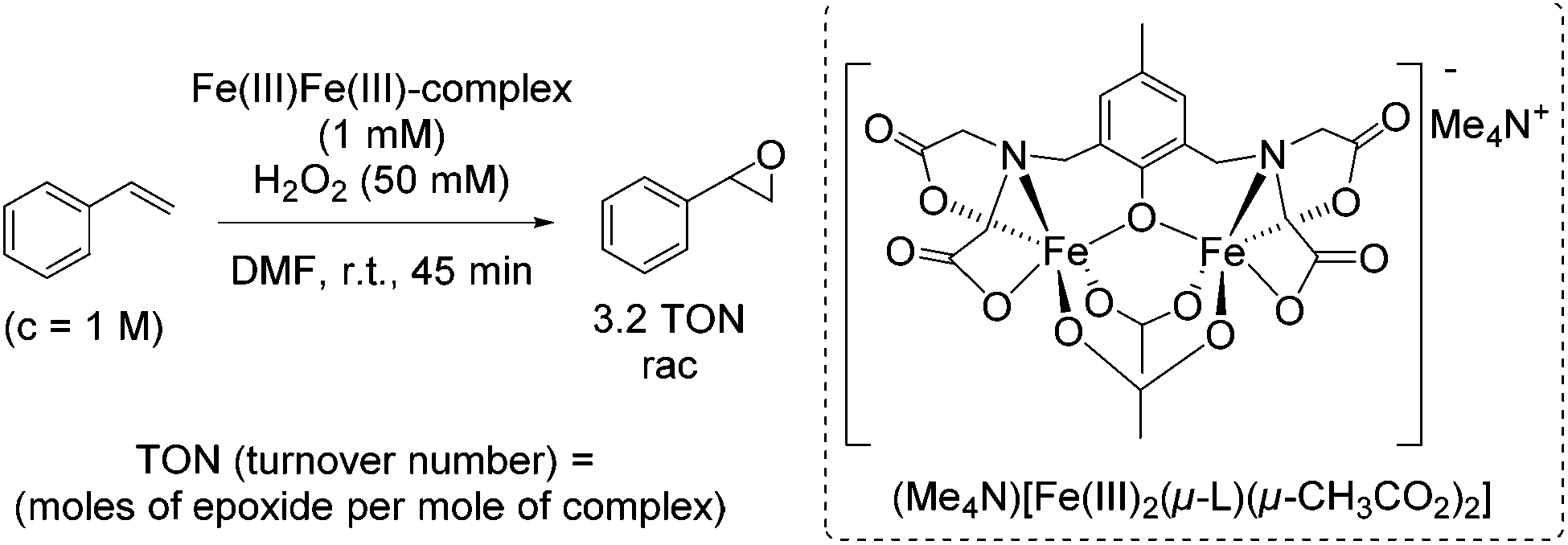 | ||
| Scheme 8 | ||
A couple of years later a further breakthrough was presented by Valentine and co-workers with a cyclam iron(II) complex.58 Acetonitrile, as a relatively less toxic solvent in comparison to DMF, was used. Further improvements could be observed applying this system compared to previously reported epoxidation systems: higher turnovers based on the complex, higher yields, tolerance for small amounts of water and stereospecificity. Furthermore, radical reactions could be excluded, because only small amounts of allylic oxidation product were formed. However, the epoxidation of the terminal alkene 1-octene gave a less promising yield of 14% (Scheme 9) compared to 40% yield of the cyclohexene oxide. Related cyclam ligands, presented in this study, showed lower activity for epoxidation reactions.
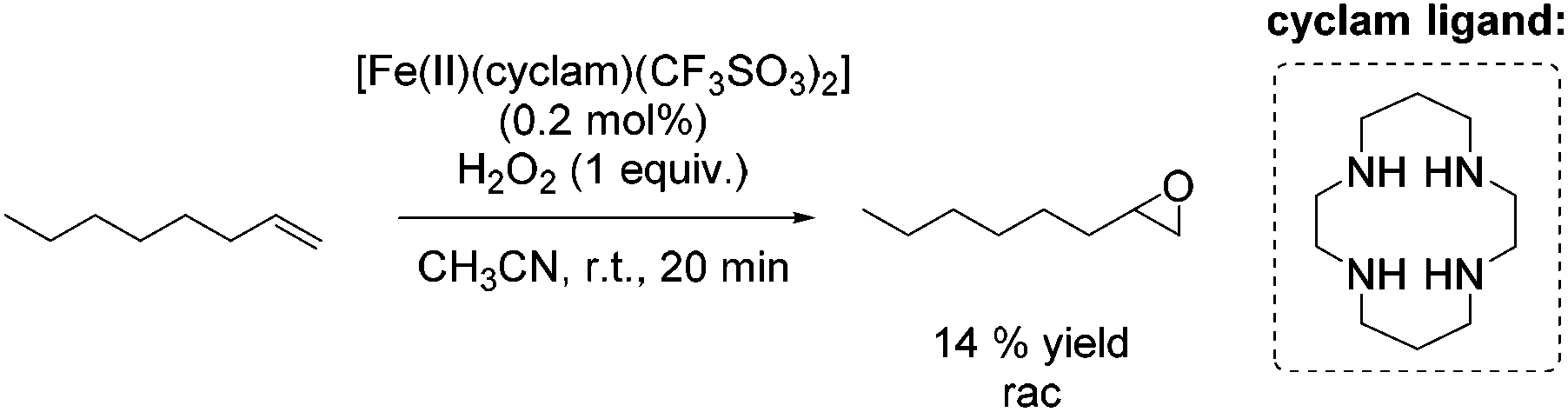 | ||
| Scheme 9 | ||
In 2001, the group of Jacobsen reported the first methane monooxygenase (MMO) model system which is applicable in preparative oxidation chemistry.59 The self-assembling μ-oxo, carboxylate bridged diiron(III) complex which is formed in situ from the mononuclear [Fe(II)(mep)(CH3CN)](SbF6)2 complex in the presence of acetic acid was proven to be highly active for a broad range of aliphatic alkenes. Notably, the ability of binuclear μ-oxo non-heme iron(III) complexes being highly active as catalyst in epoxidation reactions was previously reported in the literature.57,60 Subsequently, terminal alkenes were found by the Jacobsen group to undergo non-enantioselective epoxidation using μ-oxo, carboxylate bridged diiron(III) complex and hydrogen peroxide with yields of up to 90% within 5 minutes reaction time (Scheme 10). They demonstrated successfully the high influence of catalytic amounts of acetic acid on the selectivity of the reaction towards epoxide formation. This enabled lowering of catalyst loading to a minimum without losing selectivity. The used aminopyridine ligand represents a useful template for further chiral ligand design and is a good starting point for backbone tuning concerning rigidity. Both facts were demonstrated a few years later as we will discuss further later on in this review.
 | ||
| Scheme 10 | ||
In 2003, a μ-oxo-iron(III) dimer, [((phen)2(H2O)FeIII)2(μ-O)](ClO4)4, was suggested by Stack as an efficient achiral catalyst for epoxidation using peracetic acid as the oxidant (Scheme 11).61 The catalyst was in situ generated under aerobic conditions via dimerization of the ferric species in presence of water. The bridging oxide and the water ligands occupy the adjacent cis sites of each iron center similar to the self assembled diiron complex of Jacobsen (Scheme 10).59 The authors proposed an electrophilic nature of the active oxidant due to intermolecular competition studies. The substrates, ranging from electron poor to electron rich aromatic and aliphatic alkenes, required high concentration and very low catalyst loading in order to obtain promising yields. Aliphatic terminal alkenes allowed to obtain yields up to 96%. However, using styrene as a substrate 63% yield was obtained at full product conversion. Notably, allyl acetate was converted with only 8% yield (Scheme 11). In general, an upscale to gram quantities was successful without significant loss of efficiency.
 | ||
| Scheme 11 | ||
Four years later, Beller presented the first non-heme iron catalytic system which allows epoxidation of aliphatic and aromatic olefins with hydrogen peroxide as the terminal oxidant under neutral conditions.62,63 During their investigations, starting with a PyBOX ligand and FeCl3·6H2O as metal source for the stilbene epoxidation as a model reaction, they found a catalytic active species generated from decomposition products of the PyBOX ligand, identified as pyridine-2,6-dicarboxylic acid (H2Pydic) and an amino alcohol. Inspired by these results, several amines were screened and pyrrolidine was found to be the most effective one in presence of H2Pydic, both acting as ligand of the iron center. In presence of a radical scavenger very low epoxide yield was observed, which indicates a selective radical pathway of the reaction. The chosen simple environmentally benign in situ generated catalyst system showed activity for epoxidation of aromatic and aliphatic olefins. Styrene and styrene derivatives with electron donating and electron withdrawing substituents were oxidized with yields up to 97% with good to excellent selectivity. Furthermore, epoxidation of sterically challenging aromatic olefins like 2-vinylnaphthalene and α-methylstyrene gave up to 64% yield with moderate selectivity (Scheme 12). No terminal aliphatic alkenes were tested.
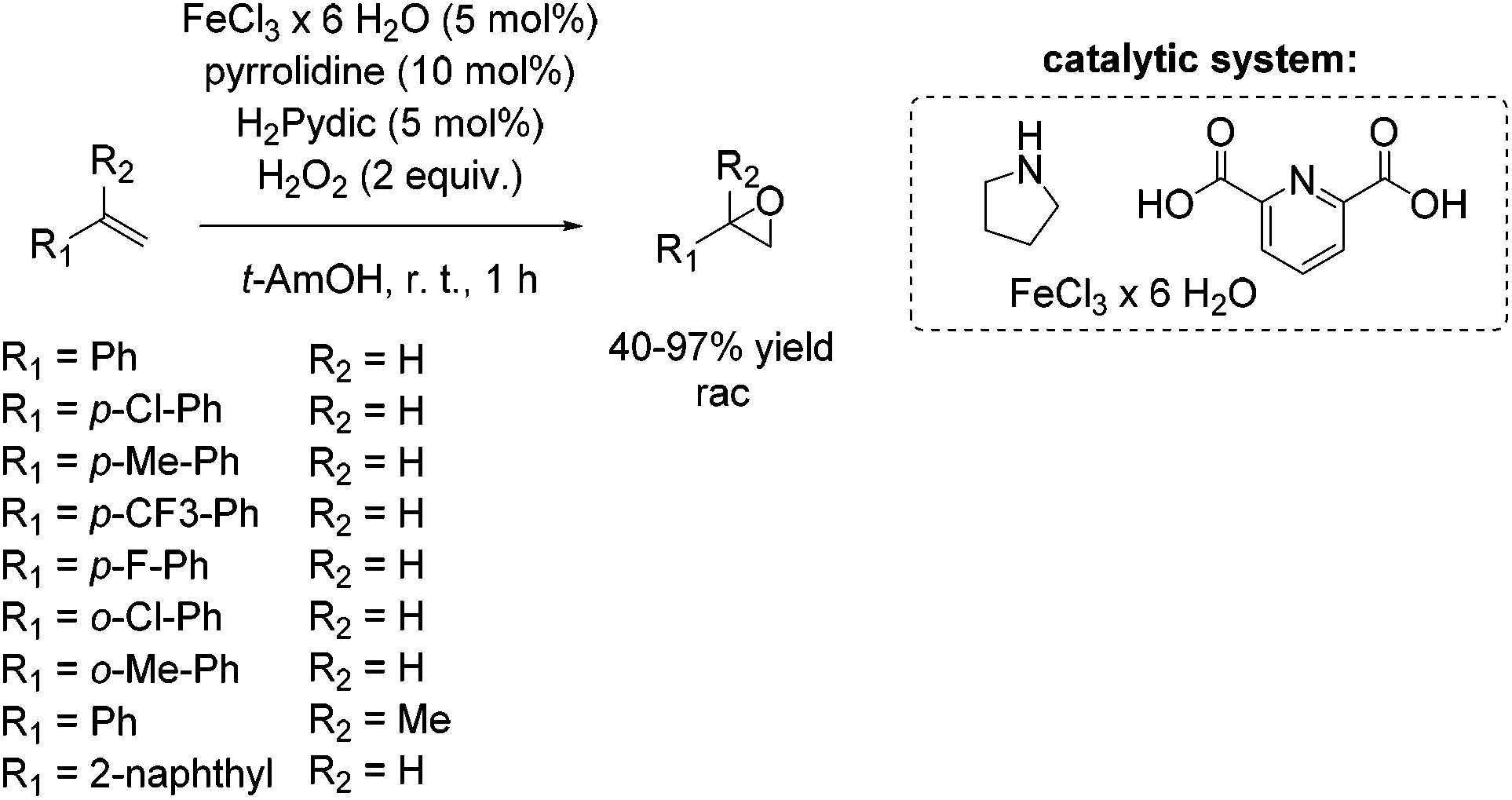 | ||
| Scheme 12 | ||
In 2008, Dilworth and co-workers presented new iron(III) complexes with asymmetric N-capped tripodal NO3 ligands and a pentadentate N2O3 ligand with catalytic activity for epoxidation reactions.64 Despite modest conversion and without transferring enantioselectivity to epoxides, the complexes were applicable for the epoxidation of styrene (Scheme 13). The different electronic and steric properties of the tripodal ligands did not lead to significant changes in conversion after even 24 h. The authors proposed a six- or higher-coordinate metastable intermediate formed during the reaction, because of the comparable results towards epoxidation applying the five coordinated complex bearing no co-ligand (1-methylimidazole) and complexes with tripodal ligands.
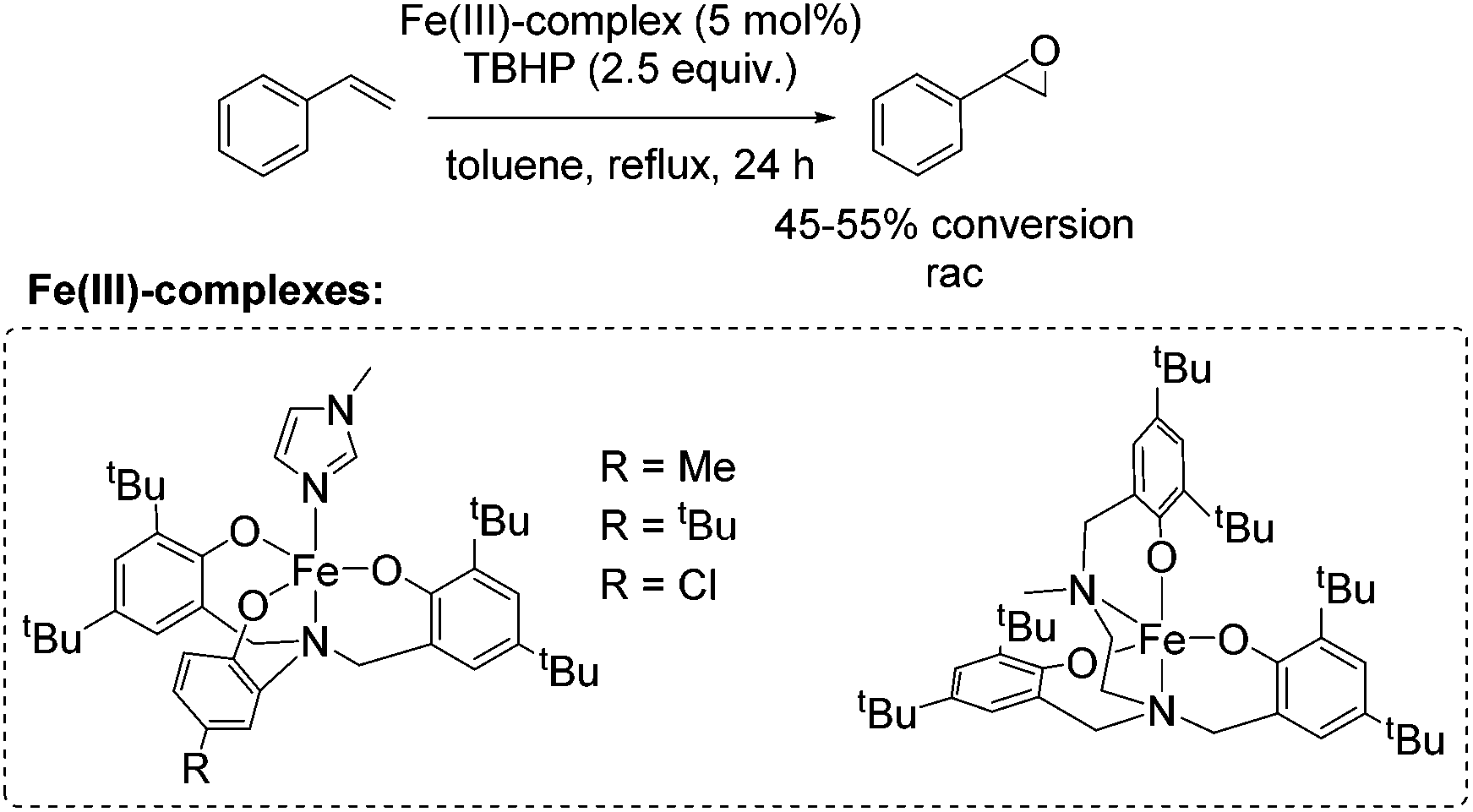 | ||
| Scheme 13 | ||
In the same year, Che and co-workers demonstrated the catalytic potential of a complex readily generated in situ from an iron(II) salt (Fe(ClO4)2·6H2O) and Cl3terpy (4,4′,4′′-trichloro-2,2′:6′,2′′-terpyridine) ligand.65 The catalyst was found to be efficient in epoxidation of alkenes using Oxone as an oxidant and CH3CN as solvent. The substrate scope ranges from aromatic and aliphatic terminal alkenes (with up to 92% yields, Scheme 14) to several symmetrical and non-symmetrical non-terminal alkenes containing an electron rich or an electron deficient double bond also with moderate to very good yields. Via ESI-MS measurements, the authors could detect reactive high-valent FeO intermediates, which are responsible for oxygen atom transfer.
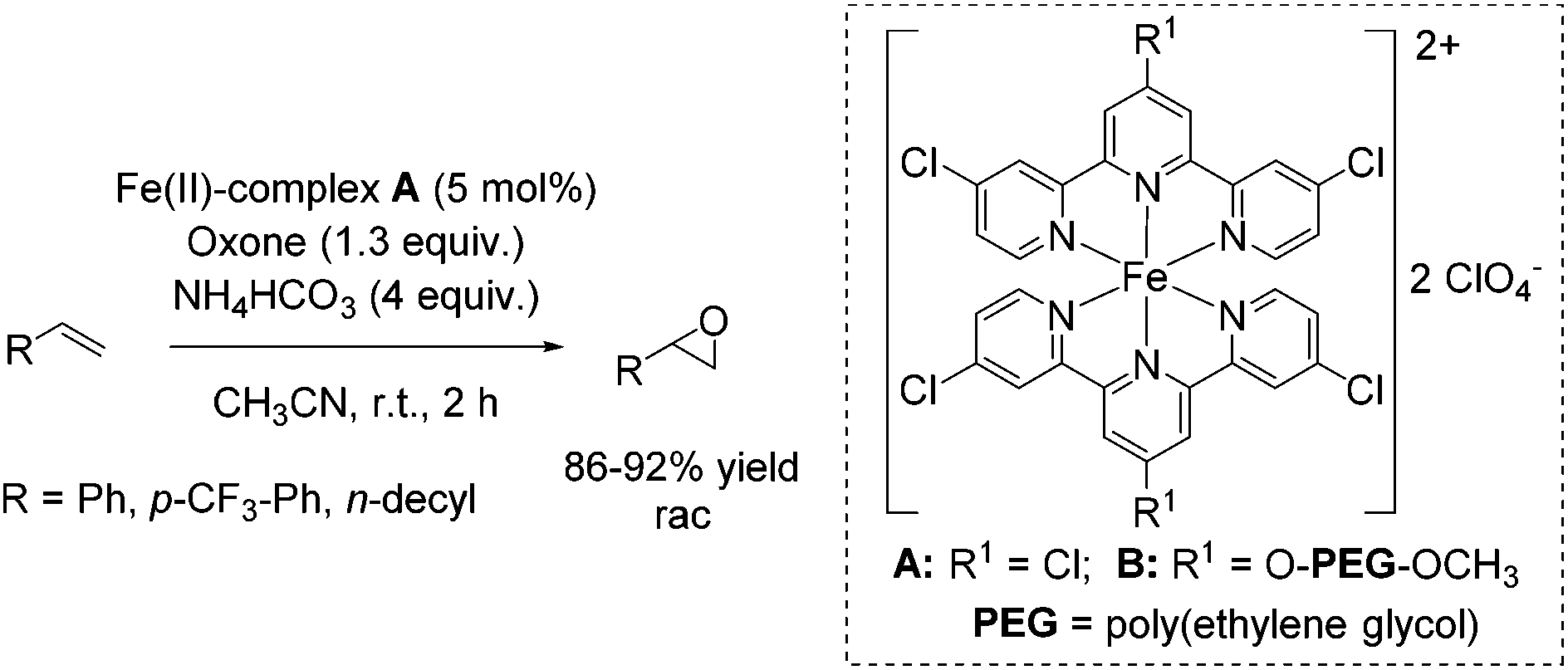 | ||
| Scheme 14 | ||
The Cl3terpy ligand was found to be oxidatively robust and could be reused (without isolation) for catalysis by addition of a new portion of Fe(ClO4)2·6H2O, as well as the oxidant and substrate to the reaction mixture. In order to make the process even “greener”, the ligand was derivatized forming a polymer-supported O-PEG-OCH3-Cl2terpy ligand (Scheme 14). Its corresponding Fe(II) complex B catalyzed the epoxidation of chalcone in 96% yield. Notably, polymer-supported ligand can be separated from the reaction product by diluting the reaction mixture with H2O followed by Et2O extraction. The O-PEG-OCH3-Cl2terpy ligand left in the aqueous layer can be further reused without a significant loss of catalytic activity.
Taking into accounts the importance of reusable catalysts and/or ligands on development of clean and recyclable synthetic methodologies, it is safe to say that the immobilization of catalysts/ligands on polymers66,67 represents an attractive and practical protocol for “Green Chemistry”.
The development of an environmentally benign non-heme iron catalyst for epoxidation which can be applied to all classes of olefins is one of the main challenges. Since the already mentioned promising pyrrolidine/iron/H2Pydic system of the Beller group (Scheme 12) showed low reactivity for aliphatic and highly substituted aromatic olefins,62,63 in 2008, Beller and co-workers reported an improved catalytic system as efficient in epoxidation of aliphatic olefins as for aromatic olefins using hydrogen peroxide oxidant.68 Pyridine-2,6-dicarboxylic acid (H2Pydic) as ligand and benzylamine derivatives as coligand play a key role in this simple and practical system regarding conversion and selectivity. In general, yields of up to 91% for aromatic olefins and 99% for the aliphatic ones could be reached and demonstrated comparable activity of the catalytic system for both olefin classes. Among di-, tri- and tetrasubstituted aromatic alkenes, styrene oxide was obtained with 91% yield at high selectivity. The poor reactive terminal aliphatic alkenes reached just yields up to 58% but at high selectivity (Scheme 15).
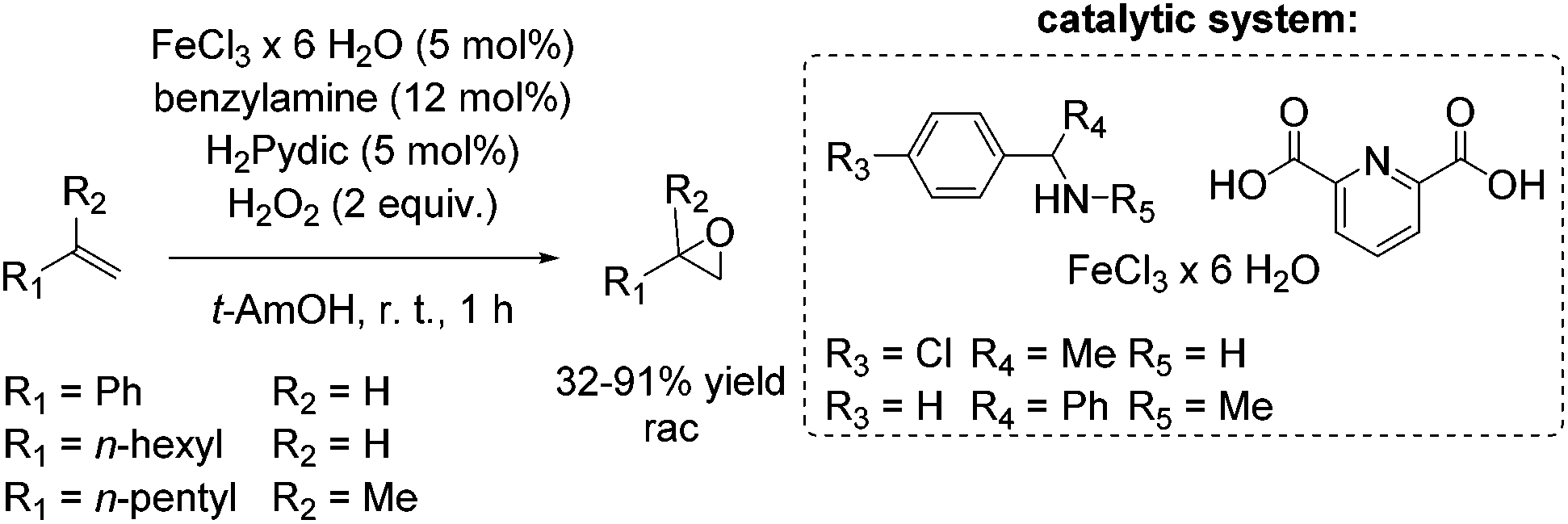 | ||
| Scheme 15 | ||
Based on previous results, Beller and co-workers aimed to improve their epoxidation system using biomimetic iron-imidazole catalysts.69 Already in 2007, the Beller group reported a selective and easily manageable epoxidation method using FeCl3·6H2O and imidazole derivatives as catalyst and hydrogen peroxide as oxidant. A screening of a broad range of aromatic substrates applying 5-chloro-1-methylimidazole brought out yields up to 87% (trans-stilbene oxide). However, aliphatic olefins remained unreacted. Yields for styrene and styrene derivatives range between 46 and 82% at high selectivities. Based on additional experiments the authors suggest the formation of a radical in the catalytic cycle of this epoxidation system.70 Several efforts have been made to develop an even more efficient iron-imidazole system and to apply the system for epoxidation of aromatic and aliphatic epoxidation with comparable results as already shown for the three component system FeCl3·6H2O/H2Pydic/benzylamine.68 Although experiments demonstrated a correlation between acidity and catalyst activity, no further additive appeared in the modified system. After screening a range of imidazole derivatives as ligand, 1-(2,6-diisopropylphenyl)imidazole was found to carry out the most promising results (Scheme 16).69 Applying 1-(2,6-diisopropylphenyl)imidazole as ligand aromatic terminal alkenes were obtained with yields up to 88% at high selectivity. The less reactive terminal aliphatic alkenes reached yields up to 53% at high selectivity. With these results, the modified biomimetic imidazole-iron system is slightly inferior to the FeCl3·6H2O/H2Pydic/benzylamine system.68 Mechanistic investigations demonstrated that the mononuclear iron(III) species, which were in situ formed, are in equilibrium with an μ-oxo diiron(III) complex in presence of water.69
 | ||
| Scheme 16 | ||
Another method towards catalytic epoxidation of olefins was reported using FeCl3·6H2O and 1-methylimidazole with hydrogen peroxide as an oxidant.71 Inspired by the results obtained by Beller and co-workers,70,72 the group of Kozak71 carried out a similar epoxidation under modified reaction conditions replacing tert-amyl alcohol with acetone as new solvent and raising the temperature to 62 °C. With these modifications Kozak and co-workers could apply 1-methylimidazole as a ligand, avoiding the use of a chlorinated imidazole derivatives. They obtained yields of trans-stilbene oxide in the same range as previously reported by Beller using 5-chloro-1-methylimidazole ligand. Noteworthy, while acetone bears some advantages over other solvents which are typically employed (like carcinogenic dichloromethane, toxic acetonitrile and more expensive tert-amyl alcohol), its combination with hydrogen peroxide can lead to explosive acetone peroxides. This reaction, therefore, could not be carried out safely on a large scale. The method allows performing non-enantioselective epoxidation of di- and trisubstituted aromatic and aliphatic alkenes and styrene derivatives on a small scale in moderate to good yields (Scheme 17).
 | ||
| Scheme 17 | ||
In 2011, Beller and co-workers described a selective non-enantioselective iron-catalyzed oxidation of olefins to epoxides using air73 as a clean and sustainable oxidant.74 Bio-inspired β-keto ester (2-oxocyclohexanecarboxylate) was employed as a sacrificial co-substrate, and oxygen transfer occurred highly selectively to produce racemic epoxides at room temperature. The mechanism of O2 activation was reported to be similar to the activation taking place in co-substrate-dependent non-heme iron oxygenases. The authors propose that the active catalytic complex corresponds to [Fe(III)(β-keto ester-H)2(imidazole)]+ (Scheme 18) and envisioned iron superoxide as the oxidizing species. Non-terminal alkenes, especially symmetric ones, gave very high conversion up to 99% with high selectivity towards epoxide formation. Although non-terminal and terminal alkenes react at comparable rates, the reaction of the latter proceeds with lower chemoselectivity leading to yields of up to 50% (Scheme 18). Noteworthy, after 20 hours reaction time the catalyst system is not deactivated and can be reused by adding further substrate and co-substrate. Nonetheless, promoting the practical application of the developed method remains a challenge because of the requirement of sacrificial co-substrate (2 equiv.) and, therefore, low atom economy of the process.
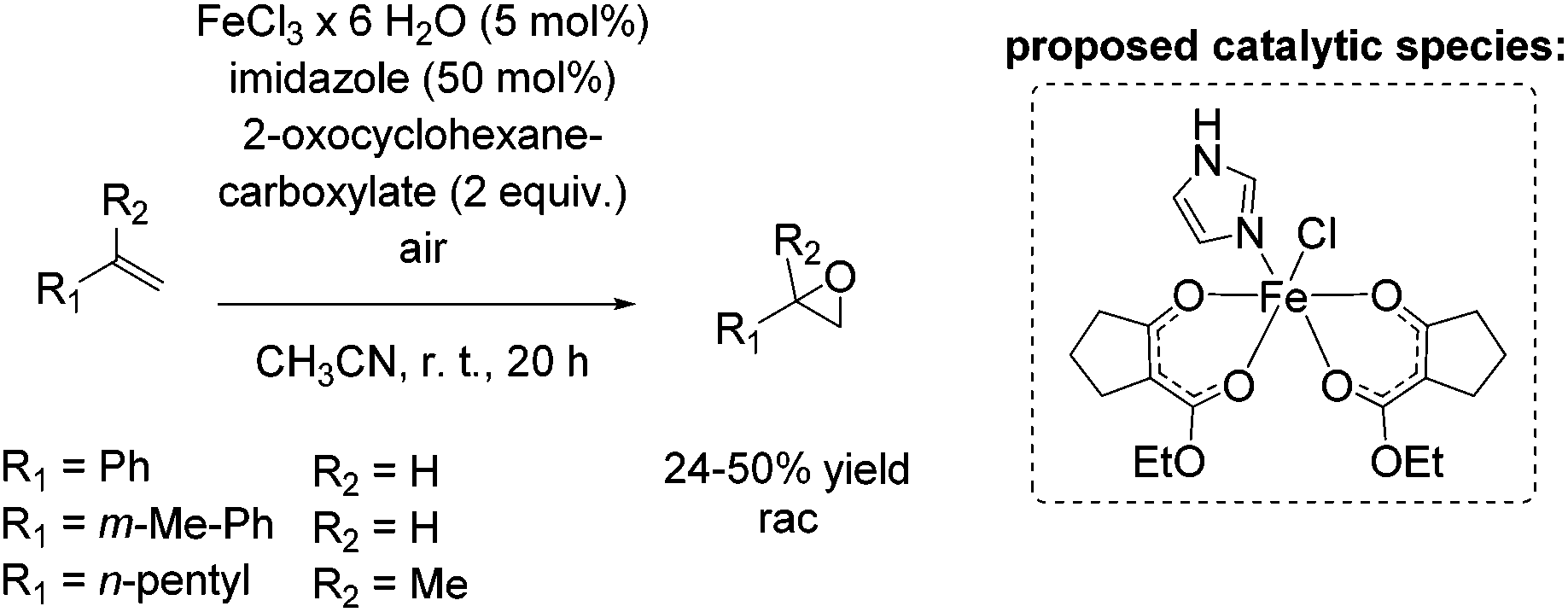 | ||
| Scheme 18 | ||
Another FeCl3 catalyzed non-enantioselective epoxidation of alkenes with hydrogen peroxide was reported by Ishizuka and co-workers.75 The authors propose an iron(III)-pyridine complex as in situ generated catalyst, which shows greater activity than for example the iron(III)-(N-methyl)imidazole complex. However, pyridine is applied in high excess (2 equiv.) to the iron salt, reducing in overall the atom efficiency of this process. The simple catalytic system was found to be applicable to aromatic and aliphatic olefins for the synthesis of the corresponding racemic epoxides in poor to very good yields. A terminal alkene was oxidized by this method to produce racemic epoxide in 70% yield (Scheme 19).
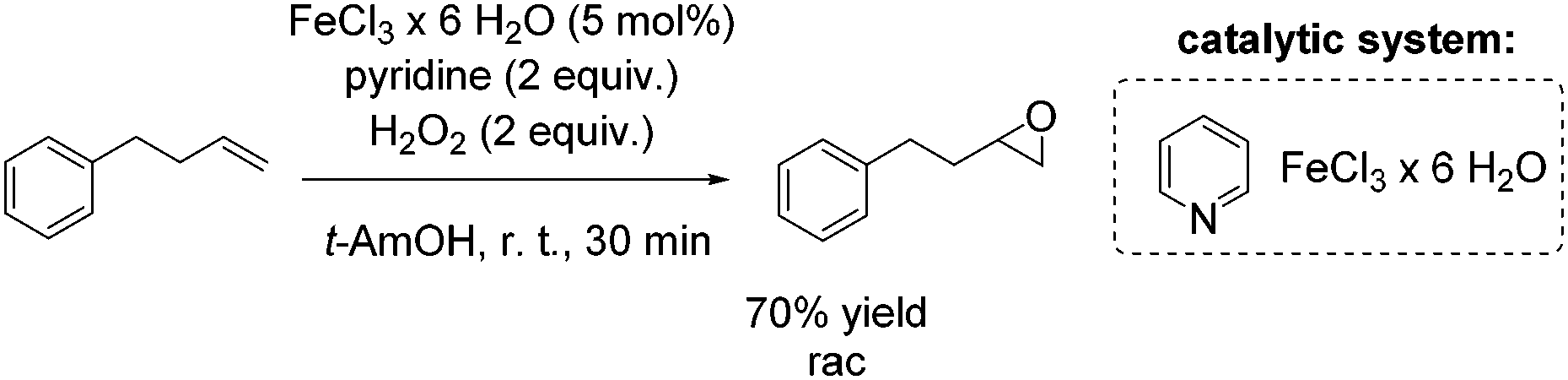 | ||
| Scheme 19 | ||
A further approach towards non-enantioselective epoxidation of terminal alkenes was demonstrated by Che and co-workers76 in 2011, based on previous studies reported in 2008 applying 4,4′,4′′-trichloro-2,2′:6′,2′′-terpyridine iron complexes for epoxidation (Scheme 14).65 They reported several oxidation reactions catalyzed by iron complexes with robust and reusable quinquepyridine, sexipyridine and septipyridine ligands. A variety of electron-rich and electron-deficient alkenes were epoxidized using oxone as oxidant. Among a wide range of substrates, using these new oligopyridine high spin iron(II) complexes (Scheme 20) the yields of tested terminal alkenes styrene, trifluoromethyl styrene derivative and 1-dodecene were located in the same area as already demonstrated with 4,4′,4′′-trichloro-2,2′:6′,2′′-terpyridine iron complexes (Scheme 14).65 Further investigations demonstrated a pH dependent epoxidation reaction tested for styrene oxidation. The highest yield of the corresponding epoxide was obtained at pH 7.
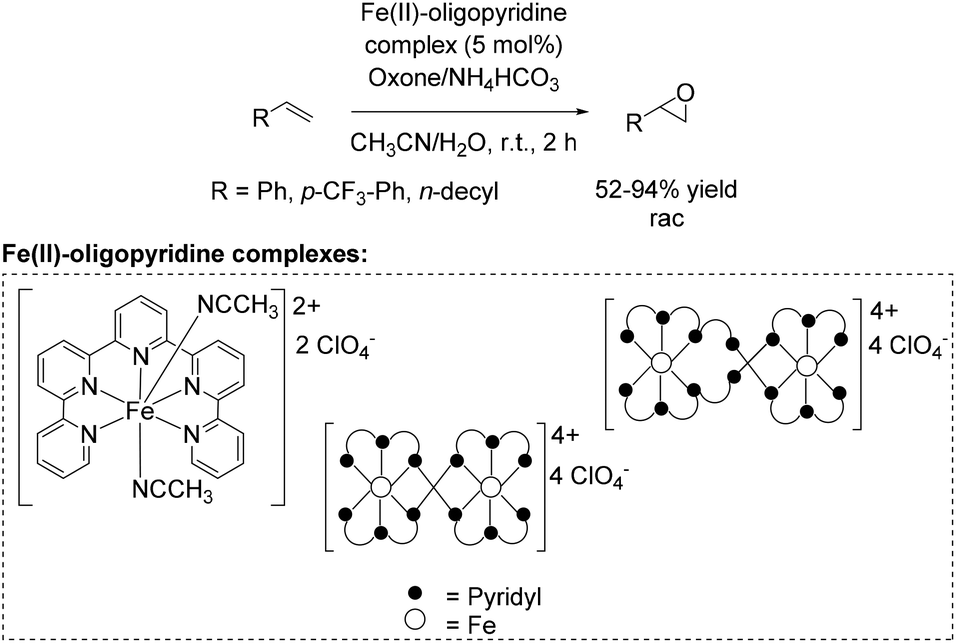 | ||
| Scheme 20 | ||
One year later, Rybak-Akimova and co-workers presented another interesting non-heme iron complex containing a new rigid racemic aminopyridine ligand, which should prevent leakage of iron and enables more regioselective C–H oxidation using hydrogen peroxide as the terminal oxidant.77 The methylamine moiety of the already described mep ligand of Jacobsen in 200159 is replaced by a 6-membered piperidine ring. The distorted octahedral iron(II) complex providing labile acetonitrile ligands in cis-position was found to be highly active in epoxidation of aliphatic alkenes in presence of acetic acid and at low catalyst loading. Beside the high conversion of aliphatic cyclic alkenes, the catalyst was less active for aliphatic terminal alkenes (Scheme 21). No aromatic alkenes were tested as substrate since the complex was found to promote aromatic hydroxylation. They demonstrated that the yield of epoxide depends on the amount of acetic acid which favors O–O cleavage in the Fe(III)–OOH intermediate forming the active oxidizing species. An electrophilic character of the oxidant was suggested due to nucleophilic reactivity of alkenes.
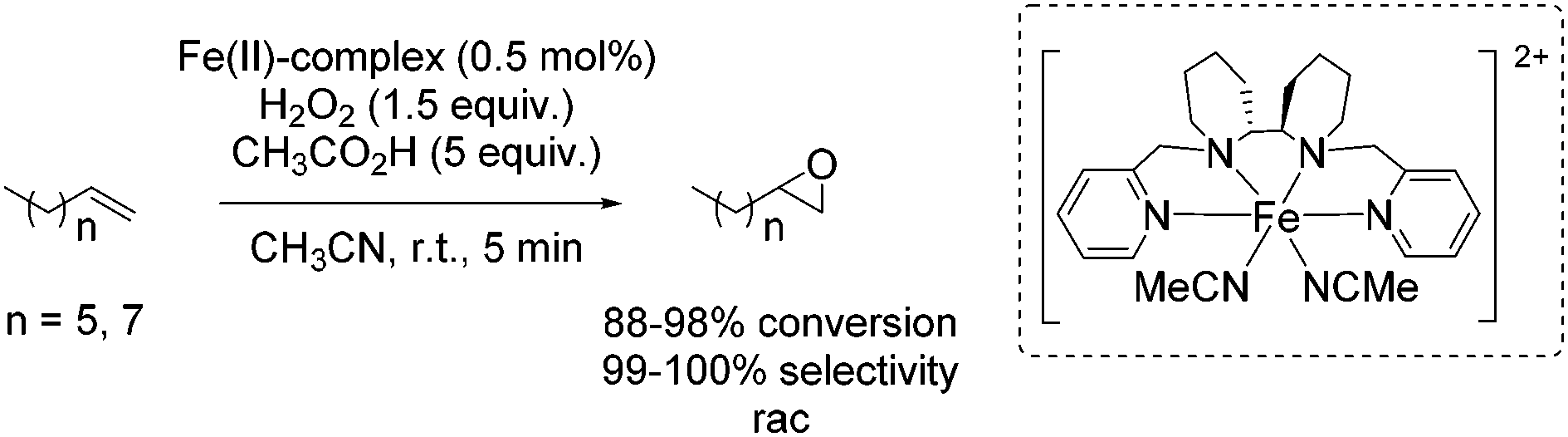 | ||
| Scheme 21 | ||
O which is formed after addition of the harsh oxidant PhIO (iodosobenzene) led to 9.3% yield and 45% ee, regarding the epoxidation of styrene (Scheme 22). However, the use of environmentally benign and inexpensive oxidizing agents (e.g. air, O2 and H2O2) is necessary wherever possible to develop a greener process.
 | ||
| Scheme 22 | ||
Almost 10 years later, an example for aerobic enantioselective epoxidation with a chiral non-heme iron catalyst [Fe(dcm)3] derived from D-camphor, was reported by You and co-workers in 2005.79 2-Ethylbutyraldehyde was found to be the best sacrificial reductant in this catalytic system focusing on epoxidation of styrene derivatives. Although the addition of the reductant minimizes the cleavage of the double bond in presence of O2, the requirement of 3 equiv. of a sacrificial reductant significantly reduces the atom economy of this method. Yields of up to 88% could be reached in combination with enantioselectivity of up to 78% (Scheme 23).
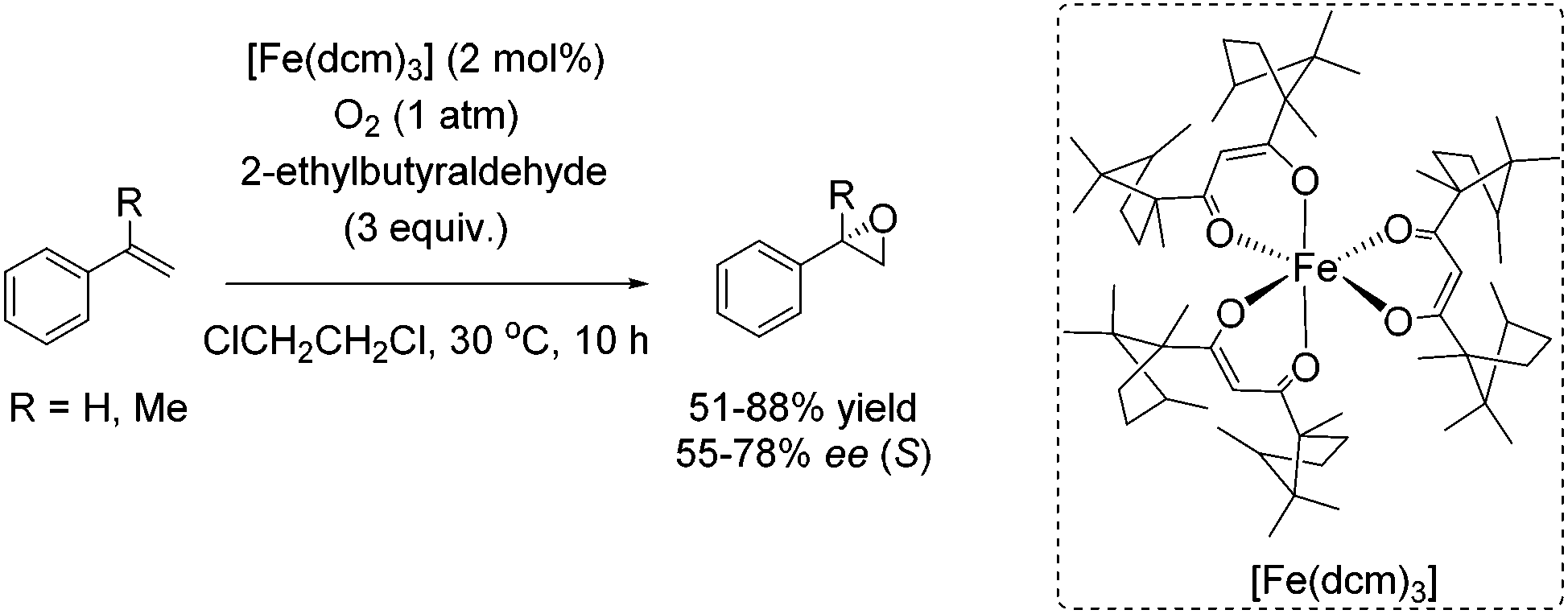 | ||
| Scheme 23 | ||
In 2007, Ménage and co-workers reported an enantioselective transformation of alkenes into epoxides catalyzed by an in situ prepared non-heme chiral diiron(III) complex (Scheme 24).80 Unsymmetrical alkenes were oxidized into epoxides in 9–63% ee, whereas the oxidation of the symmetrical alkenes led to racemic mixtures. Styrene was oxidized to the corresponding epoxide with 60% yield but low enantiomeric excess (15% ee) (Scheme 24).
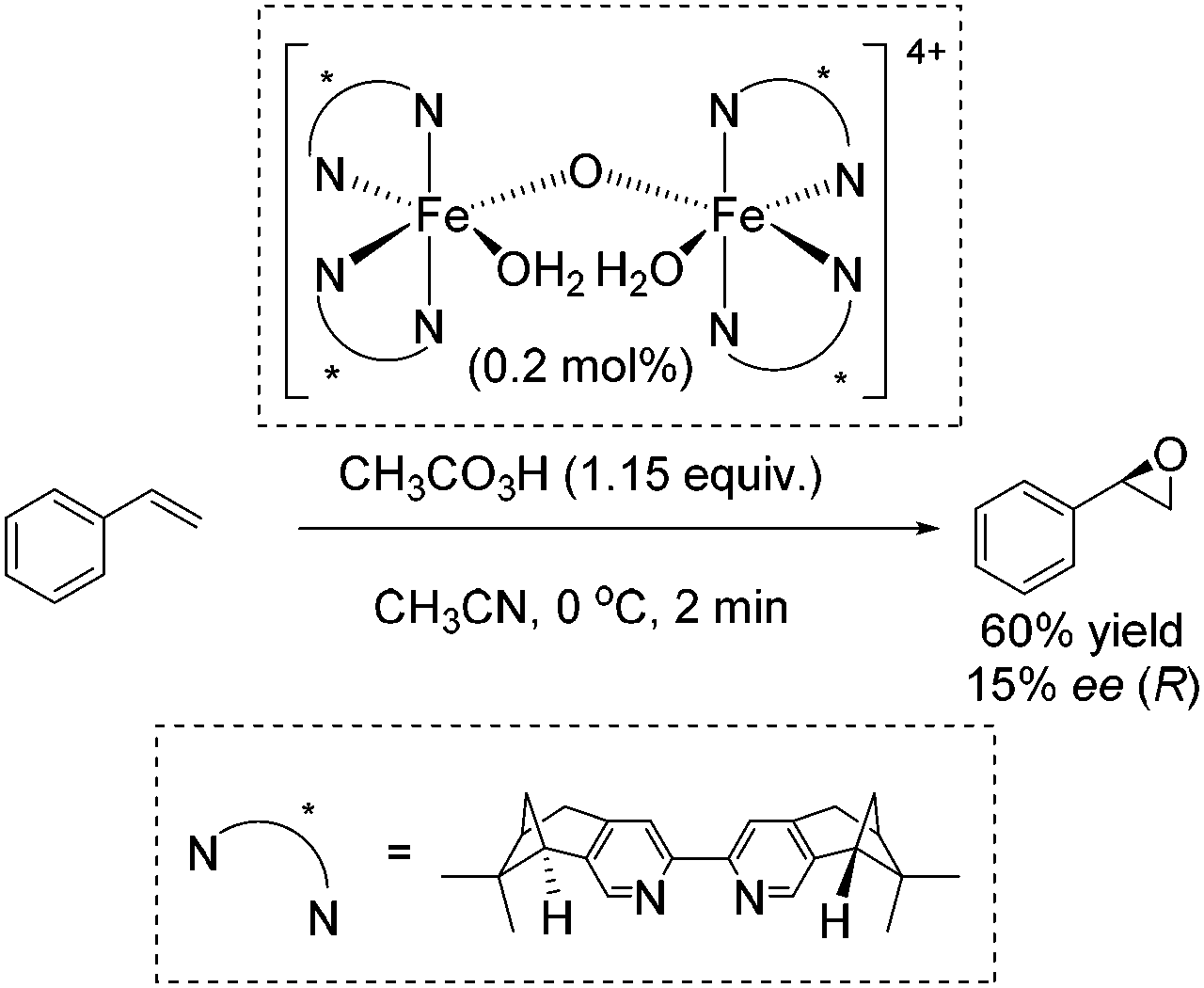 | ||
| Scheme 24 | ||
The epoxidation of (R)-carvone has been carried out as intramolecular competition experiment (Scheme 25). The regioselective epoxidation of the terminal alkene leading to a monoepoxide with a diastereomeric excess of 19% supports the electrophilic nature of the oxidizing species.80
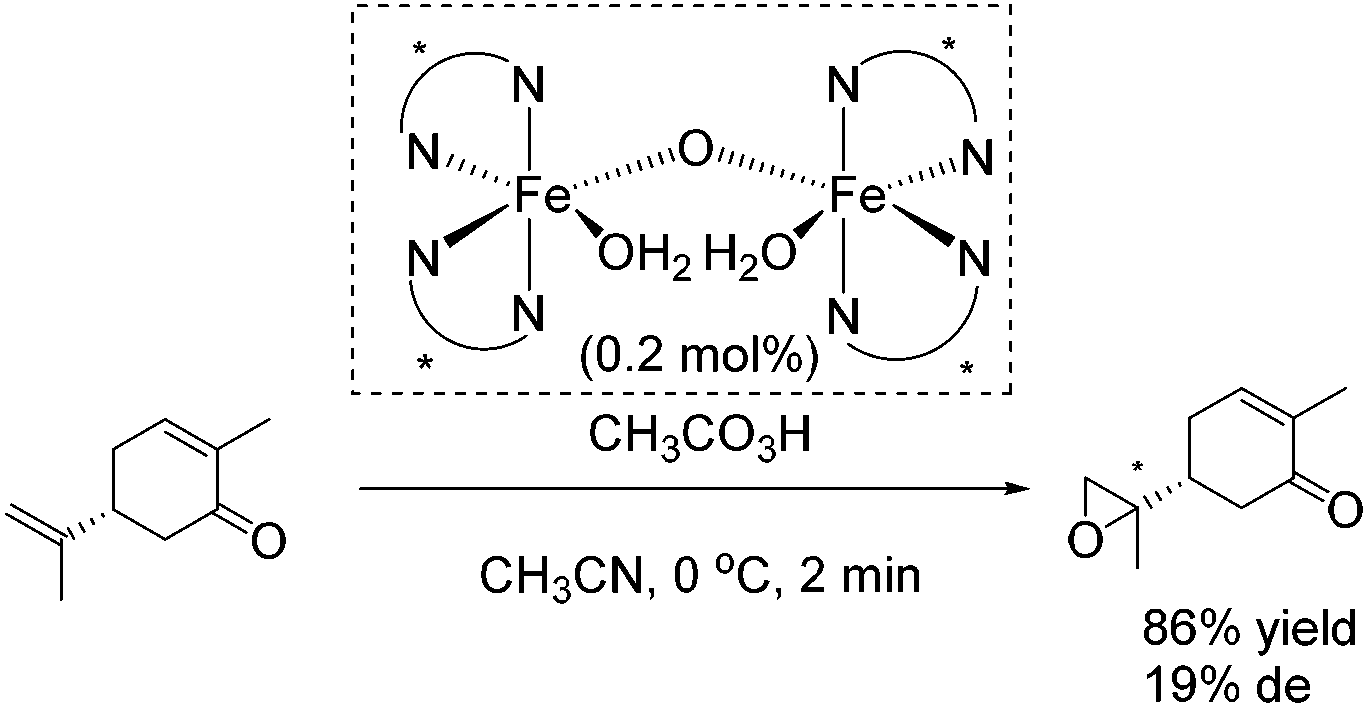 | ||
| Scheme 25 | ||
Later, Beller and co-workers developed a new method for the enantioselective epoxidation of aromatic alkenes using hydrogen peroxide as already highlighted (Scheme 2).49 The chiral iron catalyst was generated in situ from FeCl3·6H2O, the ethylenediamine based ligand, and pyridine-2,6-dicarboxylic acid (H2Pydic) as an additive (Scheme 26). Various alkenes were tested; however, only sterically hindered ones have shown high enantioselectivity. Terminal alkenes gave products with low enantiomeric excess (8–26% ee).81
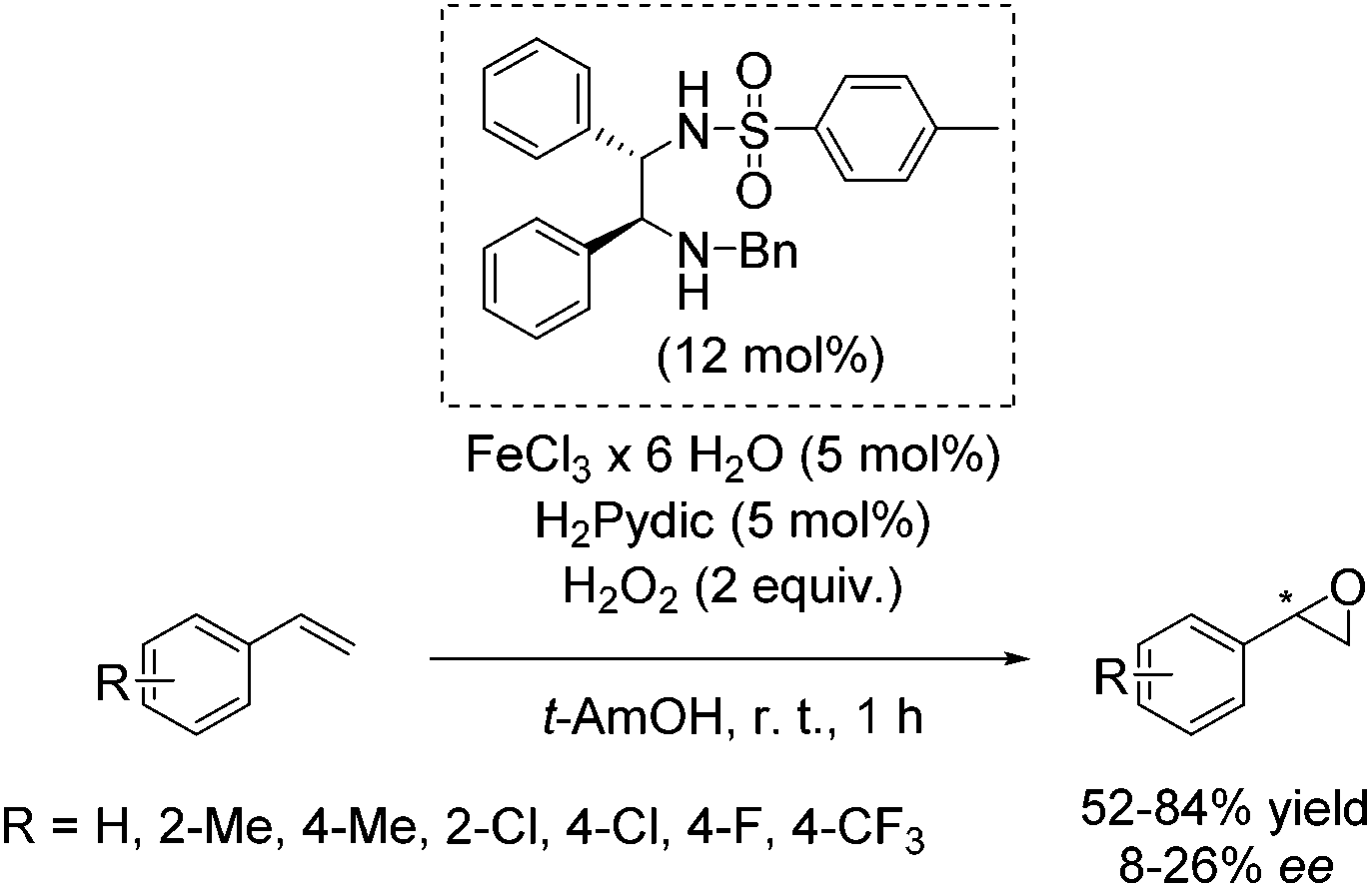 | ||
| Scheme 26 | ||
In 2008, Kwong and co-workers investigated the enantioselective epoxidation of alkenes with a chiral diiron-sexipyridine complex. Employing this complex together with hydrogen peroxide, the corresponding epoxides of aromatic terminal alkenes were obtained in 80–100% yields and 15–43% ee after 3 minutes reaction (Scheme 27).82 However, steric effects of the bulky iron catalyst may affect the enantioselective catalysis due to poor results in α-methylstyrene epoxidation (17% ee). Noteworthy, aliphatic terminal alkenes as 1-heptene remain unreacted in the system. This fact renders the here presented catalyst as a complement to Jacobsens catalyst,59 which shows high affinity to aliphatic terminal alkenes. Intermolecular competition experiments indicate the electrophilic nature of the active oxidant, which is involved in a non-radical oxidation process. Besides, further experiments demonstrated the indispensability of catalytic amounts of carboxylic acid. In order to show the practicability and sustainability of the reaction, the authors reused the ligand by simply adding new iron salt prior to the new catalytic cycle, which was carried out with 10% lower yield but the same selectivity for epoxide formation.
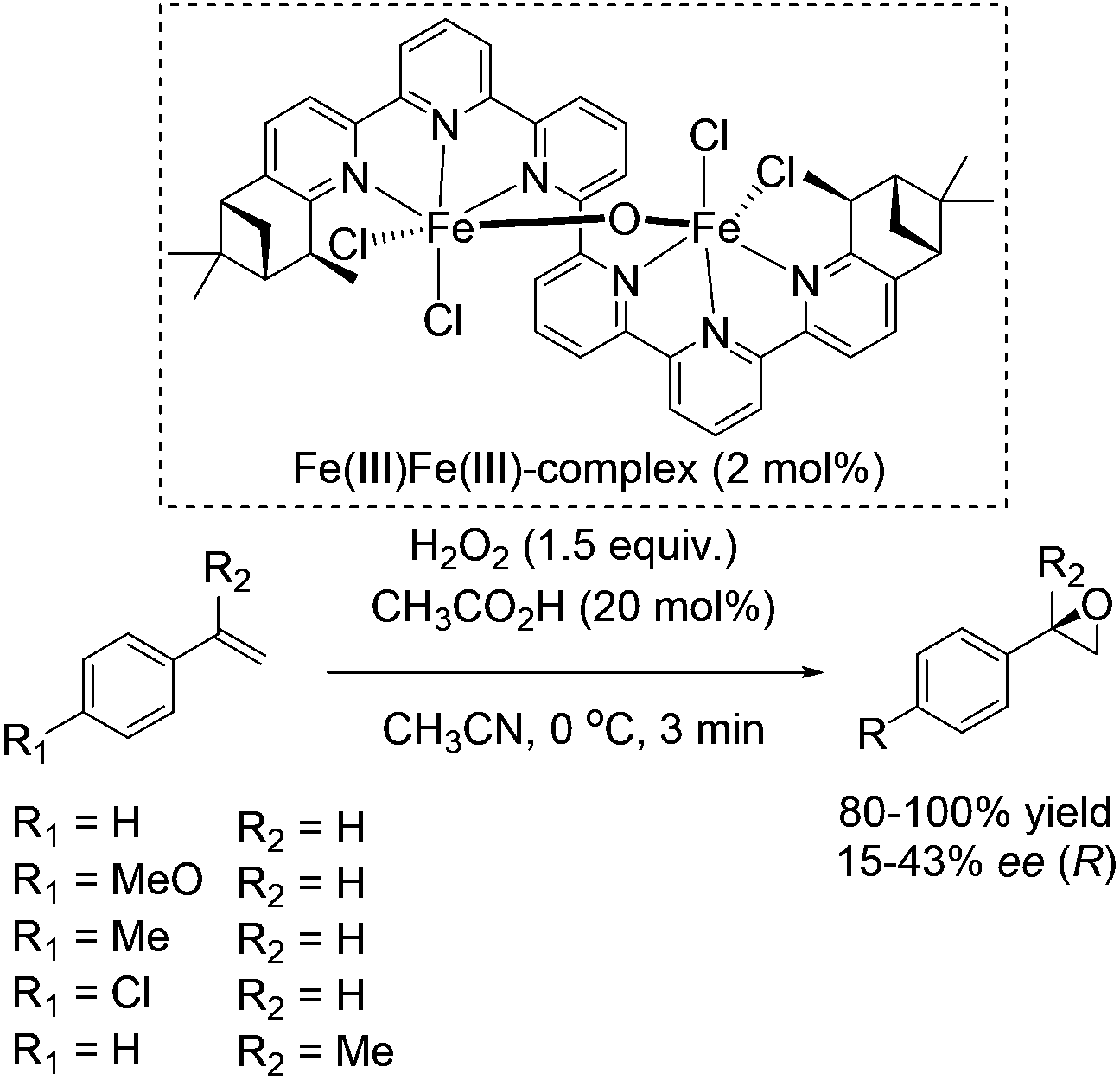 | ||
| Scheme 27 | ||
In 2012, the Tsogoeva group presented a new non-heme iron catalytic system with an in situ generated complex from FeCl3·6H2O and a primary amine-derived non-symmetrical Schiff base ligand.83 The epoxidation reaction system was applicable to different alkenes using H2O2 as oxidant. In case of trans-stilbene and derivatives, yields of up to 84% and ee values of up to 30% were observed. With styrene as representative for terminal alkenes 44% yield and 21% enantioselectivity could be obtained (Scheme 28).
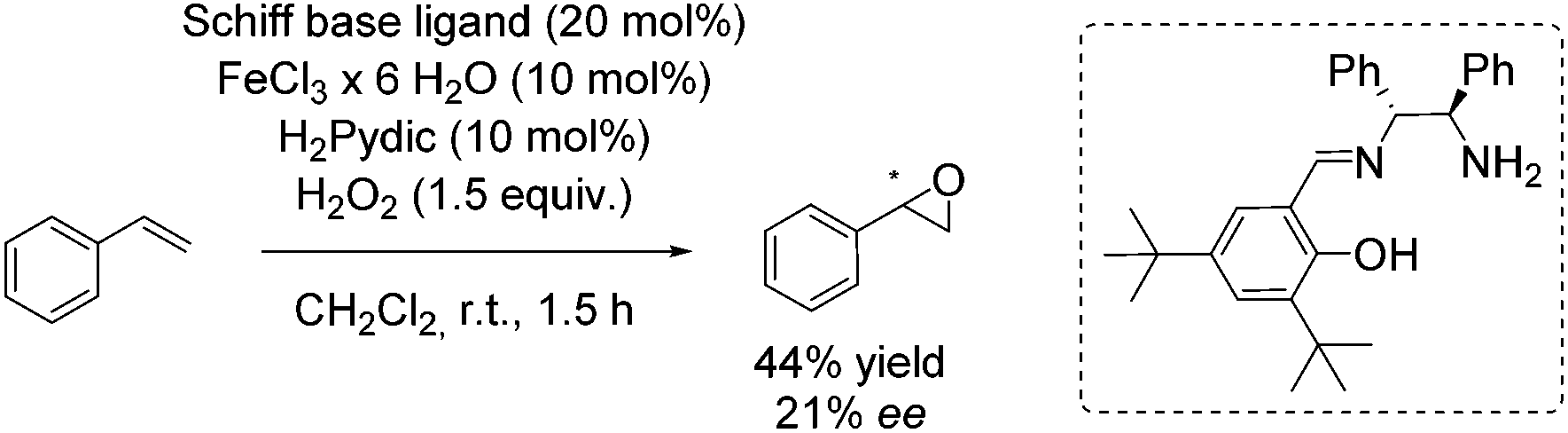 | ||
| Scheme 28 | ||
In the same year, Ménage and co-workers reported new complexes containing N2Py2 ligands with one or two internal carboxylic functionalities.84 They investigated the influence of the carboxylic function on catalytic efficiency in epoxidation reactions using hydrogen peroxide as oxidant. A stimulating effect of acetic acid, used as additive, combined with a weak effect of the carboxylic moiety from the ligand could be observed. By screening several substrates with [Fe(II)L(C3H6O)](FeCl4) as catalyst, it was demonstrated that the electron richness of the substrate affects the ee-value. The proposed electrophilic character of the oxidizing species led to selective epoxidation of the terminal alkene in case of (R)-carvone with 27% yield and 15% enantioselectivity (Scheme 29).
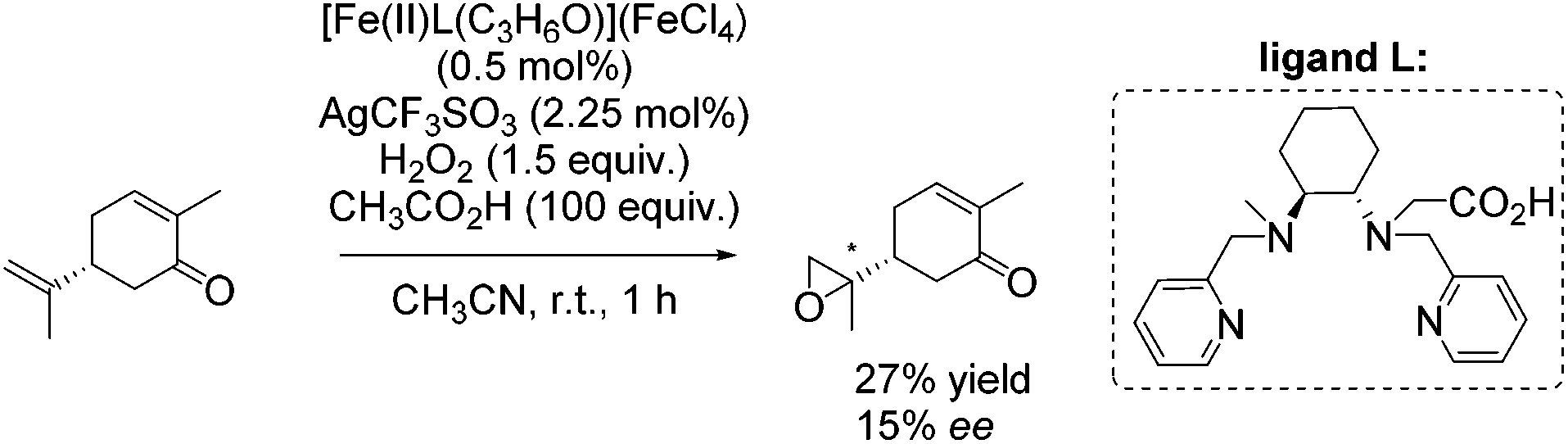 | ||
| Scheme 29 | ||
Also in 2012, Bryliakov and Talsi presented a comparative study with both manganese and iron aminopyridine complex as catalyst for enantioselective epoxidation.85 They further set the focus on the effect of diverse carboxylic acids as crucial additive ranging from acetic acid over n-caproic acid to pivalic acid. The enantioselectivity of chalcone epoxidation was raised with increasing steric bulk of the acids which demonstrates the important role of the acid in the active species at the enantioselectivity determining step. The authors assigned [(Ligand)Fe(V)O(OC(O)CH3)]2+ the role as the active species in which the acid molecule is incorporated as auxiliary ligand. However, focusing on other substrates beside chalcone, the iron catalyst is applied for a few non-terminal aromatic and aliphatic alkenes in combination with acetic acid and 2-ethylhexanoic acid. Poor to excellent yields and 26–62% enantiomeric excess were observed for these substrates, but the manganese system always shows higher efficiency and enantioselectivity. Concerning the epoxidation of a terminal alkene, like styrene, with the iron complex in combination with acetic acid, they obtained just 26% yield and 16% enantiomeric excess (Scheme 30).
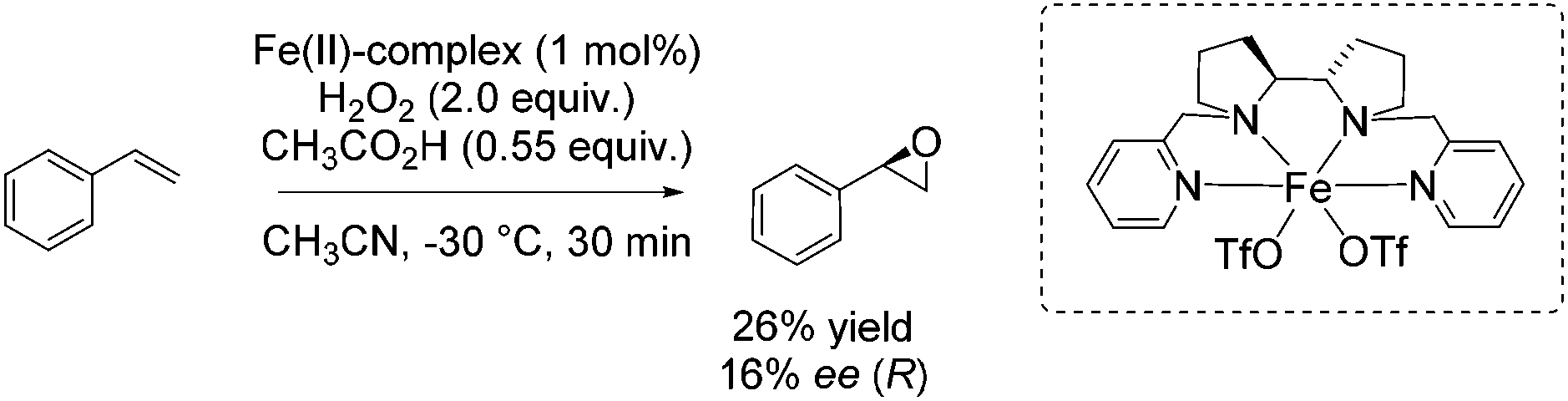 | ||
| Scheme 30 | ||
As already highlighted, in 2013 Costas and co-workers further investigated the electronic effect of aminopyridine ligands on iron(II) complexes catalyzing epoxidation reactions.53 Although the most promising catalyst was active for a wide range of substrates with excellent enantioselectivities and very good yields (Scheme 6), in case of the solely tested terminal alkene, isopropenylbenzene, 67% yield and 45% enantioselectivity could be obtained (Scheme 31).
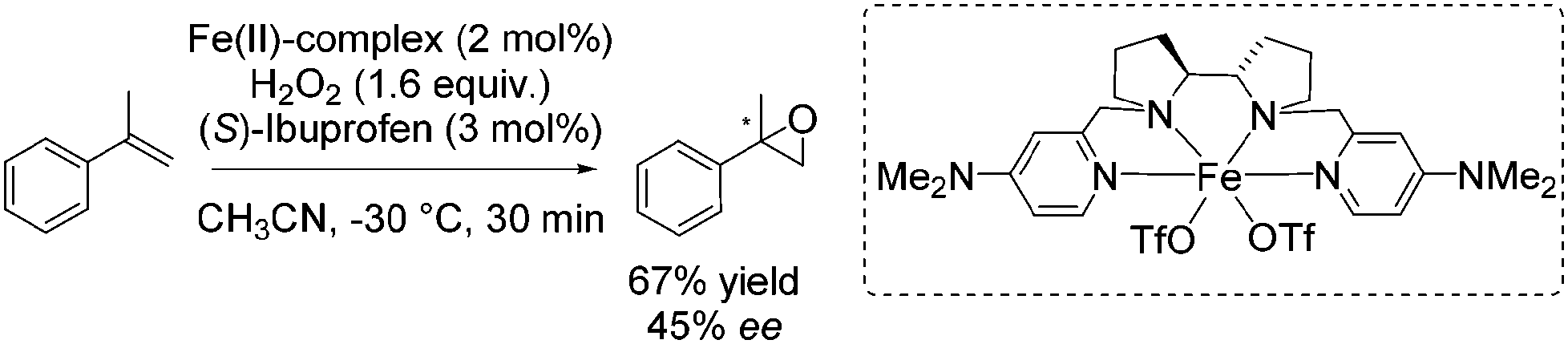 | ||
| Scheme 31 | ||
Summarizing, a few examples of chiral non-heme iron catalysts, suitable for the epoxidation of terminal alkenes, were reported recently. The major limitations of most of the oxidative procedures are low to moderate yields and enantioselectivities obtained. Furthermore, the easy synthesis of stable iron complexes is rarely possible. The challenge is currently to find out not only the highly enantioselective and easily accessible chiral iron complexes, but also to suggest a plausible rationale of the stereochemical pathway of the epoxidation to support a design of new improved chiral ligands and corresponding iron complexes. It is therefore reasonable to expect more extensive studies in this field over the coming years.
Iron-catalyzed aziridination of terminal alkenes
Among efficient catalytic systems for non-enantioselective and enantioselective aziridination, iron catalysis is the least studied, especially compared to epoxidation. Focusing on aziridination of terminal alkenes via non-heme iron based catalysis, the literature examples remain rare. To the best of our knowledge, only one example of non-heme iron-catalyzed enantioselective aziridination is described.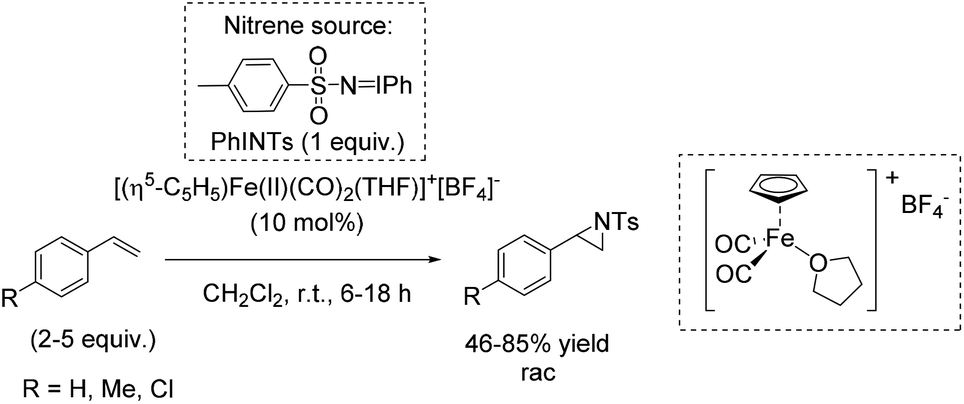 | ||
| Scheme 32 | ||
In 2004, Latour and co-workers reported a tosylamine transfer for non-enantioselective olefin aziridination catalyzed by a non-heme iron complex.87 The catalytic diiron(III) species were formed in situ from diiron mixed-valent complex of the asymmetrical hexadentate phenol ligand and PhINTs. The application of the catalyst gave aziridines in 42–69% yields adding high excess of substrate. Beside styrene as aromatic terminal alkene (69% yield) also the less active aliphatic terminal alkene 1-hexene (51% yield) could be converted to the corresponding aziridine (Scheme 33).
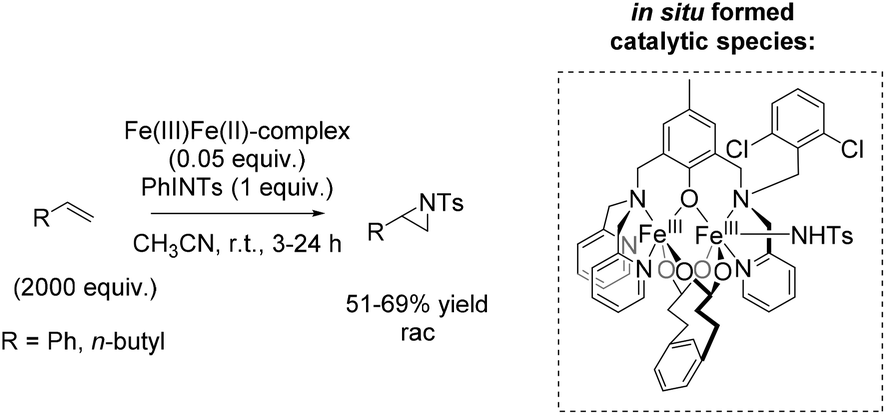 | ||
| Scheme 33 | ||
In the same year, a heterogeneous aziridination approach was presented by Chanda and co-workers using Bromamine-T as nitrene source.88 The polymer supported iron complex, synthesized by using p-phenylene diamine as a starting compound, was found to be a good catalyst for aziridination of some non-terminal alkenes (especially cis-β-methylstyrene with 75% yield), as well as for aliphatic and aromatic terminal alkenes with yields up to 66% (Scheme 34).
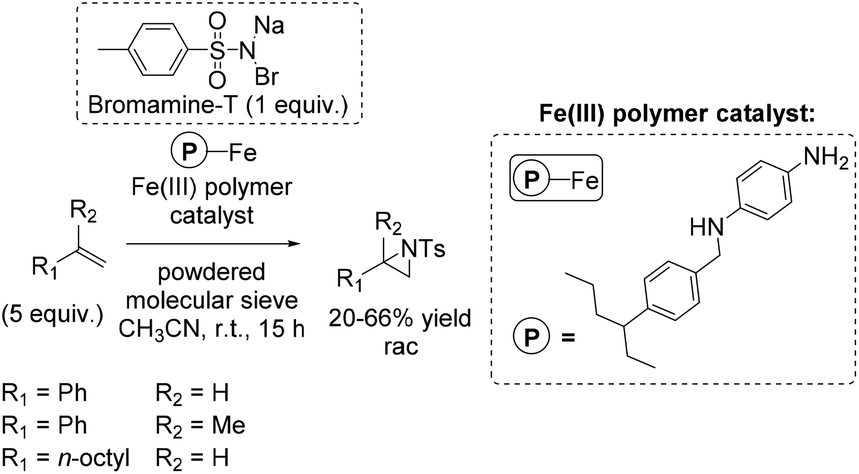 | ||
| Scheme 34 | ||
Notably, the immobilization of a homogeneous catalyst onto a polymer facilitates the separation of the catalyst and the reaction products and may also give rise to the reutilization of the catalyst in multiple subsequent cycles,66,67 making the process more economical and environmentally friendly.
The utility of mononuclear non-heme iron(II) complexes as catalysts for rapid aziridination reaction of aromatic and aliphatic alkenes using PhINTs as nitrene source was suggested by Halfen in 2007.89 It was demonstrated that neutral polyamine (macrocyclic or linear) ligands form square-pyramidal high spin iron(II) complexes containing at least one pair of cis labile coordinating sites. These complexes were found to be more active as catalysts in aziridination reactions than those with trans labile sites or a single labile site. These systems provided products in poor (aliphatic substrate) to good yields (aromatic substrate) concerning terminal alkenes requiring only small excesses of olefins for optimum reactivity (Scheme 35).
 | ||
| Scheme 35 | ||
Che and co-workers reported in 2008 a catalyst prepared from an iron(II) salt and 4,4′,4′′-trichloro-2,2′:6′,2′′-terpyridine which shows catalytic activity in non-enantioselective aziridination reaction of alkenes.65 The complex catalyzes besides intermolecular aziridination also the intramolecular aziridination with very good selectivity and high yields without substrate excess, which was required in previous examples. Via ESI-MS measurements, reactive FeNTs species could be detected. Under optimized conditions, terminal aromatic and aliphatic alkenes gave racemic aziridines in very good to excellent yields (Scheme 36). In addition, the polymer-supported O-PEG-OCH3-Cl2-terpy ligand B (already discussed for the epoxidation method of Che in Scheme 14) was found to be efficient as a reusable ligand in intramolecular aziridination of a terminal olefin.
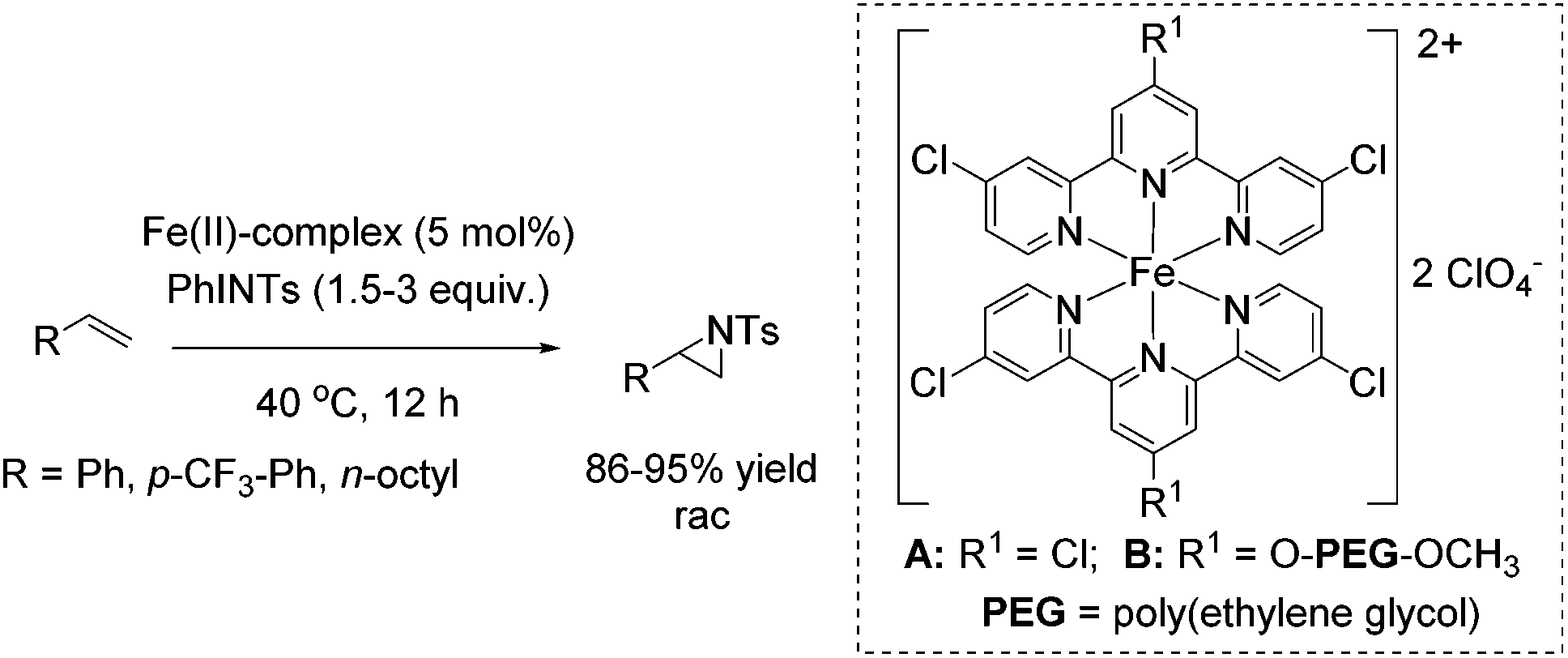 | ||
| Scheme 36 | ||
In the same year, Bolm demonstrated aziridination of styrene with in situ formed iminoiodinanes from mixtures of the corresponding sulfonamide (ArSO2NH2), iodosylbenzene (PhIO) or iodobenzene diacetate (PhI(OAc)2) in presence of magnesium oxide.54 The obtained yields of styrene aziridine reached 66% with the in situ generated nitrene source from iodobenzene diacetate and p-NO2-PhSO2NH2 and iron(II) triflate as catalyst. Further optimization of reaction conditions and a screening of previously prepared nitrogen carriers led to styrene aziridine with 90% yield obtained with PhINSO2[2-(5-methyl)pyridinyl]. The α-methylstyrene was treated with PhINTs for aziridination and the corresponding aziridine was obtained with 45% yield (Scheme 37). The authors proposed a radical intermediate during nitrogen transfer process and exclude a concerted mechanism.
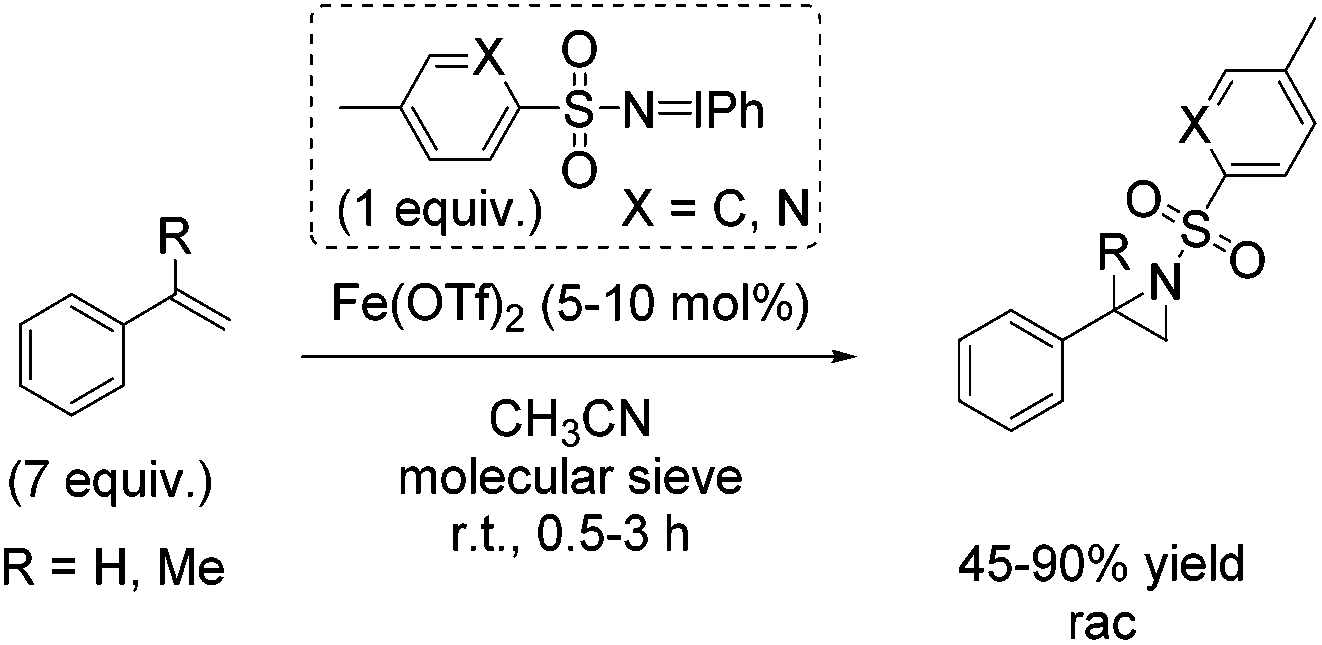 | ||
| Scheme 37 | ||
Based on these results, a few months later Bolm and co-workers presented a further catalytic system for non-enantioselective aziridination of olefins focusing mainly on terminal aromatic alkenes. The system is based on iron(II) triflate and quinaldic acid forming the active complex.90 This in situ generated complex was found to be highly efficient in combination with a hydrophobic ionic liquid contributing to the nitrene transfer and iminoiodinane with a pyridine backbone as nitrene source. Utilizing this combined catalytic system, yields of up to 95% could be obtained for styrene and styrene derivatives (Scheme 38). An excess of substrate was not necessary, in applying this catalytic system for aziridination.
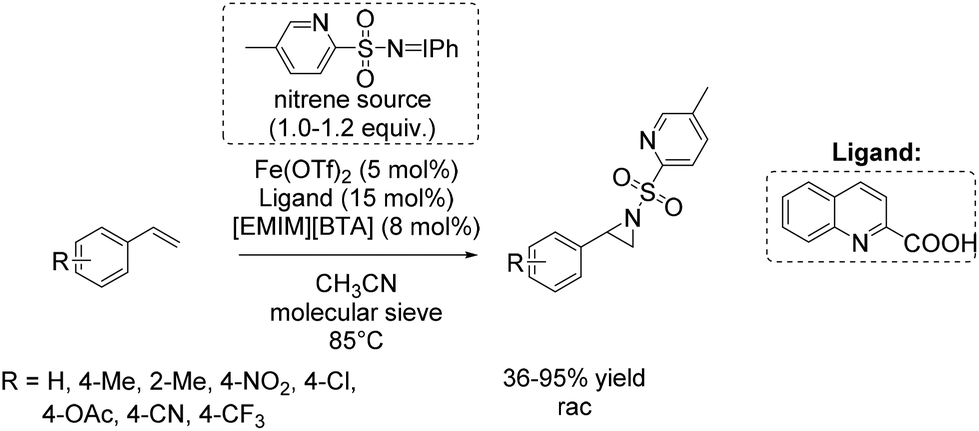 | ||
| Scheme 38 | ||
Three years later, Jenkins and co-workers presented a new macrocyclic tetracarbene iron(II) catalyst, which was found to be active in non-enantioselective aziridination of challenging aliphatic alkenes using organic azides as nitrene reagent, instead of the commonly used tosyl group containing compounds.91 The organic azides, however, often decompose explosively.92
The square planar coordination geometry of the tetracarbene ligand is essential for aziridination using organic azides because pseudo-tetrahedral iron imido complexes would not have been active for this reaction. The removal step of the tosyl group gets dispensable and improves the atom economy of the reaction. Furthermore the robust iron complex is reusable, which is an additional advantage of this reaction system. The aziridination of terminal alkenes like 1-decene and 1-octene, implying the formation of an iron(IV) imide as postulated nitrogen transferring intermediate, led to yields up to 82% (Scheme 39).91 However, a 29-fold excess of alkene is required to achieve this result, making the process less economically benign.
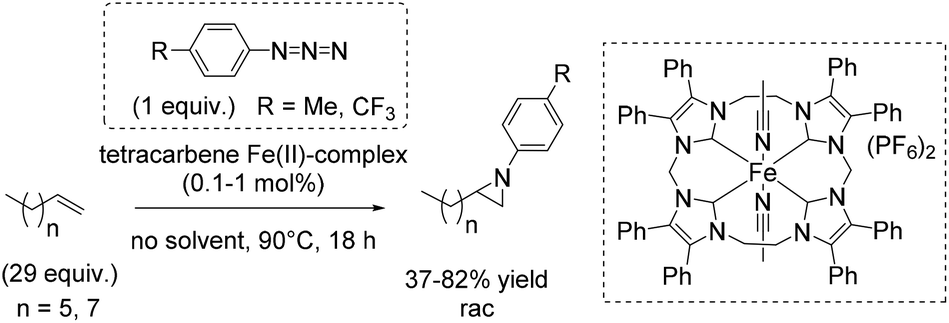 | ||
| Scheme 39 | ||
In 2012, the group of Jensen reported diverse half-sandwich scorpionates with manganese, iron, cobalt or nickel as metal center. These nitrene transfer catalysts showed activity for styrene aziridination and THF amination. The iron(II) scorpionate complex containing 3,5-dimethylpyrazoles was identified as the most active one and could carry out the aziridination reaction with 94% yield (Scheme 40).93 Aziridination of styrene derivatives bearing an electron donating or electron withdrawing substituent in para position indicated the formation of an electrophilic intermediate during the reaction, plausibly an imidoiron(IV) complex. However, the exact yields obtained for aziridination of styrene derivatives were not reported. Under optimized conditions, the catalyst was applied for aziridination of various substrates including aromatic and aliphatic olefins with promising results.
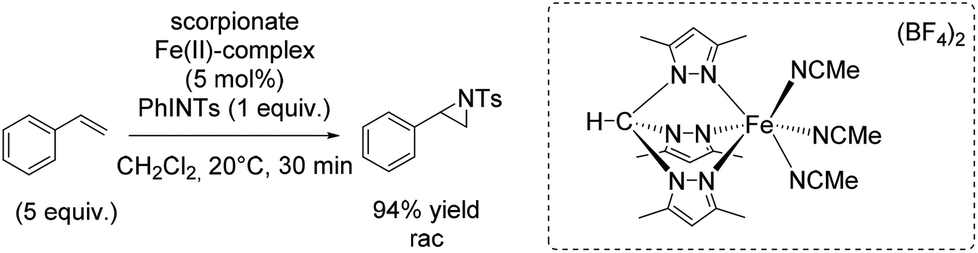 | ||
| Scheme 40 | ||
 | ||
| Scheme 41 | ||
In summary, the lack of general and simple synthetic methods towards terminal aziridines also hampered the use of achiral and chiral non-heme iron catalysts in their synthesis. The major limitation of non-heme iron catalysed aziridination of terminal alkenes appears to be achievable yields and enantioselectivities, which are mostly modest. Acceptable results can be obtained, in few cases. Thus, further development of new effective ligands and corresponding iron complexes, as well as mechanistic investigations are required and more experiments and results are therefore expected in the future.
Summary and outlook
The epoxidation and aziridation of terminal alkenes is arguably one of the most versatile and powerful tools for the preparation of terminal epoxides and aziridines, which are valuable starting compounds for the synthesis of complex molecules, in particular of biologically active compounds and pharmaceuticals. Non-enantioselective and enantioselective non-heme iron catalysis is demonstrated in this review as economically and ecologically benign method in epoxidation and aziridination reactions.Nonetheless, the existing catalytic systems provide still limitations in substrate generality. Even if the yields of terminal epoxides and aziridines, obtained through non-enantioselective methods, are high, they are inferior to yields of their non-terminal analogues. In enantioselective non-heme iron-based catalysis, the yields of chiral terminal epoxides and aziridines are predominantly lower compared to non-enantioselective transformations of terminal olefins.
From an ecological point of view several methods discussed in this review provide valuable approaches. The in situ generation of an iron-based catalyst avoids a prior catalyst synthesis and makes this chemistry greener. Even if a previously prepared well characterized catalyst is applied in epoxidation and aziridination, degradation and new arrangements during the reaction can never be excluded. This phenomenon occurred already in some presented iron-based catalytic systems.
Methods employing homogeneous non-immobilized catalysts often requires high catalyst loading to achieve high conversion in reasonable reaction time and the reusability of such catalysts is also a problem most of the time. A few successful examples of solid supported iron complexes and recycling/reusing of catalysts/ligands without losing efficiency have been demonstrated. Combining the advantages of being economically and environmentally friendly process, catalysis by solid supported iron complexes (in situ generated from iron salt and polymer supported ligand) inspire also further developments of improved methodologies for immobilization of active homogeneous ligands through both covalent and non-covalent interactions between the ligand and the support, solving also the problem of leaching.
Atom economy of most of the presented methods should be further improved employing more efficient and selective iron based catalytic systems which do not require stoichiometric amounts of ligands or sacrificial co-substrates.
Furthermore, there is still much room for improvements and a large research potential for iron-based catalysts in epoxidation and aziridination of terminal alkenes using environmentally friendly and inexpensive oxidizing agents (e.g. air, O2 and H2O2) and new non-hazardous nitrene sources, as well as non-toxic and non-expensive solvents e.g. water. Beyond using no added solvent in a reaction, water is probably the greenest alternative known.
Thus, the development of new atom economical catalytic systems and using no solvent or safer solvents as well as environmentally friendly reagents (oxidizing and aziridination agents) for these important reactions still remains an attractive and challenging task for synthetic chemists. Therefore, it seems safe to predict that many new efficient and sustainable iron based catalytic systems will be discovered in the near future.
Acknowledgements
S. B. T. is grateful to the Erlangen Catalysis Resource Center (ECRC), Interdisciplinary Center for Molecular Materials (ICMM) and Deutsche Forschungsgemeinschaft (DFG, SFB 583) for research support. Financial support by the Erika Giehrl-Stifgung (doctoral research fellowship for Anja Fingerhut) is gratefully acknowledged.Notes and references
- Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, Weinheim, 2006, p. 516 Search PubMed.
- H. Adolfsson, Modern Oxidation Methods, ed. J. E. Bäckvall, Wiley-VCH, Weinheim, 2004. pp. 21–43 Search PubMed.
- For a recent review on organocatalytic epoxidations, see: K. M. Weiß and S. B. Tsogoeva, Chem. Rec., 2011, 11, 18–39 CrossRef PubMed.
- F. E. Held, S. Wei, K. Eder and S. B. Tsogoeva, RSC Adv., 2014, 4, 32796–32801 RSC.
- E. D. Butova, A. V. Barabash, A. A. Petrova, C. M. Kleiner, P. R. Schreiner and A. A. Fokin, J. Org. Chem., 2010, 75, 6229–6235 CrossRef CAS PubMed.
- For a general review on applications of chiral aziridines, see: W. McCoull and F. A. Davis, Synthesis, 2000, 10, 1347–1365 CrossRef PubMed.
- R. T. Dorr, L. Wisner, B. K. Samulitis, T. H. Landowski and W. A. Remers, Cancer Chemother. Pharmacol., 2012, 69, 1039–1049 CrossRef CAS PubMed.
- C. D. Smith and X. Zhang, J. Biol. Chem., 1996, 271, 6192–6198 CrossRef CAS PubMed.
- J. Foulke-Abel, H. Agbo, H. Zhang, S. Mori and C. M. H. Watanabe, Nat. Prod. Rep., 2011, 28, 693–704 RSC.
- For a general review on enantioselective aziridine synthesis, see: H. M. I. Osborn and J. Sweeney, Tetrahedron: Asymmetry, 1997, 8, 1693–1715 CrossRef CAS.
- General Review about naturally occurring cyclohexane epoxides: J. Marco-Contelles, M. T. Molina and S. Anjum, Chem. Rev., 2004, 104, 2857–2899 CrossRef CAS PubMed.
- For general review about enantioselective aziridination: P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905–2920 CrossRef PubMed.
- For general review about enantioselective aziridination: H. Pellissier, Tetrahedron, 2010, 66, 1509–1555 CrossRef CAS PubMed.
- T. Katsuki, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Springer, Berlin, 1999, vol. 2, pp. 621–648 Search PubMed.
- R. A. Johnson and K. B. Sharpless, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 1993, pp. 103–158 Search PubMed.
- E. N. Jacobsen and M. H. Wu, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Springer, Berlin, 1999, vol. 2, pp. 649–677 Search PubMed.
- T. Katsuki, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty, T. J. Meyer, Elsevier, Oxford, 2003, vol. 9, pp. 207–264 Search PubMed.
- Y. Shi, Acc. Chem. Res., 2004, 37, 488–496 CrossRef CAS PubMed.
- I. Tirotta, N. L. Fifer, J. Eakins and C. A. Hutton, Tetrahedron Lett., 2013, 54, 618–620 CrossRef CAS PubMed.
- K. J. M. Beresford, N. J. Church and D. W. Young, Org. Biomol. Chem., 2006, 4, 2888–2897 CAS.
- D. Tanner and P. Somfai, Bioorg. Med. Chem. Lett., 1993, 3, 2415–2418 CrossRef CAS.
- S. Decamps, L. Sevaille, S. Ongeri and B. Crousse, Org. Biomol. Chem., 2014, 12, 6345–6348 CAS.
- I. C. Stewart, C. C. Lee, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 17616–17617 CrossRef CAS PubMed.
- S. Fioravanti, M. A. Loreto, L. Pellacani and P. A. Tardella, J. Org. Chem., 1985, 50, 5365–5368 CrossRef CAS.
- General Review about metal-mediated aziridination and amidation: J. A. Halfen, Curr. Org. Chem., 2005, 9, 657–669 CrossRef CAS.
- General review about catalytic aziridination: D. M. Jenkins, Synlett, 2012, 23, 1267–1270 CrossRef CAS PubMed.
- L. Que and R. Y. N. Ho, Chem. Rev., 1996, 96, 2607–2624 CrossRef CAS PubMed.
- J. L. McLain, J. J. Lee and T. Groves, In Biomimetic Oxidations Catalyzed by Transition Metal Complexes, ed. B. Meunier, Imperial College Press, London, 2000, pp. 91–169 Search PubMed.
- For iron catalyzed oxygen and nitrogen group transfer: T. W.-S. Chow, G.-Q. Chen, Y. Liu, C.-Y. Zhou and C.-M. Che, Pure Appl. Chem., 2012, 84, 1685–1704 CrossRef CAS.
- For a general Review: A. Correa, O. G. Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC.
- B. Marsh and D. R. Carbery, Annu. Rep. Prog. Chem., Sect. B, 2008, 104, 35–48 RSC.
- For a general Review: F. G. Gelalcha, Adv. Synth. Catal., 2014, 356, 261–299 CrossRef CAS.
- E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry – Theory and Practice, Oxford Univ. Press, Oxford, 1998 Search PubMed.
- For general Review: S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- For a general Review: C. Bolm, J. Legros, J. L. Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed.
- F. A. Cotton and G. Wilkinson, Anorg. Chem., Verlag Chemie, Weinheim, 4th edn, 1982, pp. 767 Search PubMed.
- M. W. Zettler, in Encyclopedia of Reagents of Organic Synthesis, ed. L. Paquette, Wiley, New York, 1995, vol 4, p. 2871 Search PubMed.
- A. D. White, in Encyclopedia of Reagents of Organic Synthesis, ed. L. Paquette, Wiley, New York, 1995, vol 4, p. 2873 Search PubMed.
- Encyclopedia of Inorganic Chemistry, ed. B. R. King, Wiley, New York, 1994, vol. 4 Search PubMed.
- For a general Review: L. Que Jr. and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed.
- For enantioselective catalysis using iron complexes, see: M. Darwisch and M. Wills, Catal. Sci. Technol., 2012, 2, 243–255 Search PubMed.
- Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Springer Verlag, Berlin, 1999 Search PubMed.
- Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 2nd edn, 2000 Search PubMed.
- M. B. Francis and E. N. Jacobsen, Angew. Chem., Int. Ed., 1999, 38, 937–941 CrossRef CAS.
- Green Chemistry – An Introductory Text, ed. M. Lancaster, RSC Publishing, Cambridge, 2010, p. 131 Search PubMed.
- B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2473 CrossRef CAS PubMed.
- F. M. Kerton, in Alternative Solvents for Green Chemistry, ed. J. H. Clark, G. A. Kraus, RSC Publishing, Cambridge, 2009, pp. 2–14 Search PubMed.
- F. G. Gelalcha, B. Bitterlich, G. Anilkumar, M. K. Tse and M. Beller, Angew. Chem., 2007, 119, 7431–7435 ( Angew. Chem., Int. Ed. , 2007 , 46 , 7293–7296 ) CrossRef.
- Y. Nishikawa and H. Yamamoto, J. Am. Chem. Soc., 2011, 133, 8432–8435 CrossRef CAS PubMed.
- M. Wu, C.-X. Miao, S. Wang, X. Hu, C. Xia, F. R. Kühn and W. Sun, Adv. Synth. Catal., 2011, 353, 3014–3022 CrossRef CAS.
- B. Wang, S. Wang, C. Xia and W. Sun, Chem. – Eur. J., 2012, 18, 7332–7335 CrossRef CAS PubMed.
- O. Cussó, I. Garcia-Bosch, X. Ribas, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2013, 135, 14871–14878 CrossRef PubMed.
- M. Nakanishi, A.-F. Salit and C. Bolm, Adv. Synth. Catal., 2008, 350, 1835–1840 CrossRef CAS.
- G.-S. Liu, Y.-Q. Zhang, Y.-A. Yuan and H. Xu, J. Am. Chem. Soc., 2013, 135, 3343–3346 CrossRef CAS PubMed.
- G. A. Kraus and B. S. Fulton, J. Org. Chem., 1985, 50, 1784–1786 CrossRef.
- B. P. Murch, F. C. Bradley and L. Que Jr., J. Am. Chem. Soc., 1986, 108, 5027–5028 CrossRef CAS.
- W. Nam, R. Ho and J. S. Valentine, J. Am. Chem. Soc., 1991, 113, 7052–7054 CrossRef CAS.
- M. C. White, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 7194–7195 CrossRef CAS.
- S. Ménage, J. M. Vincent, C. Lambeaux, G. Chottard, A. Grand and M. Fontecave, Inorg. Chem., 1993, 32, 4766–4773 CrossRef.
- G. Dubois, A. Murphy and T. D. P. Stack, Org. Lett., 2003, 5, 2469–2472 CrossRef CAS PubMed.
- G. Anilkumar, B. Bitterlich, F. G. Gelalcha, M. K. Tse and M- Beller, Chem. Commun., 2007, 289–291 RSC.
- B. Bitterlich, G. Anilkumar, F. G. Gelalcha, B. Spilker, A. Grotevendt, R. Jackstell, M. K. Tse and M. Beller, Chem. – Asian J., 2007, 2, 521–529 CrossRef CAS PubMed.
- L. H. Tong, Y.-L. Wong, S. I. Pascu and J. R. Dilworth, Dalton Trans., 2008, 4784–4791 RSC.
- P. Liu, E. L.-M. Wong, A. W.-H. Yuen and C.-M. Che, Org. Lett., 2008, 10, 3275–3278 CrossRef CAS PubMed.
- N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217–3274 CrossRef CAS PubMed.
- K. C. Gupta, A. K. Sutar and C.-C. Lin, Coord. Chem. Rev., 2009, 253, 1926–1946 CrossRef CAS PubMed.
- B. Bitterlich, K. Schröder, M. K. Tse and M. Beller, Eur. J. Org. Chem., 2008, 4867–4870 CrossRef CAS.
- K. Schröder, S. Enthaler, B. Bitterlich, T. Schulz, A. Spannenberg, M. K. Tse, K. Junge and M. Beller, Chem. – Eur. J., 2009, 15, 5471–5481 CrossRef PubMed.
- K. Schröder, X. Tong, B. Bitterlich, M. K. Tse, F. G. Gelalcha, A. Brückner and M. Beller, Tetrahedron Lett., 2007, 48, 6339–6342 CrossRef PubMed.
- K. Hasan, N. Brown and C. M. Kozak, Green Chem., 2011, 13, 1230–1237 RSC.
- K. Schröder, K. Junge, A. Spannenberg and M. Beller, Catal. Today, 2010, 157, 364–370 CrossRef PubMed.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS PubMed.
- K. Schröder, B. Join, A. J. Amali, K. Junge, X. Ribas, M. Costas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 1425–1429 CrossRef PubMed.
- M. Jiao, H. Matsunaga and T. Ishizuka, Chem. Pharm. Bull., 2011, 59, 799–801 CrossRef CAS.
- P. Liu, Y. Liu, E. L.-M. Wong, S. Xiang and C.-M. Che, Chem. Sci., 2011, 2, 2187–2195 RSC.
- E. A. Mikhalyova, O. V. Makhlynets, T. D. Palluccio, A. S. Filatov and E. V. Rybak-Akimova, Chem. Commun., 2012, 48, 687–689 RSC.
- J. F. Wie, X. D. Yu and D. S. Jin, Chin. Chem. Lett., 1996, 7, 962–964 Search PubMed.
- Q. F. Cheng, X. Y. Xu, W. X. Ma, S. J. Yang and T. P. You, Chin. Chem. Lett., 2005, 16, 1467–1470 CAS.
- C. Marchi-Delapierre, A. Jorge-Robin, A. Thibon and S. Ménage, Chem. Commun., 2007, 1166–1168 RSC.
- F. G. Gelalcha, G. Anilkumar, M. K. Tse, A. Brückner and M. Beller, Chem. – Eur. J., 2008, 14, 7687–7698 CrossRef CAS PubMed.
- H.-L. Yeung, K.-C. Sham, C.-S. Tsang, T.-C. Lau and H.-L. Kwong, Chem. Commun., 2008, 3801–3803 RSC.
- K. A. Stingl, K. M. Weiß and S. B. Tsogoeva, Tetrahedron, 2012, 68, 8493–8501 CrossRef CAS PubMed.
- F. Oddon, E. Girgenti, C. Lebrun, C. Marchi-Delapierre, J. Pécaut and S. Ménage, Eur. J. Inorg. Chem., 2012, 2012, 85–96 CrossRef CAS.
- O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2012, 2, 1196–1202 CrossRef CAS.
- B. D. Heuss, M. F. Mayer, S. Dennis and M. M. Hossain, Inorg. Chim. Acta, 2003, 342, 301–304 CrossRef CAS.
- F. Avenier and J. M. Latour, Chem. Commun., 2004, 1544–1545 RSC.
- B. M. Chanda, R. Vyas and S. S. Landge, J. Mol. Catal. A: Chem., 2004, 223, 57–60 CrossRef CAS PubMed.
- K. L. Klotz, L. M. Slominski, A. V. Hull, V. M. Gottsacker, R. MasBallesté, L. Que Jr. and J. A. Halfen, Chem. Commun., 2007, 2063–2065 RSC.
- A. C. Mayer, A.-F. Salit and C. Bolm, Chem. Commun., 2008, 5975–5977 RSC.
- S. A. Cramer and D. M. Jenkins, J. Am. Chem. Soc, 2011, 133, 19342–19345 CrossRef CAS PubMed.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- S. Liang and M. P. Jensen, Organometallics, 2012, 31, 8055–8058 CrossRef CAS.
Footnote |
| † Dedicated to Professor Hisashi Yamamoto on the occasion of his 72th birthday. |
| This journal is © The Royal Society of Chemistry 2015 |