
Expanding the scope of gels – combining polymers with low-molecular-weight gelators to yield modified self-assembling smart materials with high-tech applications
Daniel J.
Cornwell
and
David K.
Smith
*
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: david.smith@york.ac.uk; Fax: +44 (0)1904 324156
First published on 30th January 2015
Abstract
Combining low molecular weight gelators (LMWGs) with polymers is a broad yet relatively recent field, in a phase of rapid expansion and with huge potential for exploitation. This review provides an overview of the state-of-the-art and reflects on new technologies that might be unlocked. We divide LMWG–polymer systems into five categories: (i) polymerisation of self-assembled LMWG fibres, (ii) capture of LMWG fibres in a polymer matrix, (iii) addition of non-gelling polymer solutions to LMWGs, (iv) systems with directed interactions between polymers and LMWGs, and (v) hybrid gels containing both LMWGs and polymer gels (PGs). Polymers can have significant impacts on the nanoscale morphology and materials performance of LMWGs, and conversely LMWGs can have a major effect on the rheological properties of polymers. By combining different types of gelation system, it is possible to harness the advantages of both LMWGs and PGs, whilst avoiding their drawbacks. Combining LMWG and polymer technologies enhances materials performance which is useful in traditional applications, but it may also yield major steps forward in high-tech areas including environmental remediation, drug delivery, microfluidics and tissue engineering.
 Daniel J. Cornwell | Daniel J. Cornwell graduated with a first class MChem degree from University of York in 2012, having carried out his Masters research project at RWTH Aachen University in the group of Prof. Markus Albrecht. He is now a PhD student in the research group of Prof. David K. Smith, and his interests include developing hybrid gels including multidomain photopatterned materials with potential biological applications. In 2014, he presented his results in a lecture at the RSC Northern Universities Postgraduate Symposium. |
 David K. Smith | Professor David K. Smith carried out his DPhil at University of Oxford with Prof. Paul Beer and was then Royal Society European Research Fellow with Prof. Francois Diederich at ETH Zurich. In 1999, he was appointed Lecturer in York, and in 2006 promoted to a Professorship. His research focusses on applying a fundamental understanding of supramolecular science and self-assembly to nanomaterials and nanomedicine. In 2012 he received the RSC Corday Morgan Award for research, in 2013 was awarded a National Teaching Fellowship by the Higher Education Academy, and in 2014 was one of the RSC's 175 Faces of Chemistry. |
Introduction
Gels are soft materials with numerous roles in industrial products and well-known in everyday life. Although traditional applications of gels have often focussed on making use of their rheological performance, increasingly high-tech applications are being considered – from tissue engineering to nanoscale electronics, and drug delivery to environmental remediation.1 As such, ways of enhancing the performance of gel-phase materials are highly in demand as they may improve the scope of this fascinating family of materials. Gels are colloidal soft materials brought about by the co-existence of two different phases: a ‘liquid-like’ phase containing a sample-spanning nanoscale ‘solid-like’ network, preventing bulk flow of the liquid. The solid-like phase makes up only a small percentage of the overall material (often less than 1%), with the molecules forming it being known as gelators.We can separate gelators into two categories. The first category is that of polymer gelators (PGs),2 where long-chain polymer molecules form the sample-spanning network required for gelation, through either covalent or non-covalent3 crosslinking. PGs can be either naturally derived, such as agarose or gelatin, or synthetic, such as poly(acrylic acid) or poly(ethylene glycol). Polymer gels already have a wide variety of everyday and high-tech applications, including in foodstuffs, contact lenses and superabsorbent materials – they are the gels with which we are most likely to come into contact.2,4 PGs often form relatively robust networks (particularly those with covalent crosslinking), but as a consequence, they are sometimes unresponsive to stimuli. It can also be difficult to program desired properties into PGs.
The second category of gelator is that of the low-molecular-weight gelators (LMWGs);5 small molecules that through non-covalent interactions form a ‘supramolecular’ gel (alternatively known as a molecular or physical gel).6 Non-covalent interactions may take the form of hydrogen-bonding, van der Waals forces, π–π interactions, metal–ligand bonding, solvophobicity etc. LMWGs undergo hierarchical self-assembly, with non-covalent interactions driving the formation of fibrils, which aggregate into (nano)fibres, which ultimately entangle and interact to form a sample-spanning, solid-like network (Fig. 1). The reversible non-covalent interactions that hold this type of gelator network together can be particularly responsive to external stimuli, such as temperature or pH. The macroscopic properties of the final gel can also be tuned by structural modification or ‘synthetic programming’ of the gelators at the molecular level – changing the functional groups can, via self-assembly, lead to control over the nanoscale and materials properties/functionality of the gel.
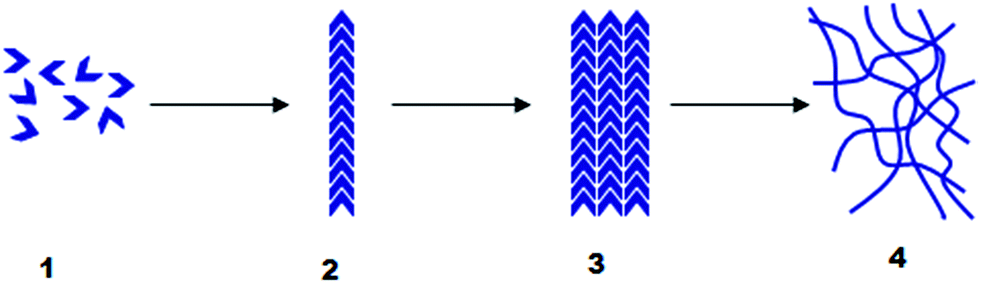 | ||
| Fig. 1 Typical hierarchical self-assembly process for a supramolecular gel; LMWG molecules (1) assemble into fibrils (2), which bundle into fibres (3), which then entangle to form a sample-spanning solid-like network (4). | ||
Low molecular weight gels are less widespread in application than PGs, but even in ancient times, gels based on fatty acids and metal salts (limestone) were used as greases on Hittite chariots, and since the mid-20th century, similar LMWGs based on lithium 12-hydroxystearate have been used in the lubrication industry.7 In more recent times, the responsive, tunable and programmable nature of supramolecular gels has captured the interest of academic chemists with high-tech applications in mind, and nanoscale electronics, sensors, biomaterials, tissue engineering and drug delivery have all received considerable interest.1,8 However, the responsiveness and tunability of LMWGs often come at a cost: because the gels are in many cases mechanically weak, they can be easily broken down and are difficult to manipulate in the solid-like form.
There has been significant interest in combining supramolecular chemistry with polymers in order to tailor gel properties.3 This review, however, focusses on the specific approach of combining an established LMWG with polymer science methods. This would seem like a logical approach to extending the scope of both these families of materials, as well as providing access to new properties and applications. Reflecting on this, we have concluded that there are currently five key categories of LMWG–polymer combination (Fig. 2):
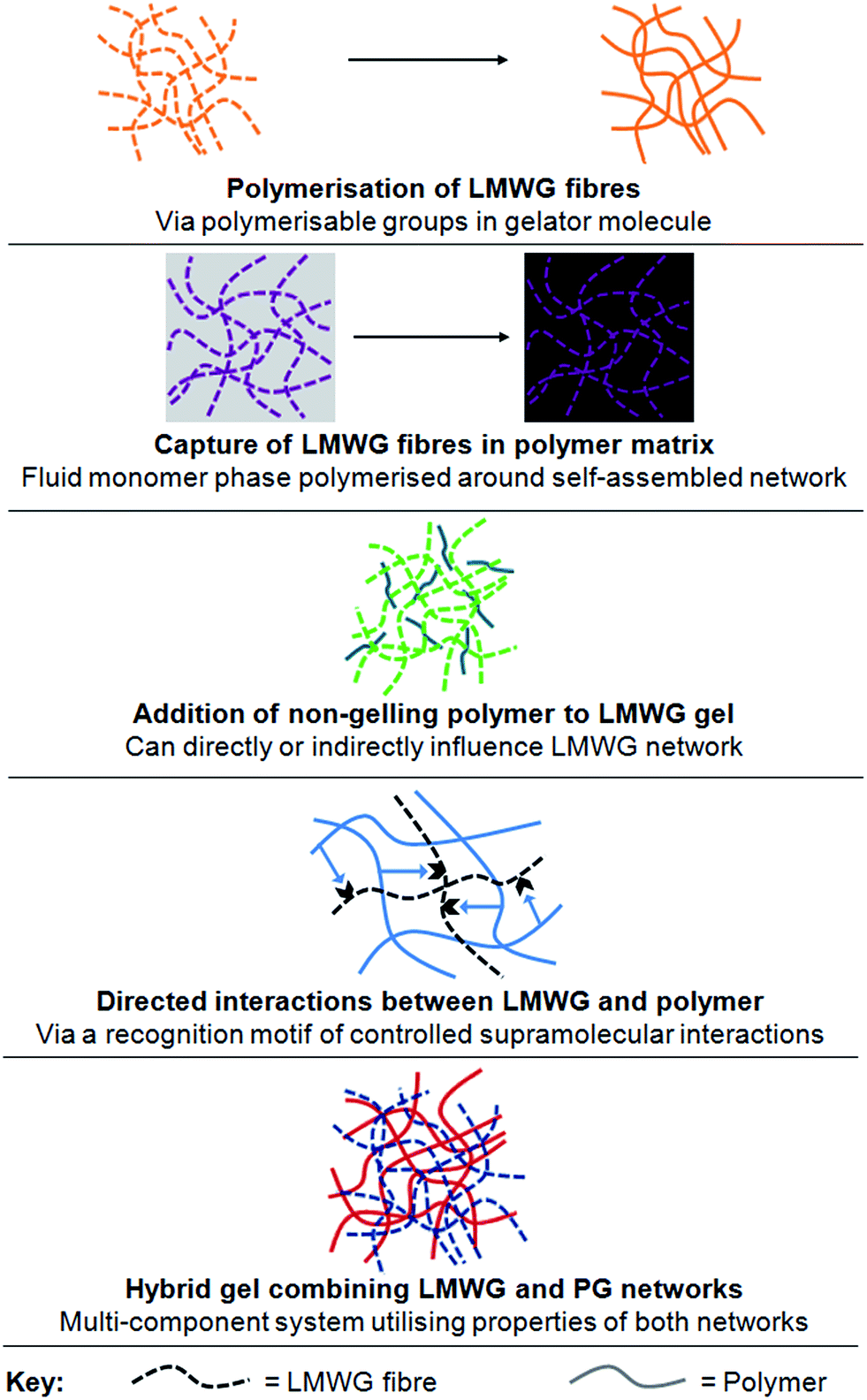 | ||
| Fig. 2 Illustrations of the five main types of LMWG–polymer combination described in this review – categories 1–5 from top to bottom. | ||
(1) Direct polymerisation of self-assembled LMWG fibres through polymerisable groups in the LMWG molecules.
(2) Embedding LMWG network in a polymerisable solvent; self-assembled LMWG fibres may subsequently be washed out of the polymer matrix giving nano-imprinted porous materials.
(3) Addition of a non-gelling polymer in solution to LMWG.
(4) Addition of a polymer in solution which is capable of directed and controlled interactions with the LMWG.
(5) Combination of LWMG with a polymer which is also capable of gelation (i.e., a PG), to yield a hybrid gel.
In this review, we present an overview of each LMWG–polymer combination, with key examples to show the development of research in each category, methods employed to access and study the combinations, and the performance and potential applications of the resulting materials.
Category 1 – polymerisation of LMWG fibres
Feringa and co-workers were the first to work with LMWG fibres which could be polymerised after self-assembly.9 Their methacrylate derivative of trans-1,2-bis(3-methylureido) cyclohexane (Fig. 3) acted as a LMWG in a variety of organic solvents, self-assembling via hydrogen-bond formation into fibres. This process provided ideal spatial arrangement of the methacrylate groups, which could be polymerised by addition of a photoinitiator and subsequent photoirradiation. The resulting gels showed an increase in thermal and temporal stability compared with the unpolymerised gel. Electron microscopy showed that while the unpolymerised gel consisted of mainly straight, occasionally intertwined fibres the polymerised gel consisted of a highly cross-linked network, explaining the observed changes in materials properties.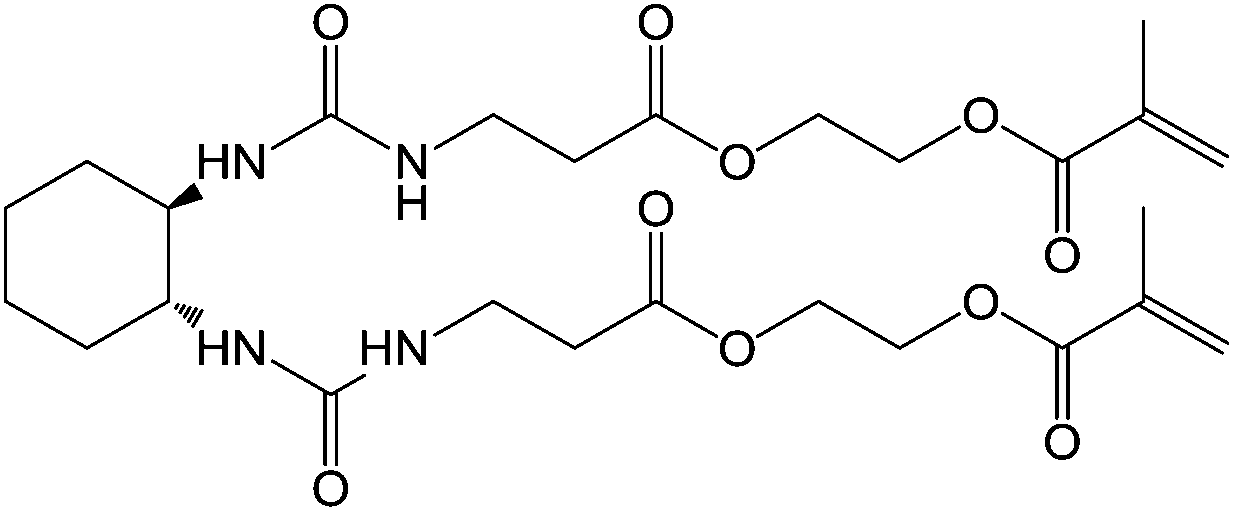 | ||
| Fig. 3 Photopolymerisable methacrylate derivative of trans-1,2-bis(3-methylureido)cyclohexane LMWG.9 | ||
More recently, acrylate functionalised gelators have been used within liquid crystals, with polymerisation of the gel fibres leading to lower driving voltages for electro-optical LC transitions.10 This clearly demonstrates the ability of covalently captured gel fibres to have a positive impact within high-tech materials.
Weiss and George studied LMWGs with conjugated diacetylene units11 – diacetylenes are able to undergo solid-state polymerisation by 1,4-addition reactions if the monomers are suitably aligned. 10,12-Pentacosadiynoic acid and derivatives (Fig. 4a) were able to gelate a variety of organic solvents, and on photoirradiation many of these gel networks polymerised. For most of the polymerised gels, microscopic phase-separation between solvent and gelator network was observed (i.e., the polymer was insoluble) – but some gel integrity was maintained. The polymerised gels appeared no more thermally or temporally stable than the unpolymerised gels – it was suggested (from X-ray diffraction) that the polymerised fibres maintained their basic morphology, with no additional crosslinking between them; this would suggest very precise translation of self-assembled structural information within the polymerisation step. Nonetheless, it remains somewhat surprising that connecting molecular units into covalent polymers did not alter gel thermal stability as this should have a significant impact on the gelation equilibria.
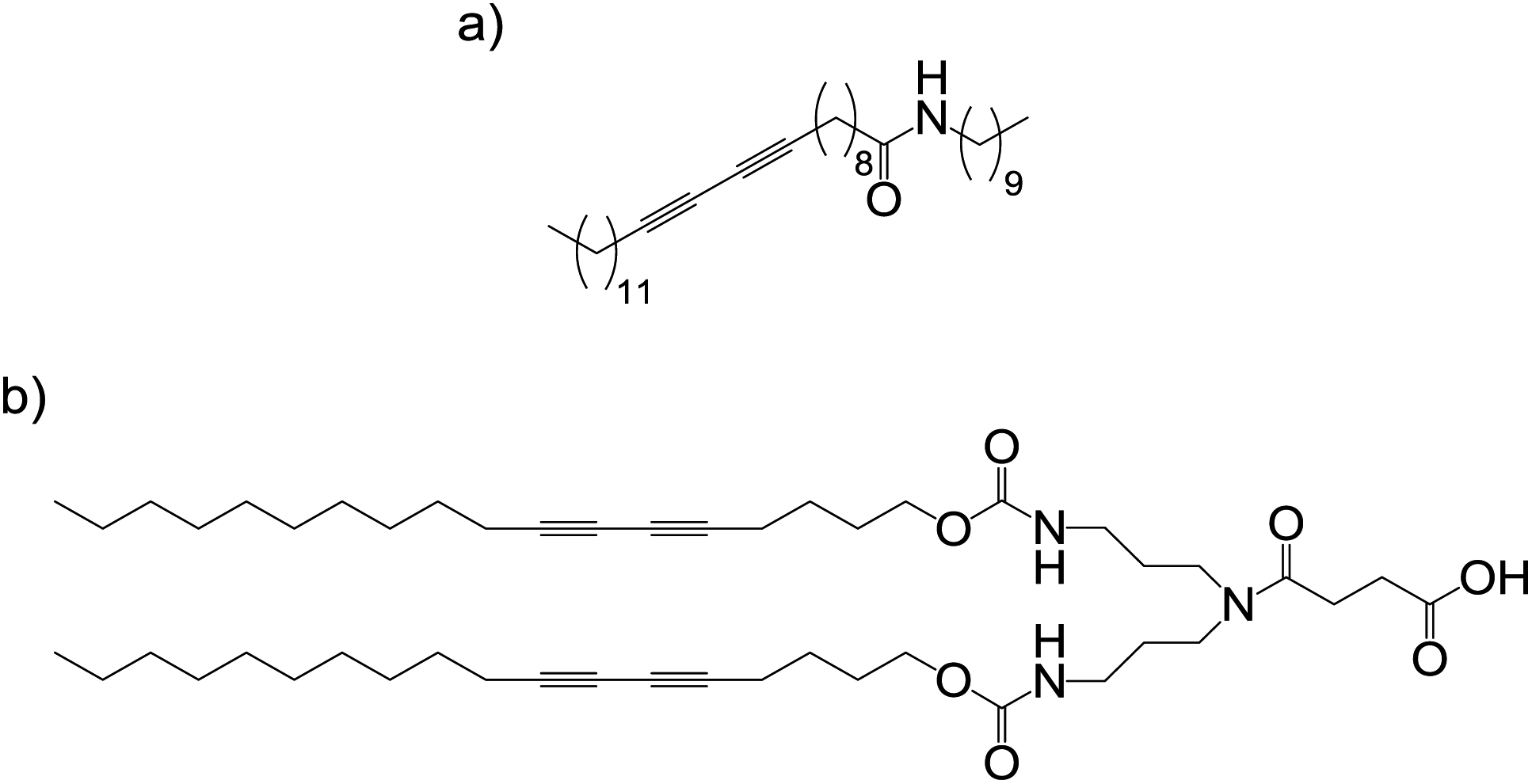 | ||
| Fig. 4 Exemplar structures of diacetylene containing LMWGs: (a) 10,12-pentacosadiynoic acid derived gelator,11 and (b) dendritic gelator.12 | ||
Kim and co-workers similarly examined the self-assembly and photopolymerisation of dendritic LMWGs containing diacetylenes (Fig. 4b).12 As the structures polymerised they became insoluble and the capture of the nano-structuring was evidenced by X-ray diffraction. Shinkai and co-workers photopolymerised diacetylene units in a copper-porphyrin LMWG; the resulting polymer was insoluble and separated from the solvent, but did retain the fibrous nanostructure of the self-assembled network.13 Many other groups have also used diacetylenes for the photopolymerisation of LMWG fibres.14 Overall, it should be noted that when polymerising self-assembled gelators, the overall solubility can change – gelators are finely balanced between solubility and precipitation, and small changes can significantly impact on gelation.15
Our research group used Grubbs' metathesis, a reversible approach to covalent capture, capable of ‘error checking’, to polymerise self-assembled networks of dendritic gelators with peripheral alkenes (Fig. 5).16 A solution of Grubbs' second generation catalyst was diffused into a pre-formed gel, whereupon alkene metathesis occurred, and the network was covalently captured as a robust, thermally stable material. This material could be dried to a solid and then re-swollen in compatible solvents. Importantly, scanning electron microscopy (SEM) showed it consisted of nanoscale fibres (Fig. 5). When other non-polymerisable low molecular weight gelators were also present, self-sorting of LMWG networks could occur. Following addition of Grubbs' catalyst, the non-polymerisable network could be washed out of the polymer to leave a more porous and highly swellable material – effectively nano-imprinted by the non-polymerised gel network.
 | ||
| Fig. 5 Lysine-based dendritic gelator with peripheral alkene groups, which can be polymerised by Grubbs' metathesis. SEM images reproduced from ref. 16b with permission of the American Chemical Society. | ||
Díaz and co-workers used copper-catalysed 1,3-dipolar cycloaddition of alkynes and azides (so-called ‘click’ chemistry) to crosslink and polymerise LMWGs based on alkyne and azide derivatives of the undecylamide of trans-1,2-diaminocyclohexane (Fig. 6), with a variety of azide or alkyne crosslinkers, in addition to direct triazole crosslinking of the gelators.17 They observed increases in thermal and mechanical stability, though it was unclear if these increases were mainly due to inter-fibre cross-linking, intra-fibre polymerisation, or both. Click chemistry has also been used to stabilise gels containing phthalocyanine, enhancing thermal stability and incorporating a photoactive addressable unit into the captured nanostructure.18 The same group also employed photochemical thiol–ene ‘click’ chemistry to tune drug release from gels, with crosslinking lowering the release rate.19
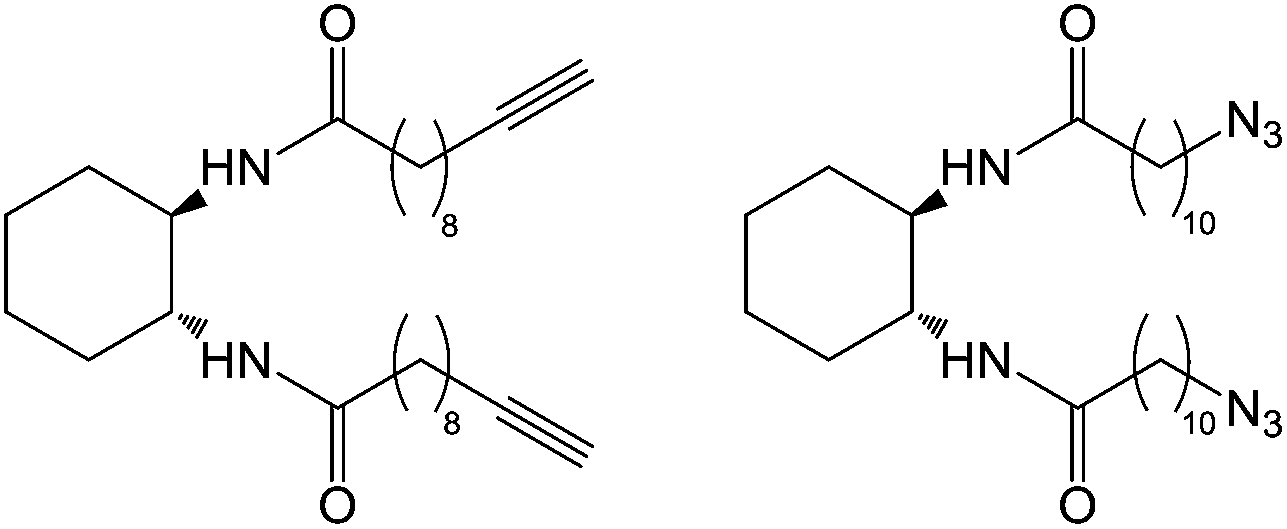 | ||
| Fig. 6 Alkyne and azide derivatives of the undecylamide of trans-1,2-diaminocyclohexane gelators.17 | ||
Steed and co-workers used hydrolysis of the triethoxysilane end groups of a bis(urea) gelator (Fig. 7) for covalent capture.20 Diffusion of (or immersion in) HCl caused hydrolysis of the SiOEt3 groups (to SiOH3), followed by condensation polymerisation, with a transparent gel becoming a white polymer block after several hours. SEM analysis showed that the polymerised network had retained its nanostructure. The polymer also had enhanced mechanical strength, presumably as a result of the embedded network.
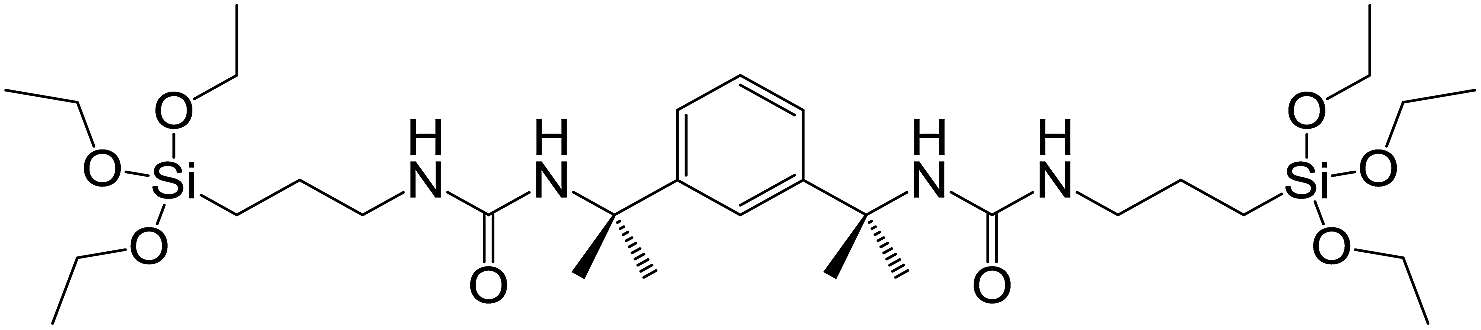 | ||
| Fig. 7 Triethoxysilane bis(urea) gelator; hydrolysis and subsequent polymerisation occurs at SiOEt3 end groups.20 | ||
In summary, molecular-scale information can be translated into nanoscale architectures by self-assembly then captured in more permanent form by polymerisation. It seems to be possible to either crosslink a wider three-dimensional network of gel nanofibres, or capture individual one-dimensional gel fibres themselves. In the former case, crosslinking the network turns these supramolecular gels into crosslinked polymer gels – but with the potential to tune molecular-scale composition much more precisely. In the latter case, this approach allows novel, well-defined one-dimensional nano-structures to be synthesised, the properties of which should reflect the molecular-scale programming. We suggest this approach could yield novel nanomaterials that may ultimately rival (e.g.) carbon nanotubes, but with much more versatility.
Category 2 – capture of LMWG fibres in a polymer matrix
Möller and co-workers were the first to capture a LMWG network inside a polymer matrix by polymerisation of a fluid monomer around the self-assembled LMWG fibres.21 They used two different LMWGs to gelate a methacrylate liquid phase, which was polymerised to a resin. After polymerisation, the LMWG fibres could be washed out to yield nanoporous membranes – the pore size depended on the LMWG and the temperature at which gelation occurred (a lower temperature led to smaller pores, as the gelators aggregated more rapidly into smaller fibres). The resulting membrane could be functionalised by charging the pore walls with anionic sites. Möller and co-workers later went on to use this polymer matrix technique to capture benzamide derived LMWGs.22,23 Embedding, then removing a LMWG can extend the function of standard polymeric materials – a key advantage of this approach.Weiss and co-workers reported similar results when using tetraoctadecylammonium bromide as LMWG in methyl methacrylate or styrene as polymerisable solvent, removing the LMWG network by simply washing with water.24 Nolte and co-workers described gluconamide gelators that could gelate a mixture of methacrylates – again, the LMWG network could be removed by washing to leave well-defined pores, as imaged by transmission electron microscopy (TEM) (Fig. 8). The helical character of the LMWG fibres was not reflected in the pores, most likely due to shrinking of the resin during polymerisation.25 The groups of John, Pina and Steed have similarly investigated the formation of nanoporous membranes of polymerised divinylbenzene and methacrylates.26–28
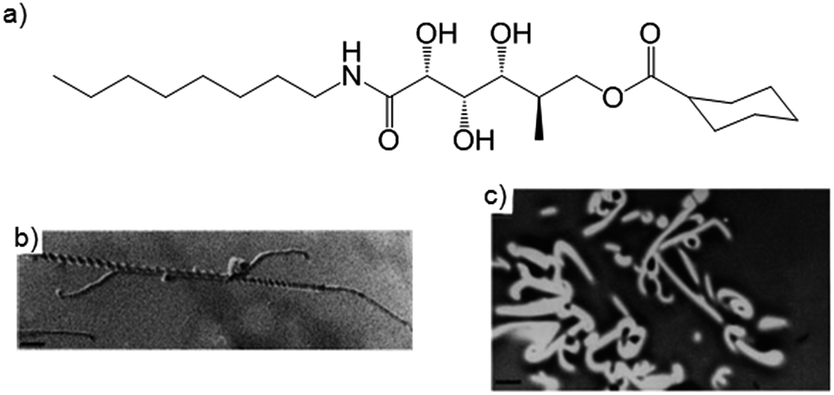 | ||
| Fig. 8 (a) Structure of a gluconamide gelator; (b) TEM image of helical fibre of gluconamide gelators (scale bar = 110 nm); (c) pores left in methacrylate polymer after gelator was removed by washing (scale bar = 1.35 μm). Images reproduced from ref. 25 with permission of the Royal Society of Chemistry. | ||
In eye-catching work, Mésini and co-workers produced polymer resins in which the pores did retain their helicity by using 3,5-bis(5-hexylcarbamoylpentyloxy) benzoic acid (BHPB), which formed helical tape-like nanostructures in polymerisable ethylene glycol diacrylate.29 Washing the polymer with DCM removed BHPB, and subsequent TEM analysis showed the presence of helical pores (Fig. 9). Clearly, the degree to which imprinting in these materials retains structural features from the gelator assemblies will depend on the polymerisation event, the structural rigidity of the self-assembled fibres and the impact of the washing conditions.
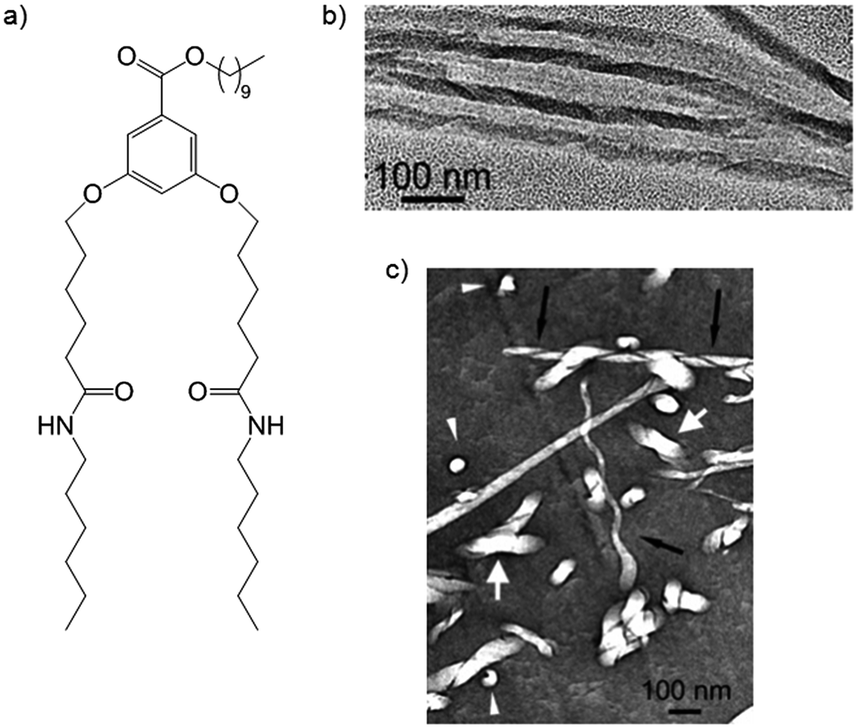 | ||
| Fig. 9 (a) BHPB gelator; (b) TEM image of helical tapes of BHPB gelators; (c) helical pores in polymer matrix after removal of BHPB – black arrows = pore viewed perpendicular to axis, white arrows = pore viewed along axis. Images reproduced from ref. 29 with permission of the American Chemical Society. | ||
In addition to producing nanoporous membranes, there has been interest in simply enhancing the materials properties of polymers by embedding LMWG networks within them. In landmark research, Stupp and co-workers toughened polystyrene through incorporation of dendron rod-coil LMWGs.30 The presence of LMWG nanoribbons directed and aligned the polymer orientation, modifying its materials properties – for example, impact strength was significantly increased (Fig. 10). This was in part due to the increased alignment of the polymer, limiting crack propagation and also due to the LMWG network acting as a nano-skeleton to dissipate impact energy. The presence of the LMWG added a degree of self-healing to the material, as the non-covalent interactions could reform after breaking on stress. Self-healing materials are useful in extending materials life and providing ability to recover after stress – potentially valuable in future transport applications.
 | ||
| Fig. 10 Left: pure polystyrene polymer cylinder; middle: hard polymer material containing both polystyrene and a LMWG network; right: rubbery polymer containing both poly(2-ethyl hexyl methacrylate) and a LMWG network. Image reproduced from ref. 30b with permission from Wiley-VCH. | ||
Our group investigated the gelation of styrene-divinylbenzene (DVB) by a dendritic gelator with terminal alkenes (Fig. 11a), and solvent polymerization.31 Although the gelator could also have polymerised under the same conditions (UV-initiated photopolymerisation), it did not do so – remaining unpolymerised and reactive. Using an osmium tetroxide (OsO4) stain, which reacted selectively with its terminal alkenes, allowed the embedded network to be visualised by TEM (Fig. 11b). This network acted as a “nano-skeleton”, enhancing the materials properties of the polymer – e.g., the modulus increased by an order of magnitude. Enhanced polymeric materials have applications as toughened films and/or coatings.32 The LMWG could be washed out of the polymer to give a nano-imprinted polymer. We also explored fluorescent pyrene-functionalised LMWGs embedded in styrene-DVB. When made as a wafer, the material had two “faces” with different properties (fluorescence and nano-texture), because the gelator migrated to the more hydrophilic surface (glass base of mould) during polymerisation.33 Bi-face materials with embedded photo-active nanostructures at one face have potential applications in device fabrication.
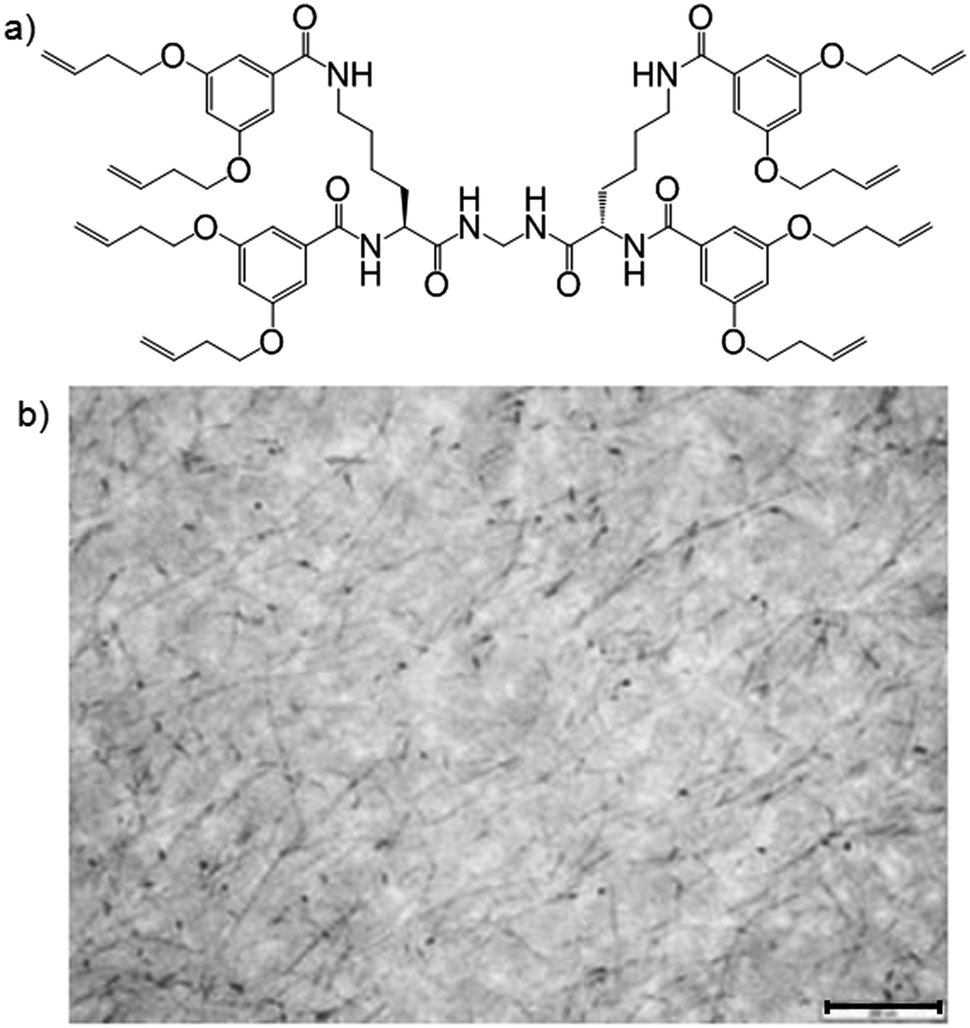 | ||
| Fig. 11 (a) Structure of dendritic gelator used to gelate polymerisable styrene-divinylbenzene mix; (b) TEM image of poly(styrene-divinylbenzene) with embedded dendritic LMWG (dark fibres and spots); the gelator network is visualised through the use of the reactive stain OsO4. Scale bar = 200 nm. Image reproduced from ref. 31 with permission of the Royal Society of Chemistry. | ||
Kim and Chang also recently combined fluorescent LMWG fibres within a highly cross-linked polymer matrix formed from ethylene glycol dimethacrylate.34 Their organogel exhibited a thermochromic gel–sol transition, a property which was maintained within the polymer matrix. It was demonstrated that self-assembly of the LMWG played a vital role in mediating thermochromism within the polymer matrix.
Wilder and co-workers examined the addition of LMWG dibenzylidene-D-sorbitol (DBS) to dental composite consisting of photopolymerisable ethoxylated bisphenol A dimethacrylate (EBPADMA) and zirconium-modified amorphous calcium phosphate (Zr-ACP) to aid remineralisation.35 Polymers used in dental applications have problems including shrinkage, biocompatibility and incomplete polymerisation. Introducing DBS led to a slightly higher rate of conversion (due to increased viscosity, limiting radical–radical termination and favouring free radical propagation). Furthermore, the nanocomposite was stronger and suffered less shrinkage. However, the addition of DBS also reduced the release of calcium and phosphate ions from the ACP. Nonetheless, this approach to dental composites is important,36 demonstrating how self-assembled LMWG networks can have benefits in polymer matrices across a wide range of applications.
Combining categories 1 and 2 – polymerised LMWG fibres within a polymer matrix
There are some cases where the two categories of LMWG–polymer combination described above have been mixed to form materials where a network of polymerised LMWG fibres are captured within a polymer matrix.Chang and co-workers produced such materials using a hetero-bifunctional gelator with both acryl and diacetylene polymerisable groups (Fig. 12a) to gelate hexyl methacrylate (HMA).37 When cured under UV light, simultaneous polymerisation of gelator and monomer took place – polydiacetylene caused the polymerised LMWG to exhibit strong fluorescence (Fig. 12b). The acryl groups of HMA and the LMWG polymerised significantly faster than the diacetylene groups (10 min versus 24 h), so by application of a mask, photopatterning took place and only in those areas exposed to UV light for a long time did polydiacetylene form; as observed by confocal fluorescence microscopy (Fig. 12c). Chang and co-workers have since investigated hetero-bifunctional gelators combined with HMA to produce polymers, and observed that in general there was an increase in both thermal and mechanical stability of poly(HMA) with the inclusion of gelator fibres.38
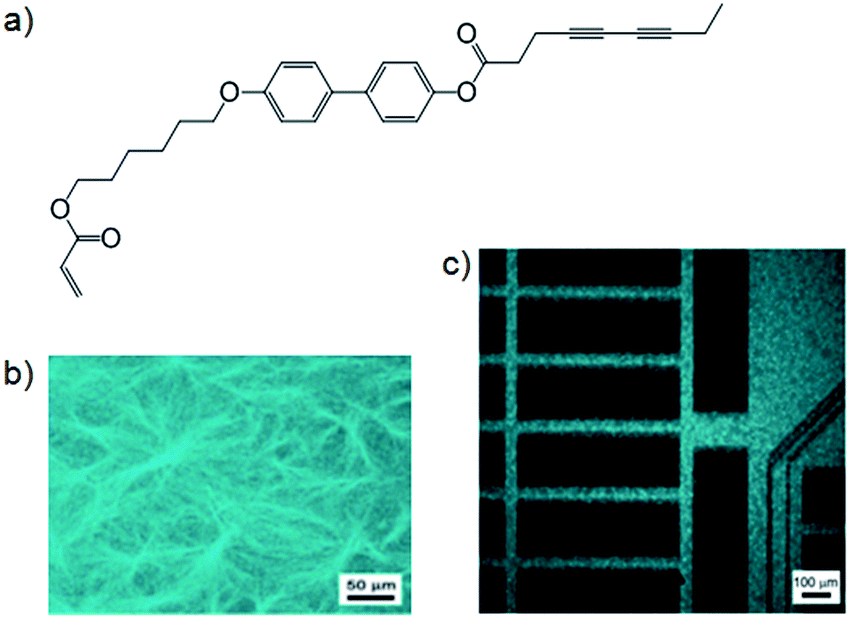 | ||
| Fig. 12 (a) Hetero-bifunctional LMWG with both acryl and diacetylene polymerisable groups; (b) fluorescent polymerised gel fibres within poly(HMA) matrix; (c) photo-patterned polymer film – fluorescing areas contain polydiacetylene. Images reproduced from ref. 37 with permission from Wiley-VCH. | ||
Yang and co-workers used N-octadecyl maleamic acid (ODMA) to gelate a mixture of polymerisable 2-hydroxyethyl methacrylate (HEMA), methacrylic acid (MAA) and poly(ethylene glycol) 200 dimethacrylate (PEG200DM).39 The gelator fibres and monomers were polymerised by UV curing, with the inclusion of L-phenylalanine ethyl ester and BOC-L-phenylalanine as templates. The templates were removed to give molecularly imprinted polymers (MIPs) which showed a high affinity for adsorption of L-phenylalanine over D-phenylalanine (Fig. 13). Such materials may have applications as a stationary phase in chromatography. The ODMA fibres also reinforced the rigidity of the polymer matrix, an approach which may offer some advantages to MIP technology.40
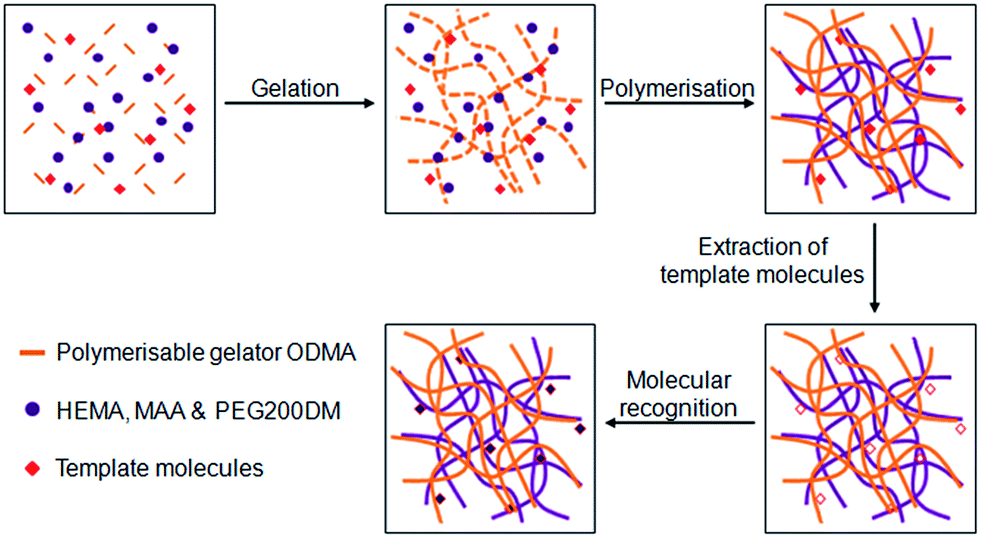 | ||
| Fig. 13 Illustration of the preparation of molecularly imprinted polymer/polymerised LMWG materials, and the process of molecular recognition. Image adapted from ref. 39 with permission from Wiley-VCH. | ||
Korley and co-workers prepared a polymer nanocomposite by gelation of an ethylene oxide–epichlorohydrin copolymer (EO–EPI) polymer solution with polymerisable diacetylene- and cholesterol-containing LMWGs.41 This was compression moulded to form a film, and curing under UV light polymerised the diacetylenes. The resulting material had significantly improved tensile storage modulus (elasticity) compared with a polymer film without the LMWG, or one containing cholesterol as a filler, once again demonstrating how incorporation of nanofibres into polymers can enhance their materials properties.
In recent work, Xu, Epstein and co-workers created fascinating oscillatory chemical gels in which a reaction can be controlled by the polymerised gel network – such systems are of interest as motile, self-switching materials.42 They polymerised self-assembled LMWG fibres with peripheral acrylamide units, in the presence of an acrylamide monomer and crosslinker, leading to a polymer hydrogel, with embedded fibrillar nanostructures. The presence of a small amount of an acrylamide-functionalised ruthenium bipyridine catalyst meant they could use these materials to direct/catalyse the famous oscillating Belousov–Zhabotinsky chemical reaction. The self-assembled nanostructures played the dominant role in controlling the periodicity of the oscillating reaction.
Category 3 – addition of a non-gelling polymer in solution to supramolecular gels
In recent years there has been increasing interest in the effects of adding a solution-phase polymer to a LMWG gel – effects may arise due to direct interaction of the LMWG fibres with the polymer (e.g., adsorption), or indirect, (e.g., polymer changing solution viscosity). Hanabusa and co-workers were, in 1999, the first to investigate adding polymer solutions to a LMWG organogel.43 They found that adding either poly(N-vinylpyrrolidone) or poly(ethylene glycol) to a 1-propanol gel of a L-valine-containing benzenedicarbonyl derivative enhanced gel strength. However, adding poly(styrene) to a similar gel in cyclohexane offered little enhancement. The authors did not comment in this early study as to the cause of this difference, or how polymer addition may increase gel strength.There were surprisingly few developments over the next ten years; in fact, all of the notable work comes from Liu and co-workers. They initially investigated a new method of forming gels from self-assembling nanostructures of lanosta-8,24-dien-3β-ol:24,25-dihydrolanosterol (L/DHL) with diisooctyl phthalate (DIOP).44,45 When L/DHL (10 wt%) was dissolved in DIOP by heating, then allowed to cool, a viscous, opaque paste was formed, shown by SEM to consist of needle-like fibres (Fig. 14a). However, on adding a very small amount (0.0006 wt%) of ethylene–vinyl acetate copolymer (EVACP), a transparent gel with interconnecting branched fibres was obtained (Fig. 14b). The dramatic change in nanostructure was reasoned to be caused by adsorption of the polymer onto the growing fibre tip, leading to what the authors termed “crystallographic mismatch branching”, in which the EVACP disrupted the structural match between the growing fibre tip and the new layers adding to it, causing branching (Fig. 14c) – a remarkable effect for such a small amount of additive.
 | ||
| Fig. 14 SEM images of L/DHL/DIOP systems: (a) separate needle-like fibres of L/DHL in DIOP; (b) addition of EVACP generated a network of interconnected branched fibres; (c) illustration of crystallographic mismatch branching. Image reproduced from ref. 44 with permission from Wiley-VCH. | ||
Liu and co-workers later used the adsorption of polymers onto growing fibres to influence the nanoscale and mechanical properties of gels formed from N-lauroyl-L-glutamic acid di-n-butylamide, which formed spherulite fibre networks in propylene glycol,46 or mixed fibre/spherulite fibre networks in benzyl benzoate.47 The addition of poly(methyl methacrylate co-methacrylic acid) (PMMMA) and EVACP slowed nucleation of the gelator. In addition to causing more branching in the gelator fibres, addition of EVACP also inhibited fibre formation of N-lauroyl-L-glutamic acid di-n-butylamide in propylene glycol, with only spherulite nanostructures being formed.
Cui, Shen and Wan studied gelator 4-(4′-ethoxyphenyl) phenyl-β-O-D-glucoside in water–1,4-dioxane, observing that the gel gradually collapsed to form crystals due to fibril aggregation.48 Such metastability and ageing effects are a relatively common feature of self-assembled dynamic LMWG gels.49 On adding poly(2-hydroxyethyl methacrylate) (PHEMA), the gel was stabilised – it was proposed that PHEMA adsorbed onto the growing fibres causing branching and preventing lateral fibril aggregation and gel collapse.
Nandi and co-workers obtained similar results by adding the biopolymer chitosan to a folic acid gel, and suggested hydrogen bond interactions between chitosan and folic acid occur.50 Increased branching once again enhanced mechanical strength. The gels could also adsorb dyes and heavy metal ions from water, suggesting such gels may have water purification applications – an area of intense activity in gel technology.51
Adams and co-workers proposed that addition of polymers can have viscosity-induced effects on the properties of LMWG gels. They added dextran biopolymers to pH-dependant naphthalene-dipeptide hydrogels.52 On changing the molecular weight or wt% of the dextrans, the viscosity prior to gelation could be altered – increased viscosity lengthened gelation time and decreased mechanical gel strength. Clearly these observations are different to those of Liu and co-workers, which might suggest that LMWG–polymer adsorptive interactions are different in the different systems. The gels with dextran were observed by TEM to have thinner fibres in a less well-defined network compared with gels without dextran (Fig. 15). This was attributed to a reduction in diffusion of the LMWG due to the increased viscosity of the solution; slowing self-assembly and limiting lateral interactions. Adams and co-workers later studied the effects of dextrans and other polymers – poly(ethylene glycol), poly(vinyl pyrrolidone), poly(ethylene oxide), and poly(acrylic acid) – on the rheology of LMWG gels.53 Again, they found mechanical strength decreased – here, the LMWGs formed spherulites, which on addition of the polymers became smaller and less interconnected – lowering gel strength. The identity of the polymer additive also played a significant role in controlling the final gel rheology. They suggested that for some of the polymers there might be adsorption onto gel fibres, as well as changes in viscosity.
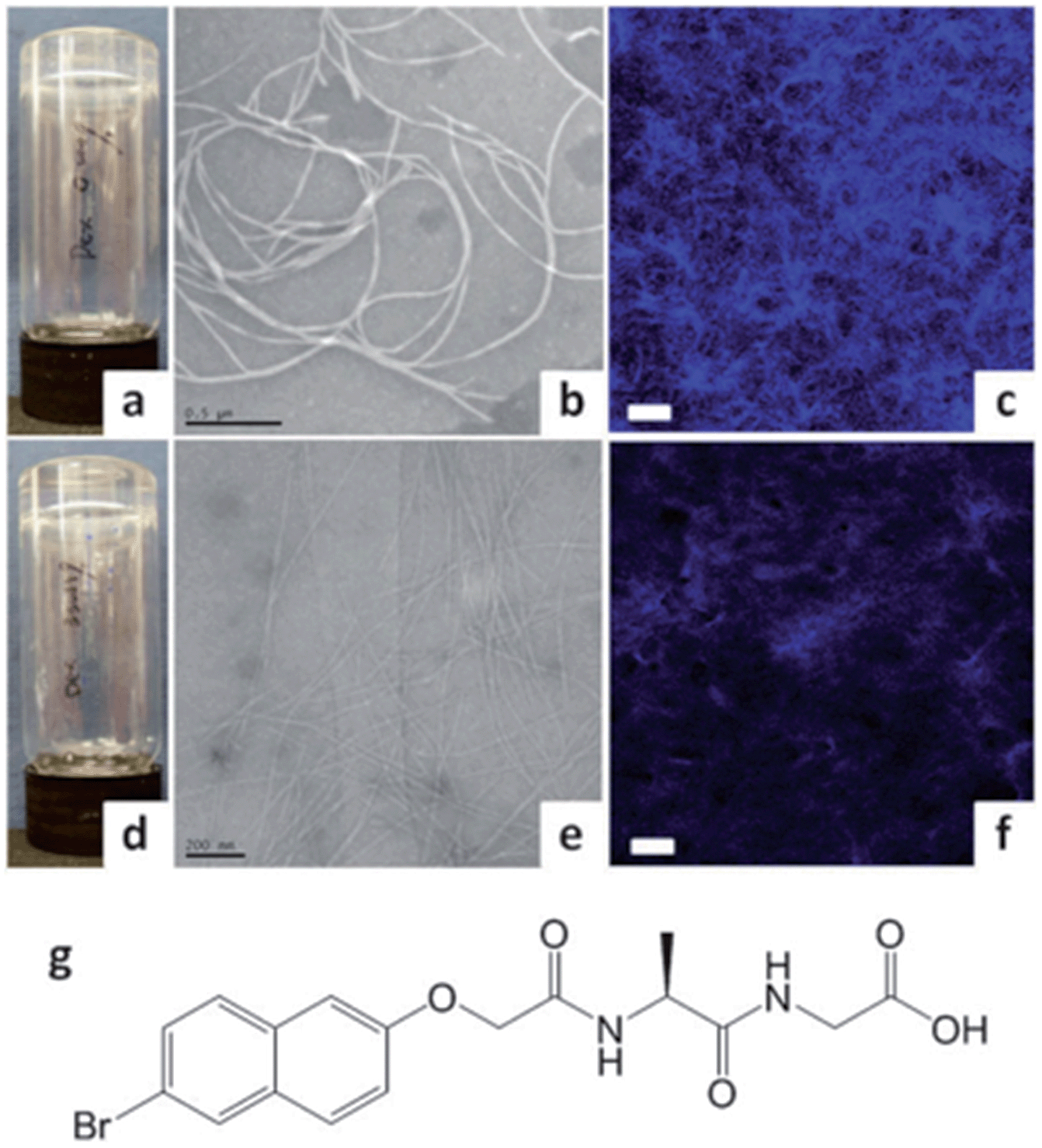 | ||
| Fig. 15 (a) Image of hydrogel of naphthalene-dipeptide; (b) TEM and (c) confocal micrograph of gel (with Nile Blue stain); (d) hydrogel of naphthalene-dipeptide with added dextran; (e) TEM and (f) confocal microscopy showing that gel fibres are significantly thinner; (g) structure of gelator. Image reproduced from ref. 52 with permission from the Royal Society of Chemistry. | ||
Building on these observations, Yang and co-workers reported the addition of hyaluronic acid (HA) polymer to a LMWG hydrogel based on succinated taxol.54 Interestingly, they found that in the presence of the polymer the hydrogel fibres were more likely to bundle, slightly enhancing the mechanical properties of the gel. However, more interestingly, they noted that the addition of more than 30% HA appeared to boost the anticancer activity of the nanofibres – they suggested this was a result of HA assisting in tumour targeting because it is a ligand for a protein overexpressed in the cancer cells. This nicely illustrates how polymeric additives may not only impact on rheology and nanostructure, but may also introduce their own functionality to such materials.
In a recent biomimetic study, Ulijn and co-workers reported that peptide gelators could self-assemble in the presence of biological polymers – specifically, clusters of proteins such as bovine serum albumin or β-lactoglobulin.55 They noted that the presence of the LMWG encouraged clustering of the proteins at much lower concentrations than normally observed. Furthermore, the presence of these protein clusters impacted on the self-assembly of the peptide gelator, changing the mechanical and morphological properties of the materials formed. Cooperative interactions between the systems gave rise to these effects, and indicate how LMWG materials can have relevance in the evolution and control of biological systems.
Overall, it is clear that the presence of polymers in the solution phase can impact on LMWG assembly either by interactions with the gel fibres or through viscosity effects. Importantly, this is a cheap and simple method of modifying LMWG nanoscale morphology and rheology, with small amounts of polymeric additive having relatively large effects. Given polymer formulation is well established in materials engineering, we suggest this approach has considerable commercial significance.
Category 4 – directed interactions between LWMGs and polymers
It is possible to design systems in which directed and controlled supramolecular interactions between LMWGs and polymers can occur in order to modify the overall gelation event in a more precise manner. We define this as category four, and although there is some overlap with category three, the adsorption interactions in that section are relatively general, whereas those described here have more complementarity.Exemplifying how a degree of specificity can be introduced to polymer-LMWG interactions, McNeil and co-workers studied the adsorption of poly(acrylic acid) (PAA) onto fibres of pyridine-based gelators (Fig. 16).56 It was proposed that PAA would be adsorbed onto the side of the fibres through acid–base interactions between the basic pyridine of the gelator and the carboxylic acid of PAA. Thinner fibres were observed by TEM due to the control over growth rate. It was proposed that this would lead to longer fibres, more fibres, or both, increasing entanglement in the gel and enhancing gel strength.
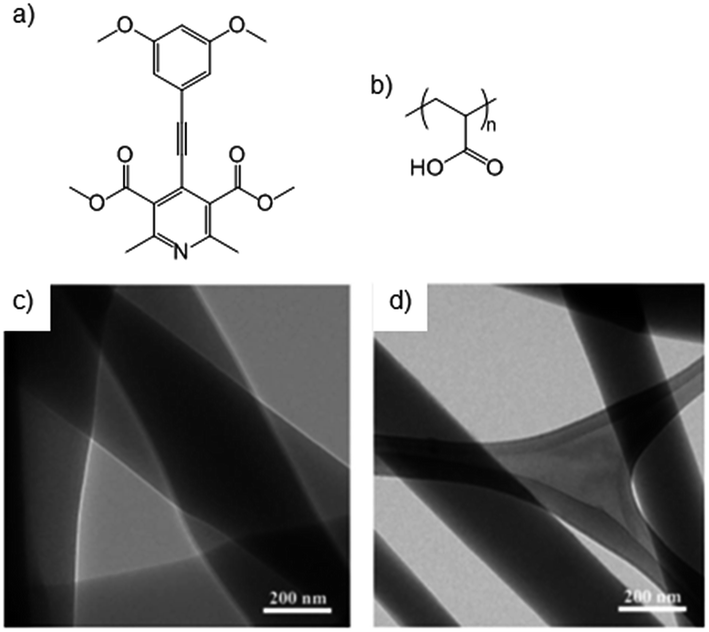 | ||
| Fig. 16 Structures of (a) pyridine-based gelator, (b) structure of poly(acrylic acid) (PAA), and TEM images of gel (c) without and (d) thinner fibres with PAA. Images reproduced from ref. 56 with permission of the Royal Society of Chemistry. | ||
Reinhoudt and co-workers studied a cholesterol–saccharide low-molecular-weight organogelator (Fig. 17b). It could not, however, gelate water–DMSO, forming instead colloidal particles/vesicles.57 However, on addition of boronic-acid appended poly(L-lysine) (Fig. 17a) a gel formed as an extended network of vesicles (Fig. 17b). The polymer partially coats the vesicles through specific reversible covalent boronic acid–glucopyranosyl interactions, causing vesicle aggregation. As such, directed interactions play a key role in mediating gelation. The physical properties of the gel could be controlled by varying the amount of polymer, and hence the level of vesicle crosslinking (Fig. 17c). Vesicle derived gels are an interesting class of gels in their own right.58
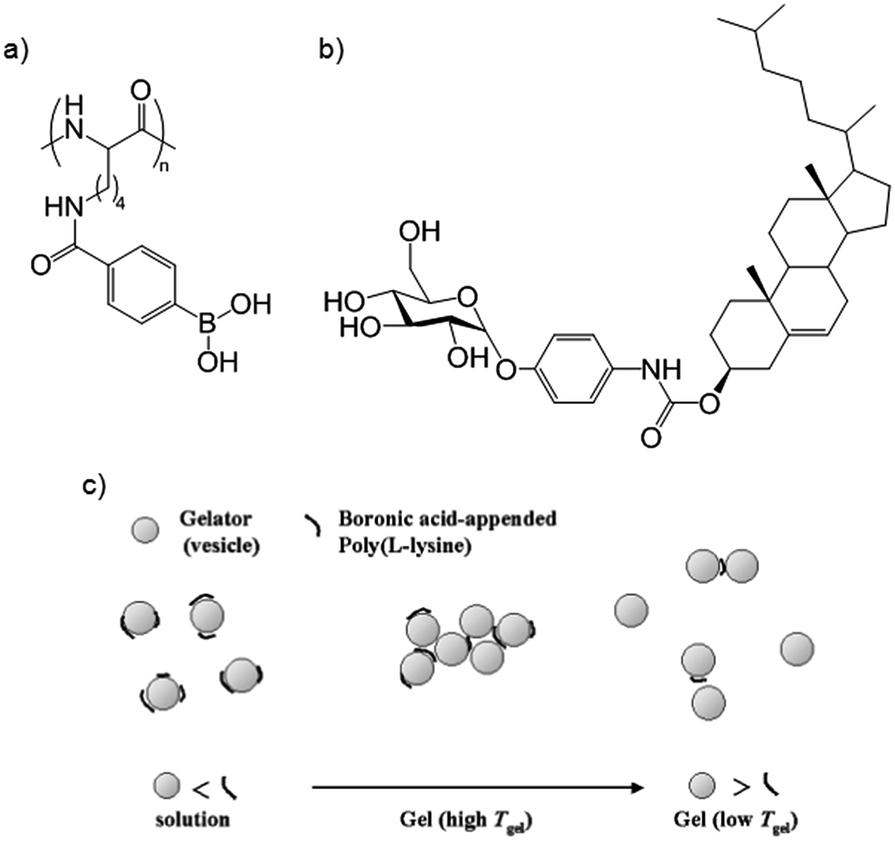 | ||
| Fig. 17 (a) Boronic acid-appended poly(L-lysine) and (b) cholesterol–saccharide LMWG produce a gel consisting of polymer “cross-linked” vesicles; (c) varying amounts of polymer control cross-linking and physical properties. Image reproduced from ref. 57 with permission from Royal Society of Chemistry. | ||
Polymers can be used as supramolecular templates for the formation of highly-ordered one-dimensional nanostructures by complexation of LMWGs. Shinkai and co-workers combined nucleobase-appended gelators with thymine as the nucleotide and complementary polynucleotides to form gels.59 A cholesterol-based nucleobase gelator (Fig. 18b), was mixed with the complementary polynucleotide (Fig. 18a) in water/n-butanol forming a non-gelling complex. When the solvent was removed and replaced with just n-butanol, a gel formed on heating – a gel was not formed when using just the gelator in this way. The authors concluded that the polynucleotide templates the LMWGs into a helical nanostructure through complementary hydrogen bonds – the polymer acts as a helical “pillar”, with the LMWG spiralling around it (Fig. 18c).
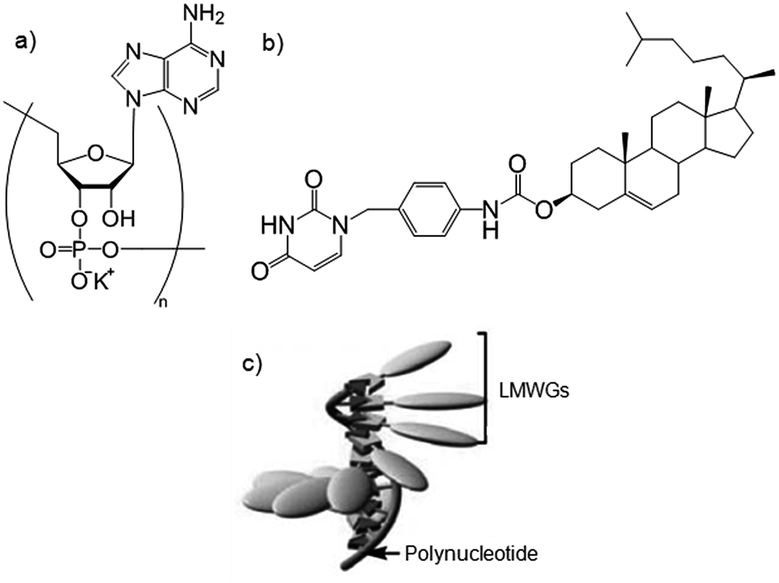 | ||
| Fig. 18 Exemplar (a) polynucleotide and (b) nucleobase-appended gelator, used to form templated polymer–LMWG complexes illustrated in (c). Images reproduced from ref. 59 with permission from the Royal Society of Chemistry. | ||
Wang and co-workers used dendritic gelators complexed with polyelectrolytes. Addition of the polymer template led to a lower minimum gelation concentration and more rapid gelation kinetics, thought to be due to improved molecular recognition brought about by pre-organising the LMWG molecules.60
In elegant work, Rowan and co-workers studied a supramolecular hydrogel produced through helical assembly of guanosine-derived gelators with sodium ions in a G-quartet assembly motif (Fig. 19). Non-gelling guanosine-derived copolymers were then added and incorporated into the helical assemblies through complementary hydrogen bonding, forming supramolecular polymeric crosslinks.61 At suitable polymer concentrations, gel strength could be dramatically improved (up to 40 times) by the effective crosslinking from the polymers.
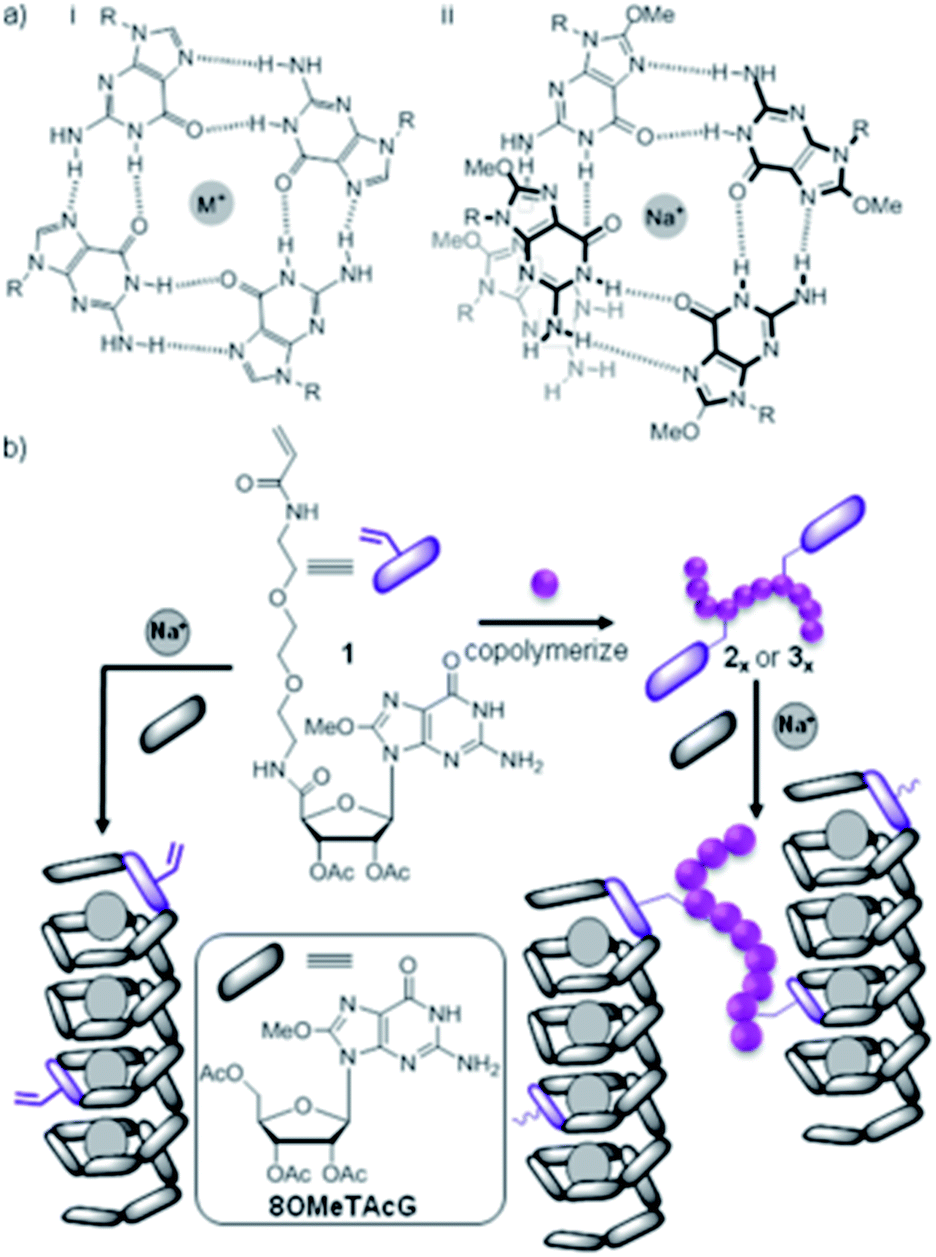 | ||
| Fig. 19 (a) Guanosine derived LMWGs assemble with Na+ as a G quartet (i) or helix (ii); (b) polymerisable guanosine derivatives and guanosine-containing polymers can be incorporated, with the latter cross-linking helices. Image reproduced from ref. 61 with permission from the American Chemical Society. | ||
These examples demonstrate how well-designed polymers can precisely interact with the recognition motifs within a LMWG, and hence impact upon the properties of the overall gel. All of these above systems bear similarities to widely-investigated two-component LMWG systems, in which two low-molecular weight components interact with one another to form a gel.62
Category 5 – hybrid gels combining low-molecular-weight and polymer gelators
A simple and potentially powerful approach to combining LMWGs with polymers is to mix them with polymers which are, in their own right, gelators (i.e. polymer gelators, PGs). LMWGs and PGs have different advantages and disadvantages, and combining the two offers the potential to significantly expand the scope of what can be achieved with gel-phase materials. We refer to these as hybrid gels – note that some authors use this term to refer to combinations of LMWGs with non-gelling polymers, but here it is used exclusively to refer to LMWG–PG combinations, in which each component is independently capable of forming a gel. Hybrid gels can be considered as a type of self-sorting multi-component gel, as they are composed of two independent gel networks.62 Given the great potential, it is surprising that hybrid gels have only recently been reported. There are few examples in the literature and we consider this area significantly under-exploited. For this review, we loosely divide hybrid gels into three categories: semi-hybrid gels, hybrid organogels and hybrid hydrogels.Semi-hybrid gels
There are a few examples in the literature where LMWGs have been combined with polymers that have the potential to be used as PGs, but are not actually being used as such. Instead, the polymers are used to form non-gelling support networks to improve the materials properties of the gel formed by the LMWGs. We term these materials semi-hybrid gels – clearly they are related to category 3 systems described above.Feng and co-workers demonstrated that the polysaccharide sodium alginate (which hydrogelates through crosslinking on addition of divalent ions or on protonation), could be added to hydrogels of 1,4-bi(phenylalanine-diglycol)-benzene (PDB) (Fig. 20).63 The polysaccharide formed a semi-interpenetrating network partially disrupting LMWG interactions – the amide NHs of PDB interacted with carboxylates of sodium alginate. The gels with sodium alginate had better mechanical and water retention properties; it was also possible to achieve controlled release of dyes from the polymer–LMWG gel, as the sodium alginate introduced attractive or repulsive electrostatic forces, meaning it could retain or release dyes respectively.
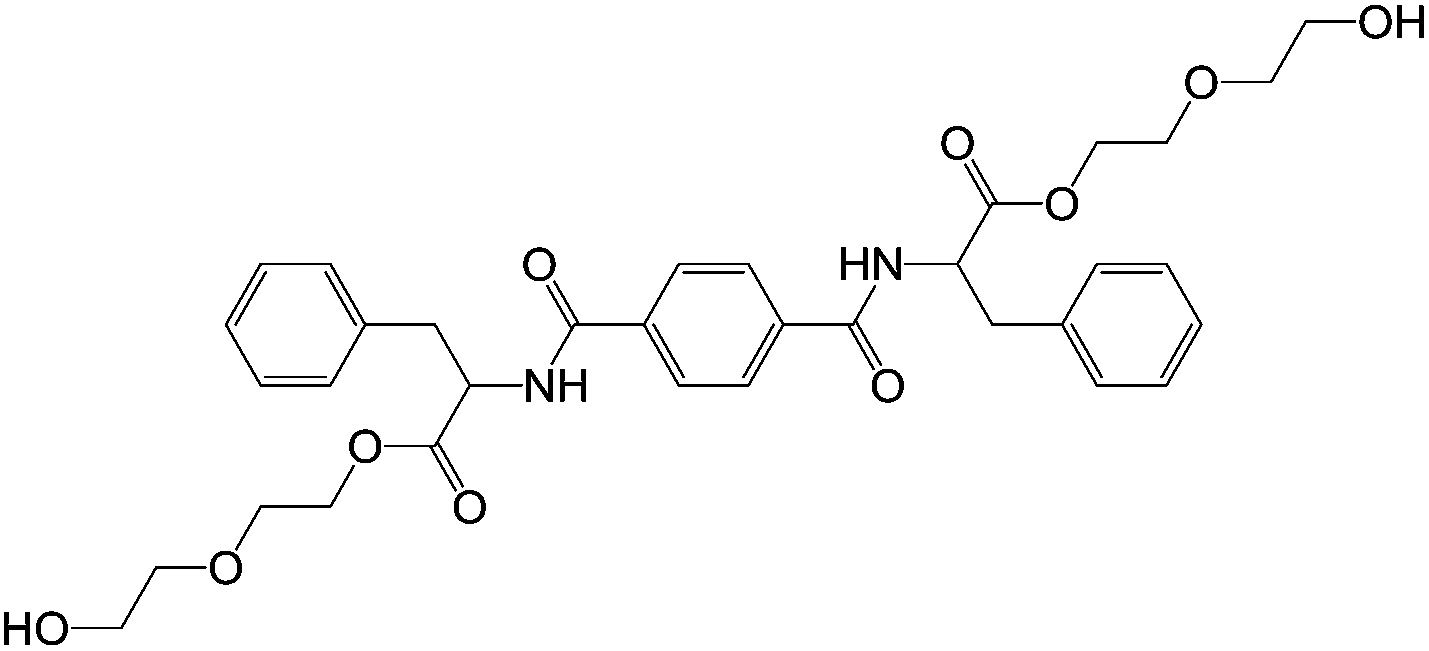 | ||
| Fig. 20 Structure of 1,4-bis(phenylalanine-diglycol)-benzene (PDB).63 | ||
Feng and co-workers also combined PDB with a non-gelling sodium hyaluronate (HA) polymer network.64 This glycosaminoglycan can form a gel by covalent crosslinking with hydrazides, or through radical polymerisation – although this was not done during this research. Addition of HA gave the dried LMWG xerogels significantly better swelling properties. The swollen PDB/HA gels had enlarged pores, providing enough room for cell migration and proliferation, enabling easier 3D growth of cells (Fig. 21). This may be useful for tissue engineering, disease modelling and drug release.
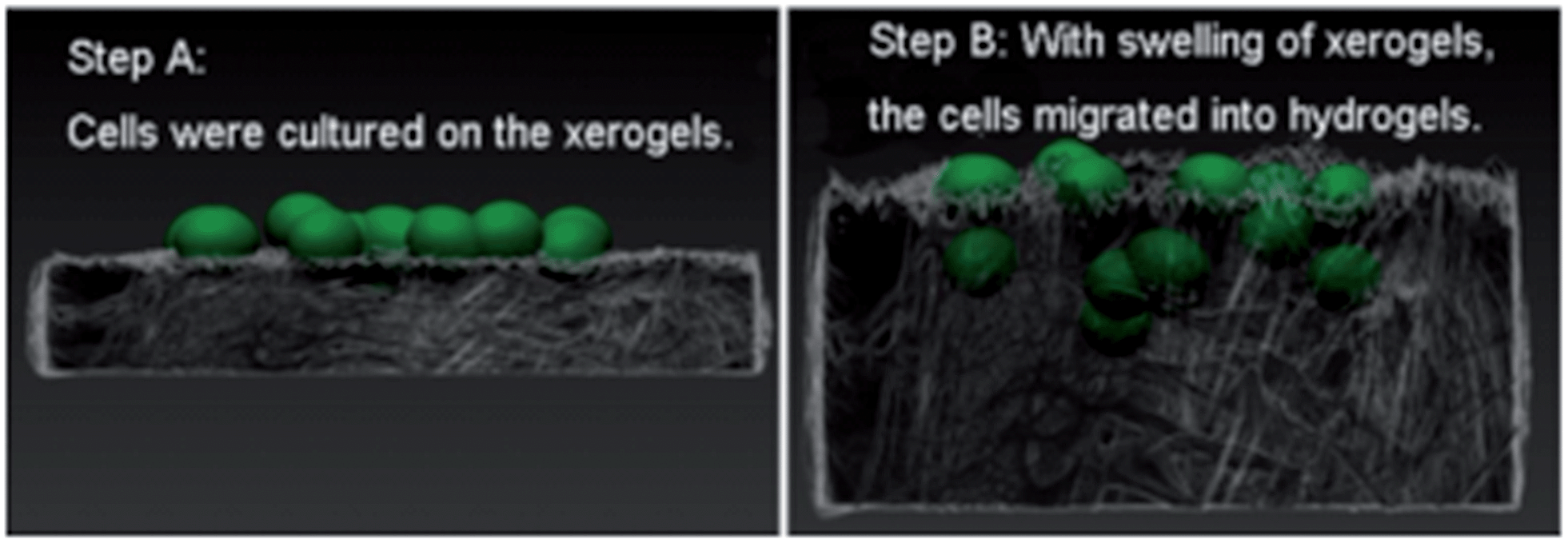 | ||
| Fig. 21 Schematic illustration of 3D cell culture strategy using PDB/HA hydrogels: cells are cultured on a xerogel (step A); and swelling of the gel, facilitated by HA then allows the cells to migrate into the bulk of the gel forming a 3D culture (step B). Image reproduced from ref. 64 with permission from American Chemical Society. | ||
Yu and co-workers also studied the assembly of a gelator within a pre-formed, non-gelling fibrillar network – in this case, a two-component oligopeptide hydrogelator assembling within a network of chitosan, alginate and chondroitin, designed to somewhat mimic the composition of soft tissue.65 The authors found there were peptide–polysaccharide interactions within the resulting LMWG–polymer combination which caused a change in the oligopeptide gel morphology – notably larger pore size.
Hybrid organogels
Guenet and co-workers combined an oligo(p-phenylenevinylene) LMWG (OPV16) (Fig. 22) with isotactic, syndiotactic or atactic polystyrene in aromatic organic solvents.66 OPV16 was highly compatible with non-gelling atactic polystyrene, with the LMWG fibres remaining unaffected. In contrast, with stereoregular iso- or syndio-tactic polystyrenes, which can form their own PG networks, hybrid organogels resulted, as imaged by atomic force microscopy (AFM). The authors postulated (but did not investigate) that the morphology of the LMWG network could be affected by changing the concentration of PG, because this PG network formed first. In hybrid gels, the order of the two individual gel-forming events can be of paramount importance, and having a degree of orthogonal control over these events can be very useful.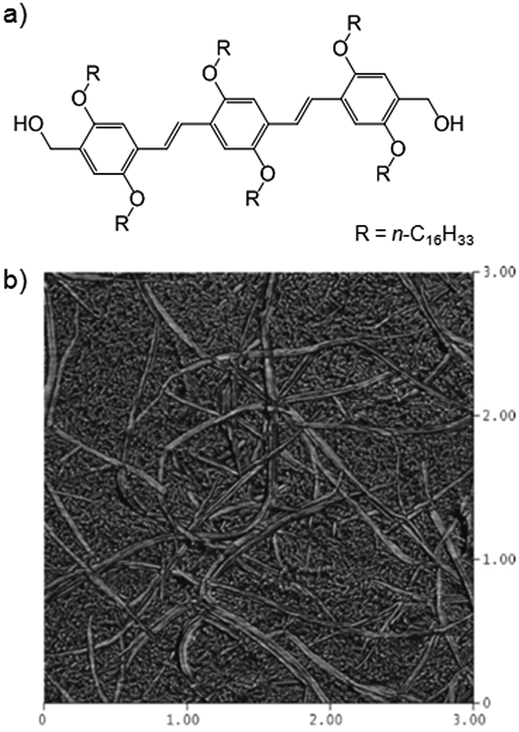 | ||
| Fig. 22 (a) Structure of oligo(p-phenylenevinylene) LMWG OPV16; (b) AFM image of hybrid organogel of OPV16 and isotactic polystyrene – two distinct sizes of fibres can be observed, larger LMWG fibres and smaller PG fibres. Image reproduced from ref. 66 with permission from American Chemical Society. | ||
Hybrid hydrogels
Hybrid hydrogels are particularly appealing as smart multifunctional materials for biomedical applications, such as tissue engineering or drug delivery, with each gelator playing its own unique role. The use of a PG to reinforce a mechanically weak LMWG network could lead to an increase in strength, and the presence of a LMWG can provide directed interactions with tissue or drugs and extra control over aspects such as growth factors or drug release rates.Yang and co-workers were the first to report hybrid hydrogels. Their materials consisted of a two-component Fmoc-peptide-based LMWG (H–lysine(Fmoc)–OH with one of three Fmoc-peptides) (Fig. 23) combined with agarose as the PG.67 Agarose provided the hybrid gels with enhanced strength when compared with either individual LMWG or PG gels. These hybrid hydrogels could incorporate additional components. Congo red was used as a model drug, with emission spectroscopy showing the presence of interactions between the dye and the LMWG nanofibres. The rate of release of Congo red could be varied depending on the LMWG selected. In follow-up work, Yang and co-workers demonstrated that a similar hybrid hydrogel could extract methyl violet from aqueous solutions more efficiently that either of its individual constituent gels,68 demonstrating the potential of such gels for environmental use.
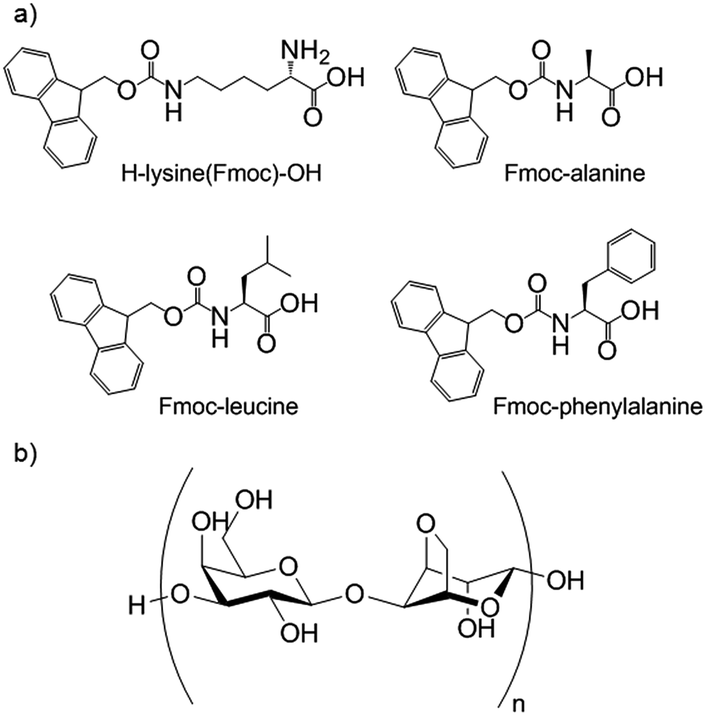 | ||
| Fig. 23 (a) The multi-component peptide-based LMWG hydrogelator used by Yang and co-workers, consisting of H–lysine(Fmoc)–OH in combination with one of three Fmoc-protected amino acids; (b) structure of PG agarose.67 | ||
Qi and co-workers investigated drug release from a hybrid hydrogel composed of Fmoc-diphenylalanine (Fmoc-FF) as LMWG and the polysaccharide konjac glucomannan (KGM) as PG.69 Again, the hybrid hydrogel was found to have a higher mechanical strength due to the nanostructure, in which the Fmoc-FF fibres were interpenetrated and interwoven with KGM polymer chains – the authors made an analogy to the structure of reinforced concrete. The Fmoc-FF fibres were also observed by SEM to be thinner than in the non-hybrid gel. This was reasoned to be a result of KGM increasing the viscosity of the solution and decreasing the rate of diffusion of the LMWG – similar arguments about the effects of polymer additives were made for category 3 materials (see above). Docetaxel was used as a drug for in vitro release studies – an increase in KGM concentration led to a decrease in drug release rate, attributed to a more stable gel. The rate could be increased by introducing β-mannanase – an enzyme able to degrade KGM. This demonstrates how responsivity can be built into hybrid hydrogels in order to achieve controlled/triggered drug release.
Feng and co-workers combined the PDB gelator (Fig. 20) with a calcium-alginate PG. PDB was used as LMWG in a solution of sodium alginate, followed by diffusion of a CaCl2 solution into the gel, causing the PG network to form.70 The fibres of PDB formed in the presence of the PG were observed by SEM to be thinner due to the increased viscosity. The presence of the PG network enhanced the mechanical stability, and increasing the concentration of calcium ions further improved this. Cells seeded onto the hybrid hydrogel showed better viability, adhesion and spreading than on the supramolecular gel alone – reasoned to result from the hybrid hydrogels being stiffer. Changing the concentration of PDB, sodium alginate or CaCl2 could tune the hybrid hydrogels to particular cell types. Control of hydrogel rheology is a powerful approach for tissue engineering, well-known for polymer gels.71 The potential to use a hybrid approach to also introduce tunable and responsive LMWG nanostructures into PG materials endows them with considerable potential.
Our group has investigated hybrid hydrogels combining the pH-responsive novel LMWG 1,3:2,4-dibenzylidene-D-sorbitol-p,p′-dicarboxylic acid (DBS-CO2H) and thermally responsive PG agarose.72 We chose these gels based on their orthogonal methods of preparation – slow pH change from basic to acidic for DBS-CO2H, and thermal cycling for agarose. This allowed us to address each gelator individually. Using circular dichroism we observed that the PG appeared to slightly slow down the initial nucleation and assembly of nanofibres but that similar nanostructures eventually formed in each case. We also demonstrated, by visual (universal indicator and tube inversion) and NMR methods that DBS-CO2H retained its pH-responsiveness within the hybrid hydrogel, and could be assembled or disassembled (by diffusion of acid or base respectively) in the presence of agarose without affecting the overall integrity of the gel (Fig. 24). This was the first example of a responsive yet robust hybrid gel, in which the LMWG network could be reversibly switched on and off using a simple trigger, in the presence of a robust PG. We believe these types of materials have potential to be used in high-tech applications such as controlled release and switchable self-healing systems.
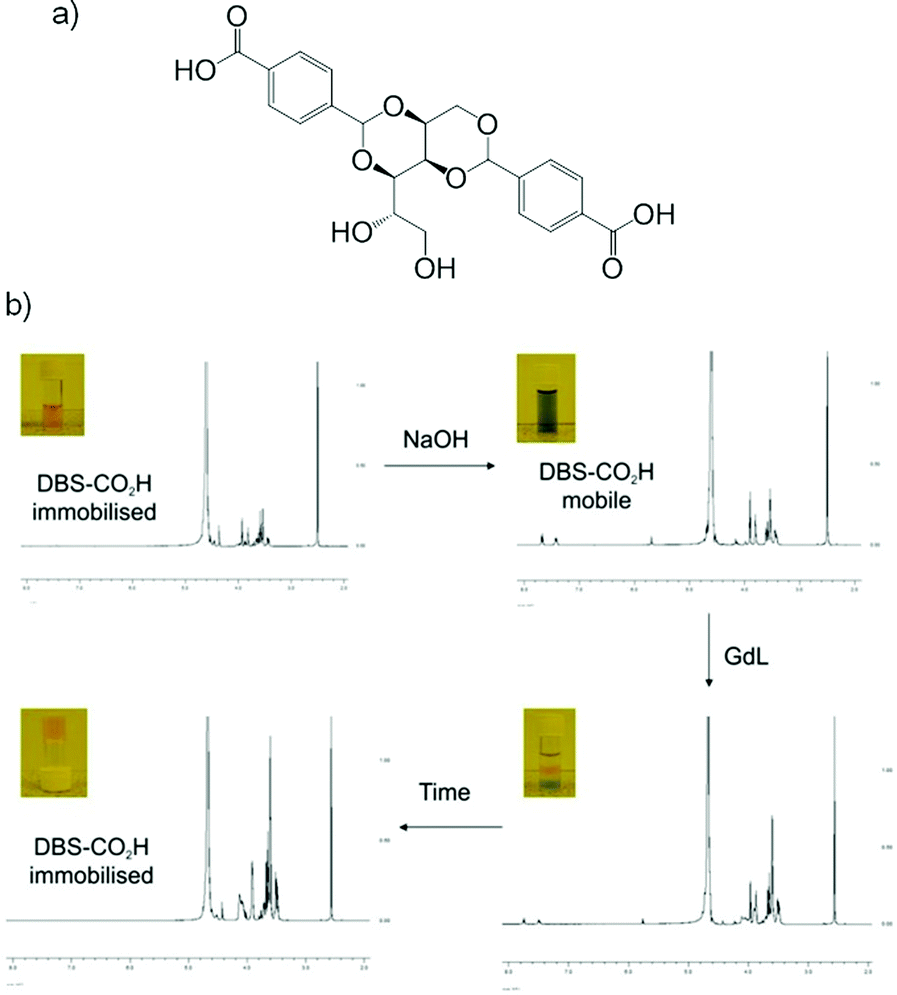 | ||
| Fig. 24 (a) Structure of LMWG DBS-CO2H; (b) progress of switchable gel: addition and diffusion of NaOH increases pH as it diffuses into the network and “switches off” the DBS-CO2H network; addition, diffusion and hydrolysis of GdL returns system to acidic pH and the DBS-CO2H network is “switched on”. Image reproduced from ref. 72 with permission of Royal Society of Chemistry. | ||
The above examples all required either a heat–cool cycle67,68,70,72 or a small volume of organic solvent69 for preparation of one or both of the gelator networks. These processes are not really suitable for encapsulating cells or biomacromolecules – a key issue if these materials are to be used in biomedical applications. Investigating this, Yang and co-workers reported an example of mild hydrogelation (enzyme or reductant triggered) using a mixture of naphthalene-peptide LMWGs (Fig. 25) and sodium alginate, followed by soaking in a CaCl2 solution to form calcium alginate PG, and hence a hybrid hydrogel.73 These hybrid gels had increased stabilities and mechanical strengths. Emission spectroscopy suggested that interactions between LMWG and alginate networks contributed towards the stability, with the authors identifying hydrogen bonding between networks. Phosphatase enzymes could be immobilized in some of the hybrids, and used at least 20 times without significant decrease in activity. In contrast, phosphatases immobilized in calcium alginate alone showed a marked decrease in activity after just one use, probably due to leaching. Whilst being an elegant approach, the relative complexity of producing these systems may limit their practical applications.
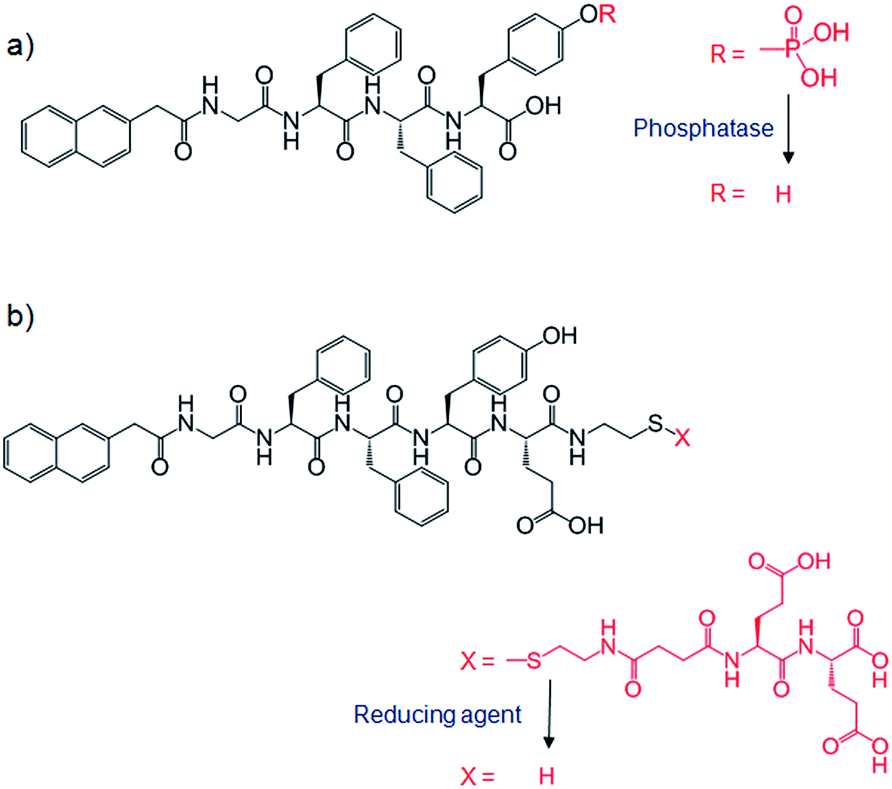 | ||
| Fig. 25 Structures for hydrogelation of naphthalene-peptide LMWGs by (a) phosphatase enzyme and (b) reducing agent (tris(2-carboxyethyl)phosphine hydrochloride). | ||
All of the above examples of hybrid hydrogels use naturally derived PGs, which are readily available, low-cost and biocompatible. However, in the search for hybrid gels which can form under simple ambient conditions, we recently reported the first example of a hybrid hydrogel which uses a synthetic PG, and this endows our hybrids with significant advantages. We chose the photo-inducible poly(ethylene glycol) dimethacrylate (PEGDM) and combined it with our LMWG, DBS-CO2H.74 Photochemical activation is a simple and practical method for gel formation, and has a number of specific advantages. When examined by SEM, we observed a mix of PEGDM and DBS-CO2H nanostructures; a result supported by CD spectroscopy, which showed the presence of chiral self-assembled DBS-CO2H nanostructures. These hybrid gels exhibited controlled release of dyes, with both networks able to play active roles, either due to network density (PG) or specific interactions with the dyes (LMWG). Such hybrid hydrogels may be able to achieve multi-kinetic release of different encapsulated drugs.
The most interesting aspect of these materials was that different regions could easily be spatially patterned into them by using a mask during UV irradiation, to produce what we have termed a multi-domain gel. These domains have either one network (LMWG fibres in a PG solution) or two networks (both LMWG and PG gels), leading to differences in materials behaviour and diffusion between domains (Fig. 26). The ability to photo-pattern, in principle down to nanoscale dimensions, provides this approach with great potential for making multi-domain devices. These types of hybrid materials have great potential for use in (i) drug delivery (regions with different release kinetics can be written into the gel), (ii) microfluidics (patterns to control diffusion can be designed), or (iii) tissue engineering (differential cell growth could be encouraged in different domains).
 | ||
| Fig. 26 (A) Patterned multi-domain gel consisting of non-hybrid region (more translucent) and hybrid dual-network Y-shaped region (less translucent). (B) The non-hybrid domain is easily deformed, whilst (C) the hybrid region is robust. (D–F) Diffusion of DR80 dye at 60 s, 3 h and 24 h – there is only minimal diffusion into hybrid region. Image reproduced from ref. 74 with permission from Wiley-VCH. | ||
Recently, Wang, Yu and co-workers also reported the combination of synthetic PGs with LMWGs using N,N,-dimethylacrylamide as PG, Fmoc-tyrosine derivatives as LMWG and dual enzymatic activation.75 A glucose oxidase mediated radical initiation activates the PG, while phosphatase catalysed cleavage of a phosphate group activates the LMWG hydrogel. These hybrid gels had strong rheology, shape persistence and self-healing characteristics. Both of the enzymes used in the gel formation were entrapped within the hybrid gel and retained their activities.
Other LMWG–polymer combinations
There are some examples which do not fit neatly into the categories described above. For example, the gelling of a liquid polymer by LMWGs to form an electrolyte,76 or the incorporation of LMWGs into polymer chains by copolymerisation,3a but they are beyond the scope and direction of interest here. Further new ideas are appearing regularly – recent examples include a folic acid gel where aniline was bound to the gel fibres and could then be polymerised to form a “sheath” around the fibres for enhanced mechanical strength,77 and a supramolecular hydrogel that incorporated polymer nanogels to improve thermal stability.78Conclusions
Combining LMWGs with polymers is a broad yet relatively recent field in a phase of rapid expansion and with huge potential for exploitation. There are an increasing number of methods for obtaining specifically tailored materials for use in a variety of applications. (i) The polymerisation of LMWG fibres can enhance mechanical strength and stability and also generate novel, tunable one-dimensional nanostructures. (ii) LMWGs can be used to scaffold polymeric materials, modifying their rheology, and on their removal can yield nano-imprinted systems. (iii) Non-gelling polymers, including biopolymers such as proteins, added in solution to LMWGs can modify their nanoscale morphologies and materials properties by polymer adsorption onto the fibres, or viscosity effects as well as introduce additional forms of activity. (iv) Controlling non-covalent interactions between PGs and LMWGs, allows precise tuning of the self-assembly gelation event to create complex yet controllable nanomaterials. (v) Hybrid gels can combine the highly tunable and responsive nature of LMWGs with the mechanical strength of PGs to yield patternable materials with great potential in environmental remediation, microfluidics and biomedical applications such as tissue engineering and drug delivery. There is significant scope for developments in all areas of LMWG–polymer combination, and by bringing these ideas together here, we hope to provide inspiration for future research and applications of this fascinating technology.Acknowledgements
We acknowledge The University of York for providing project funding to DJC through the award of a PhD Teaching Scholarship.Notes and references
- (a) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC; (b) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed; (c) S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC.
- Polymer Gels: Fundamentals and Applications, ed. H. B. Bohidar, P. Dubin and Y. Osada, American Chemical Society, Washington DC, 2002 Search PubMed.
- (a) M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2010, 39, 455–463 RSC; (b) E. A. Appel, J. Del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC; (c) X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- (a) K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed; (b) R. V. Ulijn, N. Bibi, V. Jayawarna, P. D. Thornton, S. J. Todd, R. J. Mart, A. M. Smith and J. E. Gough, Mater. Today, 2007, 10, 40–48 CrossRef CAS; (c) J. Jagur-Grodzinski, Polym. Adv. Technol., 2010, 21, 27–47 CAS; (d) I. Gibas and H. Janik, Chem. Chem. Technol., 2010, 4, 297–304 Search PubMed.
- (a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed; (b) D. K. Smith, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, 2012, vol. 7, pp. 3355–3376 Search PubMed; (c) R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- (a) Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, Netherlands, 2006 Search PubMed; (b) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed; (c) J. H. Van Esch, Langmuir, 2009, 25, 8392–8394 CrossRef CAS PubMed; (d) J. W. Steed, Chem. Commun., 2011, 47, 1379–1383 RSC.
- For an excellent overview of gels in lubrication see: C. J. Donahue, J. Chem. Educ., 2006, 83, 862–869 CrossRef CAS.
- (a) H. Cui, M. J. Webber and S. I. Stupp, Polym. Sci., 2009, 94, 1–18 Search PubMed; (b) J. B. Matson and S. I. Stupp, Chem. Commun., 2012, 48, 26–33 RSC; (c) D. M. Ryan and B. L. Nilsson, Polym. Chem., 2012, 3, 18–33 RSC; (d) K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC; (e) Y. Li, M. Qin, Y. Cao and W. Wang, Sci. China: Phys., Mech. Astron., 2014, 57, 849–858 CrossRef CAS; (f) R. Tian, J. Chen and R. Niu, Nanoscale, 2014, 6, 3474–3482 RSC.
- M. de Loos, J. Van Esch, I. Stokroos, R. M. Kellogg and B. L. Feringa, J. Am. Chem. Soc., 1997, 7863, 12675–12676 CrossRef.
- H. Eimura, M. Yoshio, Y. Shoji, K. Hanabusa and T. Kato, Polym. J., 2012, 44, 594–599 CrossRef CAS.
- M. George and R. G. Weiss, Chem. Mater., 2003, 15, 2879–2888 CrossRef CAS.
- C. Kim, S. J. Lee, I. H. Lee and K. T. Kim, Chem. Mater., 2003, 15, 3638–3642 CrossRef CAS.
- M. Shirakawa, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2005, 127, 4164–4165 CrossRef CAS PubMed.
- (a) K. Aoki, M. Kudo and N. Tamaoki, Org. Lett., 2004, 6, 429–430 CrossRef PubMed; (b) J. Nagasawa, M. Kudo and S. Hayashi, Langmuir, 2004, 6, 7907–7916 CrossRef PubMed; (c) O. J. Dautel, M. Robitzer, J. Le and J. E. Moreau, J. Am. Chem. Soc., 2006, 128, 16213–16223 CrossRef CAS PubMed; (d) X. Nie and G. Wang, J. Org. Chem., 2006, 71, 4734–4741 CrossRef CAS PubMed; (e) E. Jahnke, I. Lieberwirth, N. Severin, J. P. Rabe and H. Frauenrath, Angew. Chem., Int. Ed., 2006, 45, 5383–5386 CrossRef CAS PubMed; (f) N. Fujita, Y. Sakamoto, M. Shirakawa, M. Ojima, A. Fujii, M. Ozaki and S. Shinkai, J. Am. Chem. Soc., 2007, 129, 4134–4135 CrossRef CAS PubMed; (g) E. Jahnke, N. Severin, P. Kreutzkamp, J. P. Rabe and H. Frauenrath, Adv. Mater., 2008, 20, 409–414 CrossRef CAS; (h) J. Wang, G. Yang, H. Jiang, G. Zou and Q. Zhang, Soft Matter, 2013, 9, 9785–9791 RSC; (i) I. Levesque, S. Rondeau-Gagné, J. R. Néabo and J.-F. Morin, Org. Biomol. Chem., 2014, 12, 9236–9242 RSC.
- (a) D. K. Smith, Tetrahedron, 2007, 63, 7283–7284 CrossRef CAS PubMed; (b) K. Fan, L. Niu, J. Li, R. Feng, R. Qiu, T. Liu and J. Song, Soft Matter, 2013, 9, 3057–3062 RSC; (c) K. K. Diehn, H. Oh, R. Hashemipour, R. G. Weiss and S. R. Raghavan, Soft Matter, 2014, 10, 2632–2640 RSC.
- (a) C. S. Love, V. Chechik, D. K. Smith, I. Ashworth and C. Brennan, Chem. Commun., 2005, 5647–5649 RSC; (b) J. R. Moffat, I. A. Coates, F. J. Leng and D. K. Smith, Langmuir, 2009, 25, 8786–8793 CrossRef CAS.
- D. D. Díaz, K. Rajagopal, E. Strable, J. Schneider and M. G. Finn, J. Am. Chem. Soc., 2006, 128, 6056–6057 CrossRef PubMed.
- D. D. Díaz, J. J. Cid, P. Vázquez and T. Torres, Chem.–Eur. J., 2008, 14, 9261–9273 CrossRef PubMed.
- D. D. Díaz, E. Morin, E. M. Schön, G. Budin, A. Wagner and J.-S. Remy, J. Mater. Chem., 2011, 21, 641–644 RSC.
- M.-O. M. Piepenbrock, N. Clarke, J. A. Foster and J. W. Steed, Chem. Commun., 2011, 47, 2095–2097 RSC.
- H. Gankema, M. A. Hempenius, M. Möller, G. Johansson and V. Percec, Macromol. Symp., 1996, 102, 381–390 CrossRef CAS.
- U. Beginn, S. Keinath and M. Möller, Macromol. Chem. Phys., 1998, 119, 2379–2384 CrossRef.
- U. Beginn, S. Sheiko and M. Möller, Macromol. Chem. Phys., 2000, 201, 1008–1015 CrossRef CAS.
- W. Gu, L. Lu, G. B. Chapman and R. G. Weiss, Chem. Commun., 1997, 543–544 RSC.
- R. J. H. Hafkamp, B. P. A. Kokke, I. M. Danke, H. P. M. Geurts, A. E. Rowan, M. C. Feiters and R. J. M. Nolte, Chem. Commun., 1997, 545–546 RSC.
- G. Tan, M. Singh, J. He, V. T. John and G. L. McPherson, Langmuir, 2005, 21, 9322–9326 CrossRef CAS PubMed.
- M. I. Burguete, F. Galindo, R. Gavara, M. A. Izquierdo, J. C. Lima, S. V. Luis, A. J. Parola and F. Pina, Langmuir, 2008, 24, 9795–9803 CrossRef CAS PubMed.
- J. A. Foster, D. W. Johnson, M. O. M. Pipenbrock and J. W. Steed, New J. Chem., 2014, 38, 927–932 RSC.
- F.-X. Simon, N. S. Khelfallah, M. Schmutz, N. Díaz and P. J. Mésini, J. Am. Chem. Soc., 2007, 129, 3788–3789 CrossRef CAS PubMed.
- (a) J. C. Stendahl, L. Li, E. R. Zubarev, Y.-R. Chen and S. I. Stupp, Adv. Mater., 2002, 14, 1540–1543 CrossRef CAS; (b) E. R. Zubarev, M. U. Pralle, E. D. Stone and S. I. Stupp, Adv. Mater., 2002, 14, 198–203 CrossRef CAS; (c) J. C. Stendahl, E. R. Zubarev, M. S. Arnold, M. C. Hersam, H.-J. Sue and S. I. Stupp, Adv. Funct. Mater., 2005, 15, 487–493 CrossRef CAS.
- J. R. Moffat, G. J. Seeley, J. T. Carter, A. Burgess and D. K. Smith, Chem. Commun., 2008, 4601–4603 RSC.
- G. J. Seeley, D. K. Smith, J. R. Moffat and A. Burgess, Patent, WO2009 127885, 2009.
- J. R. Moffat and D. K. Smith, Chem. Commun., 2011, 47, 11864–11866 RSC.
- H. Kim and J. Y. Chang, Langmuir, 2014, 30, 13673–13679 CrossRef CAS PubMed.
- E. A. Wilder, K. S. Wilson, J. B. Quinn, D. Skrtic and J. M. Antonucci, Chem. Mater., 2005, 17, 2946–2952 CrossRef CAS.
- (a) N. Karim, T. D. Jones, K. M. Lewandowski, B. D. Craig. S. B. Mitra and J. Yang, US Pat. 2009 0305196, 2009; (b) N. Karim, T. D. Jones, K. M. Lewandowski, B. D. Craig. S. B. Mitra and J. Yang, US Pat. 2013 8552086, 2013.
- S. H. Kang, B. M. Jung and J. Y. Chang, Adv. Mater., 2007, 19, 2780–2784 CrossRef CAS.
- S. H. Kang, B. M. Jung, W. J. Kim and J. Y. Chang, Chem. Mater., 2008, 20, 5532–5540 CrossRef CAS.
- S. Zhang, X. Fu, H. Wang and Y. Yang, J. Sep. Sci., 2008, 31, 3782–3787 CrossRef CAS PubMed.
- G. Vasapollo, R. Del Sole, L. Mergola, M. R. Lazzoi, A. Scardino, S. Scorrano and G. Mele, Int. J. Mol. Sci., 2011, 12, 5908–5945 CrossRef CAS PubMed.
- D. A. Stone, L. Hsu, N. R. Wheeler, E. Wilusz, W. Zukas, G. E. Wnek and L. T. J. Korley, Soft Matter, 2011, 7, 2449 RSC.
- Y. Zhang, R. Zhou, J. Shi, N. Zhou, I. R. Epstein and B. Xu, J. Phys. Chem. B, 2013, 117, 6566–6573 CrossRef CAS PubMed.
- K. Hanabusa, A. Itoh, M. Kimura and H. Shirai, Chem. Lett., 1999, 767–768 CrossRef CAS.
- X. Y. Liu and P. D. Sawant, Angew. Chem., Int. Ed., 2002, 41, 3641–3645 CrossRef CAS.
- X. Y. Liu, P. D. Sawant, W. B. Tan, I. B. M. Noor, C. Pramesti and B. H. Chen, J. Am. Chem. Soc., 2002, 124, 15055–15063 CrossRef CAS PubMed.
- J.-L. Li and X.-Y. Liu, J. Phys. Chem. B, 2009, 113, 15467–15472 CrossRef CAS PubMed.
- J.-L. Li, B. Yuan, X.-Y. Liu, X.-G. Wang and R.-Y. Wang, Cryst. Growth Des., 2011, 11, 3227–3234 CAS.
- J. Cui, Z. Shen and X. Wan, Langmuir, 2010, 26, 97–103 CrossRef CAS PubMed.
- (a) J. R. Moffat and D. K. Smith, Chem. Commun., 2008, 2248–2250 RSC; (b) M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC.
- P. Chakraborty, B. Roy, P. Bairi and A. K. Nandi, J. Mater. Chem., 2012, 22, 20291–20298 RSC.
- (a) S. Dutta, D. Das, A. Dasgupta and P. K. Das, Chem.–Eur. J., 2010, 16, 1493–1505 CrossRef CAS PubMed; (b) D. M. Wood, B. W. Greenland, A. L. Acton, F. Rodríguez-Llansola, C. A. Murray, C. J. Cardin, J. F. Miravet, B. Escuder, I. W. Hamley and W. Hayes, Chem.–Eur. J., 2012, 18, 2692–2699 CrossRef CAS PubMed; (c) B. O. Okesola and D. K. Smith, Chem. Commun., 2013, 49, 11164–11166 RSC.
- L. Chen, S. Revel, K. Morris, D. G. Spiller, L. C. Serpell and D. J. Adams, Chem. Commun., 2010, 46, 6738–6740 RSC.
- G. Pont, L. Chen, D. G. Spiller and D. J. Adams, Soft Matter, 2012, 8, 7797–7802 RSC.
- C. Yang, M. Bian and Z. Yang, Biomater. Sci., 2014, 2, 651–654 RSC.
- R. N. Javid, S. Roy, M. Zelzer, Z. Yang, J. Sefcik and R. V. Ulijn, Biomacromolecules, 2013, 14, 4368–4376 CrossRef PubMed.
- Y. J. Adhia, T. H. Schloemer, M. T. Perez and A. J. McNeil, Soft Matter, 2012, 8, 430–434 RSC.
- H. Kobayashi, M. Amaike, J. H. Jung, A. Friggeri, S. Shinkai and D. N. Reinhoudt, Chem. Commun., 2001, 1038–1039 RSC.
- (a) J. H. Jung, Y. Ono, K. Sakurai, M. Sano and S. Shinkai, J. Am. Chem. Soc., 2000, 122, 8648–8653 CrossRef CAS; (b) M. Gradzielski, J. Phys.: Condens. Matter, 2003, 15, R655–R697 CrossRef CAS; (c) I. A. Coates, F. J. Leng and D. K. Smith, Chem.–Eur. J., 2009, 15, 6340–6344 CrossRef CAS PubMed.
- M. Numata, K. Sugiyasu, T. Kishida, S. Haraguchi, N. Fujita, S. M. Park, Y. J. Yun, B. H. Kim and S. Shinkai, Org. Biomol. Chem., 2008, 6, 712–718 CAS.
- (a) Z. Zhang, M. Yang, X. Zhang, L. Zhang, B. Liu, P. Zheng and W. Wang, Chem.–Eur. J., 2009, 15, 2352–2361 CrossRef CAS PubMed; (b) Z. Zhang, M. Yang, C. Hu, B. Liu, M. Hu, X. Zhang, W. Wang, Y. Zhou and H. Wang, New J. Chem., 2011, 35, 103–110 RSC.
- A. E. Way, A. B. Korpusik, T. B. Dorsey, L. E. Buerkle, H. A. von Recum and S. J. Rowan, Macromolecules, 2014, 47, 1810–1818 CrossRef CAS.
- (a) A. R. Hirst and D. K. Smith, Chem.–Eur. J., 2005, 11, 5496–5508 CrossRef CAS PubMed; (b) L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC; (c) J. Raeburn and D. J. Adams, Chem. Commun., 2014 10.1039/c4cc08626k.
- P. Li, X.-Q. Dou, Y.-T. Tang, S. Zhu, J. Gu, C.-L. Feng and D. Zhang, J. Colloid Interface Sci., 2012, 387, 115–122 CrossRef CAS PubMed.
- P. Li, Z. Yin, X.-Q. Dou, G. Zhou and C.-L. Feng, ACS Appl. Mater. Interfaces, 2014, 6, 7948–7952 CAS.
- L. L. Hyland, M. B. Taraban, Y. Feng, B. Hammouda and Y. B. Yu, Biopolymers, 2012, 97, 177–188 CrossRef CAS PubMed.
- D. Dasgupta, S. Srinivasan, C. Rochas, A. Ajayaghosh and J. M. Guenet, Langmuir, 2009, 25, 8593–8598 CrossRef CAS PubMed.
- J. Wang, Z. Wang, J. Gao, L. Wang, Z. Yang, D. Kong and Z. Yang, J. Mater. Chem., 2009, 19, 7892–7896 RSC.
- J. Wang, H. Wang, Z. Song, D. Kong, X. Chen and Z. Yang, Colloids Surf., B, 2010, 80, 155–160 CrossRef CAS PubMed.
- R. Huang, W. Qi, L. Feng, R. Su and Z. He, Soft Matter, 2011, 7, 6222–6230 RSC.
- P. Li, X.-Q. Dou, C.-L. Feng and D. Zhang, Soft Matter, 2013, 9, 3750–3757 RSC.
- P. Zorlutuna, N. Annabi, G. Camci-Unal, M. Nikkhah, J. M. Cha, J. W. Nichol, A. Manbachi, H. Bae, S. Chen and A. Khademhosseini, Adv. Mater., 2012, 24, 1782–1804 CrossRef CAS PubMed.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Soft Matter, 2013, 9, 8730–8736 RSC.
- J. Wang, X. Miao, Q. Fengzhao, C. Ren, Z. Yang and L. Wang, RSC Adv., 2013, 3, 16739–16746 RSC.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Angew. Chem., Int. Ed., 2014, 53, 12461–12465 CAS.
- Y. Mao, T. Su, Q. Wu, C. Liao and Q. Wang, Chem. Commun., 2014, 50, 14429–14432 RSC.
- W.-C. Lai and C.-C. Chen, Soft Matter, 2014, 10, 312–319 RSC.
- P. Chakraborty, P. Bairi, B. Roy and A. K. Nandi, ACS Appl. Mater. Interfaces, 2014, 6, 3615–3622 CAS.
- Q. Wang, X. Xiao, Y. Hu, H. Wang and Y. Yang, RSC Adv., 2014, 4, 22380–22386 RSC.
| This journal is © The Royal Society of Chemistry 2015 |