
Metal and metalloid determination in biodiesel and bioethanol
Raquel
Sánchez
a,
Carlos
Sánchez
a,
Charles-Philippe
Lienemann
b and
José-Luis
Todolí
*a
aDepartment of Analytical Chemistry, Nutrition and Food Sciences, P.O. Box 99, 03080, Alicante, Spain. E-mail: jose.todoli@ua.es
bIFP Energies Nouvelles, Rond-point de l'échangeur de Solaize, BP 3, F-69360 Solaize, France
First published on 9th October 2014
Abstract
Biofuel quality control involves the determination of metal and metalloid content. These species play a very important role because they may modify the efficiency of biofuel production as well as the stability of these products. Furthermore, some metals are toxic and generate environmental concerns whereas others are used as additives. Normally, products such as biodiesel and bioethanol are mixed with conventional fossil fuels (diesel and gasoline, respectively). Therefore, metals come from the raw product employed for biofuel production (seeds, sugars…) as well as from the production and storage process or even from the added fuels. The determination of the final metal and metalloid concentration in biofuels is a challenging subject because of several reasons. On the one hand, their content is usually low (i.e., from several μg L−1 to mg L−1) and, hence, sensitive techniques should be used. Besides all these, calibration with organic complex matrices becomes more difficult and degrades the accuracy of the determination. Several approaches have been evaluated to carry out this kind of analysis going from spectrochemical to electroanalytical techniques. Within the first group, Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) and Mass Spectrometry (ICP-MS) are often employed together with atomic absorption methods. The different procedures applied will be discussed in the present review emphasizing the most widely employed ones. On this subject, fundamental as well as applied studies related to the biofuel analysis through ICP-OES and ICP-MS will be shown to illustrate the current difficulties associated with these determinations. Comments regarding the possible solutions proposed to overcome the drawbacks encountered will be made.
 Raquel Sánchez | Raquel Sánchez-Romero obtained her PhD under the supervision of Prof. José Luis Todolí (University of Alicante) and Dr.Charles-Philippe Lienemann (IFP-Energies Nouvelles, Lyon, France). Her work was focused in the development and evaluation of new methods for fuel and biofuel analysis based on the use of ICP-AES. She also collaborated with other projects focused on the characterization of environmental and pharmaceutical products. Since March 2012, she has a postdoctoral fellowship at the Institute for Reference Materials and Measurements, Joint Research Center, European Commission (Geel, Belgium) working as a project manager for the development, the production and the validation of certified reference materials in environmental science. |
 Carlos Sánchez | Carlos Sánchez obtained his B.Sc. in chemistry in 2012 from University of Alicante (Spain) with extraordinary award and he obtained his M.Sc. from the same university in 2013. He is currently carrying out his Ph.D. under the supervision of Dr. José Luis Todolí Torró and Dr. Charles Philippe Lienemann with a FPU grant from Spanish Ministry of Education. His thesis research involves the development of new analytical methods based in ICP techniques for the determination of metals and metalloids in fuels, biofuels and related products and their speciation through hyphenated techniques. He also collaborated with other projects focused on the characterization of aqueous samples and pharmaceutical products. |
 Charles-Philippe Lienemann | Charles-Philippe Lienemann graduated from the University of Geneva, Switzerland, in 1993 and obtained his Ph.D. from the University of Lausanne, Switzerland, in 1997. He then worked at SGS in Lyon (France) as Lab Head for environmental analysis, before joining IFP New Energy in 2000. A new research area was then investigated with all the elemental analysis needed by the petroleum industry (petroleum product, catalyst, renewable fuel). With more than 30 papers in this new area, his actual research is focused on the behaviour, fate and analysis of trace metals in petroleum and related samples. |
 José-Luis Todolí | José-Luis Todolí is full professor of Analytical Chemistry at the University of Alicante since April 2012. His fundamental research has been focused on the development of new instrumentation for Inductively Coupled Plasma techniques as well as new chromatographic separation methods and their coupling to ICP. His research has been applied to the analysis of environmental samples, foodstuff, fuels and clinical samples. He has coauthored about 100 papers, book chapters and research books and he has about 200 contributions to international meetings among them over 20 invited lectures. |
1. General introduction
Nowadays, the interest in the development of energy sources alternative to fossil fuels has increased significantly. The most widely used biofuels are bioethanol and biodiesel and their increasing demand involves the development of new methods to ensure the quality of the final products. In this sense, the determination of metals and metalloids plays a fundamental role. Within this category one can find alkaline and alkaline earth elements (Na, K, Ca, Mg), heavy metals (Cd, Zn, Cr, Fe, Mn, etc.), metalloids (As, B) and non-metals such as S or P. These elements are present at variable concentrations depending on factors such as raw materials, production processes and post-production pollution, among others. Because the presence of these elements may affect the quality of the biofuel, official specifications have appeared. For example, ASTM D6751 in the USA and EN 14214 in Europe are specifications related to biodiesel quality requirements. Table 1 shows that both standards differ in some points. In the case of bioethanol, some specifications refer to the so-called ethanol fuel that corresponds to an ethanol–gasoline blend. In general terms, it can be stated that there is no information regarding the maximum allowable level of heavy metals in biodiesel and bioethanol.| Biofuel | Element(s) | Content | Standardb | Year |
|---|---|---|---|---|
| a Applies only to Fatty Acid Methyl Esters (FAME). b References for test methods are given in the case of bioethanol. | ||||
| Biodiesel | Na + K (Group I metals) | 5 mg kg−1 | ASTM D6751/EN 14214a | 2012/2014 |
| Ca + Mg (Group II metals) | 5 mg kg−1 | ASTM D6751/EN 14214a | 2012/2014 | |
| S | Two grades: | ASTM D6751/EN 14214a | 2012/2014 | |
| S15 (15 mg kg−1) | ||||
| S500 (0.05%) | ||||
| P | 0.001% (w/w) | ASTM D6751/EN 14214a | 2012/2014 | |
| Ethanol fuel | S | 30 mg kg−1 | ASTM D4806 | 2014 |
| Bioethanol | Cu | 0.1 mg kg−1 | EN 15488/ASTM D1688/JIS K0101 | 2007/2012/1998 |
| P | 0.5 mg L−1 | EN 15487/ASTM D3231 | 2007/2013 | |
| S | 10 mg kg−1 | EN 15487/ASTM D3231 | 2007/2013 | |
The quantification of metals and metalloids in bioethanol and biodiesel has several associated difficulties: (i) some of them are present at very low concentrations (μg L−1); (ii) there are limited certified reference materials, see Table 2; (iii) commercially available bioethanol, for instance, exists in a large variety of matrices with different water content; (iv) several sources of raw materials can be employed affecting the characteristics of the final product; and, (v) bioethanol and biodiesel contain around 300 different organic compounds depending on their origin and treatment.1,2
| Matrix | Element | Concentration | Source | Web |
|---|---|---|---|---|
| a Reformulated gasoline has a 10% of ethanol content (fuel ethanol E10). | ||||
| Biodiesel | Na, K | 2.5–50 μg g−1 | LGC | http://www.lgcstandards.com |
| Biodiesel B100 | Ca, K, Mg, Na, P | 2.5–50 μg g−1 | LGC | http://www.lgcstandards.com |
| Biodiesel B100 | S | 5–500 μg g−1 | LGC | http://www.lgcstandards.com |
| Biodiesel B5 | S | 5–500 μg g−1 | LGC | http://www.lgcstandards.com |
| Biodiesel B20 | S | 5–500 μg g−1 | LGC | http://www.lgcstandards.com |
| Biodiesel | Ca, Mg | 2.5–50 μg g−1 | LGC | http://www.lgcstandards.com |
| B100 biodiesel (Soy-based) SRM-2772 | Ca | 0.5 mg kg−1 | National Institute of Standards & Technology | http://www.nist.gov/ |
| Cu | <0.2 mg kg−1 | |||
| Fe | <0.2 mg kg−1 | |||
| Mg | <0.2 mg kg−1 | |||
| P | <0.4 mg kg−1 | |||
| K | <0.1 mg kg−1 | |||
| Na | 0.07 mg kg−1 | |||
| B100 biodiesel (Animal-based) SRM-2773 | Ca | 0.1 mg kg−1 | National Institute of Standards & Technology | http://www.nist.gov/ |
| Cu | <0.2 mg kg−1 | |||
| Fe | <0.2 mg kg−1 | |||
| Mg | 0.05 mg kg−1 | |||
| P | <0.4 mg kg−1 | |||
| K | <0.1 mg kg−1 | |||
| Na | 0.9 mg kg−1 | |||
| Reformulated gasolinea | S | 13.6 μg g−1 | LGC | http://www.lgcstandards.com |
| Reformulated gasolinea | S | 13.8 mg kg−1 | National Institute of Standards & Technology | http://www.nist.gov/ |
For all these reasons, it is obvious that sensitive techniques are required to carry out the determination of metals and metalloids in these types of samples. In addition, it is necessary to develop analytical methods able to compensate for matrix effects due to a large variety of matrices found in bioethanol and biodiesel samples. Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) and Mass Spectrometry (ICP-MS) appear as the most appropriate techniques to perform elemental determination in biofuels, although alternative techniques have also been applied for this purpose.
The fundamentals, applications and latter developments of biodiesel and bioethanol analysis through ICP techniques are revisited in the present work. The use of alternative analytical techniques for this purpose is also mentioned.
2. Fundamental studies
In order to understand the phenomena occurring when organic samples such as biodiesel and bioethanol are introduced into inductively coupled plasma, fundamental studies are required. These kinds of samples may interfere with each step of sample analysis from aerosol production to signal recording. Additionally, due to the high viscosity of biodiesel, for instance, a pretreatment step of the sample is usually required. The dilution with a suitable solvent is the most extended procedure, and ethanol, kerosene and xylene are usually employed for this purpose.3–5 Bioethanol, in turn, may contain variable proportions of water, propanol, butanol and other low molecular weight alcohols.1 Therefore, the physico-chemical properties of the sample will change thus causing an intensification of the matrix effects.2.1. Aerosol generation
When a pneumatic nebulizer is used to generate the aerosol, the solution physical properties will affect the characteristics of the produced mist. For this kind of nebulization device, the most important properties are the surface tension and viscosity. Organic samples, such as those included in bioethanol and biodiesel, have a quite wide range of viscosities and surface tension values. Table 3 summarizes the density, viscosity and surface tension of representative FAME and biodiesel samples. Moreover, two synthetic solutions usually prepared to simulate the blanks also included. In this case, the portion of biodiesel was replaced by an Element Stock Oil (75 Viscosity, Conostan, Ponca City, Oklahoma, USA). As it may be observed, viscosity is different according to the particular solution considered.| Sample | Viscosity (cP) | Density (g cm−3) | Surface tension (mN m−1) |
|---|---|---|---|
FAME–xylene 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
2.5 | 0.83 | n.a. |
| Stock oil–xylene 1:1 |
1.5 | 0.84 | 30.0 |
| FAME–kerosene 1:1 |
3.2 | 0.84 | n.a. |
| Stock oil–kerosene 1:1 |
1.9 | 0.84 | 29.5 |
| Xylene | 0.6 | 0.85 | 27.5 |
| Biodiesel | 5.1 | 0.84 | 31.4 |
| Biodiesel–xylene 1:10 |
0.7 | 0.85 | 28.8 |
| Ethanol | 1.14 | 0.79 | 22.3 |
| Water | 1.00 | 1.00 | 72.8 |
| Bioethanol | n.a. | 0.82 | 23.3 |
In order to evaluate the influence of solution physical properties on the nebulizer performance, the aerosols produced in the first instance (i.e., primary aerosols) can be measured. Farino and Browner6 studied the effect of sample surface tension on aerosol properties. As this physical property decreases, the energy required to generate a droplet from the solution bulk goes down. In addition, in solvents with low surface tension, the waves generated on the liquid surface have a short wavelength and the gas penetrates easily into the liquid bulk. As a result, the liquid and gas interaction becomes highly efficient, thus favouring aerosols with low droplet diameters. For example, when a pneumatic concentric nebulizer is operated under typical conditions (i.e., 1 mL min−1 liquid flow rate and 0.7 L min−1 nebulizer gas flow rate) the median of the aerosol volume drop size distribution (D50) for primary aerosols is 17 and 11 μm for water and ethanol, respectively. It is worth mentioning that the surface tension of ethanol (21.4 dyn cm−1) is approximately three times lower than that of water, whereas both solvents have similar viscosity values.7
Regarding viscosity, as Sharp studied, the instabilities generated on the liquid surface during the nebulization event are attenuated for liquids with high viscosity values,8 thus promoting the generation of coarse aerosols. As revealed in Table 3, the final viscosity depends on the solvent employed to dilute the sample, thus affecting the primary aerosol characteristics. Thus, for instance, for a pneumatic concentric nebulizer, when xylene is used to dilute the samples, all the primary aerosol liquid volume is contained in droplets with diameters below 13 μm, whereas this maximum diameter increases up to 17 μm when the employed solvent is kerosene.
In the case of biodiesel, the D50 takes values of 11, 63 and 23 μm for xylene, biodiesel and 1:10 diluted biodiesel, respectively. As expected, compounds with low viscosities promote the production of fine aerosols.8,9 It is also worth noting the poor nebulization yield observed for a pure biodiesel sample. Due to the high D50 value, the sensitivity finally obtained will be extremely low. The proposed solution to generate finer aerosols is, thus, to dilute the sample with an appropriate solvent.
In the case of bioethanol, the final sample composition may vary as a function of several factors among them the water content or the additives present. This fact is illustrated in Fig. 1 in which the Sauter mean diameter, D3,2, significantly changes as a function of the sample considered.
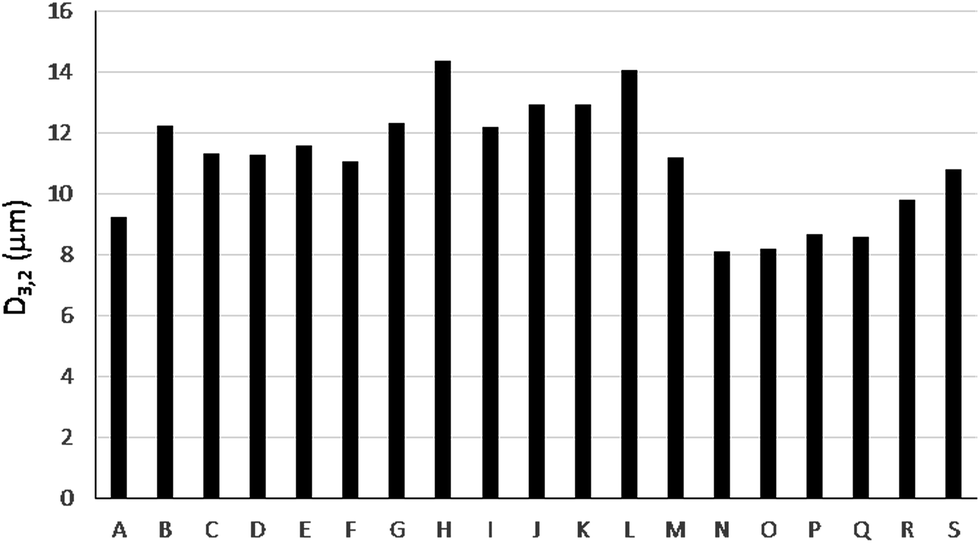 | ||
| Fig. 1 Sauter mean diameter (D3,2) for primary aerosols generated by a conventional pneumatic concentric nebulizer working with 19 different bioethanol samples (A–S). | ||
2.2. Aerosol transport
Once the aerosol is generated, several phenomena take place inside the spray chamber that lead to a modification in its characteristics. These are the so-called aerosol transport phenomena and they are responsible for analyte losses inside the spray chamber. The most influencing events are: (i) solvent evaporation; (ii) droplet coalescence, and; (iii) droplet inertial impacts. The major changes in the primary aerosol characteristics are caused by the nebulizer gas flow rate and the design of the spray chamber.10 However, the primary aerosol characteristics together with sample physical properties, mainly density and volatility, affect the extent of all these processes.In the case of organic samples, the solvent volatility is the most relevant property precluding the mass of solution delivered to the plasma. The solvent evaporation takes place mostly just after the aerosol generation until the gas becomes saturated in solvent. The high solvent volatility together with the fineness of the organic aerosols contribute to an enhancement in the mass of analyte and solvent delivered to the plasma.11,12 Under these circumstances, nebulization conditions (liquid and gas flow rates) have a more determining effect for organic solvents than for aqueous solutions. Thus, for volatile solvents, the solvent transport efficiency may reach values close to 100%. Therefore, the selection of the appropriate experimental conditions is a more challenging issue for the formers.13
The fineness of the aerosol leaving the spray chamber (tertiary aerosol) and the mass of solvent and analyte transported to the plasma are indicators of the quality of the primary aerosol transport. In fact, the drop size distribution of the tertiary aerosol is proposed by several authors as the property that plays a major role in terms of plasma thermal state because it determines the amount of energy required to vaporize the matrix.14 On this subject, finer aerosols are found when working with 50% (v/v) ethanol–water mixtures than for water alone. These results are independent of the spray chamber considered.15 A stirred tank methodology has been used to thoroughly study the effect of increasing the ethanol concentration on the characteristics of the aerosols leaving the spray chamber.16 The results proved that the median of the tertiary aerosol volume drop size distribution decreased significantly as the concentration of this alcohol went up to 5%. Then the decrease in this statistical parameter with the ethanol content became less pronounced. The intensification of the solvent evaporation inside the spray chamber and the fineness of the generated aerosols as the ethanol proportion grows appear to be the dominating phenomena.
The evolution of drop diameter versus time as a result of the solvent evaporation is a function of the so-called evaporation factor12 which is defined as the volume of solvent evaporated per unit of time. This magnitude can be calculated according to:
| E = 48DvσPsM2(∂RT)−2 | (1) |
As Boorn et al.17 reported, the solvent evaporation factor for ethanol is about three times higher than the evaporation factor for water (Eethanol = 45.6 μm3 s−1vs. Ewater = 13.1 μm3 s−1).
As a result of the finer aerosols and the higher evaporation factor, the total mass of solvent transport rate leaving the chamber for a pneumatic nebulizer adapted to a double pass spray chamber was 6 times higher for ethanol as compared to water. Note that the relative volatility values were 0.1 and 0.7 for water and ethanol, respectively.7 As the solvent evaporation becomes more significant and finer aerosols are generated for ethanol than for water, droplets decrease their diameters and they have more chance to be transported through the spray chamber. The net result is an increase in the analyte transport rate for the former. In the particular case of ethanol samples, this parameter was about five times higher than for water.
2.3. Plasma effects
When carrying out the analysis of bioethanol or biodiesel samples, plasma effects should be carefully considered. These effects are related to the plasma energy consumed for the solvent vaporization and dissociation. Obviously, the operating nebulization conditions (i.e., the liquid flow rate and nebulizer gas flow rate) play a fundamental role, because they dictate the aerosol mass reaching the plasma. For instance, it is sometimes advisable to lower both variables so as to reduce the solvent plasma load, simultaneously increasing the residence time of the analyte in the plasma.18 Nevertheless, if these variables (especially the nebulizer gas flow rate) are excessively decreased, the analyte mass transported towards the plasma and, hence, the sensitivity may be too low. Plasma degradation caused by the presence of bioethanol or biodiesel becomes less pronounced at high R.F. power values. Under these conditions, sensitivities may be higher for organic samples than for aqueous matrices. In contrast, if plasma effects are not taken into consideration, organic solvents may cause a decrease in the sensitivity.19Several studies have been conducted to understand the effects caused by the presence of an organic matrix on the plasma performance. When an organic sample (e.g. ethanol, biodiesel) is introduced into the ICP, specific effects take place such as: (i) molecular emission of solvent pyrolysis products; (ii) modifications in the plasma geometry; (iii) generation of a vortex in the plasma; (iv) changes in electron number density, hydrogen density and excitation temperature; and, (v) formation of carbon or soot deposits somewhere in the spectrometer.
The introduction of an organic solvent may increase the thermal conductivity hence accelerating the heat conduction away from the plasma. As a result, the peripheral zones of the plasma cool rapidly thus causing a reduction in the plasma volume. This is the so-called plasma thermal pinch that has been observed when introducing solvents such as methanol and can be extrapolated to ethanol20 and ethanol–water solutions.14,23
Of course, hydrogen emission and electron density depended on the operating conditions. As it has been reported, the effect of ethanol concentration on H emission intensity is more pronounced at low than at high R.F. power. At 1.02 kW the emission signal of hydrogen (434.05 nm) for 10% of ethanol was around 3 times higher than that obtained for water while at 1.36 kW this enhancement factor was only 2 times.23
Besides electron number density, plasma excitation temperature also changes when an organic solvent is delivered to the excitation cell. Several authors reported decreases in this parameter as compared to aqueous solutions.17,19,22,23,26–29 However, a maximum pattern in the excitation temperature was reported as the ethanol content increased.23,30
A change in the hydrogen content can be claimed in order to try to explain the eventual increase in plasma fundamental parameters found when introducing ethanol. The effect of adding molecular hydrogen to the plasma has been previously described and its beneficial role in both ne and excitation temperature has been demonstrated.23,26,31–35 The increase in hydrogen generation and, hence, the rise in the plasma thermal conductivity, in the presence of ethanol with respect to water are based on the fact that the energy requirements to induce its dissociation are very low in comparison with those for water.
A parameter widely studied to monitor the plasma thermal state and its robustness is the magnesium ionic to atomic net emission intensity ratio (MgII/MgI). According to previous studies it has been indicated that this ratio increased with ethanol concentration up to 25%.14 This trend was confirmed by the experiments done with a stirred tank setup. For a less robust plasma, it was found that the MgII/MgI ratio peaked at about 8% ethanol and then decreased.16 Possible explanations could be based on the increased plasma thermal conductivity and/or thermal pinch. Once the ethanol plasma load becomes too high, a degradation in its excitation conditions is produced.
2.4. Spectral interference
Spectral interference caused by organic samples in ICP-OES is due to solvent pyrolysis products. In the presence of an organic solvent, the most abundant species in plasma are C2, CN, and C. Furthermore, depending on the solvent nature, other molecules may be present such as CS, CH, NO and CO. Fig. 2 shows the evolution of the background emission spectrum versus the plasma observation height. When an alcohol is introduced into the plasma, spectral interference is strongest at its base. Note that plasma operating conditions can alter the distribution of pyrolysis products.39 Moreover, it is very important to take into account the physical form in which the solvent reaches the plasma because a large fraction of it is in vapor form.19 Pan et al.19 demonstrated that the main impact of desolvation with organic solvents is to reduce the C2 species population in the plasma, which in turn strongly influences plasma temperatures.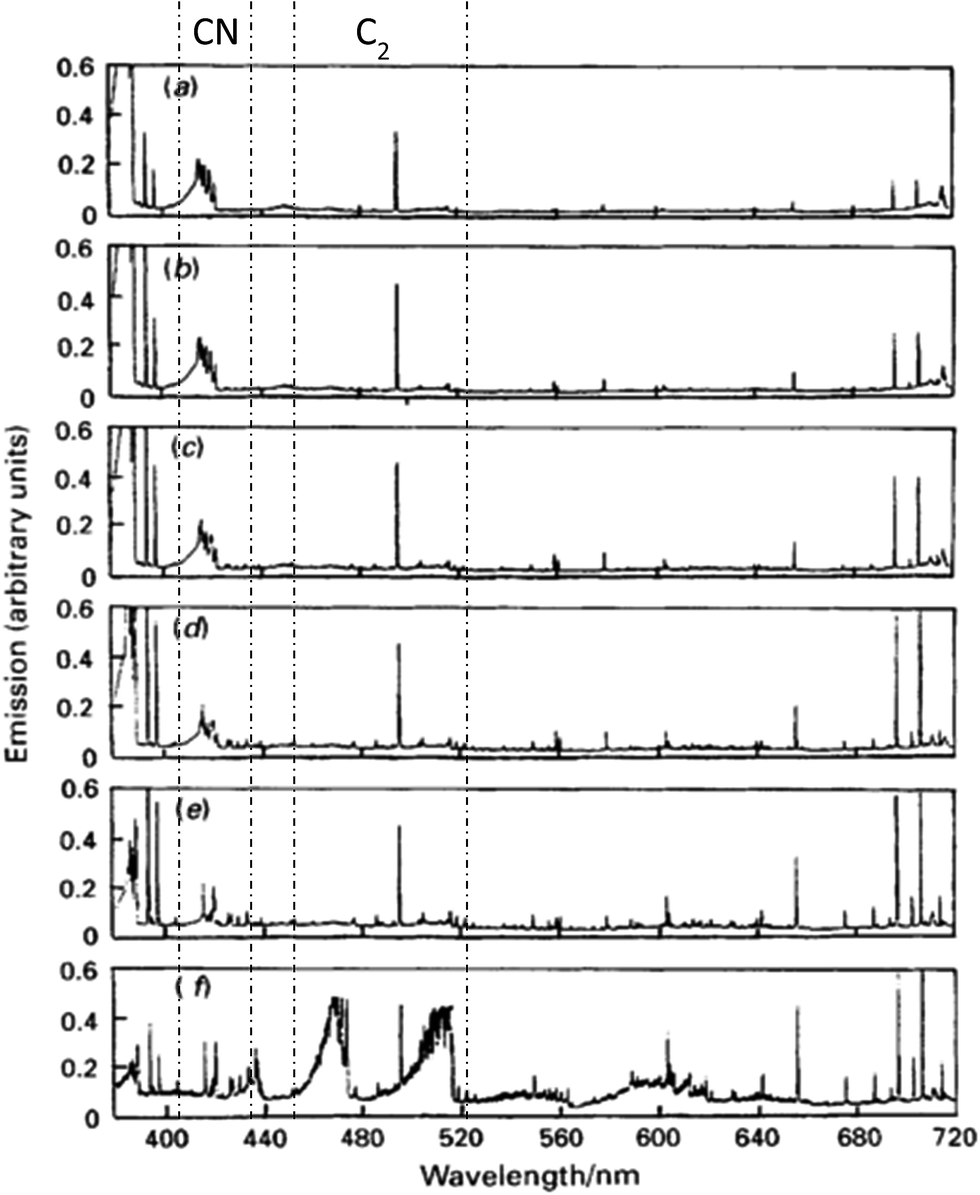 | ||
| Fig. 2 Spectral survey of the visible emission from de ICP loaded with methanol for several observation heights: (a) 21 mm; (b) 18 mm; (c) 15 mm; (d) 12 mm; (e) 9 mm; (f) 6 mm. Cyanide radical (410–430 nm) and diatomic carbon (450–520 nm) taken from ref. 20. | ||
Alcohols also induce ICP-MS spectral interference.40–42 They can be explained in terms of: (i) charge transfer reactions from C species to the analyte ions;43 (ii) enhancement of the aerosol transport efficiency through the sample introduction system;44 and (iii) shifts in the plasma zone of maximum ion density.45,46 The presence of ethanol47 can lead to an increase in the sensitivity for some isotopes because of polyatomic interference. Also, for this technique, the interference could be due to the formation of pyrolysis products.48 In order to avoid these phenomena, collision and reaction cells can be employed. Woods et.al.49 applied an ICP-MS fitted with an octopole reaction system (ORS) to the direct determination of the inorganic content of several biodiesel samples. Intense plasma-based species such as 14N2 on 28Si, 38Ar1H on 39K and 40Ar on 40Ca were removed by reaction mode; in this case with H2 cell gas. Sulfur, in turn, was measured removing the O2 interference by reaction with Xe cell gas.
3. Biodiesel
Nowadays, there is an increasing demand for biodiesel production. In fact, the European Directive RED 2009/28/EC50 promotes the use of substitute fuels coming from renewable, non-edible origin. Among biodiesel, Fatty Acid Methyl Ester (FAME) is available as directly blended with diesel from fossil origin. In the last decade, the number of papers focused on biodiesel production has increased from 31, in 2003, to 1296 in 2013.Generally speaking, biodiesel comprises a mix of mono-alkyl esters of long chain fatty acids produced mainly by transesterification.51 However, there are four primary ways to produce biodiesel: direct use and blending, microemulsification, thermal cracking (pyrolysis) and transesterification.52–57 For all these processes the resulting product shows a high combustion efficiency. In the case of pyrolysis, the obtained fuel is chemically similar to petroleum products. However, the main drawback of pyrolysis is the high amount of energy consumed in the cracking step. Meanwhile, in the case of the transesterification process, the main disadvantage is the formation of reaction by-products, such as glycerol and wastewater.
Alternative processes have been developed, such as hydrogenation of fat towards kerosene/diesel, as well as FT synthesis. The most employed process is currently based on transesterification.
3.1. Synthesis and presence of metals. Importance of their determination
Most of the metals present in biofuel come from the raw material (e.g., seeds) or are introduced during the processing or storage of the final product. Several inorganic contaminants may occur in the raw materials, mainly due to the absorption of some minerals from the soil where the plant was grown, other sources such as pesticides and fertilizers could be considered. Seeds, commonly employed for biodiesel production, with different origin were analyzed: castor bean, cotton seed, curcas bean, fodder turnip, sunflower, soybean and tung. After digestion of the seed, element concentrations were determined by ICP OES (Ca, K, Mg, Na and P) and by ICP-MS, using external calibration with aqueous standard solutions.58 As it was expected the elements, whose concentration limit is regulated by international organizations,59,60 presented the highest concentration in the seeds. Regarding minor elements, Al, Fe, Mn and Zn concentration was strongly related to the soil characteristics. The concentration of Al in the tung sample, about 200 μg g−1, was at least 4 times higher than in the other seed samples. The maximum Fe concentration was found in the fodder turnip, about 130 μg g−1. Zinc was more concentrated in the sunflower and in the castor bean samples, around 45 μg g−1. The concentrations of Mn varied from about 7 μg g−1 in tung seeds to about 27 μg g−1 for curcas bean.58 Paredes et al. have recently proposed the use of normalized ratios of mass fractions found for B, Fe, Cu, Zn, P and S as markers of the biological origin of raw materials of 1st generation biodiesels.61 However, a clear relationship between metal fingerprints and sample origin has not been established. Pillay et al.62 demonstrated that sharp differences could exist due to the nature of the feedstock ensuing from differences in cultivation techniques, soil conditions and plant parts used for obtaining the biofuel.Generally speaking, transesterification compromises the reaction between fats or oils, triglycerides and an alcohol, usually methanol or ethanol, in the presence of a catalyst to produce glycerine and methyl esters or biodiesel.63 When methanol is employed the biodiesel is called FAME (fatty acid methyl esters), whereas for ethanol it is called FAEE (fatty acid ethyl ester).The catalysts employed could be classified mainly into four groups: (i) basic homogeneous; (ii) acid homogeneous; (iii) heterogeneous; and (iv) lipases. Basic catalysts are the most widely employed as they provide better reaction efficiencies. Among the basic catalysts sodium and potassium hydroxides, carbonates and sodium and potassium alkoxides, such as methoxide, epoxide, and nitrous dioxide, are included. Sodium and potassium hydroxides are the most common basic catalysts in industry.64,65 However, in order to be able to use these catalysts, the raw material to obtain biodiesel, must be purified so as to remove free acids. This is because the basic catalyst neutralizes free fatty acids, which may cause the formation of soaps thus promoting the formation of stable emulsions. These emulsions do not allow separation of biodiesel and glycerine affecting the purification of esters.66 Moreover, the separation of the catalysts from the reaction products in the purification steps is technically difficult precluding the quality of the final product.4,67 The use of a suitable heterogeneous catalyst has been suggested by several research groups. The main advantage incorporated by heterogeneous catalysts is that they can be separated from the reaction products by filtration.63,68–89
In addition, it is important to note that the commercial biodiesel is a blend of the pure biodiesel (e.g. FAME, FAEE) and diesel. The European Union legislation established the maximum blend ratio in B7.5 (7.5% biodiesel, 92.5% diesel) for technical reasons.90 Whereas, in certain non-European countries a percentage blend is mandatory. In Brazil, which has the world's most developed biofuel industry, a 25% blend is mandatory. On the other side, blends of 20% biodiesel and lower can be used in diesel equipment with no, or only minor modifications.
For “pure biodiesel”, metal content determination is important to ensure the quality of the final product. Some metals, especially sodium and potassium, could be incorporated into the final product during the transesterification reaction. Sodium and potassium compounds promote the formation of insoluble and abrasive solids contributing to the degradation of engine parts or to deposit formation in vehicles filters.91–93 Moreover, “pure biodiesel” may contain additional elements. For example some elements such as Cu, Cd, Ni, etc. could be absorbed from the soil by the plant itself. In addition, the fingerprint in terms of metal in the “industrial biodiesel” gives an indication of the environmental risk. Moreover, some metallic species are incorporated into the product as additives: anti-knock agents, anti-oxidants, burn improvers, metal deactivators, anti-rust agents, anti-icing agents, upper-cylinder lubricants, and detergents. In some instances, elements are incorporated into the product during transportation and/or production or storage.92,94–96
Finally, the presence of some metals can affect the stability of the biodiesel.97–99 Sarin et al.100 studied the influence of metal contaminants on oxidation stability of Jatropha biodiesel. The induction period of the biodiesel decreased drastically with small concentrations (mg kg−1) of metal contaminants. The biodiesel exhibited oxidation stability of 3.95 h in the Rancimat test, according to the EN 14112.101 The biodiesel standard EN 1421459 required the oxidation stability determination at 110 °C with a minimum induction time of 6 h by the Rancimat method101 whereas the ASTM standard D-675160 recently introduced a limit of 3 h. The stability of biodiesel is critical to ensure fuel quality at all points along the distribution chain. Among the metals investigated, copper appears to have the strongest detrimental effect. Additional elements such as Co, Cu, Fe, Mn, and Ni can promote oxidative degradation, whereas some elements such as Pb, and Zn can also catalyze the biodiesel oxidation.102,103
3.2. Analysis by ICP techniques
Because the metal concentration in biodiesel is usually low; the selection of the determination technique should be strongly related to the target metal and to its concentration.104 The main techniques employed are flame atomic absorption spectrometry (FAAS), graphite furnace atomic absorption spectrometry (ETAAS), inductively coupled plasma atomic emission spectroscopy (ICP-OES) and inductively coupled plasma mass spectrometry (ICP-MS).4,49,91–94,103–110Sample preparation. Dilution has been widely recommended as a sample treatment method for the analysis of biodiesel. The selection of the solvent could influence the quality of the analytical results. Xylene and kerosene have been widely used to perform routine analysis of these kinds of samples.111–118 In the case of phosphorous determination by ICP-OES Sanchez et al.119 employed two sample introduction systems: (i) a concentric micronebulizer fitted to a glass cyclonic spray chamber; and, (ii) the same nebulizer coupled to a glass single pass spray chamber (Torch Integrated Sample Introduction System, TISIS).119–121 For the conventional cyclonic spray chamber, the signal enhancement factor observed for xylene with respect to kerosene ranged from 2.9 to 3.9. Similar trends were found for the TISIS although the influence of the solvent was less marked than that observed for the cyclonic spray chamber.
Ethanol was proposed as an alternative solvent by dos Santos et al.3 for simultaneous determination of Ca, P, Mg, K and Na in biodiesel by ICP-OES. Dilution with ethanol enabled the use of aqueous standards, leading to accurate and precise results. An oxygen flow was used to decrease the background and non-spectral interference was compensated for by employing yttrium as an internal standard. The maximum allowed concentration59,60 was higher than the limits of detection obtained with this procedure. The obtained LODs, considering 2.5 g of sample in a final volume of 25 mL, were: 0.03, 0.5, 0.005, 0.3 and 0.1 μg g−1, for Ca, P, Mg, K and Na, respectively. Moreover, the validity of the method was evaluated throughout the analysis of four biodiesel samples produced from different raw materials. The samples were spiked with 5 μg g−1 of the analytes. Calibration was carried out with standard solutions containing an ethanol–water mixture as a solvent. All recoveries were in the 82 to 114% range for all analytes, demonstrating the accuracy of the proposed procedure. Moreover, Chaves et al.5 evaluated alternative solvents, such as ethanol and 1-propanol, for the determination of Ca, Cu, Fe, K, Mg, Na, P, S and Zn in biodiesel and vegetable oils by ICP-OES. Calibration was carried out against inorganic standards diluted in ethanol or 1-propanol, while yttrium was used as an internal standard, correcting for non-spectral interference and sensitivity drift. Recovery tests yielded figures included within the 87 to 116% range. The measured precision expressed as relative standard deviation (n = 3) was lower than 5% and limits of detection were at the low μg g−1 level.
While dilution of samples is one of the most widespread approaches, other alternatives have been explored (e.g., emulsification) so as to reduce the mass of organic solvent introduced into the plasma.122 The emulsification involves the addition of an aqueous phase containing an acid and/or surfactant in an appropriate proportion.123 De Souza et al.124 developed a simple and rapid method for the simultaneous determination of seven trace elements in biodiesel by axial and radial viewed ICP OES. The sample was emulsified with Triton X-100 and water and yttrium was employed as an internal standard. One of the advantages of the emulsification was that aqueous standards could be used. Good recoveries, in the range of 90 to 109%, were achieved for all the studied analytes. Moreover, the LODs obtained in the axial mode increased from 0.007 to 0.660 μg g−1. Young et al.125 developed a method for the determination of sulphur in biodiesel samples based on the sample micro-emulsification. Microemulsions were prepared using 0.5 mL of 20% v/v HNO3, 0.5 mL of Triton X-100, 2–3 mL of biodiesel sample, and diluted with n-propanol to a final volume of 10 mL. The novelty of the method was the summation of the emission intensities of multiple sulphur lines to increase the accuracy and sensitivity. The recoveries obtained ranged from 72 to 119%. Recently, the same emulsifier was used by Lisboa et al.126 and, as in the previous work, external calibration with aqueous standard solutions was applied. The LOD was in the sub-mg kg−1 range and recoveries increased from 91 to 107%.
Moreover, the sample digestion was explored as an alternative sample preparation method by Korn et al.127 Two digestion procedures were evaluated for the determination of Ca, P, Mg, K and Na in biodiesel by ICP OES: (i) an open system with conventional heating using concentrated nitric and sulfuric acids and the addition of hydrogen peroxide to complete the digestion; and, (ii) a microwave-assisted closed system using concentrated nitric acid and hydrogen peroxide. The analytical performances were evaluated through the residual carbon contents. These contents were 0.358 ± 0.012% with the open system with conventional heating and 0.614 ± 0.023% with the microwave-assisted closed vessel system, demonstrating the high efficiency of both proposed procedures. The closed system was preferred because the process was faster and safer. Moreover, the accuracy determined by a recovery test was better than for the open systems. In both cases the LOD was in the sub-μg g−1 range. Inductively Coupled Plasma Mass Spectrometry (ICP-MS) can be an alternative approach for the determination of trace elements in biodiesel. Woods and Fryer49 explored the use of an ICP-MS instrument fitted with an octopole reaction system (ORS) for the elemental determination in several biofuel materials. Dilution with kerosene was used as a sample preparation procedure. The ORS removed matrix- and plasma-based spectral interference reducing the LOD. In fact the LODs found were 0.0109 μg kg−1 and 0.0293 mg kg−1 for Be and S, respectively. Moreover, the rapeseed FAME sample was spiked with a multielemental solution and recoveries for all elements increased from 90 to 120%, although the majority fell within 5% of the target value, indicating reliable interference removal for the spiked matrices.
As in ICP-OES, microemulsification has been explored as an alternative sample preparation method. Amais et al.110 developed a method for the determination of Cd, Co, Cu, Mn, Ni, Pb, Ti, and Zn in microemulsified biodiesel samples by ICP-MS. Microemulsions were prepared using 0.25 mL Triton X-100, 0.25 mL 20% v/v HNO3, 0.50 mL biodiesel sample and 4.0 mL n-propanol. The accuracy of the method was evaluated by recovery experiments. Recoveries found were in the range of 76.5 to 116.2% for all analytes and LODs were in the of 9.63 × 10−3 to 19.5 μg L−1 range. It is important to note, that an oxygen gas flow was additionally incorporated, and as a consequence, the noise of the blank signal increased. In fact, LODs without the additional oxygen gas flow were lower.
Alternative sample introduction systems. The sample introduction systems employed in the studies mentioned so far consisted of a nebulizer operating at liquid flow rates on the order of mL min−1 adapted to a spray chamber. Microsample introduction systems have been considered as suitable devices for the analysis or organic samples through ICP techniques. The main advantages of these devices are: (i) low sample volume required to perform the analysis; (ii) high analyte transport efficiency; (iii) low plasma solvent load; (iv) reduction in the volume of waste generated.128 De Souza et al.129 compared the performance of a parallel path micronebulizer with that of a concentric micronebulizer for the elemental determination in biodiesel and other oils by ICP-OES. The main advantage of the parallel path micronebulizers over the conventional ones is the low risk of blockage, thus allowing the introduction of samples with high contents of dissolved solids. Moreover, limits of detection for the parallel flow nebulizer were lower than for the concentric one.
Additional systems have been used to carry out biodiesel analysis. Thus, cross-flow micronebulizers have been modified with an additional channel for the introduction of an extra liquid flow.130,131 In this way, the organic sample is continuously introduced through one channel of the nebulizer, while aqueous calibration standards are sequentially nebulized through the other one. Aerosol droplets generated by both channels are mixed in the spray chamber and the resulting mixture reaches the ICP, thus allowing the analysis of organic samples by on-line standard addition calibration using aqueous calibration solutions. Concentric nebulizers were also used for this purpose.132 The accuracy of the system was tested by the recovery test, for all the analytes, the results were included in the range of 96–101%.
On the other hand, the spray chamber has been modified to promote the complete transport of the sample to the plasma. In this sense, Sanchez et al.121 employed a 350 °C heated low inner volume single pass spray chamber to mitigate the matrix effects in the analysis of biofuel samples by ICP-OES. The results have proved that the higher the chamber wall temperature, the higher the sensitivity. As a result, limits of detection decreased below 7 μg L−1 for elements such as manganese, vanadium and silicon. Furthermore, memory effects were less severe as the temperature raised. Another benefit of increasing the TISIS chamber wall temperature was that matrix effects became less pronounced as compared to a cyclonic chamber.119–121 Thus, at 350 °C non-spectral interference was eliminated likely because the analyte transport efficiency to the plasma was close to 100% irrespective of the sample analyzed. The developed procedure was applied to the analysis of biodiesel with recoveries close to 100% for four biodiesel samples. The TISIS spray chamber and flow injection was used for the determination of nickel, vanadium and manganese in fuel and biofuel samples by ICP-MS.120 In this case, the amount of sample injected was only 2.5 μL. Moreover, the chamber temperature was optimized in terms of sensitivity and mitigation of matrix effects. It was found that sensitivity peaked at 110 °C heating temperature. However, non-spectral interference caused by differences in the matrix composition became less severe as this variable was increased and it was virtually eliminated at 200 °C. As a consequence, a single xylene based standard could be used as a universal standard.133,134
Another approach explored for the analysis of biodiesel has been to decrease the temperature of the spray chamber, thus reducing the amount of organic matter reaching the plasma. Chaves et al.5 demonstrated that cooling a cyclonic spray chamber at −5 °C reduced sufficiently the amount of organic solvent introduced into the plasma. Therefore, it was not necessary to introduce oxygen using ethanol and 1-propanol as solvents. For this device, the relative standard deviation was lower than 5% and limits of detection were at the low μg g−1 level (Table 4).
| Element | Technique | Conditions | LOD | Range concentration (min–max)b | Ref. |
|---|---|---|---|---|---|
| a n.d.: not determined in real samples. b This range corresponds to the minimum and maximum concentrations found for a given analyte and analytical method for several samples. A single figure is included when only a sample was analyzed. | |||||
| Ag | ICP-MS | EC (kerosene, 1:3) |
0.149 μg kg−1 | 0.257–3.15 μg kg−1 | 49 |
| Al | ETAAS | EC (ethanol, 1:5 m:v) |
0.013 μg g−1 | 0.038–0.443 μg g−1 | 109 |
| Pd(NO3)2 + Mg(NO3)2 as modifier | |||||
| As | ICP-MS | EC (kerosene, 1:3) |
0.066 μg kg−1 | 1.02–1.29 μg kg−1 | 49 |
| ETAAS | EC (direct sampling) | 5.1 μg kg−1 | n.d. | 151 | |
| Pd + Mg + Triton X-100 as modifier | |||||
| B | ICP-MS | EC (kerosene, 1:3) |
6.57 μg kg−1 | 40.3–334 μg kg−1 | 49 |
| Ba | ICP-MS | EC (kerosene, 1:3) |
0.0990 μg kg−1 | 4.64–55.8 μg kg−1 | 49 |
| Be | ICP-MS | EC (kerosene, 1:3) |
0.0109 μg kg−1 | 0.0202–0.0609 μg kg−1 | 49 |
| Ca | ICP-OES | EC (kerosene, 1:4) |
0.4–9 μg kg−1 | 2–10 mg kg−1 | 4 |
| ICP-OES | EC (kerosene, 1:10) |
0.003 mg kg−1 | 0.603–401.2 mg kg−1 | 113 | |
| ICP-OES | EC (kerosene, 1:10) |
0.05 mg kg−1 | 0.06–7.4 mg kg−1 | 115 | |
| ICP-OES | EC (kerosene, 1:10) |
0.04 mg kg−1 | 0.17–36.3 mg kg−1 | 117 | |
| ICP-OES | EC (ethanol, 1:10) |
0.03 μg g−1 | 0.38–0.56 μg g−1 | 3 | |
| IS: Y | |||||
| ICP-OES | EC (ethanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.08 μg g−1 | 0.4–28.5 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (1-propanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.05 μg g−1 | 0.4–28.5 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (aqueous standards) | 0.05 μg g−1 | 0.19–1.09 μg g−1 | 124 | |
| Emulsification | |||||
| ICP-OES | EC (aqueous standards) | 0.121 mg kg−1 | 0.27–0.32 mg kg−1 | 126 | |
| Emulsification | |||||
| IS: Y | |||||
| ICP-OES | EC (open digestion) | 0.78 μg g−1 | n.d. | 127 | |
| IS: Y | |||||
| ICP-OES | EC (microwave close digestion) | 0.40 μg g−1 | n.d. | 127 | |
| IS: Y | |||||
| ICP-MS | EC (kerosene, 1:3) |
6.40 μg kg−1 | 20.8–135 μg kg−1 | 49 | |
| FAAS | EC (ethanol, 1:10 m:v) |
0.31 mg kg−1 | 0.37–1.30 mg kg−1 | 147 | |
| FAAS | EC microemulsification | 0.11 mg L−1 | n.d. | 148 | |
| FAAS | EC microemulsification | 0.1 μg g−1 | 0.10–5.34 μg g−1 | 108 | |
| IC | EC (Ca2+) | 0.23 mg kg−1 | 0.42–6.64 mg kg−1 | 159 | |
| CE + diode array detector | IS | 0.3 mg kg−1 | 1.9–3.4 mg kg−1 | 161 | |
| Liquid–liquid extraction (Ca2+) | |||||
| CE + coupled contactless conductivity detector | Liquid–liquid extraction (Ca2+) | 0.12 mg L−1 | 0.12–0.23 mg kg−1 | 162 | |
| Squarewave voltammetry | Glassy carbon electrode | 1.6 10−3 μmol L−1 | 0.34–2.84 μmol L−1 | 168 | |
| Sample digestion | |||||
| Standard addition (Ca2+) | |||||
| HR-CS FAAS | EC (xylene, 1:10 m:v) |
0.34 mg kg−1 | 2.09–2.11 mg kg −1 | 144 | |
| LS FAAS | EC (xylene, 1:10 m:v) |
0.52 mg kg−1 | 2.09–2.11 mg kg −1 | 144 | |
| Cd | ICP-MS | EC (kerosene, 1:3) |
0.108 μg kg−1 | 0.304–0.589 μg kg−1 | 49 |
| ICP-MS | EC (aqueous standards) | 9.63 × 10−3 to 7.77 × 10−2 μg L−1 | 0.14–0.25 μg L−1 | 110 | |
| Microemulsification | |||||
| ETAAS | EC microemulsification | 0.1 μg L−1 | n.d. | 92 | |
| W as modifier | |||||
| ETAAS | Standard addition | 0.3 μg kg−1 | 4.83 μg kg−1 | 150 | |
| Emulsification | |||||
| Pd–Mg mixture as modifier | |||||
| ETAAS | EC (direct sampling) | 0.2 μg kg−1 | n.d. | 151 | |
| 0.1% Pd + 0.06% Mg + 0.06% Triton X-100 as modifier | |||||
| Anodic stripping voltammetry | Bismuth film electrode | 2 ng L−1 | 0.17–0.65 mg kg−1 | 165 | |
| Sample digestion (Cd2+) | |||||
| Cl | ICP-OES | EC (kerosene, 1:4) |
400–950 μg kg−1 | n.d. | 4 |
| Co | ICP-MS | EC (kerosene, 1:3) |
0.0337 μg kg−1 | 0.0449–0.124 μg kg−1 | 49 |
| ICP-MS | EC (aqueous standards) | 9.23 × 10−2 μg L−1 | 5.87–6.11 μg L−1 | 110 | |
| Microemulsification | |||||
| ETV-ICP-MS (Pd as modifier) | EC | 0.5 ng g−1 | n.d. | 94 | |
| Cr | ICP-OES | EC (kerosene, 1:10) |
0.011 mg kg−1 | 0.269 mg kg−1 | 113 |
| ICP-MS | EC (kerosene, 1:3) |
0.0224 μg kg−1 | 0.376–1.36 μg kg−1 | 49 | |
| WCAES | Tungsten coil atomizer | 70–300 μg kg−1 | n.d. | 169 | |
| Standard addition | |||||
| Cu | ICP-OES | EC (kerosene, 1:10) |
0.003 mg kg−1 | 0.118–0.869 mg kg−1 | 113 |
| ICP-OES | EC (ethanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.01 μg g−1 | 0.14–1.62 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (1-propanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.008 μg g−1 | 0.14–1.62 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (aqueous standards) | 0.03 μg g−1 | 0.99–1.09 μg g−1 | 124 | |
| Emulsification | |||||
| ICP-OES | EC (aqueous standards) | 0.008 mg kg−1 | <0.008–0.303 mg kg−1 | 126 | |
| Emulsification | |||||
| IS: Y | |||||
| ICP-MS | EC (kerosene, 1:3) |
0.0264 μg kg−1 | 0.730–11.5 μg kg−1 | 49 | |
| ICP-MS | EC (kerosene, 1:3) |
0.101 μg kg−1 | 0.730–11.5 μg kg−1 | 49 | |
| ICP-MS | EC (aqueous standards) | 5.13–5.47 μg L−1 | n.d. | 110 | |
| Microemulsification | |||||
| ETV-ICP-MS (Pd as modifier) | EC | 1.5 ng g−1 | 13.8–142 ng g−1 | 94 | |
| ETAAS | EC (treatment with tetramethylammonium hydroxide) | 15 ng g−1 | 130–182 ng g−1 | 106 | |
| 0.1% Pd + 0.06% Mg + 0.06% Triton X-100 as modifier | |||||
| ETAAS | EC (ethanol, 1:5 m:v) |
0.009 μg g−1 | 0.010–0.194 μg g−1 | 109 | |
| Pd(NO3)2 + Mg(NO3)2 as modifier | |||||
| Anodic stripping voltammetry | Mercury-film electrode | 4.69 × 10−9 mol L−1 | n.d. | 164 | |
| Microemulsification | |||||
| Standard addition (Cu2+) | |||||
| Anodic stripping voltammetry | Bismuth film electrode | 12 ng L−1 | 0.37–1.10 mg kg−1 | 165 | |
| Sample digestion (Cu2+) | |||||
| Fe | ICP-OES | EC (kerosene, 1:10) |
0.011 mg kg−1 | 0.104–17.12 mg kg−1 | 113 |
| ICP-OES | EC (ethanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.01 μg g−1 | 0.78–21.2 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (1-propanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.01 μg g−1 | 0.78–21.2 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (aqueous standards) | 0.01 μg g−1 | 0.04–1.09 μg g−1 | 124 | |
| Emulsification | |||||
| ICP-OES | EC (aqueous standards) | 0.006 mg kg−1 | 0.029–2.200 mg kg−1 | 126 | |
| Emulsification | |||||
| IS: Y | |||||
| ICP-MS | EC (kerosene, 1:3) |
0.0869 μg kg−1 | 4.61–50.8 μg kg−1 | 49 | |
| ETV-ICP-MS (Pd as modifier) | EC | 3 ng g−1 | 120–375 ng g−1 | 94 | |
| ETAAS | EC (treatment with tetramethylammonium hydroxide) | 24 ng g−1 | 86–4940 ng g−1 | 106 | |
| 0.1% Pd + 0.06% Mg + 0.06% Triton X-100 as modifier | |||||
| ETAAS | EC (ethanol, 1:5 m:v) |
0.006 μg g−1 | 0.023–5.18 μg g−1 | 109 | |
| Pd(NO3)2 + Mg(NO3)2 as modifier | |||||
| Hg | ICP-MS | EC (kerosene, 1:3) |
0.123 μg kg−1 | 0.396–0.791 μg kg−1 | 49 |
| ETAAS | Standard addition | 10.2 μg kg−1 | 23.2 μg kg−1 | 150 | |
| Emulsification | |||||
| Pd–Mg mixture as modifier | |||||
| FI-CV-AAS | Emulsification | 0.2 μg kg−1 | 0.5–3.7 μg kg−1 | 96 | |
| K | ICP-OES | EC (kerosene, 1:4) |
7.1 μg kg−1 | 5–10 mg kg−1 | 4 |
| ICP-OES | EC (kerosene, 1:10) |
0.070 mg kg−1 | 2.059–32.46 mg kg−1 | 113 | |
| ICP-OES | EC (kerosene, 1:10) |
0.1 mg kg−1 | n.d. | 115 | |
| ICP-OES | EC (kerosene, 1:10) |
0.8 mg kg−1 | n.d. | 117 | |
| ICP-OES | EC (ethanol, 1:10) |
0.3 μg g−1 | 1.3–6.0 μg g−1 | 3 | |
| IS: Y | |||||
| ICP-OES | EC (ethanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.4 μg g−1 | 17.5–189 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (1-propanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.2 μg g−1 | 17.5–189 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (aqueous standards) | 0.241 mg kg−1 | n.d. | 126 | |
| Emulsification | |||||
| IS: Y | |||||
| ICP-OES | EC (open digestion) | 0.11 μg g−1 | 2.1–7.3 μg g−1 | 127 | |
| IS: Y | |||||
| ICP-OES | EC (microwave close digestion) | 0.16 μg g−1 | 2.1–7.3 μg g−1 | 127 | |
| IS: Y | |||||
| ICP-MS | EC (kerosene, 1:3) |
2.10 μg kg−1 | 15.4–50.6 μg kg−1 | 49 | |
| FAES | EC microemulsification | 0.06–0.09 μg g−1 | 2.00–63.76 μg g−1 | 143 | |
| FAES | EC (ethanol, 1:10 and 1:20) |
0.60 mg kg−1 (1:10) |
n.d. | 105 | |
| 1.08 mg kg−1 (1:20) |
|||||
| FAAS | EC (ethanol, 1:10 m:v) |
0.17 mg kg−1 | 2.7–7.2 mg kg−1 | 147 | |
| FAAS | EC (aqueous standards) | 0.06 μg g−1 | 0.71–36.2 mg kg−1 | 91 | |
| Microemulsification | |||||
| FAAS | EC microemulsification | 0.01 μg g−1 | 0.13–2.30 μg g−1 | 108 | |
| IC | EC (K+) | 0.42 mg kg−1 | 0.35–0.91 mg kg−1 | 159 | |
| CE + diode array detector | IS | 0.3 mg kg−1 | 1.1–16.8 mg kg−1 | 161 | |
| Liquid–liquid extraction (K+) | |||||
| CE + coupled contactless conductivity detector | Liquid–liquid extraction (K+) | 0.12 mg L−1 | 0.46–0.61 mg kg−1 | 162 | |
| Voltammetry | Glassy carbon electrode modified with nickel(II) hexacyanoferrate nanoparticles | 5.0 × 10−5 mol L−1 | 12.9 mg kg−1 | 166 | |
| Microemulsification | |||||
| Standard addition (K+) | |||||
| Voltammetry | Nickel hexacyanoferrate-modified electrode | 1.9 × 10−5 mol L−1 | 0.96 mg kg−1 | 167 | |
| Liquid–liquid extraction (K+) | |||||
| Standard addition | |||||
| HR-CS FAAS | EC (xylene, 1:10 m:v) |
0.023 mg kg−1 | 9.20–10.00 mg kg −1 | 144 | |
| LS FAAS | EC (xylene, 1:10 m:v) |
0.57 mg kg−1 | 9.20–10.00 mg kg −1 | 144 | |
| WCAES | Tungsten coil atomizer | 70–80 μg kg−1 | 10.8–95.6 mg kg−1 | 169 | |
| Standard addition | |||||
| Mg | ICP-OES | EC (kerosene, 1:4) |
0.9–39 μg kg−1 | 1–10 mg kg−1 | 4 |
| ICP-OES | EC (kerosene, 1:10) |
0.009 mg kg−1 | 0.353–27.31 mg kg−1 | 113 | |
| ICP-OES | EC (kerosene, 1:10) |
0.01 mg kg−1 | 0.63–3.6 mg kg−1 | 115 | |
| ICP-OES | EC (kerosene, 1:10) |
0.02 mg kg−1 | 0.10–22.1 mg kg−1 | 117 | |
| ICP-OES | EC (ethanol, 1:10) |
0.005 μg g−1 | 0.058–5.9 μg g−1 | 3 | |
| IS: Y | |||||
| ICP-OES | EC (ethanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.001 μg g−1 | 0.06–33.80 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (1-propanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.001 μg g−1 | 0.06–33.80 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (aqueous standards) | 0.002 μg g−1 | 0.007–1.08 μg g−1 | 124 | |
| Emulsification | |||||
| ICP-OES | EC (aqueous standards) | 0.006 mg kg−1 | 0.030–0.033 mg kg−1 | 126 | |
| Emulsification | |||||
| IS: Y | |||||
| ICP-OES | EC (open digestion) | 0.04 μg g−1 | n.d. | 127 | |
| IS: Y | |||||
| ICP-OES | EC (microwave close digestion) | 0.03 μg g−1 | n.d. | 127 | |
| IS: Y | |||||
| ICP-MS | EC (kerosene, 1:3) |
8.65 μg kg−1 | 6.16–12.1 μg kg−1 | 49 | |
| FAAS | EC (ethanol, 1:10 m:v) |
0.05 mg kg−1 | 0.068 mg kg−1 | 147 | |
| FAAS | EC microemulsification | 0.03 mg L−1 | n.d. | 148 | |
| FAAS | EC microemulsification | 0.004 μg g−1 | 0.041–0.52 μg g−1 | 108 | |
| IC | EC (Mg2+) | 0.36 mg kg−1 | 0.06–0.93 mg kg−1 | 159 | |
| CE + diode array detector | IS | 0.3 mg kg−1 | n.d. | 161 | |
| Liquid–liquid extraction (Mg2+) | |||||
| CE + coupled contactless conductivity detector | Liquid–liquid extraction (Mg2+) | 0.07 mg L−1 | 0.28 mg kg−1 | 162 | |
| HR-CS FAAS | EC (xylene, 1:10 m:v) |
0.057 mg kg−1 | 0.47–0.59 mg kg−1 | 144 | |
| LS FAAS | EC (xylene, 1:10 m:v) |
0.11 mg kg−1 | 0.47–0.59 mg kg −1 | 144 | |
| Mn | ICP-OES | EC (aqueous standards) | 0.005 μg g−1 | 1.00–1.08 μg g−1 | 124 |
| Emulsification | |||||
| ICP-OES | EC (aqueous standards) | 0.001 mg kg−1 | 0.001 mg kg−1 | 126 | |
| Emulsification | |||||
| IS: Y | |||||
| ICP-MS | EC (kerosene, 1:3) |
0.0563 μg kg−1 | 0.114–0.450 μg kg−1 | 49 | |
| ICP-MS | EC (aqueous standards) | 7.51 × 10−1 μg L−1 | <0.75–1.23 μg L−1 | 110 | |
| Microemulsification | |||||
| ICP-MS | EC | Room temperature (spray chamber): 0.31 ng mL−1 | 0.22–0.24 μg L−1 | 120 | |
| 110 °C (spray chamber): 0.06 ng mL−1 | |||||
| 200 °C (spray chamber): 0.23 ng mL−1 | |||||
| ETV-ICP-MS (Pd as modifier) | EC | 0.3 ng g−1 | 4.9–76 ng g−1 | 94 | |
| ETAAS | EC (ethanol, 1:5 m:v) |
0.003 μg g−1 | 0.004–0.037 μg g−1 | 109 | |
| Pd(NO3)2 + Mg(NO3)2 as modifier | |||||
| Mo | ICP-MS | EC (kerosene, 1:3) |
0.371 μg kg−1 | n.d. | 49 |
| Na | ICP-OES | EC (kerosene, 1:4) |
1.4–1.6 μg kg−1 | 2–10 mg kg−1 | 4 |
| ICP-OES | EC (kerosene, 1:10) |
0.019 mg kg−1 | 1.414–21.59 mg kg−1 | 113 | |
| ICP-OES | EC (kerosene, 1:10) |
0.1 mg kg−1 | 0.6–23 mg kg−1 | 115 | |
| ICP-OES | EC (kerosene, 1:10) |
0.2 mg kg−1 | 0.23–13.8 mg kg−1 | 117 | |
| ICP-OES | EC (ethanol, 1:10) |
0.1 μg g−1 | 1.4–44.3 μg g−1 | 3 | |
| IS: Y | |||||
| ICP-OES | EC (ethanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.1 μg g−1 | 0.9–29.0 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (1-propanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.1 μg g−1 | 0.9–29.0 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (aqueous standards) | 0.04 μg g−1 | 0.14–1.08 μg g−1 | 124 | |
| Emulsification | |||||
| ICP-OES | EC (aqueous standards) | 0.071 mg kg−1 | 0.022–1.490 mg kg−1 | 126 | |
| Emulsification | |||||
| IS: Y | |||||
| ICP-OES | EC (open digestion) | 0.56 μg g−1 | 6.5–7.6 μg g−1 | 127 | |
| IS: Y | |||||
| ICP-OES | EC (microwave close digestion) | 0.16 μg g−1 | 6.5–7.6 μg g−1 | 127 | |
| IS: Y | |||||
| ICP-MS | EC (kerosene, 1:3) |
1.19 μg kg−1 | 127–1430 μg kg−1 | 49 | |
| FAES | EC microemulsification | 0.08–0.10 μg g−1 | 3.60–3.73 μg g−1 | 143 | |
| FAES | EC (ethanol, 1:10 and 1:20) |
0.65 mg kg−1 (1:10) |
n.d. | 105 | |
| 1.20 mg kg−1 (1:20) |
|||||
| FAAS | EC (ethanol, 1:10 m:v) |
0.14 mg kg−1 | 0.60–2.70 mg kg−1 | 147 | |
| FAAS | EC (aqueous standards) | 0.1 μg g−1 | 0.5–39.7 mg kg−1 | 91 | |
| Microemulsification | |||||
| FAAS | EC microemulsification | 0.1 μg g−1 | 1.18–1.51 μg g−1 | 108 | |
| IC | EC (Na+) | 0.11 mg kg−1 | 0.99–3.56 mg kg−1 | 159 | |
| CE + diode array detector | IS | 0.3 mg kg−1 | 2.3–39.6 mg kg−1 | 161 | |
| Liquid–liquid extraction (Na+) | |||||
| CE + coupled contactless conductivity detector | Liquid–liquid extraction (Na+) | 0.14 mg L−1 | 0.97 mg kg−1 | 162 | |
| HR-CS FAAS | EC (xylene, 1:10 m:v) |
0.10 mg kg−1 | 0.54–0.98 mg kg−1 | 144 | |
| LS FAAS | EC (xylene, 1:10 m:v) |
0.23 mg kg−1 | 0.54–0.98 mg kg−1 | 144 | |
| WCAES | Tungsten coil atomizer | 20 μg kg−1 | 6.08–41.3 mg kg−1 | 169 | |
| Standard addition | |||||
| Ni | ICP-OES | EC (kerosene, 1:10) |
0.006 mg kg−1 | 0.220–0.948 mg kg−1 | 113 |
| ICP-MS | EC (kerosene, 1:3) |
0.126 μg kg−1 | 0.397–3.64 μg kg−1 | 49 | |
| ICP-MS | EC (aqueous standards) | 19.3–19.5 μg L−1 | n.d. | 110 | |
| Microemulsification | |||||
| ICP-MS | EC | Room temperature (spray chamber): 0.22 ng mL−1 | 1.15–1.17 μg L−1 | 120 | |
| 110 °C (spray chamber): 0.07 ng mL−1 | |||||
| 200 °C (spray chamber): 0.18 ng mL−1 | |||||
| ETV-ICP-MS microemulsification (Pd as modifier) | EC | 0.5 ng g−1 | 6.5–14.1 ng g−1 | 94 | |
| ETAAS | EC microemulsification | 0.9 μg L−1 | 0.2–2.4 μg g−1 | 92 | |
| W as modifier | |||||
| P | ICP-OES | EC (kerosene, 1:4) |
32–67 μg kg−1 | n.d. | 4 |
| ICP-OES | EC (kerosene, 1:10) |
0.023 mg kg−1 | 0.799–223.8 mg kg−1 | 113 | |
| ICP-OES | EC (kerosene, 1:10) |
0.09 mg kg−1 | 1.2–7.6 mg kg−1 | 115 | |
| ICP-OES | EC (kerosene, 1:10) |
0.4 mg kg−1 | 0.07–26.3 mg kg−1 | 117 | |
| ICP-OES | EC (ethanol, 1:10) |
0.5 μg g−1 | 2.8–7.9 μg g−1 | 3 | |
| IS: Y | |||||
| ICP-OES | EC (ethanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.1 μg g−1 | 0.6–321.0 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (1-propanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.1 μg g−1 | 0.6–321.0 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (aqueous standards) | 0.20 μg g−1 | 0.96–1.09 μg g−1 | 124 | |
| Emulsification | |||||
| ICP-OES | EC (open digestion) | 0.22 μg g−1 | n.d. | 127 | |
| IS: Y | |||||
| ICP-OES | EC (microwave close digestion) | 0.40 μg g−1 | n.d. | 127 | |
| IS: Y | |||||
| ICP-MS | EC (kerosene, 1:3) |
22.7 μg kg−1 | 21.4–2120 μg kg−1 | 49 | |
| ETV-ICP-MS (tungsten coil electrothermal matrix decomposition) | EC | 0.4 mg kg−1 | 0.51–5.75 mg kg−1 | 138 | |
| ETAAS | EC (direct sampling) | 1.2 μg g−1 | 2.4–4.5 μg g−1 | 155 | |
| 20 μL of Pd (1000 μg mL−1) in 0.1% HNO3 and 0.025% Triton X-100 as modifier | |||||
| ETAAS | EC (direct sampling) | 0.5 μg g−1 | 4.2–4.86 mg kg−1 | 157 | |
| 30 μg Pd(NO3)2 + 20 μg Mg(NO3)2 mixture dissolved in 0.2% HNO3 and 0.1% Triton X-100 as modifier | |||||
| IC | EC (PO43−) | 0.1 mg kg−1 | 33–417 mg kg−1 | 160 | |
| Cyclic voltammetry | 1:12 Phosphomolybdic modified electrode |
8.7 × 10−6 mol L−1 | 1.36 mg kg−1 | 163 | |
| Liquid–liquid extraction | |||||
| Standard addition | |||||
| Pb | ICP-MS | EC (kerosene, 1:3) |
0.0226 μg kg−1 | 0.0450–0.385 μg kg−1 | 49 |
| ICP-MS | EC (aqueous standards) | 1.49 × 10−1 μg L−1 | <0.15–0.401 μg L−1 | 110 | |
| Microemulsification | |||||
| Anodic stripping voltammetry | Mercury-film electrode | 2.91 × 10−9 mol L−1 | n.d. | 164 | |
| Microemulsification | |||||
| Standard addition (Pb2+) | |||||
| Anodic stripping voltammetry | Bismuth film electrode | 8 ng L−1 | 0.39–2.20 mg kg−1 | 165 | |
| Sample digestion (Pb2+) | |||||
| S | ICP-OES | EC (kerosene, 1:10) |
0.01 mg kg−1 | 0.6–0.9 mg kg−1 | 115 |
| ICP-OES | EC (ethanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.4 μg g−1 | 1.4–817 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (1-propanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.3 μg g−1 | 1.4–817 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (aqueous standards) | 0.21–0.80 mg L−1 | 2–7 mg L−1 | 125 | |
| Microemulsification | |||||
| ICP-MS | EC (kerosene, 1:3) |
0.0293 μg kg−1 | 1.29–18.9 mg kg−1 | 49 | |
| SF-ICP-MS | ID | 7.42 mg kg−1 | 0.7 mg kg−1 | 140 | |
| Sample digestion | |||||
| ICP-MS/MS | ID | 0.5–2.0 μg kg−1 | 7.231 μg g−1 | 141 | |
| Dilution (ethanol) | |||||
| Sb | ICP-MS | EC (kerosene, 1:3) |
0.0395 μg kg−1 | 0.0528–0.399 μg kg−1 | 49 |
| Si | ICP-OES | EC (aqueous standards) | 0.024 mg kg−1 | 0.34–0.40 mg kg−1 | 126 |
| Emulsification | |||||
| IS: Y | |||||
| ICP-MS | EC (kerosene, 1:3) |
7.44 μg kg−1 | 6.02–8220 μg kg−1 | 49 | |
| ICP-OES | EC | Room temperature (spray chamber): 3–26 μg L−1 | n.d. | 121 | |
| 200 °C (spray chamber): 4.2 μg L−1 | |||||
| 350 °C (spray chamber): 1.3–3 μg L−1 | |||||
| ETV-ICP-MS (tungsten coil electrothermal matrix decomposition) | EC | 0.1 mg kg−1 | 0.22–0.57 mg kg−1 | 138 | |
| MIP-OES | EC sample digestion | 20–240 μg L−1 | n.d. | 158 | |
| MIP-OES | EC (aqueous standards) | 5 μg L−1 | n.d. | 158 | |
| Microemulsification | |||||
| Sn | ICP-MS | EC (kerosene, 1:3) |
0.411 μg kg−1 | 0.138–131 μg kg−1 | 49 |
| Sr | ICP-MS | EC (kerosene, 1:3) |
0.0631 μg kg−1 | 0.339–4.59 μg kg−1 | 49 |
| Ti | ICP-MS | EC (kerosene, 1:3) |
0.706 μg kg−1 | 0.342–8.08 μg kg−1 | 49 |
| ICP-MS | EC (aqueous standards) | 12.8 μg L−1 | 145.8–180 μg L−1 | 110 | |
| Microemulsification | |||||
| V | ICP-MS | EC (kerosene, 1:3) |
0.0409 μg kg−1 | 0.186–1.36 μg kg−1 | 49 |
| ICP-MS | EC | Room temperature (spray chamber): 0.17 ng mL−1 | 1.30–1.40 μg L−1 | 120 | |
| 110 °C (spray chamber): 0.06 ng mL−1 | |||||
| 200 °C (spray chamber): 0.08 ng mL−1 | |||||
| ETV-ICP-MS (Pd as modifier) | EC | 1 ng g−1 | n.d. | 94 | |
| WCAES | Tungsten coil atomizer | 90–500 μg kg−1 | n.d. | 169 | |
| Standard addition | |||||
| W | ICP-MS | EC (kerosene, 1:3) |
0.0177 μg kg−1 | 0.0181–0.121 μg kg−1 | 49 |
| Zn | ICP-OES | EC (kerosene, 1:10) |
0.011 mg kg−1 | 0.099–2.4 mg kg−1 | 113 |
| ICP-OES | EC (ethanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.08 μg g−1 | 1.0–9.1 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-OES | EC (1-propanol, 1:10 for vegetable oil and 1:20 biodiesel) |
0.05 μg g−1 | 1.0–9.1 μg g−1 | 5 | |
| IS: Y | |||||
| ICP-MS | EC (kerosene, 1:3) |
0.211 μg kg−1 | 2.8–27.4 μg kg−1 | 49 | |
| ICP-MS | EC (aqueous standards) | 4.22–4.25 μg L−1 | 64.7–184.3 μg L−1 | 110 | |
| Microemulsification | |||||
| FAAS | EC microemulsification | 0.08 mg L−1 | 0.49–0.68 mg L−1 | 148 | |
| Anodic stripping voltammetry | Bismuth film electrode | 18 ng L−1 | 2.3–4.0 mg kg−1 | 165 | |
| Sample digestion (Zn2+) | |||||
Electrothermal vaporization (ETV) can be used as an alternative approach to minimize the problems related to the use of conventional sample introduction systems.135,136 The main advantage of this device is the separation of the analyte from the matrix sample in the pyrolysis step. As a result carbon deposit formation and some polyatomic interference is mitigated.137 Moreover, due to the low amount of sample introduced (ca., 20 μL) the problems related to the degradation of the plasma ionization or excitation capability are avoided. Besides, this sample introduction system allows performing a preconcentration procedure from several consecutive sample injections on the surface of the vaporizer thus improving the analytical figures of merit. Chaves et al.94 developed a methodology for the determination of Co, Cu, Fe, Mn, Ni and V in emulsions of diesel and biodiesel samples by ETV-ICP-MS. Pd played two main roles: as a chemical modifier it stabilized the analytes and as a carrier this element improved the transport of the analytes from the ETV to the plasma. The LODs were, in ng g−1, 0.5 for Co, 1.5 for Cu, 3 for Fe, 0.3 for Mn, 0.5 for Ni, and 1 for V. Moreover, recovery tests were carried out to evaluate the accuracy of the method. This parameter was in the 80–120% range. Recently, a tungsten filament has been employed to vaporize the analyte.138 Advantages of this approach over conventional graphite ovens are: (i) it is simpler and less expensive, since it requires a single low power source; and, (ii) carbide formation is minimized, what is highly interesting for the determination of some elements such as silicon. In fact, this element, together with phosphorous were accurately determined. Limits of detection of 0.4 and 0.1 mg kg−1 were obtained for P and Si, respectively. The main drawback of ETV is the transient nature of the signal, which reduces the amounts of elements determined simultaneously.139
To overcome problems related to isotopic equilibration, a pre-treatment step such as the digestion of the sample is required. Recently, Amais et al.140 developed a method for the determination of sulphur in biodiesel by sector field inductively coupled plasma mass spectrometry (SF-ICP-MS) after sample digestion. The applied procedure involved pre-digestion and spiking of approximately 0.25 g aliquots of the samples with 34S (approximately 0.25 g of a nominal 10 μg g−1 34S solution). For the digestion of the sample, a diluted nitric acid and hydrogen peroxide decomposition medium was used. Furthermore, medium resolution mode was employed to eliminate isobaric interference at 32S and 34S caused by polyatomic phosphorus and oxygen species, as well as sulfur hydride ions. The accuracy and the precision of the method were tested by analysing a diesel certified reference material. Despite the favourable accuracy and precision of the proposed method, it did not have a limit of detection low enough to conduct S determination below 0.6 mg kg−1. This was due to the magnitude of the instrument background.
One of the main drawbacks of the SF-ICP-MS is a 10-fold reduction in ion transmission efficiency and hence, in signal intensity. Moreover, the cost of this type of instrument is higher than for a quadrupole-based ICP-MS. This is why Balcaen et.al.141 used a triple quadrupole (ICP-QQQ) instrument for the determination of S by isotope dilution. This system consisted of an octopole-based collision/reaction cell located between two quadrupole analyzers. The major advantage of the ICP-QQQ is the enhanced spectral resolution owing to the double mass selection in MS/MS mode and the production of chemical reactions. Thus S was detected after the conversion of S+ ions into SO+ ions through reaction with oxygen. As a proof-of-concept, the technique was successfully applied to the S determination in a biodiesel reference material. Moreover, the LOD for this approach was in the range of μg kg−1.
3.3. Analysis by additional techniques
Alternative techniques have been proposed for the determination of trace elements in biodiesel samples. In this way, FAES,59,105,142,143 FAAS,91,108,144–148 and ETAAS92,94,104,106,107,109,112,149–157 have been used to quantify some alkaline metals in these samples. Other alternative techniques have been MIP-OES,158 ionic chromatography,159,160 capillary electrophoresis161,162 and voltammetry.163–168Flame Atomic Emission Spectrometry (FAES) could be considered as a low cost alternative for the determination of the four major elements (Ca, Na, K and Mg) in biodiesel samples. Alkalines are easily and efficiently atomized in flames.142 A method for the determination of Na and K in biodiesel, from different vegetable oils, was proposed by Chaves et al.143 Microemulsions were prepared by mixing biodiesel samples with n-propanol and an aqueous acid solution, which allowed the use of aqueous standards for the calibration. Sample introduction through discrete aspiration was compared with that by continuous aspiration (CA), moreover, the results obtained with ICP-OES were taken as a reference. Na and K concentrations were determined and for the employed methods, the values obtained were not significantly different for a 95% confidence level. Furthermore, by comparing LODs for discrete and continuous aspiration modes, values lower than 0.1 μg g−1 were obtained. The direct dissolution of the sample into ethanol was proposed by Barros et al.105 aqueous standards were again employed. Two different sample:solvent proportions were evaluated, 1:10 and 1:20. The limits of quantification (LOQ) in biodiesel–ethanol solution (1:20, w/v) were 4.00 and 3.60 mg kg−1 for Na and K, respectively. In 1:10 (w/v) biodiesel–ethanol solutions the LOQs were 2.16 and 2.00 mg kg−1 for Na and K, respectively. In both cases LOQs were lower than the limit established by the EN 14214 (5 mg kg−1).59 Moreover, the feasibility of the use of aqueous standards was studied by recovery tests. For both metals, the recoveries were in the range of 91–108%.
Flame Atomic Absorption Spectrometry (FAAS) has been explored as an alternative to ICP techniques for the determination of K, Na, Ca and Mg in biodiesel samples. The advantage of this technique is that it is simpler, cheaper and more tolerant to organic matrices than ICP. The dilution of biodiesel samples in xylene or n-hexane is widely used as a pretreatment sample step. In fact, it has been recommended by international legislation.144–146 However, the main drawback of this technique is that organometallic standards are required, which have a low stability in solution and are expensive. Ethanol147 was compared with xylene as a solvent for the determination of Ca, K, Na and Mg in biodiesel. It was observed that the ethanolic medium provided greater sensitivity for K and Mg; whereas, for Ca and Na, similar sensitivities were obtained using both media. Because the surface tension of ethanol is lower than for xylene, the nebulization process is favored, thus increasing the mass of analyte reaching the flame. Moreover, since a higher proportion of ethanol reached the flame, compared to xylene, the flame temperature increased. In the case of Ca, a different flame was used (N2O/C2H2), and the temperature increase due to the presence of the organic solvents became less significant.
As in ICP techniques, microemulsification of the sample has been applied allowing the use of aqueous standards. De Jesus et.al.91 used n-pentanol, Triton X-100 and water for the microemulsion preparation. Microemulsified aqueous standards were employed. The flame composition was optimized in terms of sensitivity and the optimal C2H2/air ratio was 0.131. For these experimental conditions the limits of detection obtained were 0.1 μg g−1 and 0.06 μg g−1 for sodium and potassium, respectively. The LODs obtained were compared with those obtained following the European Standards145,146 and higher values were found for the dilution procedure (0.2 μg g−1 and 0.1 μg g−1 for sodium and potassium, respectively). The same emulsifier was used by Amais et al.,148 whereas a different flame composition was employed. LODs were in the same range as in the previous work. Additional emulsifiers have been applied for the alkaline metals in these kinds of samples. Lyra et al.108 prepared the microemulsions by using HNO3 and CsCl, for sodium and potassium determination, KCl, for calcium and magnesium determination, and n-propanol.
Electrothermal atomic absorption spectrometry (ETAAS) sensitivity is 2–3 orders of magnitude higher than that of FAAS. For this reason it has been used for the analysis of some metals in petroleum samples.104,149 The field of application of this determination technique has been extended to biodiesel samples by several research groups. Lobo et al.92 developed a method for the Ni and Cd analysis using microemulsification as a sample preparation procedure. Tungsten was employed as a chemical modifier. In a previous study, two chemical modifiers (Pd + Mg and W) and two distinct sample preparation procedures (microemulsification and wet digestion in a focused microwave system) were investigated107 and the optimum experimental conditions corresponded to microemulsion preparation and use of W. Recoveries were measured varying from 93% to 108% for Ni and from 98% to 116% for Cd. Therefore, the accuracy was good enough for the routine analysis of these samples. The improvement of the sensitivity achieved by ETAAS and the advantages of the emulsion sample preparation, were taken for the determination of Cd and Hg in these kinds of samples at the μg kg−1 level.150 Ghisi et al.106 developed a method for the determination of Cu and Fe. The procedure for the sample preparation was its treatment with tetramethylammonium hydroxide (TMAH) as an alternative to sample dilution and emulsification. The main advantage was that the analyte was not diluted. Moreover, this treatment of the sample allowed the use of higher pyrolysis temperature eliminating the majority of the matrix before atomization thus mitigating interference.
In order to improve LODs, de Jesus et al.151 proposed direct sampling graphite furnace atomic absorption spectrometry for the determination of As and Cd. The samples were weighed directly on the solid sampling platforms and introduced into the graphite tube for analysis, thus reducing the contamination problems and increasing the sensitivity. The chemical modifier used was a mixture of 0.1% Pd + 0.06% Mg + 0.06% Triton X-100. The suitability of Pd and Mg as modifiers was previously established for petroleum.152–154 However, the main drawback of this technique is its relatively high uncertainty (5–20% RSD). This result was explained in terms of the heterogeneity of natural samples and the small amount of sample (8 and 10 mg for As and Cd, respectively) which was introduced into the atomizer.151
Phosphorous determination is not commonly carried out by AAS, since its three resonance lines are in the ultraviolet vacuum (UV). Therefore, non-resonance lines (213.6 nm and 214.9 nm) should be used. As a result, poor limits of detection may be found. In order to reduce the LOD, phosphorous was stabilized by adding chemical modifiers thus avoiding the formation of volatile molecular species.155 Several modifiers were evaluated: Pd, Pd + Ca and Pd + Mg. The results showed that Pd was the best option in terms of sensitivity. The suitability of the method was evaluated by comparison with the EN 14107.112 No significant differences were observed between the results afforded by the proposed and the standard procedures. Another important issue in the P determination affecting the LOD is that the P hollow cathode lamps are among the least intense ones. This problem could be solved by means of high-resolution continuum source graphite furnace atomic absorption spectrometry (HR-CS AAS). Moreover, HR-CS AAS allowed the simultaneous observation of the P atomic lines and PO molecular bands. Again Pd-based modifiers enhanced the formation of P atoms, whereas inhibited the formation of PO molecules.156 The advantages of this technique were demonstrated by the decrease of the LOD (0.5 μg g−1) in comparison with the conventional ETAAS.157 The unsurpassed background correction systems, the visualization of the entire analytical spectrum and the improvement on the LODs due to the HR-CS AAS were advantageous for the determination of Al, Cu, Fe and Mn.109 For the improvement of the Al analytical figures of merit, a platform pre-treated with Zr as a permanent chemical modifier was employed to prevent the formation of aluminium carbide. Furthermore, different calibration approaches were used depending on the analyte. For Cu, Fe and Mn, the calibration was carried out using aqueous standards, whereas, ethanolic ones were used for Al. LODs for Cu, Fe and Mn obtained with this approach were similar to those found for ETV-ICP-MS.94
A procedure for total and inorganic mercury determination in biodiesel by CV-AFS was developed by Aranda et al.96 The samples were introduced directly as oil-in-water emulsions in a flow injection manifold. Mercury vapour was generated using an acidic SnCl2 solution in a continuous flow system what gave a 0.2 μg kg−1 LOD.
Dancsak et al.169 have recently employed tungsten filaments extracted from microscope light bulbs to decompose the biodiesel matrix, and atomize and excite the analytes to determine sodium, potassium, chromium and vanadium by tungsten coil atomic emission spectrometry (WCAES). The accuracy was checked by determining Na and K in a biodiesel reference sample and carrying out spike experiments for Cr and V. No statistically significant differences were observed between reference and determined values for all analytes at a 95% confidence level.
Microwave-induced plasma optical emission spectrometry (MIP-OES) and a flow blurring nebulizer were used to determine silicon in diesel and biodiesel samples by Amais et al.158 A simple dilution with ethanol was used as a sample preparation procedure. Two additional sample preparation methods were also evaluated for comparison: closed-vessel microwave-assisted acid digestion and microemulsification. Limits of detection (LOD) vary from 5 to 20 μg L−1 and relative standard deviations (RSD) were lower than 2% in all cases.
Ion chromatography has been employed for the determination of elements such as Na, Ca, K, Mg159 and P.160 De Caland et al.159 developed a method for the quantitative determination of Na+, K+, Mg2+ and Ca2+. The proposed method employed water extraction, heating and ultrasound as a pre-treatment sample procedure. For comparison, the samples were also analyzed using ICP-OES with similar accuracy and precision results. Zhang et al.160 developed a method to measure the content of inorganic phosphate in oil samples, by direct injection of solvent extracted oil into ion chromatography. Biodiesel oils were dissolved in acetone and an ion chromatography system with sample matrix elimination function was applied to directly determine their phosphate content against acetone based standards.
Capillary electrophoresis equipped with a diode array detector was used for the determination of Na+, K+, Ca2+, Mg2, using barium (Ba2+) as an internal standard.161 Separation was conducted in a fused-silica capillary column with indirect UV detection at 214 nm. The method presented a good linearity in the concentration range of 0.5–20 mg kg−1. The same separation technique coupled to a conductivity detector was used for the determination of the four main cations among other species (i.e. sulfate, phosphate, formate, acetate, propionate and glycerol).162
Moreover, voltammetry has been used for the determination of metal contents in biodiesel samples. A method for P quantification in the form of phosphate using a 1:12 phosphomolybdic film modified glassy carbon electrode in cyclic voltammetry was developed by Zezza et al.163 Anodic stripping voltammetry (ASV) has been also applied for biodiesel analysis. Pinto et al.165 optimized the determination of trace levels of Cd2+, Cu2+, Pb2+ and Zn2+via ASV using a bismuth film electrode. The deposition time and voltage step were the most important factors identified. The optimized method was applied to the determination of these elements in biodiesel samples after microwave digestion with diluted acid, presenting satisfactory values for accuracy and precision. A mercury film electrode was used by Martiniano et al.164 to determine directly and simultaneously Pb2+ and Cu2+. De Souza et al.167 used a nickel hexacyanoferrate-modified electrode for K+. The modified electrodes exhibited a linear response in the concentration range of 4.0 × 10−5 to 1.0 × 10−2 mol L−1, with a detection limit of 1.9 × 10−5 mol L−1. A chemically modified electrode with nanoparticles of nickel hexacyanoferrate was employed for the determination of potassium ions in a microemulsion of biodiesel.114 An alternative method for the determination of calcium in biodiesel samples using square-wave voltammetry and a glassy carbon electrode in a solution containing EDTA was proposed by Almeida et al.168 A microwave assisted acid digestion of the biodiesel samples was carried out before analysis. In addition, good reproducibility (CV maximum of 0.70%) and accuracy (recovery around 102%) were obtained making the method suitable for the determination of Ca2+ in biodiesel samples.
3.4. Comparison among techniques
As it has been discussed throughout the previous sections, different approaches have been developed for the determination of trace elements in biodiesel samples. Because metal concentration in biodiesel samples (Table 4) is usually low, the selection of the determination technique could be considered as one of the most challenging steps.104Fig. 3 summarizes the techniques employed for the determination of several metals in biodiesel samples. | ||
| Fig. 3 Techniques employed for the determination of several metals in biodiesel samples (bars) and the number of studies dealing with the determination of each one of the elements (red line). | ||
The included data have been calculated taking into account the data shown in Table 4 according to:
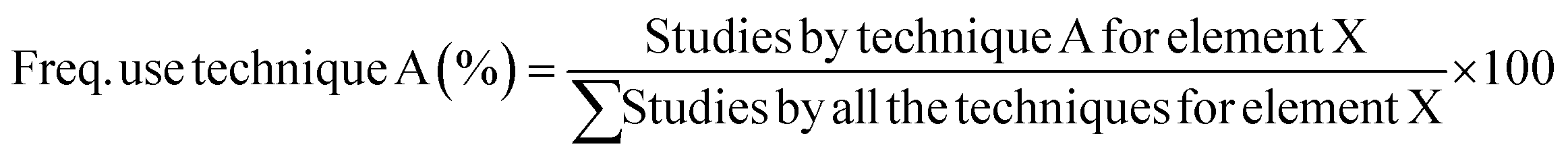 | (2) |
As shown in Fig. 3, ICP-OES and ICP-MS are the most employed techniques in the research related to biodiesel analysis. Some studies dealing with FAAS determination also appeared. Spectral interference for AAS is minimized compared with other techniques, whereas sample throughput and limits of detection are favourable for ICP techniques. This figure also shows the total number of studies related to the determination of each one of the elements considered. As expected, the most studied ones are Na, K, Ca, Mg, P and Cu.
Chromatographic techniques have been developed for the determination of the alkaline elements. However, because LODs are slightly higher than those found using spectrometric techniques they have been mainly used for the determination of alkalines. Cu, Fe, Mn and Zn are expected to be found at the sub-mg kg−1 or even μg kg−1 level, for that reason, ICP techniques have been extensively applied. It is important to note that since Cu and Zn are redox active, voltammetry has been proposed as an alternative.
3.5. Standards for the analysis of biodiesel
Several test methods have been proposed to perform the elemental determination in biodiesel samples. For example, the European Standard EN 1421459 describes the requirements and test methods for FAME analysis, the most common type of biodiesel, whereas, the ASTM D6751-0860 details specifications for biodiesels blended with middle distillate fuels. Both standards establish the determination of Ca, Mg, K, Na, S and P.
Table 5 summarizes the standards dealing with the elemental determination in biodiesel samples. This table also includes the analytical techniques recommended by each one of those standards. If this information is compared with that included in Fig. 3 it may be concluded that ICP-MS is not yet considered. This is likely due to the fact that the elements determined by the standards are the most abundant in biodiesel samples (Na, K, Ca and Mg) at levels that fit perfectly with the LODs afforded by techniques such as FAAS or ICP-OES. Because sulfur determination through ICP-OES and ICP-MS presents problems related to the different response as a function of the analyte chemical form, XRF techniques are often recommended by the corresponding standards.
| Standard reference | Standard title/scope | Determined elements | Analytical technique | Year |
|---|---|---|---|---|
| ASTM D7039 | Standard test method for sulfur in gasoline, diesel fuel, jet fuel, kerosene, biodiesel, biodiesel blends, and gasoline–ethanol blends by monochromatic wavelength dispersive X-ray fluorescence spectrometry | S | MWDXRF | 2013 |
| EN 14107 | Fat and oil derivatives – Fatty Acid Methyl Esters (FAME) – determination of phosphorus content by inductively coupled plasma (ICP) emission spectrometry | P | ICP-OES | 2003 |
| EN 14108 | Fat and oil derivatives – Fatty Acid Methyl Esters (FAME) – determination of sodium content by atomic absorption spectrometry | Na | FAAS | |
| EN 14109 | Fat and oil derivatives – Fatty Acid Methyl Esters (FAME) – determination of potassium content by atomic absorption spectrometry | K | FAAS | 2003 |
| EN 14538 | Fat and oil derivatives – fatty acid methyl ester (FAME) – determination of Ca, K, Mg and Na content by optical emission spectral analysis with inductively coupled plasma (ICP OES) | Ca, Mg, Na, K | ICP-OES | 2006 |
| ASTM D6751 | Standard specification for biodiesel fuel blend stock (B100) for middle distillate fuels | Specifications* | 2012 | |
| ASTM D7467 | Standard specification for diesel fuel oil, biodiesel blend (B6 to B20) | Specifications* | 2013 | |
| prEN16709 | Automotive fuels – high FAME diesel fuel (B20 or B30) – requirements and test methods | Specifications* | 2014 | |
| *These standards refer to: | ||||
| ASTM D4294 (S by EDXRF) | S | EDXRF | 2010 | |
| ASTM D2622 (S by WDXRF) | S | WDXRF | 2010 | |
| ASTM D7039 (S by MWDXRF) | S | MWDXRF | 2013 | |
| ASTM D4951 (P by ICP-OES) | P | ICP-OES | 2009 | |
| EN 14538 (Ca, Mg, K and Na by ICP-OES) | Ca, Mg, Na, K | ICP-OES | 2006 | |
4. Bioethanol
Bioethanol is referred to as ethanol obtained through fermentation of carbohydrates from a wide range of renewable feedstocks (e.g. sugar cane, corn and switchgrass) using various types of microorganisms.1,170Bioethanol can be employed directly or mixed in several concentrations with unlead gasoline (e.g. E85 ethanol fuel is a mixture of 85% of bioethanol and 15% of gasoline).170 This kind of mixture bioethanol–gasoline is known as fuel ethanol. Modifications in the engine are not required up to E10, whereas higher concentrations of ethanol are appropriate for flex-fuel engines.170
Bioethanol and fuel ethanol show several advantages over fossil fuels such as: (i) a reduction of greenhouse emissions down to 65% lower than petroleum products;170,171 (ii) ethanol is an oxygenated additive which improves the octane rating of fuels; and, (iii) burning is clean and therefore the toxicity of the generated compounds is low.170 For these reasons the production of fuel ethanol and bioethanol is growing with the simultaneous increase in the research related to the production and characterization of these new fuels. The research in fuel ethanol production and characterization was developed in the 70s whereas the production of research documents dealing with bioethanol virtually started at the beginning of the XXI century. In both cases the number of papers per year has exponentially increased over the last 15 years up to more than 1000 research documents a year.
4.1. Synthesis and presence of metals. Importance of their determination
Several materials have been employed to produce bioethanol.171–173 The synthesis process depends strongly on the raw material. First generation bioethanol is produced from foodstuffs such as beet, sugarcane, cereal grain or corn, among others.1 Meanwhile, second generation bioethanol generates from wood or straw and it is also known as “lignocellulosic bioethanol”.1,171The production of bioethanol includes four main steps (Fig. 4): (i) physico-chemical structure break up of the raw material; (ii) enzymatic hydrolysis of cellulose to monomeric sugars; (iii) conversion of these sugars to ethanol by fermentation; (iv) separation of ethanol from the fermentation broth by distillation generally followed by a final dehydratation.171
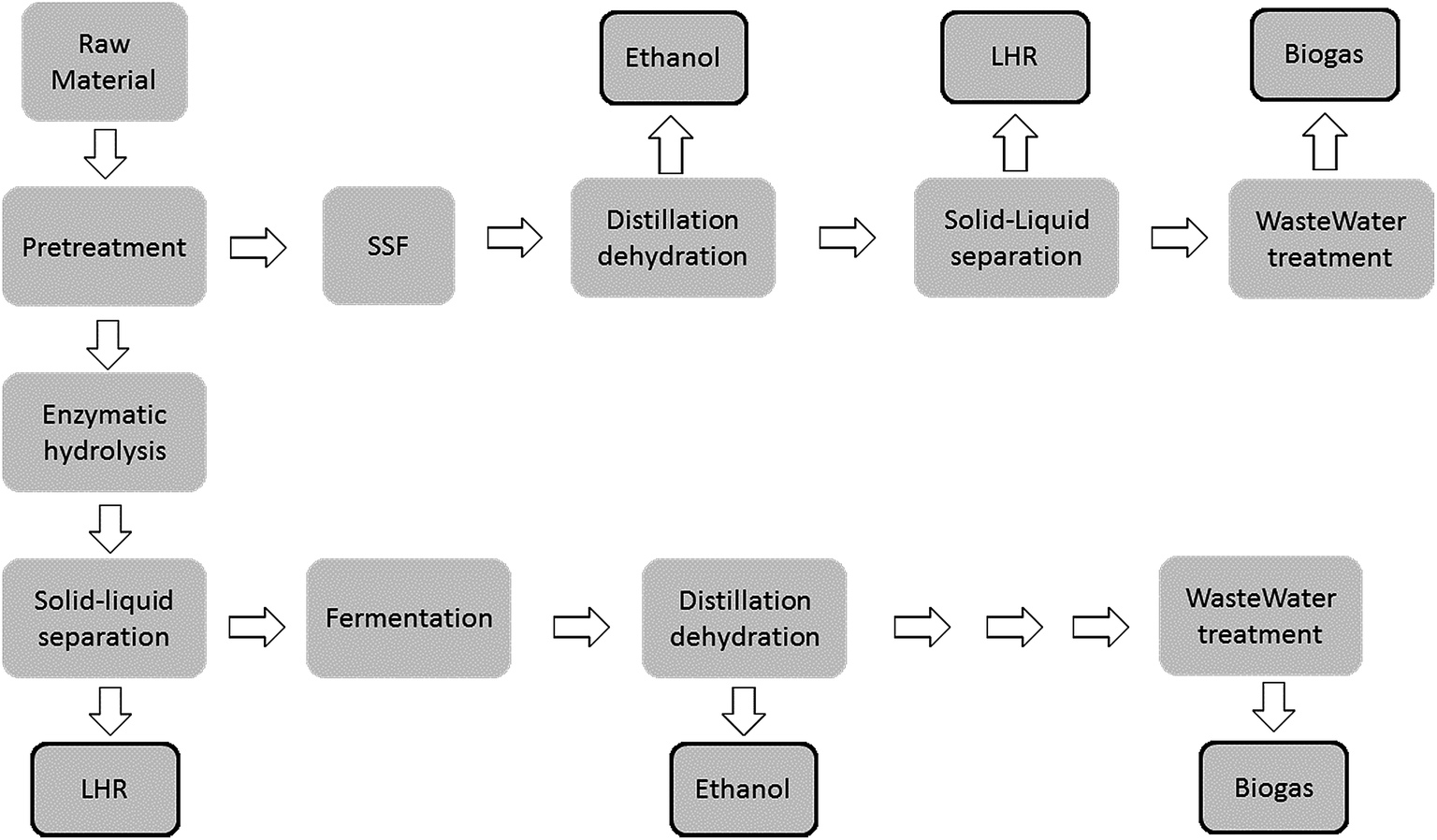 | ||
| Fig. 4 General flow chart of the bioethanol production process from lignocellulosic biomass (second generation). SSF: Simultaneous Saccharification and Fermentation; LHR: solid Lignin Hydrolysate Residue. Taken from ref. 171. | ||
At the end of the process either anhydrous ethanol (content of water lower than 0.7%) or hydrated ethanol (content of water from 2 to 7%) can be obtained.174 The final product also may contain up to roughly 300 compounds depending on the origin of the raw material and the applied treatment.1 Compounds such as alcohols (methanol, 1-propanol, isopropanol, 1-butanol, 2-butanol, etc.), esters (ethyl formate, ethyl acetate, etc.), ketones, aldehydes can be present. This fact together with the low content of metals can hinder their quantification in bioethanol or fuel ethanol samples.
It is difficult to establish the source of metals in bioethanol. The first one can be the raw material.2,175–177 Thus the metal content depends on the soil where the raw material has grown as well as on the atmospheric pollution.177 Concentrations on the order of mg kg−1 have been found in biomass for 26 elements (Sr, Ba, F, Cl, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Mo, Zn, Cd, Hg, Al, Sn, Pb, B, As, Sb, S, Se, Te and P). Meanwhile, the content of elements such as Na, K, Ca, Mg and Si in products frequently used to obtain bioethanol can be as high as g kg−1.2 Moreover, bioethanol may be contaminated with metals during its synthesis.175,176,178 Several metals can also appear during the fuel storage and transport in metallic containers.175–179 Finally, some metallic species can be used as additives to promote the combustion process.180 Obviously, when a blend is considered (e.g., ethanol fuel) metals and metalloids come mainly from gasoline.119
As it has been previously mentioned, metals and other trace elements are present at very low concentrations in bioethanol and fuel ethanol. However, their determination is important for several reasons: (i) they can cause catalyst deactivation in the bioethanol transformation process1 (e.g. sulfur impurities) and in the industrial process;175,177 (ii) some metals such as As, Cd, Hg, Tl or Pb cause health problems even at low concentrations;177,181 (iii) others, Fe and Cu, cause damage of the vehicle engine;175,177,178,182–185 (iv) heavy metals have an environmental risk;178,180,185,186 and, (v) some elements preclude the stability of the bioethanol or fuel ethanol (e.g. Cu can catalyze the oxidation of gasoline in the presence of alcohol).177,178,183,187
4.2. Analysis by ICP techniques
In another study developed by the same authors the LODs, sensitivity and the background equivalent concentration (BEC) were determined for Pb, Cd, Al, Cr, Fe, Na, Mn, Mo and V in the presence and in the absence of ethanol. They observed that the operating conditions played a very important role. For a 0.6 L min−1 nebulizer gas flow rate and 1.36 kW R.F. power, the sensitivity for all analytes increased with ethanol concentration by a factor that depended on the element. In contrast, for a 0.4 L min−1 flow rate the addition of ethanol did not improve the sensitivity for almost all the analytes and it decreased for some elements such as Na and Al.23
On the other hand, some authors reported that the presence of ethanol in the plasma caused an increase in the background signal.20,21,23 According to McCrindle et al.14,23 the enhancement in terms of sensitivity was similar to that in terms of background intensity.23 The same effect was observed for 95% ethanol solutions, the LOD was similar to that of pure water although the sensitivity for 95% ethanol solution was between 2 and 5 times higher than for water.14
Saint'Pierre et al.186 studied the effect of ethanol on sensitivity for 15 elements (V, Mn, Co, Cu, Zn, Ga, As, Se, Rb, Sr, Mo, Cd, Tl, Pb and Bi) in ICP-MS and they reported that the signal in the presence of ethanol was from 15 to 25 times higher than the signal for plain water solutions depending on the isotope. These findings were in concordance with the results obtained by Dressler et al.41 who evaluated the effect of methanol, ethanol and isopropanol in ICP-MS for 13 elements (As, Ba, Bi, Cd, Ce, Cu, Hg, In, Pb, Rh, Se, Tl and U). On the other hand, Rocha et al.184 reported that for copper and iron 7% of water in hydrated fuel ethanol (HFE) induced a 30% signal decrease with respect to anhydrous fuel ethanol (AFE).
Sample treatment methods. Several studies recommend ethanol or fuel ethanol dilution with an appropriate solvent.37,38,186,190 For these kinds of samples, water is the most widely employed,37,38,186,190 although other solvents can be employed to dilute these types of samples.37 The choice of the solvent may affect the method sensitivity, precision and accuracy. Thus, using a programmable temperature spray chamber, it has been verified that the sensitivity in isopropanol is from three to four times higher than that in methanol.191 However, an obvious limitation of sample dilution is that LODs and sensitivities are severely degraded.
Overcoming non-spectral interference. Besides sample dilution, several methods have been developed in order to overcome non-spectral interference caused by ethanol, among them: (i) matrix matching; (ii) internal standardization; and, (iii) isotopic dilution.
As regards matrix matching Rocha et al.184 prepared the standards in ethanol with 0.5% of water to analyze AFE and ethanol with 7% of water to analyze HFE. They did not find significant differences between found concentrations by matrix matching in ICP-OES and isotope dilution in ICP-MS. Additional studies have tried to minimize non-spectral interference in the analysis of metals in bioethanol and fuel ethanol through matrix matching.38,180,181,184,190 Unfortunately, this method is time consuming and inaccurate in many cases because normally the matrix of the sample is very complex and/or unknown.
Internal standardization can be applied in order to carry out an accurate and precise analysis of ethanol and ethanol fuel.184,190 This methodology shows, as the most important concern, the correct selection of the best internal standard. Tormen et al.186 evaluated yttrium, rhodium and iridium as internal standards to analyze 19 elements by ICP-MS. External calibration was taken as a reference. The authors concluded that yttrium or even rhodium could be satisfactorily employed as an IS in the routine analysis of fuel ethanol. However, provided that the samples were diluted the found concentrations for V, Ni, Ga, Sr, Cd, Sn and Tl were below the limit of quantification.186
Isotopic dilution, in turn, has been applied in order to minimize or remove non-spectral interference in ICP-MS with good results.38,181,186 This method shows several advantages against internal standard calibration because it is very simple, fast and clean.181
Alternative sample introduction systems. The spray chamber can be cooled in order to minimize the mass of the organic material reaching the plasma.172,173 Thus, for instance, a cyclonic spray chamber was operated at 10 °C with the aim of determining Cu, P and S in ethanol through ICP-OES with recoveries between 93.5 and 107.3%.172 An alternative approach is to cool a spray chamber using a Peltier effect based system making it possible to introduce pure ethanol into the plasma.
Desolvation systems are based on a previous aerosol heating step followed by either a membrane or a condenser. The first stage promotes the efficient solvent evaporation from the aerosol whereas the second one removes the generated vapor. This device is also appropriate to overcome matrix effects. Alcohols have been analyzed using an ICP-OES with a membrane desolvator193 or cryogenic desolvation.194 Rocha et al.184 reported a method allowing the determination of Cu and Fe in AFE and HFE through ICP-OES by direct sample introduction using an ultrasonic nebulizer and a membrane desolvator (USN-MD-ICP-OES) and they obtained LODs similar to those previously reported in ICP-MS.38,181,186 Saint'Pierre et al.38 employed a flow injection system coupled to an ultrasonic nebulizer and a desolvator to carry out the analysis of AFE and HFE in ICP-MS. The obtained LODs for Ag, Cd, Co, Cu, Fe, Mn, Ni and Pb were higher than those found by ETV-ICP-MS.38,180
Additional systems employed for bioethanol and ethanol fuel samples analysis include low sample consumption systems. A microconcentric nebulizer (MCN) was used by Tormen et al.186 to carry out the determination of Cu, Cd, Ni, Pb, Tl and Sn in fuel ethanol through ICP-MS. Compared with conventional nebulizers, MCN showed lower limits of detection and better precision even at lower sample consumption rates. This was due to the finer primary aerosols and higher analyte transport efficiencies as compared to conventional nebulization systems.195 External calibration and internal standardization were applied and the results were in concordance with those found with isotopic dilution.186 With this device it was possible to introduce in the plasma 70% ethanol solutions. For higher ethanol concentrations carbon deposits appeared in the ICP-MS interface cones.186
Electrothermal vaporization (ETV) is a good approach to remove non-spectral interference when ethanol and fuel ethanol are analyzed.180,181,192 Nonetheless, a few authors have reported methods to carry out the determination of some elements in these kinds of samples through ETV coupled to ICP-MS.180,181 Saint'Pierre et al.181 reported a method to determine trace metals in ethanol fuel by isotopic dilution ETV-ICP-MS. In this study Ag, Cd, Cu, Pb and Tl were determined in fuel alcohol with LODs of 0.02, 0.08, 0.1, 0.05, and 0.001 μg L−1, respectively (Table 6). For Cd, Pb and Tl that evaporated at lower temperatures, the use of Pd aqueous solution as a chemical modifier was necessary. However, in the Ag and Cu determination it was not necessary to use chemical modifiers because these elements showed lower volatilities than Cd, Pb and Tl.181 In another study, the determination of Ag, As, Cd, Cu, Co, Fe, Mn, Ni, Sb, Sn, and Tl in ethanol fuel was successfully done through ETV-ICP-MS using external calibration with ethanolic solutions and Pd as a chemical modifier.180 Recoveries for all elements were between 80 and 120% without a modifier and from 60 to 140% with palladium.
| Element | Technique | Conditions | LOD (μg L−1) | Found concentration (μg L−1) range (min–max) | Ref. |
|---|---|---|---|---|---|
| a ID: Isotopic Dilution; EC: External Calibration; EC (W): External Calibration with water; EC (MM): External calibration with matrix matching. b n.d.: not determined in real samples; n.a.: not available data. c These ranges correspond to minimum and maximum values of LOD obtained with the four types of calibration employed by the authors (external calibration and internal standardization using Ir, Rh and Y). d Concentration values have been obtained employing Y as the internal standard. | |||||
| Ag | ETV-ICP-MS | ID | 0.02 | <0.02–0.079 | 181 |
| EC | 0.02 | <0.02–0.072 | |||
| ETV-ICP-MS | EC (Pd as modifier) | 0.013 | 0.041–0.102 | 180 | |
| ETV-ICP-MS | EC | 0.015 | <0.015–0.072 | ||
| FI-USN-ICP-MS | EC (W) | 0.1 | n.d. | 38 | |
| EC (MM) | 0.07 | n.d. | |||
| ID | 0.02 | n.d. | |||
| ICP-OES | EC | 0.47 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Ag 328.028 | |||||
| Al | CRI-ICP-MS | IS (Y) | 0.20 | 33–411 | 190 |
| CRI (H2 through skimmer) | |||||
| ETAAS | EC (ethanol 1:1) |
1.2 | n.a | 202 | |
| THGA with Pd(NO3)2 + Mg (NO3)2 | |||||
| ETAAS | EC (ethanol 1:1) |
1.9 | n.a | 201 | |
| W–Rh permanent modifier and Pd(NO3)2 + Mg(NO3)2 | |||||
| ICP-OES | EC | 0.15 | n.a | 173 | |
| Cooled spray chamber | |||||
| Al 167.020 | |||||
| ICP-OES | EC | 2.68 | n.a | 173 | |
| Cooled spray chamber | |||||
| Al 396.152 | |||||
| As | ETV-ICP-MS | EC (Pd as modifier) | 0.02 | 0.23–2.84 | 180 |
| ETV-ICP-MS | EC | 0.04 | <0.04–2.03 | ||
| ETAAS | EC (ethanol 1:1) |
2.5 | n.d. | 202 | |
| THGA with Pd(NO3)2 + Mg (NO3)2 | |||||
| ETAAS | EC (ethanol 1:1) |
2.9 | n.d. | 201 | |
| W–Rh permanent modifier and Pd(NO3)2 + Mg(NO3)2 | |||||
| ETAAS | EC | 0.7 | n.d. | 203 | |
| Ru as modifier | |||||
| ETAAS | EC | 2.0 | <2.0–2.7 | 204 | |
| Ir + Rh as modifier | |||||
| ICP-OES | EC | 2.22 | n.d. | 173 | |
| Cooled spray chamber | |||||
| As 189.042 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.01–0.03c | 1.13–3.62d | 186 | |
| B | ICP-OES | EC | 1.42 | n.d. | 173 |
| Cooled spray chamber | |||||
| B 249.773 | |||||
| Ba | ICP-OES | EC | 0.04 | n.d. | 173 |
| Cooled spray chamber | |||||
| Ba 455.403 | |||||
| CRI-ICP-MS | IS (Y) | 0.11 | <0.11 | 190 | |
| CRI (H2 through skimmer) | |||||
| Be | ICP-OES | EC | 0.21 | n.d. | 173 |
| Cooled spray chamber | |||||
| Be 313.042 | |||||
| Bi | MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.02c | <0.02–0.17d | 186 |
| Ca | ICP-OES | EC | 1.56 | n.d. | 173 |
| Cooled spray chamber | |||||
| Ca 317.933 | |||||
| Cd | ETV-ICP-MS | ID (Pd as modifier) | 0.08 | <0.08–0.53 | 181 |
| EC (Pd as modifier) | 0.07 | <0.07–0.54 | |||
| ETV-ICP-MS | EC (Pd as modifier) | 0.07 | <0.07–1.15 | 180 | |
| ETV-ICP-MS | EC | 0.13 | <0.13–1.05 | ||
| FI-USN-ICP-MS | EC (W) | 0.2 | n.d. | 38 | |
| EC (MM) | 0.03 | n.d. | |||
| ID | 0.02 | n.d. | |||
| FAAS | EC | 5.50 | n.d. | 185 | |
| Using Moringa oleifera seeds as a biosorbent | |||||
| ETAAS | EC | 0.05 | <0.05–3.0 | 204 | |
| Ir + Rh as modifier | |||||
| ETAAS | EC with pure ethanol | 0.1 | <0.1–0.83 | 205 | |
| Filter-ETAAS | |||||
| ICP-OES | EC | 0.17 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Cd 228.802 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.03–0.05c | <0.05d | 186 | |
| Co | ETV-ICP-MS | EC | 0.002 | 0.011–0.094 | 180 |
| FI-USN-ICP-MS | EC (W) | 0.04 | n.d. | 38 | |
| EC (MM) | 0.5 | n.d. | |||
| FAAS | Preconcentration with 2,2′-dipyridylamine bonded silica | 0.44 | n.d. | 176 | |
| ICP-OES | EC | 0.32 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Co 228.616 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.03c | <0.1d | 186 | |
| CRI-ICP-MS | IS (Y) | 0.05 | 5.6–26.1 | 190 | |
| CRI (H2 through skimmer) | |||||
| Cr | FAAS | Preconcentration with 2,2′-dipyridylamine bonded silica | 0.33 | n.d. | 176 |
| MIP-OES | EC ethanol 10% | 9 | <9 | 177 | |
| ICP-OES | EC | 0.35 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Cr 267.716 | |||||
| USN-CD-ICP-OES | EC | 0.8 | n.d. | 194 | |
| CRI-ICP-MS | IS (Y) | 0.18 | 12.3–77.2 | 190 | |
| CRI (H2 through skimmer) | |||||
| Cu | ETV-ICP-MS | ID | 0.1 | 1.96–14.44 | 181 |
| EC | 0.2 | 1.80–14.98 | |||
| ETV-ICP-MS | EC | 0.22 | 1.80–14.98 | 180 | |
| ETAAS | EC | n.a. | 2.15–13.93 | ||
| FAAS | 5-Amino-1,3,4-thiadiazole-2-thiol modified silica gel preconcentrated | n.a. | 52–78 | 207 | |
| FAAS | Preconcentrated by evaporation | n.a. | 49–76 | 207 | |
| FAAS | Preconcentrated by 2,5-dimercapto-1,3,4-thiadiazole | n.a. | 11–190a | 208 | |
| 5000 for a sample in a copper distillation columna | |||||
| FI-USN-ICP-MS | EC (W) | 0.4 | n.d. | 38 | |
| EC (MM) | 0.8 | n.d. | |||
| ID | 0.2 | n.d. | |||
| FAAS | Preconcentrated with 2-aminothiazole modified silica gel | 1.7 | 5.4–7.3 | 179 | |
| FAAS | Preconcentrated with 2-aminothiazole modified silica gel | n.a. | 5.4–7.3 | 179 | |
| FAAS | Preconcentration with 2,2′-dipyridylamine bonded silica | 0.40 | 51–66 | 176 | |
| FAAS | Preconcentrated by evaporation | n.a. | 49–57 | 176 | |
| ETAAS | EC (ethanol 1:1) |
0.22 | n.d. | 202 | |
| THGA with Pd(NO3)2 + Mg (NO3)2 | |||||
| ETAAS | EC (ethanol 1:1) |
0.57 | n.d. | 201 | |
| W–Rh permanent modifier and Pd(NO3)2 + Mg(NO3)2 | |||||
| ETAAS | EC without modifier | 0.6 | 2.15–13.93 | 203 | |
| GFASS | W permanent modifier + co-injection of Ir | 0.086 | 8.0–47 | 175 | |
| FAAS | EC | n.a. | 6.9–7.2 | 206 | |
| Preconcentration with N-acyl-N′-benzoylthiourea modified silica gel | |||||
| ASV | IS | 0.120 | 13.3–20.1 | 183 | |
| Evaporation of ethanol and redissolution in aqueous media | |||||
| HPCIC | EC (Cu2+) | 7.4 | n.d. | 178 | |
| ICP-OES | EC | 1.5 | n.d. | 172 | |
| Cooled spray chamber | |||||
| Cu 324.754 nm | |||||
| ICP-OES | EC | 0.28 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Cu 324.754 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.09–0.2c | 3.1–24.1d | 186 | |
| USN-MD-ICP-OES | EC (MM) in AFE | 0.10 | <0.10–2.20 | 184 | |
| USN-MD-ICP-OES | EC (MM) in HFE | 0.23 | 2.58–2.75 | 184 | |
| USN-CD-ICP-OES | EC | 0.3 | n.d. | 194 | |
| CRI-ICP-MS | IS (Y) | 0.33 | 23–205 | 190 | |
| CRI (H2 through skimmer) | |||||
| Fe | ETV-ICP-MS | EC | 0.72 | 6.55–42.99 | 180 |
| ETAAS | EC | n.a. | 6.88–29.43 | ||
| FAAS | 5-Amino-1,3,4-thiadiazole-2-thiol modified silica gel preconcentrated | n.a. | 12–23 | 207 | |
| FAAS | Preconcentrated by evaporation | n.a. | 11–21 | 207 | |
| FAAS | Preconcentrated by 2,5-dimercapto-1,3,4-thiadiazole | n.a. | n.d. – 7 | 208 | |
| FI-USN-ICP-MS | EC (W) | 27 | n.d. | 38 | |
| EC (MM) | 10 | n.d. | |||
| FAAS | Preconcentration with 2,2′-dipyridylamine bonded silica | 0.28 | 10–25 | 176 | |
| FAAS | Preconcentrated by evaporation | n.a. | 11–21 | 176 | |
| ETAAS | EC (ethanol 1:1) |
1.6 | n.d. | 202 | |
| THGA with Pd(NO3)2 + Mg (NO3)2 | |||||
| ETAAS | EC (ethanol 1:1) |
1.3 | n.d. | 201 | |
| W–Rh permanent modifier and Pd(NO3)2 + Mg(NO3)2 | |||||
| ETAAS | EC without modifier | 1.4 | 6.88–29.43 | 203 | |
| HPCIC | EC (Fe3+) | 8.9 | n.d. | 178 | |
| ICP-OES | EC | 0.52 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Fe 259.940 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.7–4c | <4–18d | 186 | |
| USN-MD-ICP-OES | EC (MM) in AFE | 0.20 | <0.20–13.95 | 184 | |
| USN-MD-ICP-OES | EC (MM) in HFE | 0.50 | 5.34–5.80 | 184 | |
| ID-ICP-MS | ID in AFE | n.a. | 14.20 | 184 | |
| ID-ICP-MS | ID in HFE | n.a. | 5.30–5.75 | 184 | |
| USN-CD-ICP-OES | EC | 0.6 | n.d. | 194 | |
| CRI-ICP-MS | IS (Y) | 0.10 | 6–124 | 190 | |
| CRI (H2 through skimmer) | |||||
| Ga | MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.06–0.2c | <0.2d | 186 |
| Hg | ICP-OES | EC | 1.80 | n.d. | 173 |
| Cooled spray chamber | |||||
| Hg 194.163 | |||||
| K | ICP-OES | EC | 29.67 | n.d. | 173 |
| Cooled spray chamber | |||||
| K 766.490 | |||||
| Li | ICP-OES | EC | 0.65 | n.d. | 173 |
| Cooled spray chamber | |||||
| Li 670.784 | |||||
| Mg | ICP-OES | EC | 4.01 | n.d. | 173 |
| Cooled spray chamber | |||||
| Mg 279.806 | |||||
| CRI-ICP-MS | IS (Y) | 0.24 | 17–204 | 190 | |
| CRI (H2 through skimmer) | |||||
| Mn | ETV-ICP-MS | EC | 0.025 | 0.884–1.306 | 180 |
| FI-USN-ICP-MS | EC (W) | 0.7 | n.d. | 38 | |
| EC (MM) | 0.8 | n.d. | |||
| ETAAS | EC (ethanol 1:1) |
0.20 | n.d. | 202 | |
| THGA with Pd(NO3)2 + Mg (NO3)2 | |||||
| ETAAS | EC (ethanol 1:1) |
0.40 | n.d. | 201 | |
| W–Rh permanent modifier and Pd(NO3)2 + Mg(NO3)2 | |||||
| ICP-OES | EC | 0.13 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Mn 257.610 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.02–0.4c | 0.77–1.25d | 186 | |
| CRI-ICP-MS | IS (Y) | 0.02 | 1.7–15.4 | 190 | |
| CRI (H2 through skimmer) | |||||
| Mo | ICP-OES | EC | 0.45 | n.d. | 173 |
| Cooled spray chamber | |||||
| Mo 202.030 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.03c | <0.03–0.46d | 186 | |
| CRI-ICP-MS | IS (Y) | 0.05 | <0.05 | 190 | |
| CRI (H2 through skimmer) | |||||
| Na | ICP-OES | EC | 4.96 | n.d. | 173 |
| Cooled spray chamber | |||||
| Na 589.592 | |||||
| CRI-ICP-MS | IS (Y) | 0.80 | 54–184 | 190 | |
| CRI (H2 through skimmer) | |||||
| Ni | ETV-ICP-MS | EC | 0.026 | 0.096–0.477 | 180 |
| FAAS | 5-Amino-1,3,4-thiadiazole-2-thiol modified silica gel preconcentrated | n.a. | 8–14 | 207 | |
| FAAS | Preconcentrated by evaporation | n.a. | 10–13 | 207 | |
| FAAS | Preconcentrated by 2,5-dimercapto-1,3,4-thiadiazole | n.a. | 5–45 | 208 | |
| FI-USN-ICP-MS | EC (W) | 0.4 | n.d. | 38 | |
| EC (MM) | 2.5 | n.d. | |||
| FAAS | Preconcentrated with 2-aminothiazole modified silica gel | 2.3 | 4.4–5.6 | 179 | |
| ETAAS | EC | n.a. | 4.1–6.1 | 179 | |
| FAAS | Preconcentration with 2,2′-dipyridylamine bonded silica | 0.51 | 9–15 | 176 | |
| FAAS | Preconcentrated by evaporation | n.a. | 10–13 | 176 | |
| ETAAS | EC (ethanol 1:1) |
1.1 | n.d. | 202 | |
| THGA with Pd(NO3)2 + Mg (NO3)2 | |||||
| ETAAS | EC (ethanol 1:1) |
1.3 | n.d. | 201 | |
| W–Rh permanent modifier and Pd(NO3)2 + Mg(NO3)2 | |||||
| MIP-OES | EC ethanol 10% | 300 | <300 | 177 | |
| ICP-OES | EC | 0.30 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Ni 221.647 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.1–0.5c | <0.5d | 186 | |
| CRI-ICP-MS | IS (Y) | 0.17 | 14–73 | 190 | |
| CRI (H2 through skimmer) | |||||
| P | ICP-OES | EC | 11 | n.d. | 172 |
| Cooled spray chamber | |||||
| P 177.495 nm | |||||
| ICP-OES | EC | 4.92 | n.d. | 173 | |
| Cooled spray chamber | |||||
| P 177.440 | |||||
| ICP-OES | EC | 4.32 | n.d. | 173 | |
| Cooled spray chamber | |||||
| P 178.229 | |||||
| ICP-OES | EC | 2.63 | n.d. | 173 | |
| Cooled spray chamber | |||||
| P 213.618 | |||||
| Pb | ETV-ICP-MS | ID (Pd as modifier) | 0.05 | 0.62–1.58 | 181 |
| EC (Pd as modifier) | 0.02 | 0.51–1.51 | |||
| ETV-ICP-MS | EC (Pd as modifier) | 0.02 | 0.57–1.50 | 180 | |
| ETV-ICP-MS | EC | 0.03 | 0.39–1.51 | ||
| FI-USN-ICP-MS | EC (W) | 0.2 | n.d. | 38 | |
| EC (MM) | 0.1 | n.d. | |||
| ID | 0.04 | n.d. | |||
| FAAS | Preconcentration with 2,2′-dipyridylamine bonded silica | 0.55 | n.d. | 176 | |
| ETAAS | EC | 0.7 | n.d. | 203 | |
| Ru as modifier | |||||
| ETAAS | EC | 0.7 | n.d. | 203 | |
| NH4H2PO4 as modifier | |||||
| ETAAS | EC | 1.1 | <1.1–6.4 | 204 | |
| Ir + Rh as modifier | |||||
| ETAAS | EC with pure ethanol | 0.3 | <0.3–1.16 | 205 | |
| Filter-ETAAS | |||||
| GFASS | W permanent modifier + co-injection of Ir | 2.47 | <2.47 | 175 | |
| ASV | IS | 0.235 | <0.235–1.43 | 183 | |
| Evaporation of ethanol and redissolution in aqueous media | |||||
| MIP-OES | EC ethanol 10% | 500 | <500 | 177 | |
| ICP-OES | EC | 1.68 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Pb 220.353 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.03c | <0.03–1.08d | 186 | |
| USN-CD-ICP-OES | EC | 5 | n.d. | 194 | |
| CRI-ICP-MS | IS (Y) | 0.01 | 5.6–38 | 190 | |
| CRI (H2 through skimmer) | |||||
| Rb | MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.03c | <0.1d | 186 |
| S | ICP-OES | EC | 21 | n.d. | 172 |
| Cooled spray chamber | |||||
| S 180.731 | |||||
| ICP-OES | EC | 5.13 | n.d. | 173 | |
| Cooled spray chamber | |||||
| S 180.676 | |||||
| Sb | ETAAS | EC | 1.8 | n.d. | 203 |
| Ru as modifier | |||||
| ICP-OES | EC | 2.30 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Sb 206.833 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.02c | n.d. | 186 | |
| CRI-ICP-MS | IS (Y) | 0.11 | <0.11 | 190 | |
| CRI (H2 through skimmer) | |||||
| Se | ICP-OES | EC | 39.63 | n.d. | 173 |
| Cooled spray chamber | |||||
| Se 196.026 | |||||
| MCN-ICP-MS | IS (Y) | 0.6 | 1.8–3.3 | 186 | |
| Si | ICP-OES | EC | 1.84 | n.d. | 173 |
| Cooled spray chamber | |||||
| Si 251.611 | |||||
| CRI-ICP-MS | IS (Y) | 14 | <14 | 190 | |
| CRI (H2 through skimmer) | |||||
| Sn | ETV-ICP-MS | EC (Pd as modifier) | 0.010 | <0.010–0.062 | 180 |
| ETV-ICP-MS | EC | 0.007 | <0.007–0.067 | ||
| ETAAS | EC | 3.8 | n.d. | 203 | |
| Ru as modifier | |||||
| ICP-OES | EC | 2.83 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Sn 189.989 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.09c | <0.09d | 186 | |
| Sr | ICP-OES | EC | 0.01 | n.d. | 173 |
| Cooled spray chamber | |||||
| Sr 407.771 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.01–0.03c | <0.03d | 186 | |
| Ti | ICP-OES | EC | 0.13 | n.d. | 173 |
| Cooled spray chamber | |||||
| Ti 337.280 | |||||
| Tl | ETV-ICP-MS | ID (Pd as modifier) | 0.001 | <0.001–0.0047 | 181 |
| EC (Pd as modifier) | 0.0008 | <0.0008–0.0045 | |||
| ETV-ICP-MS | EC (Pd as modifier) | 0.0008 | <0.0008–0.0045 | 180 | |
| ETV-ICP-MS | EC | 0.0009 | <0.0009–0.0045 | ||
| ICP-OES | EC | 2.66 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Tl 190.864 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.01c | <0.01d | 186 | |
| V | MIP-OES | EC ethanol 10% | 4 | <4 | 177 |
| ICP-OES | EC | 3.59 | n.d. | 173 | |
| Cooled spray chamber | |||||
| V 292.402 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.06–0.5c | <0.5d | 186 | |
| CRI-ICP-MS | IS (Y) | 0.41 | <0.4 | 190 | |
| CRI (H2 through skimmer) | |||||
| Zn | FAAS | 5-Amino-1,3,4-thiadiazole-2-thiol modified silica gel preconcentrated | n.a. | 6–8 | 207 |
| FAAS | Preconcentrated by evaporation | n.a. | 8–11 | 207 | |
| FAAS | Preconcentrated by 2,5-dimercapto-1,3,4-thiadiazole | n.a. | 3–4.5 | 208 | |
| FAAS | Preconcentrated with 2-aminothiazole modified silica gel | 0.34 | 6.3–8.3 | 179 | |
| ETAAS | EC | n.a. | 7.1–8.1 | 179 | |
| FAAS | Preconcentration with 2,2′-dipyridylamine bonded silica | 0.58 | 6–10 | 176 | |
| FAAS | Preconcentrated by evaporation | n.a. | 8–11 | 176 | |
| FAAS | EC | n.a. | 1.0–2.4 | 206 | |
| Preconcentration with N-acyl-N′-benzoylthiourea modified silica gel | |||||
| HPCIC | EC (Zn2+) | 2.0 | n.d. | 178 | |
| ICP-OES | EC | 0.53 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Zn 206.200 | |||||
| ICP-OES | EC | 0.60 | n.d. | 173 | |
| Cooled spray chamber | |||||
| Zn 213.856 | |||||
| MCN-ICP-MS | EC and Y, Ir and Rh as IS | 0.4–0.6c | 14.4–36.1d | 186 | |
| USN-CD-ICP-OES | EC | 0.3 | n.d. | 194 | |
| CRI-ICP-MS | IS (Y) | 0.26 | 17–400 | 190 | |
| CRI (H2 through skimmer) | |||||
Spectral interference. Spectral interference in ICP techniques when organic samples (e.g. ethanol) are introduced has been extensively described by several authors.119,196 In ICP-OES this interference is related to peak overlapping.197 Polyatomic interference occurs in ICP-MS when ethanol is injected into the plasma. For example, 12C2+, 12C14N+, 13C14N+, 12C16O+, 13C16O+, 12C17O+, 40Ar12C+, and 40Ar13C+ may interfere with 24Mg+, 26Mg+, 27Al+, 28Si+, 29Si+, 52Cr+ and 53Cr+ determination, respectively.198 Recently, Neves et al.190 verified that flow-rates below 112.5 mL min−1 were insufficient to remove the formed carbon compounds when ethanol concentration was higher than 80%. On the other hand, an excess of oxygen in the plasma could cause the formation of metal oxide polyatomic species.110
Additional possibilities to remove spectral interference due to polyatomic species in ICP-MS when ethanol and fuel ethanol are analyzed are the use of a dynamic collision cell (DCC), a dynamic reaction cell (DRC) or a collision–reaction interface (CRI). Kishi et al.199 reported a reduction of carbon-based interference in alcohols using a DRC with pure ammonia as a reaction gas. Neves et al.190 evaluated the use of He or H2 as collision and reaction gases in a CRI system and they observed that the introduction of either two gases through the sampling cone was inefficient whereas an opposite effect was observed when H2 was introduced through the skimmer cone. The signal at m/z = 56 due to 40Ar16O+ was around 12-fold lower when 60 mL min−1 H2 or He were introduced through the skimmer cone in comparison with the signals without insertion of these gases. A similar behavior was observed for 24Mg+ (12C2+), 28Si+ (12C16O+) and 52Cr+ (40Ar12C+) showing the capability of this device to reduce isobaric interference when ethanol was analyzed. In the same study it was verified that reaction mode using H2 was more effective than collision mode with He.
4.3. Analysis by other techniques
Although ICP based techniques have been the most widely used to carry out the determination of metals and metalloids in ethanol fuel, alternative methods have been explored such as ETAAS,175,200–205 FAAS,176,179,185,206–209 voltammetry,183,210,211 ionic chromatography (IC)178 and microwave plasma optical emission spectrometry (MIP-OES).177Several modifiers have been used to carry out the determination of metals in bioethanol and ethanol fuel by ETAAS. The most used one corresponds to a mixture of Pd(NO3)2 and Mg(NO3)2200–202 although permanent modifiers such as the W–Rh mixture,201 Ru–Zr,203 Ir–Rh204 or W–Ir (co-injected) have also been evaluated.175 De Oliveira et al.200 carried out a comparative study of chemical modifiers employed to determine metals in ethanol fuel. Three possibilities were studied for six elements, Pd(NO3)2 + Mg(NO3)2, W/Rh and W + co-injection of Pd(NO3)2 + Mg(NO3)2. The last one was the modifier providing the best recoveries.
De Oliveira et al.201,202 developed two methods to carry out the determination of metals in fuel ethanol through ETAAS. In the first method they determined Al, As, Cu, Fe, Mn and Ni in ethanol fuel using a transversely heated graphite atomizer (THGA) and a Pb(NO3)2 and Mg(NO3)2 mixed modifier. The recoveries obtained increased from 73 to 116% and the RSD for all elements was lower than 6%.202 In the second method they used the W–Rh permanent modifier together with Pd(NO3)2 + Mg(NO3)2. The values of RSD and LOD (Table 6) were similar to those achieved without a permanent modifier and recoveries were between 81 and 109%.201
Saint'Pierre et al.203 evaluated several modifiers in order to perform the direct determination of As, Cu, Fe, Pb, Sb and Sn in ethanol fuel by ETAAS. Finally, they proposed to determine Cu and Fe without a chemical modifier whereas Ru was selected as the modifier to determine As, Sb, Sn and Pb. In the case of Pb, NH4H2PO4 could be employed as an alternative modifier. Recoveries were included within the 89.3 to 103.8% range.203 Giacomelli et al.204 studied the use of Ir together with Rh as a permanent modifier to determine As, Cd and Pb in pure ethanol by ETAAS. In this case, the obtained recoveries were between 94 and 96.7%. Saint'Pierre et al.205 reported a method to determine Cd and Pb in fuel ethanol by ETAAS. The standards were prepared in ethanol and the recoveries ranged from 90 to 120%.
Recently, Santos et al.175 have developed a method for simultaneous determination of Cu and Pb in ethanol fuel by ETAAS using a transversely heated graphite atomizer with W as the permanent modifier and co-injection of Ir. The recovery was between 93 and 103% for Cu and from 96 to 110% for Pb while the RSD was below 1% in all the cases.
Because FAAS provides higher limits of detection than techniques described previously (ICP-OES, ICP-MS and ETAAS), the use of this technique to perform ethanol fuel analysis involves a previous preconcentration stage.176,179,185,207–209 Thus Alves et al.185 developed a method to determine Cd in ethanol fuel through FAAS using Moringa oleifera seeds as a on-line biosorbent to carry out sample preconcentration. The recoveries of three samples were from 97.5 to 100% and the LOD was 5.50 μg L−1. The same authors had developed a similar study using vermicompost as the adsorbent material and acceptable results in terms of precision and accuracy were obtained.209 Several authors have reported on the determination of different metals through FAAS using modified silica gel as a preconcentration medium.176,179,206–208 De Melo et al.207 employed a column with 5-amino-1,3,4-thiadiazole-2-thiol modified silica gel to preconcentrate Cd(II), Co(II), Fe(III), Ni(II), Pb(II) and Zn(II). The recoveries obtained were between 98 and 99%. A column with 2,5-dimercapto-1,3,4-thiadiazole modified silica gel was used to determine Cu(II), Zn(II), Cd(II), Ni(II), Pb(II), Co(II) and Fe(III) in ethanol fuel. Recoveries close to 100% were found for binary mixtures whereas they were lower for mixtures of all elements (20–30% for Cd).208 Additional adsorbing media have been described such as 2-aminothiazole179 modified silica gel or N-acyl-N′-benzoylthiourea modified silica gel.206 Recently, Vieira et al.176 have used 2,2′-dipyridylamine bonded silica as a preconcentration system to determine Fe(III), Cr(III), Cu(II), Co(II), Pb(II), Ni(II) and Zn(II) in fuel ethanol through FAAS. The recovery obtained for all the analytes was close to 100% and the accuracy was good with RSD for all elements lower than 3%. The concentrations found and LODs for all methods proposed176,179,185,207,208 are shown in Table 6.
Donati et al.177 have developed a method to determine Cr, Ni, Pb and V in ethanol fuel through MIP-OES. The samples have been diluted in an aqueous nitric acid medium. The method provided good precision and accuracy, the recoveries were in the 92 to 108% range.
Voltammetry can also be useful to determine metals in ethanol fuel and water–ethanol mixtures. A method to determine Cd in alcohol–water mixtures using an ion-selective electrode was developed by Motonaka et al.210 It was found that cadmium ion-selective electrodes could be used to determine Cd ions in an alcohol–water mixture. Nevertheless, the response time became longer and the dynamic range was narrower as the ethanol content increased. Kamenev et al.211 carried out the determination of Pb(II) in water–alcohol mixtures by stripping voltammetry with a modified carbon–glass–ceramic electrode. The procedure was based on electrochemical and chemical modification of the surface and provided reproducible results. Another method was based on anodic stripping voltammetry (ASV)183 with the aim of determine Cu and Pb simultaneously. Two different procedures were applied: the first one was the direct quantification of metals in alcohol–water mixtures whereas the second one involved the evaporation of organic solvent and re-suspension of ions in water + electrolyte. The results obtained with two methods were in good agreement.
A high-performance chelation ionic chromatography method was used to quantify Fe2+, Fe3+, Cu2+, Mn2+, Pb2+, Cd2+, Co2+, Zn2+ and Ni2+ in fuel ethanol through post-column reaction with 4-(2-pyridylazo)resorcinol and spectrophotometric detection at 510 nm.178
4.4. Comparison among techniques
As it has been previously discussed, several techniques have been employed to quantify metals in bioethanol and fuel ethanol samples. Generally speaking the elemental concentration in these kinds of samples is very low (Table 6) and, hence, it is necessary to select a sensitive enough technique. For this reason ICP-OES and ICP-MS are widely used10,14,20,22–24,30,37,38,41,172,173,180,181,184,186,189–195,199 because it is possible to carry out the sample analysis without any pre-concentration step. Unfortunately, these techniques are quite sensitive to spectral as well as non-spectral interference that could be circumvented by applying suitable approaches.38,172,173,180,181,192–195,199 Another technique that has been frequently used to determine metals in fuel ethanol is ETAAS.175,200–205 Meanwhile, techniques such as voltammetry,183,210,211 chromatographic techniques178 or MIP-OES177 are less frequently employed.Fig. 5 shows the percentage of studies carried out with each technique for all the elements studied in the literature. These data have been obtained from data collected in Table 6. The data of the Y-axis have been obtained according to eqn (2).
 | ||
| Fig. 5 Techniques employed for the determination of several metals in biodiesel samples (bars) and the number of studies dealing with the determination of each one of the elements (red line). | ||
As shown in Fig. 5, ICP-MS is the most widely employed technique. ICP-OES, in turn, has been used for the determination of 7 elements present at concentrations of around a few μg L−1. However, because ICP-based techniques are very sensitive to organic solvents, ETAAS has been used as a good alternative. On the other hand, FAAS and some chromatographic techniques have been applied to the determination of major elements (Fig. 5) in bioethanol and fuel ethanol.
4.5. Standards for the analysis of bioethanol
Table 7 gathers the existing standards for the elemental determination in ethanol employed for fuel applications. It is interesting to notice that in some instances, methods such as colorimetry or potentiometry are recommended. This situation does not correspond to that presented in Fig. 5 issued from the research articles included in the present review. As regards ICP-OES and ICP-MS these techniques are seldom considered. A similar comment can be made regarding atomic absorption techniques.| Standard reference | Standard title | Determined elements | Analytical technique | Year |
|---|---|---|---|---|
| EN 15485 | Ethanol as a blending component for petrol – determination of sulfur content – wavelength dispersive X-ray fluorescence spectrometric method | S | WDXRF | 2007 |
| EN 15486 | Ethanol as a blending component for petrol – determination of sulfur content – ultraviolet fluorescence method | S | UVF | 2007 |
| EN 15487 | Ethanol as a blending component for petrol – determination of phosphorus content – ammonium molybdate spectrometric method | P | Colorimetry | 2007 |
| EN 15488 | Ethanol as a blending component for petrol – determination of copper content – graphite furnace atomic absorption spectrometric method | Cu | ETAAS | 2007 |
| EN 15492 | Ethanol as a blending component for petrol – determination of inorganic chloride and sulfate content – ion chromatographic method | Cl, S | Ionic chromatography | 2012 |
| EN 15837 | Ethanol as a blending component for petrol – determination of phosphorus, copper and sulfur content – direct method by inductively coupled plasma optical emission spectrometry (ICP OES) | P, Cu, S | ICP-OES | 2010 |
| ASTM D7319 | Standard test method for determination of existent and potential sulfate and inorganic chloride in fuel ethanol and butanol by direct injection suppressed ion chromatography | Cl, S | Ionic chromatography | 2013 |
| ASTM D7039 | Standard test method for sulfur in gasoline, diesel fuel, jet fuel, kerosene, biodiesel, biodiesel blends, and gasoline–ethanol blends by monochromatic wavelength dispersive X-ray fluorescence spectrometry | S | MWDXRF | 2013 |
| ASTM D7328 | Standard test method for determination of existent and potential inorganic sulfate and total inorganic chloride in fuel ethanol by ion chromatography using aqueous sample injection | Cl, S | Ionic chromatography | 2013 |
| ASTM D7318 | Standard test method for existent inorganic sulfate in ethanol by potentiometric titration | S | Potentiometry | 2013 |
| ASTM D5798 | Standard specification for fuel ethanol (Ed75-Ed85) for automotive spark-ignition engines | Specifications* | 2014 | |
| ASTM D4806 | Standard specification for denatured fuel ethanol for blending with gasolines for use as automotive spark-ignition engine fuel | Specifications* | 2014 | |
|
||||
| *These standards refer to : | ||||
| ASTM D5453 (S by UVF) | S | UVF | 2012 | |
| ASTM D2622 (S by XRF) | S | WDXRF | 2010 | |
| ASTM D5059 (Pb by XRF) | Pb | WDXRF | 2014 | |
| ASTM D3231 (P by colorimetry) | P | Colorimetry | 2013 | |
In Brazil, where bioethanol and ethanol fuel are widely used, the quality of fuel ethanol is carefully regulated by the National Agency of Petroleum (ANP).178,179,183,184 However, only a standard for sulfur and copper (D4806-07a;172 D3237177) and another for iron (D1688-07184) have been established by ASTM. Besides, in 2009 a European standard for Cu, P and S was published.188 The most widely employed techniques to quantify metals in biofuel products are ICP-OES, ICP-MS, ETAAS and FAAS.
5. Conclusions
Summarizing the results obtained in the literature, Fig. 6 shows the elements found in biofuel samples. Data from Tables 4 and 6 have been employed. The considered data correspond to ‘pure’ biodiesel and fuel ethanol samples. It is important to note that metal concentration in blend biodiesel has not been taken into account. Since, there are no clear data regarding the metal concentration in only bioethanol, the results corresponding to fuel ethanol are included in Fig. 6. A code indicating the metal content is also applied in order to distinguish major elements from trace elements. | ||
Fig. 6 Main elements found in real biodiesel and ethanol fuel samples.  : biodiesel; : biodiesel;  : bioethanol. (1) Present at concentrations on the order of mg L−1; (2) present at concentrations higher than 10 μg L−1 and lower than 1 mg L−1; (3) present at concentrations lower than 10 ng mL−1. : bioethanol. (1) Present at concentrations on the order of mg L−1; (2) present at concentrations higher than 10 μg L−1 and lower than 1 mg L−1; (3) present at concentrations lower than 10 ng mL−1. | ||
The results concerning biodiesel characterization are more abundant than those corresponding to bioethanol analysis. Thus, in the first case, official directives have been developed so as to ensure the quality of the employed fuel. This is in clear contrast to the situation found when bioethanol samples are considered. In that case, the studies provide information about the concentration of metals in the blend corresponding to bioethanol and gasoline (fuel ethanol). As a result, it is difficult to discern among the different sources of metallic species. It is also interesting to notice that there are no data regarding organometallic speciation in these kinds of products. Additional data regarding isotopic analysis are also scarce. This information would provide a better insight into the toxic potential of the different fuels. Furthermore, they would also give information about the geographical origin as well as the raw materials employed for production.
According to the information reviewed in the present work, it is obvious that additional work is required based on the development of more sensitive methods and less prone to interference than the existing ones. In this sense, the work related to new ICP liquid sample introduction systems able to mitigate non-spectral interference while increasing the sensitivity is highly promising. Likewise, the use of robust ICP-MS equipped with collision and/or reaction cells to overcome spectral interference should be encouraged. Standards must adapt to the new developments in this field and propose ICP analytical tools because they can provide multielemental information in a quick fashion and they afford suitable analytical figures of merit. Simple, fast and chip pre-treatment methods for biodiesel and bioethanol analysis aimed at pre-concentrating the sample while removing the matrix are extremely useful and more effort is needed in this field.
Acronyms
| AAS | Atomic absorption spectrometry |
| AFE | Anhydrous fuel ethanol |
| ANP | National Agency of Petroleum |
| ASTM | American Society for Testing and Materials |
| ASV | Anodic stripping voltammetry |
| BEC | Background equivalent concentration |
| CA | Continuous aspiration |
| CDA | Chelidamic acid |
| CRI | Collision–reaction interface |
| CV-AFS | Cold vapour atomic fluorescence spectroscopy |
| ETAAS | Electrothermal atomic absorption spectroscopy |
| D 3,2 | Sauter mean diameter |
| D 50 | Median of the aerosol volume drop size distribution |
| DCC | Dynamic collision cell |
| DPA | Diphenylamine |
| DRC | Dynamic reaction cell |
| ETV | Electrothermal vaporization |
| FAAS | Flame atomic absorption spectrometry |
| FAEE | Fatty acid ethyl esters |
| FAES | Flame atomic emission spectrometry |
| FAME | Fatty acid methyl esters |
| HFE | Hydrated fuel ethanol |
| HR-CS-AAS | High-resolution continuum source graphite furnace atomic absorption spectrometry |
| IC | Ionic chromatography |
| ICP | Inductively coupled plasma |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| ICP-OES | Inductively coupled plasma optical emission spectrometry |
| ICP-QQQ | Inductively coupled plasma triple quadrupole |
| ID | Isotopic dilution |
| LHR | Solid lignin hydrolysate residue |
| LOD | Limit of detection |
| LOQ | Limit of quantitation |
| MCN | Microconcentric nebulizer |
| MIP-OES | Microwave-induced plasma optical emission spectrometry |
| n e | Electron number density |
| ORS | Octopole reaction system |
| PAR | 4-(2-Pyridylazo)resorcinol |
| RSD | Relative standard deviation |
| SF-ICP-MS | Sector field inductively coupled plasma mass spectrometry |
| SSF | Simultaneous saccharification and fermentation |
| TEA | Triethylamine |
| THGA | Transversely heated graphite atomizer |
| TISIS | Torch Integrated Sample Introduction System |
| TMAH | Tetramethylammonium hydroxide |
| USN-MD-ICP-OES | Ultrasonic nebulizer and membrane desolvator inductively coupled plasma optical emission spectrometry |
| UV | Ultraviolet vacuum |
| WCAES | Tungsten coil atomic emission spectrometry |
Acknowledgements
The authors would like to thank Dr Vincent Coupard for his useful comments on biodiesel production.References
- H. Habe, T. Shinbo, T. Yamamoto, S. Sato, H. Shimada and K. Sakaki, J. Jpn. Pet. Inst., 2013, 56, 414–422 CrossRef CAS.
- D. Chiche, C. Diverchy, A. C. Lucquin, F. Porcheron and F. Defoort, Oil Gas Sci. Technol., 2013, 68, 707–723 CrossRef CAS PubMed.
- E. J. dos Santos, A. B. Herrmann, E. S. Chaves, W. W. D. Vechiatto, A. C. Schoemberger, V. L. a. Frescura and A. J. Curtius, J. Anal. At. Spectrom., 2007, 22, 1300 RSC.
- M. Edlund, H. Visser and P. Heitland, J. Anal. At. Spectrom., 2002, 17, 232–235 RSC.
- E. S. Chaves, M. T. C. De Loos-Vollebregt, A. J. Curtius and F. Vanhaecke, Spectrochim. Acta, Part B, 2011, 66, 733 CrossRef CAS PubMed.
- J. Farino and R. F. Browner, Anal. Chem., 1984, 56, 2709–2714 CrossRef CAS.
- J. L. Todolí, A. Canals and V. Hernandis, J. Anal. At. Spectrom., 1996, 11, 949–956 RSC.
- B. L. Sharp, J. Anal. At. Spectrom., 1988, 3, 613–652 RSC.
- R. Sánchez, J. L. Todolí, C. P. Lienemann and J. M. Mermet, J. Anal. At. Spectrom., 2010, 25, 178–185 RSC.
- R. I. McCrindle and C. J. Rademeyer, J. Anal. At. Spectrom., 1994, 9, 1087–1091 RSC.
- L. Ebdon, E. H. Evans and N. W. Barnett, J. Anal. At. Spectrom., 1989, 4, 505–508 RSC.
- A. W. Boorn, M. S. Cresser and R. F. Browner, Spectrochim. Acta, Part B, 1980, 35, 823–832 CrossRef.
- A. C. Lazar and P. B. Farnsworth, Anal. Chem., 1997, 69, 3921–3929 CrossRef CAS PubMed.
- R. I. McCrindle and C. J. Rademeyer, J. Anal. At. Spectrom., 1996, 11, 437–444 RSC.
- J.-L. Todoli and J.-M. Mermet, J. Anal. At. Spectrom., 2002, 17, 211–218 RSC.
- E. Paredes, S. E. Maestre and J. L. Todoli, Spectrochim. Acta, Part B, 2006, 61, 326 CrossRef PubMed.
- A. W. Boorn and R. F. Browner, Anal. Chem., 1982, 54, 1402–1410 CrossRef CAS.
- R. F. Browner, A. Canals and V. Hernandis, Spectrochim. Acta, Part B, 1992, 47, 659–673 CrossRef.
- C. K. Pan, G. X. Zhu and R. F. Browner, J. Anal. At. Spectrom., 1990, 5, 537–542 RSC.
- D. G. Weir and M. W. Blades, J. Anal. At. Spectrom., 1994, 9, 1311–1322 RSC.
- D. G. Weir and M. W. Blades, J. Anal. At. Spectrom., 1996, 11, 43–52 RSC.
- R. I. McCrindle and C. J. Rademeyer, Anal. Bioanal. Chem., 1996, 355, 264–266 CrossRef CAS PubMed.
- R. I. McCrindle and C. J. Rademeyer, J. Anal. At. Spectrom., 1995, 10, 399–404 RSC.
- D. G. Weir and M. W. Blades, J. Anal. At. Spectrom., 1996, 11, 1011–1018 RSC.
- G. Kreuning and F. Maessen, Spectrochim. Acta, Part B, 1989, 44, 367–384 CrossRef.
- M. W. Blades and B. L. Caughlin, Spectrochim. Acta, Part B, 1985, 40, 579–591 CrossRef.
- P. W. J. M. Boumans and M. C. Lux-Steiner, Spectrochim. Acta, Part B, 1982, 37, 97–126 CrossRef.
- G. Kreuning and F. J. M. J. Maessen, Spectrochim. Acta, Part B, 1987, 42, 677–688 CrossRef.
- V. M. Goldfarb and H. V. Goldfarb, Spectrochim. Acta, Part B, 1985, 40, 177–194 CrossRef.
- H. Benli, Spectrochim. Acta, Part B, 1983, 38, 81–91 CrossRef.
- K. Visser, F. M. Hamm and P. B. Zeeman, Appl. Spectrosc., 1976, 30, 34–38 CrossRef CAS.
- Y. Q. Tang and C. Trassy, Spectrochim. Acta, Part B, 1986, 41, 143–150 CrossRef.
- P. E. Walters and C. A. Barnardt, Spectrochim. Acta, Part B, 1988, 43, 325–337 CrossRef.
- M. Murillo and J. M. Mermet, Spectrochim. Acta, Part B, 1989, 44, 359–366 CrossRef.
- L. Ebdon and P. Goodall, J. Anal. At. Spectrom., 1992, 7, 1111–1116 RSC.
- S. J. Kumar and S. Gangadharan, J. Anal. At. Spectrom., 1999, 14, 967–971 RSC.
- E. McCurdy and D. Potter, Agil. Technol., 2002, 5988–6190E Search PubMed.
- T. D. Saint'Pierre, L. Tormen, V. L. Frescura and A. J. Curtius, J. Anal. At. Spectrom., 2006, 21, 1340 RSC.
- P. C. Hauser and M. W. Blades, Appl. Spectrosc., 1988, 42, 595–598 CrossRef CAS.
- J. Goossens, F. Vanhaecke, L. Moens and R. Dams, Anal. Chim. Acta, 1993, 280, 137–143 CrossRef CAS.
- V. L. Dressler, D. Pozebon and A. J. Curtius, Anal. Chim. Acta, 1999, 379, 175 CrossRef CAS.
- R. M. Olivas, C. R. Quetel and O. F. X. Donard, J. Anal. At. Spectrom., 1995, 10, 865–870 RSC.
- A. S. Al-Ammar, E. Reitznerová and R. M. Barnes, Spectrochim. Acta, Part B, 1999, 54, 1813–1820 CrossRef.
- Z. Hu, S. Hu, S. Gao, Y. Liu and S. Lin, Spectrochim. Acta, Part B, 2004, 59, 1463 CrossRef PubMed.
- S. C. K. Shum, S. K. Johnson, H. M. Pang and R. S. Houk, Appl. Spectrosc., 1993, 47, 575–585 CrossRef CAS.
- F. Vanhaecke, R. Dams and C. Vandecasteele, J. Anal. At. Spectrom., 1993, 8, 433–438 RSC.
- H. P. Longerich, J. Anal. At. Spectrom., 1989, 4, 665 RSC.
- E. H. Evans and L. Ebdon, J. Anal. At. Spectrom., 1990, 5, 425–430 RSC.
- G. D. Woods and F. I. Fryer, Anal. Bioanal. Chem., 2007, 389, 753–761 CrossRef CAS PubMed.
- European Parliament & Council, Directive 2009/28/EC on the promotion of the use of energy from renewable sources and amending and subsequently repealing directives 2001/77/EC and 2003/30/EC.
- D. E. López, G. G. J. James, D. A. Bruce and E. Lotero, Appl. Catal., A, 2005, 295, 97–105 CrossRef PubMed.
- Y. C. Sharma, B. Singh and S. N. Upadhyay, Fuel, 2008, 87, 2355–2373 CrossRef CAS PubMed.
- M. Zabeti, W. M. A. W. Daud and M. K. Aroua, Appl. Catal., A, 2009, 366, 154–159 CrossRef CAS PubMed.
- D. Y. C. Leung, X. Wu and M. K. H. Leung, Appl. Energy, 2010, 87, 1083–1095 CrossRef CAS PubMed.
- F. A. C. Amorim, B. Welz, A. C. S. Costa, F. G. Lepric, M. G. R. Vale and S. L. C. Ferreira, Talanta, 2007, 72, 349–359 CrossRef CAS PubMed.
- S. P. Singh and D. Singh, Renewable Sustainable Energy Rev., 2010, 14, 200–216 CrossRef CAS PubMed.
- H. Fukuda, A. Kondo and H. Noda, J. Biosci. Bioeng., 2001, 92, 405–416 CrossRef CAS.
- E. S. Chaves, E. J. dos Santos, R. G. O. Araujo, J. V. Oliveira, V. L. a. Frescura and A. J. Curtius, Microchem. J., 2010, 96, 71–76 CrossRef CAS PubMed.
- European Committee for Standardization, UNE-EN 14214:2013: Liquid petroleum products – Fatty acid methyl esters (FAME) for use in diesel engines and heating applications – Requirements and test methods, 2013.
- American Society for Testing and Materials (A.S.T.M.), ASTM D6751–12: Standard Specification for Biodiesel Fuel Blend Stock (B100) for Middle Distillate Fuels, 2012.
- E. Paredes, S. Z. Can and C. R. Quétel, Fuel, 2014, 123, 248–255 CrossRef CAS PubMed.
- E. Pillay, M. Elkadi, S. C. Fok, S. Stephen, J. Manuel, M. Z. Khan and S. Unnithan, Fuel, 2012, 97, 385–389 CrossRef PubMed.
- J. M. Marchetti, V. U. Miguel and A. F. Errazu, Renewable Sustainable Energy Rev., 2007, 11, 1300–1311 CrossRef CAS PubMed.
- L. C. Meher, V. S. S. Dharmagadda and S. N. Naik, Bioresour. Technol., 2006, 97, 1392–1397 CrossRef CAS PubMed.
- T. Furusawa, F. Kurayama, H. Handa, R. Kadota, M. Sato and N. Suzuki, Appl. Catal., A, 2014, 475, 69–75 CrossRef CAS PubMed.
- F. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef CAS.
- T. D. Saint'Pierre, L. F. Dias, S. M. Maia and A. J. Curtius, Spectrochim. Acta, Part B, 2004, 59, 551–558 CrossRef PubMed.
- M. L. Granados, M. D. Z. Poves, D. M. Alonso, R. Mariscal, F. C. Galisteo, R. Moreno-Tost, J. Santamaría and J. L. G. Fierro, Appl. Catal., B, 2007, 73, 317–326 CrossRef CAS PubMed.
- A. Kawashima, K. Matsubara and K. Honda, Bioresour. Technol., 2009, 100, 696–700 CrossRef CAS PubMed.
- X. Liu, H. He, Y. Wang, S. Zhu and X. Piao, Fuel, 2008, 87, 216–221 CrossRef CAS PubMed.
- N. Santiago-Torres, I. C. Romero-Ibarra and H. Pfeiffer, Fuel Process. Technol., 2014, 120, 34–39 CrossRef CAS PubMed.
- W. Xie and L. Zhao, Energy Convers. Manage., 2014, 79, 34–42 CrossRef CAS PubMed.
- W. Liu, P. Yin, X. Liu, W. Chen, H. Chen, C. Liu, R. Qu and Q. Xu, Energy Convers. Manage., 2013, 76, 1009–1014 CrossRef CAS PubMed.
- W. Xie and L. Zhao, Energy Convers. Manage., 2013, 76, 55–62 CrossRef CAS PubMed.
- M. Li, Y. Zheng, Y. Chen and X. Zhu, Bioresour. Technol., 2014, 154, 345–348 CrossRef CAS PubMed.
- M. Farooq, A. Ramli and D. Subbarao, J. Cleaner Prod., 2013, 59, 131–140 CrossRef CAS PubMed.
- Y.-L. Meng, S.-J. Tian, S.-F. Li, B.-Y. Wang and M.-H. Zhang, Bioresour. Technol., 2013, 136, 730–734 CrossRef CAS PubMed.
- A. Islam, Y. H. Taufiq-Yap, C.-M. Chu, E.-S. Chan and P. Ravindra, Process Saf. Environ. Prot., 2013, 91, 131–144 CrossRef CAS PubMed.
- S. Semwal, A. K. Arora, R. P. Badoni and D. K. Tuli, Bioresour. Technol., 2011, 102, 2151–2161 CrossRef CAS PubMed.
- A. K. Endalew, Y. Kiros and R. Zanzi, Biomass Bioenergy, 2011, 35, 3787–3809 CrossRef CAS PubMed.
- M. Takase, M. Zhang, W. Feng, Y. Chen, T. Zhao, S. J. Cobbina, L. Yang and X. Wu, Energy Convers. Manage., 2014, 80, 117–125 CrossRef CAS PubMed.
- J. M. Dias, M. C. M. Alvim-Ferraz, M. F. Almeida, J. D. M. Díaz, M. S. Polo and J. R. Utrilla, Fuel, 2012, 94, 418–425 CrossRef CAS PubMed.
- B.-X. Peng, Q. Shu, J.-F. Wang, G.-R. Wang, D.-Z. Wang and M.-H. Han, Process Saf. Environ. Prot., 2008, 86, 441–447 CrossRef CAS PubMed.
- D. M. Alonso, R. Mariscal, M. L. Granados and P. Maireles-Torres, Catal. Today, 2009, 143, 167–171 CrossRef CAS PubMed.
- P.-L. Boey, G. P. Maniam and S. A. Hamid, Chem. Eng. J., 2011, 168, 15–22 CrossRef CAS PubMed.
- H. Wu, J. Zhang, Q. Wei, J. Zheng and J. Zhang, Fuel Process. Technol., 2013, 109, 13–18 CrossRef CAS PubMed.
- C. S. MacLeod, A. P. Harvey, A. F. Lee and K. Wilson, Chem. Eng. J., 2008, 135, 63–70 CrossRef CAS PubMed.
- A. P. S. Chouhan and A. K. Sarma, Renewable Sustainable Energy Rev., 2011, 15, 4378–4399 CrossRef CAS PubMed.
- S. Yan, M. Kim, S. O. Salley and K. Y. S. Ng, Appl. Catal., A, 2009, 360, 163–170 CrossRef CAS PubMed.
- Commission of the European Communities, EC (2006) Communication from the commission: an EU strategy for biofuels, Brussels, 2006.
- A. de Jesus, M. M. Silva and M. G. R. Vale, Talanta, 2008, 74, 1378–1384 CrossRef CAS PubMed.
- F. A. Lobo, D. Goveia, A. P. Oliveira, L. P. C. Romão, L. F. Fraceto, N. L. D. Filho and A. H. Rosa, Fuel, 2011, 90, 142–146 CrossRef CAS PubMed.
- A. P. de Oliveira, R. D. Villa, K. C. P. Antunes, A. de Magalhães and E. C. Silva, Fuel, 2009, 88, 764–766 CrossRef PubMed.
- E. S. Chaves, F. G. Lepri, J. S. a. Silva, D. P. C. de Quadros, T. D. Saint'Pierre and A. J. Curtius, J. Environ. Monit., 2008, 10, 1211 RSC.
- J. S. A. Silva, E. S. Chaves, E. J. Santos, T. D. Saint'pierre, V. L. Frescura and A. J. Curtius, J. Braz. Chem. Soc., 2010, 21, 620–626 CrossRef CAS PubMed.
- P. R. Aranda, P. H. Pacheco, R. A. Olsina, L. D. Martinez and R. A. Gil, J. Anal. At. Spectrom., 2009, 24, 1441 RSC.
- J. Paligova, L. Joríkova and J. Cvengros, Energy Fuels, 2008, 22, 1991–1996 CrossRef CAS.
- G. Knothe, Fuel Process. Technol., 2007, 88, 669–677 CrossRef CAS PubMed.
- G. Knothe and R. O. Dunn, J. Am. Oil Chem. Soc., 2003, 80, 1021–1026 CrossRef CAS.
- A. Sarin, R. Arora, N. P. Singh, M. Sharma and R. K. Malhotra, Energy, 2009, 34, 1271–1275 CrossRef CAS PubMed.
- European Committee for Standardization, UNE-EN 14112:2003: Fat and oil derivatives. Fatty Acid Methyl Esters (FAME). Determination of oxidation stability (accelerated oxidation test), 2003.
- A. Gonzálvez, M. E. Ghanjaoui, M. El Rhazi and M. de la Guardia, Food Sci. Technol. Int., 2010, 16, 65–71 CrossRef PubMed.
- F. G. Lepri, E. S. Chaves, M. A. Vieira, A. S. Ribeiro, A. J. Curtius, L. C. C. De Oliveira and R. C. De Campos, Appl. Spectrosc. Rev., 2011, 46, 175 CrossRef.
- M. D. G. A. Korn, D. S. S. dos Santos, B. Welz, M. G. R. Vale, A. P. Teixeira, D. D. C. Lima, S. L. C. Ferreira, M. das Graças Andrade Korn, D. de Castro Lima and D. S. S. Dos Santos, Talanta, 2007, 73, 1–11 CrossRef CAS PubMed.
- A. I. Barros, A. P. de Oliveira, M. R. L. de Magalhães and R. D. Villa, Fuel, 2012, 93, 381–384 CrossRef CAS PubMed.
- M. Ghisi, E. S. Chaves, D. P. C. Quadros, E. P. Marques, A. J. Curtius and A. L. B. Marques, Microchem. J., 2011, 98, 62 CrossRef CAS PubMed.
- F. A. Lobo, D. Goveia, A. P. de Oliveira, E. R. Pereira-Filho, L. F. Fraceto, N. L. D. Filho and A. H. Rosa, Fuel, 2009, 88, 1907–1914 CrossRef CAS PubMed.
- F. H. Lyra, M. T. W. D. Carneiro, G. P. Brandão, H. M. Pessoa and E. V. de Castro, Microchem. J., 2010, 96, 180–185 CrossRef CAS PubMed.
- D. P. C. Quadros, M. Rau, M. Idrees, E. S. Chaves, A. J. Curtius and D. L. G. Borges, Spectrochim. Acta, Part B, 2011, 66, 373–377 CrossRef CAS PubMed.
- R. S. Amais, E. E. Garcia, M. R. Monteiro, A. R. A. Nogueira and J. A. Nóbrega, Microchem. J., 2010, 96, 146–150 CrossRef CAS PubMed.
- European Committee for Standardization, UNE-EN 14538:2006: Fat and oil derivatives – Fatty acid methyl ester (FAME) – Determination of Ca, K, Mg and Na content by optical emission spectral analysis with inductively coupled plasma (ICP OES), 2006.
- European Committee for Standardization, UNE EN 14107: 2003: Fat and oil derivatives. Fatty Acid Methyl Esters (FAME). Determination of phosphorus content by inductively coupled plasma (ICP) emission spectrometry, 2003.
- Leeman-Labs, Determination of Trace Elements in Biodiesel Feedstocks using Inductively Coupled Plasma Optical Emission Spectrometry.
- Leeman-Labs, The Determination of Phosphorous, Sulfur, Sodium, Potassium, Calcium and Magnesium in Biodiesel using the Teledyne FuelPro.
- Z. A. Grosser, L. J. Davidowski and P. Wee, The analysis of biodiesel for inorganic contaminants, including sulfur, by ICP-OES, Perkin Elmer Instruments, 2009 Search PubMed.
- R. Knoll and M. Knopp, Phosphorus, Calcium and Magnesium Analysis of Soybean Oil-feedstock for Biodiesel Production Using the Optima Inductively Coupled Plasma Optical Emission Spectrometer, 2007 Search PubMed.
- P. Sarojam, Quality Control of Biofuels Using a Inductively Coupled Plasma Optical Emission Spectrophotometer (ICP-OES) for Metals Determination, 2009 Search PubMed.
- D. Johnson, Determination of metals in oils by ICP-AES, 1993, vol. ICP-13 Search PubMed.
- R. Sanchez, J. L. Todolí, C.-P. Lienemann and J.-M. Mermet, Spectrochim. Acta, Part B, 2013, 88, 104–126 CrossRef CAS PubMed.
- R. Sanchez, C. Sanchez, J. L. Todoli, C.-P. Lienemann and J.-M. Mermet, J. Anal. At. Spectrom., 2014, 29, 242–248 RSC.
- R. Sanchez, J. L. Todoli, C.-P. Lienemann and J.-M. Mermet, J. Anal. At. Spectrom., 2012, 27, 937 RSC.
- T. L. Thiem and J. D. Watson, Microchem. J., 1997, 57, 245–250 CrossRef CAS.
- J. L. Burguera and M. Burguera, Talanta, 2004, 64, 1099–1108 CrossRef CAS PubMed.
- R. M. de Souza, L. G. Leocádio and C. L. P. da Silveira, Anal. Lett., 2008, 41, 1615–1622 CrossRef.
- C. G. Young, R. S. Amais, D. Schiavo, E. E. Garcia, J. A. Nóbrega and B. T. Jones, Talanta, 2011, 84, 995–999 CrossRef CAS PubMed.
- M. T. Lisboa, C. D. Clasen, D. C. de S. Vellar, E. Q. Oreste, T. D. Saint'Pierre, A. S. Ribeiro and M. A. Vieira, J. Braz. Chem. Soc., 2014, 25, 143–151 CAS.
- M. G. A. Korn, D. C. M. B. Santos, M. A. B. Guida, I. S. Barbosa, M. L. C. Passos, M. L. M. F. S. Saraiva and J. L. F. C. Lima, J. Braz. Chem. Soc., 2010, 21, 2278–2284 CrossRef CAS PubMed.
- J. L. Todolí and J. M. Mermet, TrAC, Trends Anal. Chem., 2005, 24, 107–116 CrossRef PubMed.
- J. R. de Souza, E. F. dos Santos, C. B. Duyck and T. D. Saint'Pierre, Spectrochim. Acta, Part B, 2011, 66, 356–361 CrossRef CAS PubMed.
- M. Bauer and J. A. C. Broekaert, Spectrochim. Acta, Part B, 2007, 62, 145–154 CrossRef PubMed.
- M. Bauer and J. A. C. Broekaert, J. Anal. At. Spectrom., 2008, 23, 479–486 RSC.
- M. A. Aguirre, N. Kovachev, M. Hidalgo and A. Canals, J. Anal. At. Spectrom., 2012, 27, 2102–2110 RSC.
- R. I. Botto, J. J. Zhu and O. N. Cetac Technol Inc, in 1994 Winter Conference on Plasma Spectrochemistry, Royal Soc Chemistry, San Diego, Ca, 1994, vol. 9, pp. 905–912 Search PubMed.
- R. I. Botto, J. J. Zhu and O. N. Cetac Technol Inc, in 1996 Winter Conference on Plasma Spectrochemistry, Royal Soc Chemistry, Ft Lauderdale, Fl, 1996, vol. 11, pp. 675–681 Search PubMed.
- R. Wennrich, A. Mroczek, K. Dittrich, G. Werner, R. Wennrich, A. Mroczek, K. Dittrich and G. Werner, Fresenius. J. Anal. Chem., 1995, 352, 461–469 CrossRef CAS.
- M. Aramendia, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2009, 648, 23–44 CrossRef CAS PubMed.
- S. M. Maia, M. G. R. Vale, B. Welz and A. J. Jose, Spectrochim. Acta, Part B, 2001, 56, 1263–1275 CrossRef.
- G. L. Donati, R. S. Amais and J. A. Nobrega, J. Anal. At. Spectrom., 2013, 28, 280–287 RSC.
- T. D. Saint'Pierre, L. F. Dias, D. Pozebon, R. Q. Aucelio, A. J. Curtius and B. Welz, Spectrochim. Acta, Part B, 2002, 57, 1991–2001 CrossRef.
- R. S. Amais, S. E. Long, J. A. Nóbrega and S. J. Christopher, Anal. Chim. Acta, 2014, 806, 91–96 CrossRef CAS PubMed.
- L. Balcaen, G. Woods, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2013, 28, 33–39 RSC.
- J. V. Sokolnikova, I. E. Vasilyeva and V. I. Menshikov, Spectrochim. Acta, Part B, 2003, 58, 387–391 CrossRef.
- E. S. Chaves, T. D. Saint'Pierre, E. J. Dos Santos, L. Tormen, V. L. A. F. Bascuñan and A. J. Curtius, J. Braz. Chem. Soc., 2008, 19, 856 CrossRef CAS PubMed.
- L. C. C. de Oliveira, M. A. Vieira, A. S. Ribeiro, P. M. Baptista, R. A. Gonsalves and R. C. de Campos, J. Braz. Chem. Soc., 2012, 23, 1400–1408 CrossRef CAS PubMed.
- European Committee for Standardization, UNE-EN 14108:2003: Fat and oil derivatives. Fatty Acid Methyl Esters (FAME). Determination of sodium content by atomic absorption spectrometry, 2003.
- European Committee for Standardization, UNE-EN 14109:2003: Fat and oil derivatives. Fatty Acid Methyl Esters (FAME). Determination of potassium content by atomic absorption spectrometry, 2003.
- M. R. L. de Magalhães, A. I. Barros, A. P. de Oliveira, A. da Silva and R. D. Villa, Curr. Anal. Chem., 2014, 10, 166–171 CrossRef.
- R. S. Amais, E. E. Garcia, M. R. Monteiro and J. A. Nóbrega, Fuel, 2012, 93, 167–171 CrossRef CAS PubMed.
- R. E. Santelli, M. A. Bezerra, A. S. Freire, E. P. Oliveira and M. D. B. de Carvalho, Fuel, 2008, 87, 1617–1622 CrossRef CAS PubMed.
- P. R. Aranda, J. A. Gásquez, R. A. Olsina, L. D. Martinez and R. A. Gil, Talanta, 2012, 101, 353–356 CrossRef CAS PubMed.
- A. de Jesus, A. V. Zmozinski, I. C. F. Damin, M. M. Silva and M. G. R. Vale, Spectrochim. Acta, Part B, 2012, 71–72, 86–91 CrossRef CAS PubMed.
- G. P. Brandao, R. C. de Campos, E. V. R. de Castro and H. C. de Jesus, Spectrochim. Acta, Part B, 2008, 63, 880–884 CrossRef PubMed.
- R. J. Cassella, B. Barbosa, R. E. Santelli and A. T. Rangel, Anal. Bioanal. Chem., 2004, 379, 66–71 CrossRef CAS PubMed.
- E. Becker, R. T. Rampazzo, M. B. Dessuy, M. G. R. Vale, M. M. da Silva, B. Welz and D. A. Katskov, Spectrochim. Acta, Part B, 2011, 66, 345–351 CrossRef CAS PubMed.
- F. H. Lyra, M. Carneiro, G. P. Brandao, H. M. Pessoa and E. V. R. de Castro, J. Anal. At. Spectrom., 2009, 24, 1262–1266 RSC.
- F. G. Lepri, M. B. Dessuy, M. G. R. Vale, D. L. G. Borges, B. Welz and U. Heitmann, Spectrochim. Acta, Part B, 2006, 61, 934–944 CrossRef PubMed.
- R. C. De Campos, C. L. T. Correia, F. Vieira, T. D. Saint'Pierre, A. C. Oliveira and R. Gonçalves, Spectrochim. Acta, Part B, 2011, 66, 352 CrossRef PubMed.
- R. S. Amais, G. L. Donati, D. Schiavo and J. A. Nóbrega, Microchem. J., 2013, 106, 318–322 CrossRef CAS PubMed.
- L. B. de Caland, E. L. C. Silveira and M. Tubino, Anal. Chim. Acta, 2012, 718, 116–120 CrossRef PubMed.
- Y. Zhang, P. Thepsithar, X. Jiang and J. H. Tay, Ind. Crops Prod., 2013, 44, 459–464 CrossRef CAS PubMed.
- M. Piovezan, A. C. O. Costa, A. V. Jager, M. A. L. de Oliveira and G. A. Micke, Anal. Chim. Acta, 2010, 673, 200–205 CrossRef CAS PubMed.
- T. Nogueira and C. L. Do Lago, Microchem. J., 2011, 99, 267–272 CrossRef CAS PubMed.
- T. R. C. Zezza, M. de Souza Castilho and N. R. Stradiotto, Fuel, 2012, 95, 15–18 CrossRef CAS PubMed.
- L. C. Martiniano, V. R. Abrantes, S. Y. Neto, E. P. Marques, T. C. O. Fonseca, L. L. Paim, A. G. Souza, N. R. Stradiotto, R. Q. Aucélio, G. H. R. Cavalcante and A. L. B. Marques, Fuel, 2013, 103, 1164–1167 CrossRef CAS PubMed.
- L. Pinto and S. G. Lemos, Microchem. J., 2013, 110, 417–424 CrossRef CAS PubMed.
- G. C. Sedenho, L. L. Paim and N. R. Stradiotto, Anal. Methods, 2013, 5, 4145–4151 RSC.
- M. de Souza Castilho and N. R. Stradiotto, Talanta, 2008, 74, 1630–1634 CrossRef PubMed.
- J. M. S. Almeida, R. M. Dornellas, S. Yotsumoto-Neto, M. Ghisi, J. G. C. Furtado, E. P. Marques, R. Q. Aucélio and A. L. B. Marques, Fuel, 2014, 115, 658–665 CrossRef CAS PubMed.
- S. E. Dancsak, S. G. Silva, J. A. Nóbrega, B. T. Jones and G. L. Donati, Anal. Chim. Acta, 2014, 806, 85–90 CrossRef CAS PubMed.
- G. M. Walker, Bioethanol: Science and Technology of Fuel Alcohol, Ventus Publishing ApS, 2010 Search PubMed.
- F. Monot, A. Margeot, B. Hahn-Hägerdal, J. Lindstedt and R. Slade, Oil Gas Sci. Technol., 2013, 68, 693–705 CrossRef CAS PubMed.
- M. Cassap, I. C. P. A. Specialist, T. F. Scientific, and S. Instruments.
- A. Cosnier, S. Lebouil, S. Vélasquez and H. J. Yvon, ICP At. Emiss. Spectrosc. Application Note, http://www.horiba.com/fileadmin/uploads/Scientific/Documents/Emission/ICPPETRO02.pdf, 2014 Search PubMed.
- F. Rosillo-Calle and A. Walter, Energy Sustainable Dev., 2006, 10, 20–32 CrossRef.
- L. N. Santos, J. A. G. Neto and N. M. Caldas, Fuel, 2012, 99, 9–12 CrossRef CAS PubMed.
- E. G. Vieira, I. V. Soares, N. L. Dias Filho, N. C. da Silva, E. F. Garcia, A. C. Bastos, S. D. Perujo, T. T. Ferreira, A. H. Rosa and L. F. Fraceto, J. Colloid Interface Sci., 2013, 391, 116–124 CrossRef CAS PubMed.
- G. L. Donati, R. S. Amais, D. Schiavo and J. a. Nóbrega, J. Anal. At. Spectrom., 2013, 28, 755 RSC.
- J. C. Dias, L. T. Kubota, P. N. Nesterenko, G. W. Dicinoski and P. R. Haddad, Anal. Methods, 2010, 2, 1565 RSC.
- P. S. Roldan, I. L. Alcântara, G. R. Castro, J. C. Rocha, C. C. F. Padilha and P. M. Padilha, Anal. Bioanal. Chem., 2003, 375, 574 CAS.
- T. D. Saint'Pierre, T. D. A. Maranhão, V. L. A. Frescura and A. J. Curtius, Spectrochim. Acta, Part B, 2005, 60, 605 CrossRef PubMed.
- T. D. Saint'Pierre, V. L. A. Frescura and A. J. Curtius, Talanta, 2006, 68, 957–962 CrossRef PubMed.
- M. F. De Oliveira, A. A. Saczk, L. L. Okumura, A. P. Fernandes, M. De Moraes and N. R. Stradiotto, Anal. Bioanal. Chem., 2004, 380, 135–140 CrossRef PubMed.
- R. A. A. Munoz and L. Angnes, Microchem. J., 2004, 77, 157–162 CrossRef CAS PubMed.
- M. S. Rocha, M. F. Mesko, F. F. Silva, R. C. Sena, M. C. B. Quaresma, T. O. Araújo and L. A. Reis, J. Anal. At. Spectrom., 2011, 26, 456 RSC.
- V. N. Alves, R. Mosquetta, N. M. M. Coelho, J. N. Bianchin, K. C. Di Pietro Roux, E. Martendal and E. Carasek, Talanta, 2010, 80, 1133–1138 CrossRef CAS PubMed.
- L. Tormen, E. S. Chaves, T. D. Saint'Pierre, V. L. a. Frescura and A. J. Curtius, J. Anal. At. Spectrom., 2008, 23, 1300 RSC.
- D. B. Taylor and R. E. Synovec, Talanta, 1993, 40, 495–501 CrossRef CAS.
- European Committee for Standardization, EN 15837:2009: Ethanol as a blending component for petrol – Determination of phosphorous, copper and sulfur content – Direct method by inductively coupled plasma optical emission spectrometry (ICP-OES), 2009.
- B. Huang, J. Yang, A. Pei, X. Zeng and P. W. J. M. Boumans, Spectrochim. Acta, Part B, 1991, 46, 407–416 CrossRef.
- D. R. Neves, R. S. Amais, J. A. Nóbrega and J. A. G. Neto, Anal. Lett., 2012, 45, 1111–1121 CrossRef CAS.
- A. Smith, Application Note Improved Accuracy in the Analysis of Precious Metals by ICP-OES.
- R. E. Sturgeon and J. W. Lam, J. Anal. At. Spectrom., 1999, 14, 785–791 RSC.
- J. S. Lee and H. B. Lim, Bull. Korean Chem. Soc., 1999, 20, 1040–1044 CAS.
- D. R. Wiederin, R. S. Houk, R. K. Winge and A. P. D'Silva, Anal. Chem., 1990, 62, 1155–1160 CrossRef CAS.
- J. A. McLean, M. G. Minnich, L. A. Iacone and H. Liu, J. Anal. At. Spectrom., 1998, 13, 829–842 RSC.
- E. H. Evans and J. J. Giglio, J. Anal. At. Spectrom., 1993, 8, 1–18 RSC.
- Y. Cheung, G. C.-Y. Chan and G. M. Hieftje, J. Anal. At. Spectrom., 2013, 28, 241 RSC.
- T. W. May and R. H. Wiedmeyer, At. Spectrosc., 1998, 19, 150–155 CAS.
- Y. Kishi, K. Kawabata, H. Shi and R. Thomas, Spectroscopy, 2004, 19, 14–23 CAS.
- A. P. De Oliveira, J. A. G. Neto and M. M. C. Ferreira, Eclet. Quim., 2006, 31, 7–12 CrossRef CAS PubMed.
- A. P. De Oliveira, M. De Moraes, J. A. Gomes Neto and E. C. Lima, At. Spectrosc., 2002, 23, 190 Search PubMed.
- A. P. De Oliveira, M. De Moraes, J. A. Gomes Neto and E. C. Lima, At. Spectrosc., 2002, 23, 39 Search PubMed.
- T. Saint'Pierre, R. Q. Aucélio and A. J. Curtius, Microchem. J., 2003, 75, 59–67 CrossRef.
- M. B. O. Giacomelli, J. B. B. Da Silva, T. D. Saint'Pierre, A. J. Curtius and J. B. B. Da Silva, Microchem. J., 2004, 77, 151 CrossRef CAS PubMed.
- T. D. Saint'Pierre, T. D. A. Maranhão, V. L. Frescura, A. J. Curtius, R. Q. Aucélio and T. D. Saint'Pierre, Quim. Nova, 2008, 31, 1626–1630 CrossRef PubMed.
- C. Pesco, E. A. De Campos, C. Maieru and M. Costa, Mikrochim. Acta, 1997, 232, 229–232 CrossRef.
- L. A. de M. Gomes, P. de M. Padilha, J. C. Moreira, N. L. D. Filho and Y. Gushikem, J. Braz. Chem. Soc., 1998, 9, 494–498 CrossRef CAS PubMed.
- P. Lessi, N. L. D. Filho, C. Moreira and J. T. S. Campos, Anal. Chim. Acta, 1996, 327, 183 CrossRef CAS.
- J. N. Bianchin, E. Martendal, R. Mior, V. N. Alves, C. S. T. Araújo, N. M. M. Coelho and E. Carasek, Talanta, 2009, 78, 333–336 CrossRef CAS PubMed.
- J. Motonaka, H. Konishi, S. Ikeda and N. Tanaka, Bull. Chem. Soc. Jpn., 1986, 59, 737–740 CrossRef CAS.
- A. I. Kamenev and M. A. Kovalenko, J. Anal. Chem., 2000, 55, 594–597 CrossRef CAS.
- J. C. Dias, L. T. Kubota, P. N. Nesterenko and P. R. Haddad, Chromatographia, 2012, 75, 867–873 CAS.
| This journal is © The Royal Society of Chemistry 2015 |