
Manganese, the stress reliever
J.-M.
Latour
Université Grenoble Alpes, LCBM, and, CEA, DSV, iRTSV, LCBM, PMB, and CNRS UMR 5249, LCBM 38054 Grenoble, France. E-mail: jean-marc.latour@cea.fr
First published on 12th November 2014
Abstract
Convergent evidence has emerged over the past decade to highlight the role of manganese as a key player in the defenses that many organisms are building to fight oxidative stress. For redox processes replacing iron by manganese requires adaptation at different levels. The aim of this perspective is to summarize recent important observations and to analyze the implications of the present knowledge for resolving future issues.
Introduction
The generation of Reactive Oxygen Species (ROS) is the price to pay for an aerobic life style and aerobic organisms are equipped with a battery of enzymic defenses able to keep them at a low level. Oxidative stress is due to an increase in the amount of ROS which then overwhelm the basal cell defenses.1 One major drawback is the generation of the hydroxyl radical through reaction of hydrogen peroxide with Fe2+ ions, the so-called “Fenton reaction” (see Scheme 1 below). Fe thus appears directly implicated in the formation of various kinds of damages to cell constituents. In a seminal paper, in the search for identifying cellular processes inhibited by H2O2 in Escherichia coli, Sobota and Imlay identified the Fe enzyme ribulose-5-phosphate 3-epimerase (Rpe) as a major target whose damage led to disturbance of the pentose-phosphate pathway.2 Other enzymes like peptide deformylase, cytosine deaminase and threonine dehydrogenase were similarly affected, showing that the whole class of non-redox mononuclear nonheme iron enzymes constitutes a major oxidative stress target.3 Interestingly, Mn supplementation maintains Rpe activity through replacing Fe2+ by Mn2+ in the active site during H2O2 stress.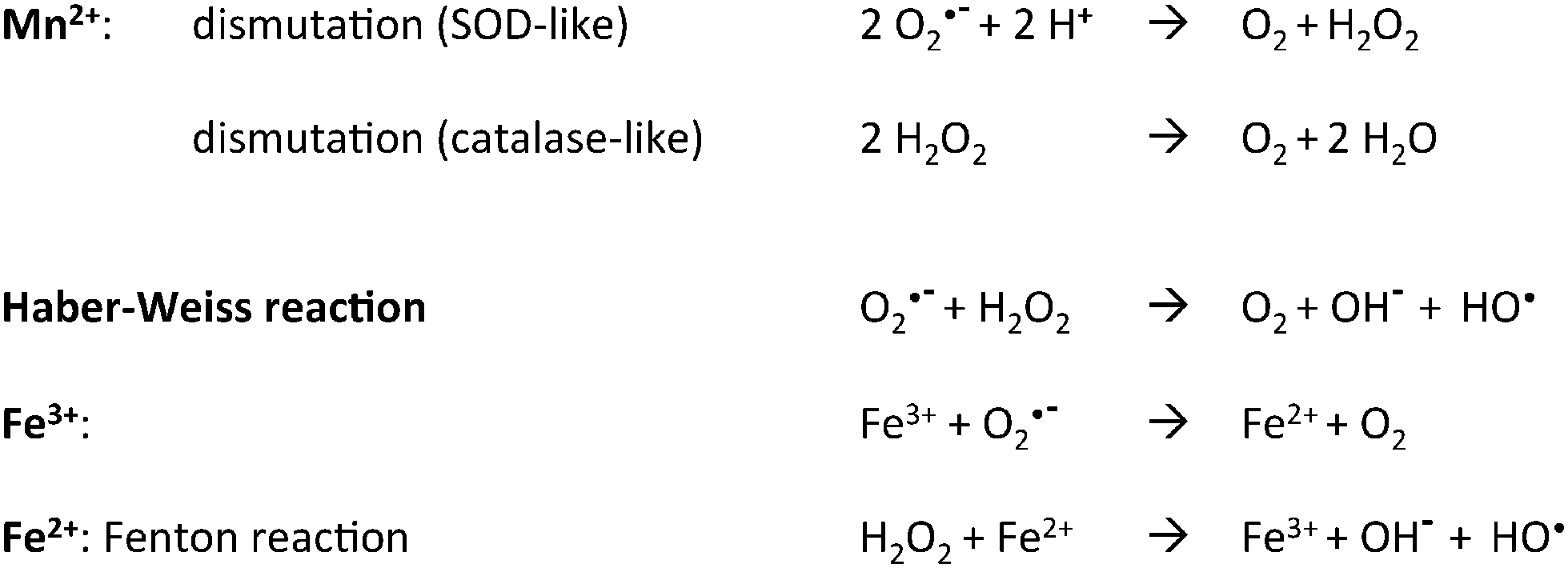 | ||
| Scheme 1 Reaction of H2O2 with Mn2+ and Fe2+. | ||
Further involvement of Mn as an Fe substitute under conditions of oxidative stress is shown by the fact that expression of the Mn transporter MntR is under the control of the peroxide sensor OxyR4 as is the miniferritin protein DPS, an Fe2+ scavenger. It thus appears that to fight a peroxide stress, Escherichia coli initiates both Fe scavenging and Mn import. The case of the Bacillus subtilis peroxide regulator PerR is of special interest in this discussion. Indeed, it is a functional homolog of OxyR and is activated by binding either Fe2+ or Mn2+.5,6 Interaction of the Fe form with H2O2 causes the derepression of a regulon including peroxide defense enzymes (KatA, AhpCF) and an Fe sequestration protein (MrgA). By contrast, the Mn form does not respond to H2O2 and controls the Mn/Fe ratio by repressing the ferric uptake regulator Fur.7 This further illustrates the link between Fe, Mn and peroxide stress.
These observations appear totally consistent with previous data for other organisms such as Lactobacillus plantarum and Borrelia burgderfori which are partially or totally devoid of Fe and possess high Mn content.8 They are also in line with the finding that radioresistant organisms such as the bacterium Deinococcus radiodurans and the alga Coccomyxa actinobiotis possess high Mn/Fe content ratios, 0.24 and 0.33 respectively.9 Indeed, radiation exposure of water media generates various ROS that may further react with Fe2+.
Evidence thus abounds supporting Fe replacement by Mn in organisms facing an oxidative stress. This may appear to be an obvious choice owing to their neighboring in the Mendeleev table and the overall similarities in their charge and size properties (see Table 1).
| Mn | Fe | |
|---|---|---|
| Ionic radius M2+ (Å) | 0.83 | 0.78 |
| Ionic radius M3+ (Å) | 0.66 | 0.64 |
| pKa M[(H2O)5(H2O)]2+ | 10.7 | 9.5 |
| pKa M[(H2O)5(H2O)]2+ | 0.7 | 2.3 |
| k exchange M[(H2O)6]2+ (s−1) | 1 × 106 | 5 × 106 |
| Redox potential M[(H2O)6]2+/M[(H2O)6]3+ (VNHE) | 1.52 | 0.77 |
These properties are indeed key determinants of the acidic character of the element which roots its capacity to catalyze hydrolysis or isomerization reactions. In this respect, it is known for more than twenty years that in the class of purple acid phosphatases several equally active isoenzymes exist that rely on FeIIIMII pairs where MII = MnII, FeII or ZnII.10 This is a general feature of hydrolytic enzymes for which identifying the true physiological metal can be problematic. The situation is quite different for redox enzymes owing to the 750 mV difference in the respective MII/MIII potentials of the two hexa aqua ions (Mn[(OH2)6]2+/Mn[(OH2)6]3+, E = 1.52 VNHE and Fe[(OH2)6]2+/Fe[(OH2)6]3+, E = 0.77 VNHE).11 As a consequence of this difference, FeII, but not MnII, reacts readily with dioxygen and the interactions of the two ions with O2˙− and H2O2 differ in nature, disproportionation for MnII as opposed to hydroxyl production for FeII (Scheme 1).12 Replacement of Fe2+ by Mn2+ to perform redox functions thus requires some kind of adaptation to maintain comparable activity.13
Mechanisms of adaptation to Mn substitution of Fe
1. Selective H bonding
How this is achieved in superoxide dismutase has been elucidated.14,15 By providing extra-stabilization of the MnIII–OH form through H-bonding with a glutamine from the N-terminal domain, Mn(SOD) compensates for ca. two thirds of the 750 mV difference in the redox potentials of the aqua ions (Fe(SOD) which has a glutamine in the C-terminal domain lacks this H-bond). As a result, the MnII(OH2)/MnIII(OH) potential of Mn(SOD) and the FeII/FeIII potential of Fe(SOD) differ only by ca. 200 mV and the two enzymes are equally effective in superoxide disproportionation. This shows that a change in the protein sequence can tune the redox potential of the metal through specific H-bonding and enhance the activity of the protein to make it physiologically significant.2. Substrate activation
However, the results obtained for ribonucleotide reductase in the past decade are the most enlightening on the way Nature operates to replace Fe enzymes when needed to face a stress situation. Indeed, it was commonly known16 that class Ia of these enzymes rely on a dinuclear FeII unit which upon reaction with dioxygen generates a tyrosyl radical which in turn promotes the oxidation of the active cysteine residue (Scheme 2) located 35 Å apart through an electron-transport chain that has been identified.17 By contrast, the identity of the metal pair at the enzyme active site of class Ib RNR has been a matter of controversy for several decades: it has eventually been settled recently and the presence of a dimanganese center has been ascertained.16,18 But this finding immediately gave rise to the question: how can a MnIIMnII pair activate dioxygen? Indeed, although Mn2 pairs are active towards H2O2, as in Mn-catalases, they are generally not reductive enough to activate O2. The answer to this question was provided by the seminal work of J. Stubbe and coworkers. In a series of papers culminating with the determination of the X-ray structures,19 they showed that the RNR enzyme NrdF works in concert with another enzyme NrdI, the role of which is to activate O2 through reduction to O2−˙ (Scheme 2).20 Hence, the reduced RNR itself reacts with O2−˙ to produce a high-valent MnIIIMnIV pair able to generate the tyrosyl radical. This illustrates how to cope with the change in the metal pair, i.e. to compensate for its inability to activate the “usual” substrate O2, an activation enzyme (dioxygen reductase) changes the substrate into a more reactive form. | ||
| Scheme 2 Activation of ribonucleotide reductases. | ||
3. Radical change
It was recently found that RNR enzymes from a few organisms including the pathogen Chlamydia trachomatis lack the active tyrosine present in classes Ia and Ib, which is replaced by phenylalanine.21 How these class Ic enzymes function has been a matter of debate but the groundbreaking paper of Bollinger, Krebs and coworkers established that these RNR enzymes operate thanks to a FeMn center.22 Oxidation of the FeIIMnII center by dioxygen generates an oxidized form FeIIIMnIV that is oxidizing enough to play the role of the tyrosyl radical and promotes the oxidation of the active cysteine residue (Scheme 2). Thus replacing one Fe by Mn changes the initial step of the mechanism but the function is retained.23 The question is why this change? The reason that has been proposed resides in the high sensitivity of the active tyrosyl radical to nitric oxide produced by macrophages.24 By contrast, the FeIIIMnIV center does not react with NO and hence the Fe/Mn substitution provides these pathogens with a way to sustain macrophage aggression.4. Cascade effect
Very recently, the same Fe/Mn substitution was identified at the ferroxidase center of ferritin of a radiotolerant organism Kineococcus radiotolerans.25 Ferroxidase centers of ferritin are composed of a diiron unit similar to the active site of many O2 activating enzymes.26 It is responsible for the oxidation of the trapped Fe2+ ion allowing the ferrihydrite core to be formed. One can imagine that in K. radiotolerans, owing to the small concentration of Fe2+, the Fe2 ferroxidase site of ferritin may become difficult to assemble therefore precluding further scavenging of Fe2+ ions. An active FeMn ferroxidase site favored by the increased concentration of Mn2+ may pursue this process. It is thus of interest that in organisms which must lower their Fe content to survive, Mn can come to rescue to further scavenge the remaining Fe ions.The few examples outlined above illustrate how enzymes can adapt to Mn replacement of Fe to provide an answer to stress situations of many organisms. They also show that although this is easily achieved for maintaining hydrolase and isomerase functions, maintaining equally efficient redox activities requires adaptation of the metal center or the activation of the oxygen cofactor.
Reshaping Mn and Fe homeostasis
For these adaptations to be fully operative, metallation of the enzymes must be achieved with the required metal, which requires adaptation at the cellular level. Indeed, the cellular concentrations of metal ions are tightly controlled to fulfill the specific needs of every system in compliance with the intrinsic properties of each metal ion.27 The latter are reflected in the Irving–Williams series of metal affinities which state that Mn2+ < Fe2+ ≪ Zn2+ to focus on ions of major interest here. As an illustration of this property, metallation studies of Escherichia coli Fur by these metals provided KD values of 2.4 × 10−5, 1.2 × 10−6 and 1.4 × 10−10 M, respectively. The ca. 20 times stronger affinity of His/Glu or Asp binding sites for Fe2+ over Mn2+ is also documented for Bs PerR, albeit with one order of magnitude higher affinities (KD values: Mn2+, 2.8 × 10−6 M vs. Fe2+, 10−7 M). Owing to its strong binding ability, the concentration of Zn2+ is buffered at values at least 104 times lower than that of Fe2+, 10−12 to 10−10 M vs. 10−6 M. These data clearly show that protein metallation with Mn2+ over Fe2+ will require a substantial increase in the Mn/Fe ratio to compensate for the protein's intrinsic lower affinity for Mn2+. These mononuclear Fe or Mn enzymes with a moderate metal affinity get the metal ions from “pools” where they are exchanging between proteins and molecules in a dynamic way. The concentration of each metal is globally regulated by specific sensors and import and efflux systems28 and the homeostasis of Fe and Mn will thus have to be substantially adapted to fit the new needs. Moreover, inserting the right metal in the right binding site as is required for the MnFe heterodimetallic sites mentioned above is not trivial as revealed by numerous examples of proteins29,30 and model complexes31,32 and their biosynthesis may be challenging. This brief survey emphasizes that the metal homeostasis machinery of the cell will need to be markedly reshaped in favor of Mn. In addition, mismetallation by Zn2+ may become a risk. In eukaryotes, compartmentalization will play a central role in these processes.33The above described adaptation is only a part of the overall profound metabolic reshaping that the cell will endeavor to minimize Fe-dependent ways, keeping those inescapably Fe-dependent to the smallest possible level and promoting alternative ones. It is likely that these processes may bear some resemblance to the Fe-sparing mechanisms used by organisms to grow in Fe-limited media.34,35
Future developments
Whereas the way how Mn replacement of Fe can operate at the enzymic level has started to emerge, the understanding at the cellular level is less advanced and constitutes the most important issue and the next challenge.36 Progress on this matter will require analyses at a global level, including both quantitative and qualitative (speciation and compartmentalization) descriptions. In this respect, well established techniques to perform metallomic analyses of the organisms are available.37–39 It is worth noting that the research on Mn toxicity has fueled many analytical developments associating highly sensitive techniques (i.e. ICP-AES and ICP-MS) with separation methods (i.e. capillary zone electrophoresis and size exclusion chromatography). These techniques proved to be instrumental to identify low molecular mass Mn species and identified Mn(citrate) as a potential major player able to circulate in various human body fluids and cross neuronal barriers causing neuronal injury.39 Several Mn enzymes were also identified using these techniques.37 Of special interest in the present context is the fact that these studies have also revealed the Mn/Fe interplay and link to oxidative stress.Contrasting with speciation techniques, global studies of the metalloproteome are still in their infancy.40 In this respect, the development of magnetic resonance methods (EPR/ENDOR for Mn41–44 and Mössbauer for Fe44,45) may contribute essential information by providing both identification and quantification of Fe and Mn proteins involved in the adjustments.
The profound knowledge accumulated over the past few decades on Fe and Mn enzymes and homeostasis constitutes a sound starting base to explore the cellular processes allowing Mn replacement of Fe. The alga C. actinobiotis,46 which has been to date the only known radioresistant eukaryote, is certainly an organism of choice to investigate these processes. Understanding more in-depth the Mn/Fe interplay is of potential strong interest owing to its close association with host–pathogen interactions8 and the development of many diseases involving oxidative stress.47
Acknowledgements
Dr S. Ollagnier-de-Choudens is acknowledged for careful reading of the manuscript. J.-M. L. acknowledges the partial support from Labex ARCANE (ANR-11-LABX-0003-01).References
- J. A. Imlay, Nat. Rev. Microbiol., 2013, 11, 443–454 CrossRef CAS PubMed.
- J. M. Sobota and J. A. Imlay, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5402–5407 CrossRef CAS PubMed.
- J. Imlay, J. Biol. Chem., 2014, 289, 28121–28128 CrossRef CAS PubMed.
- A. Anjem, S. Varghese and J. A. Imlay, Mol. Microbiol., 2009, 72, 844–858 CrossRef CAS PubMed.
- M. Faulkner and J. Helmann, Antioxid. Redox Signaling, 2011, 15, 175–189 CrossRef CAS PubMed.
- V. Duarte and J.-M. Latour, Mol. BioSyst., 2010, 6, 316–323 RSC.
- J. D. Helmann, J. Biol. Chem., 2014, 289, 28112–28120 CrossRef CAS PubMed.
- J. Lisher and D. Giedroc, Front. Cell. Infect. Microbiol., 2013, 3, 1–14 Search PubMed.
- T. Leonardo, E. Farhi, A. M. Boisson, J. Vial, P. Cloetens, S. Bohic and C. Rivasseau, Metallomics, 2014, 6, 316–329 RSC.
- N. Mitic, S. J. Smith, A. Neves, L. W. Guddat, L. R. Gahan and G. Schenk, Chem. Rev., 2006, 106, 3338–3363 CrossRef CAS PubMed.
- M. Carboni and J.-M. Latour, Coord. Chem. Rev., 2011, 255, 186–202 CrossRef CAS PubMed.
- Superoxide dismutation is well established for Mn2+ which plays a main role in oxidative stress defense. The situation is more ambiguous for Fe2+ which can either disproportionate (Fe(SOD)) or reduce (SuperOxide Reductase) superoxide.15.
- J. A. Cotruvo and J. Stubbe, Metallomics, 2012, 4, 1020–1036 RSC.
- A. F. Miller, Acc. Chem. Res., 2008, 41, 501–510 CrossRef CAS PubMed.
- Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A.-F. Miller, M. Teixeira and J. S. Valentine, Chem. Rev., 2014, 114, 3854–3918 CrossRef CAS PubMed.
- J. A. Cotruvo and J. Stubbe, in Annual Rev. Biochem., ed. R. D. Kornberg, C. R. H. Raetz, J. E. Rothman and J. W. Thorner, 2011, vol. 80, pp. 733–767 Search PubMed.
- E. C. Minnihan, D. G. Nocera and J. Stubbe, Acc. Chem. Res., 2013, 46, 2524–2535 CrossRef CAS PubMed.
- N. Cox, H. Ogata, P. Stolle, E. Reijerse, G. Auling and W. Lubitz, J. Am. Chem. Soc., 2010, 132, 11197–11213 CrossRef CAS PubMed.
- A. K. Boal, J. A. Cotruvo, J. Stubbe and A. C. Rosenzweig, Science, 2010, 329, 1526–1530 CrossRef CAS PubMed.
- J. A. Cotruvo, T. A. Stich, R. D. Britt and J. Stubbe, J. Am. Chem. Soc., 2013, 135, 4027–4039 CrossRef CAS PubMed.
- C. Roshick, E. R. Iliffe-Lee and G. McClarty, J. Biol. Chem., 2000, 275, 38111–38119 CrossRef CAS PubMed.
- W. Jiang, D. Yun, L. Saleh, E. W. Barr, G. Xing, L. M. Hoffart, M. A. Maslak, C. Krebs and J. M. Bollinger, Science, 2007, 316, 1188–1191 CrossRef CAS PubMed.
- J. M. Bollinger, Jr., W. Jiang, M. T. Green and C. Krebs, Curr. Opin. Struct. Biol., 2008, 18, 650–657 CrossRef PubMed.
- M. Hogbom, P. Stenmark, N. Voevodskaya, G. McClarty, A. Graslund and P. Nordlund, Science, 2004, 305, 245–248 CrossRef PubMed.
- M. Ardini, A. Fiorillo, M. Fittipaldi, S. Stefanini, D. Gatteschi, A. Ilari and E. Chiancone, Biochim. Biophys. Acta, 2013, 1830, 3745–3755 CrossRef CAS PubMed.
- E. C. Theil, R. K. Behera and T. Tosha, Coord. Chem. Rev., 2013, 257, 579–586 CrossRef CAS PubMed.
- A. Foster, D. Osman and N. Robinson, J. Biol. Chem., 2014, 289, 28095–28103 CrossRef CAS PubMed.
- Z. Ma, F. E. Jacobsen and D. P. Giedroc, Chem. Rev., 2009, 109, 4644–4681 CrossRef CAS PubMed.
- C. S. Andersson, M. Ohrstrom, A. Popovic-Bijelic, A. Graslund, P. Stenmark and M. Hogbom, J. Am. Chem. Soc., 2012, 134, 123–125 CrossRef CAS PubMed.
- L. M. K. Dassama, A. K. Boal, C. Krebs, A. C. Rosenzweig and J. M. Bollinge, J. Am. Chem. Soc., 2012, 134, 2520–2523 CrossRef CAS PubMed.
- M. Carboni, M. Clémancey, F. Molton, J. Pécaut, C. Lebrun, L. Dubois, G. Blondin and J. M. Latour, Inorg. Chem., 2012, 51, 10447–10460 CrossRef CAS PubMed.
- S. J. Tereniak, R. K. Carlson, L. J. Clouston, V. G. Young, E. Bill, R. Maurice, Y. S. Chen, H. J. Kim, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2014, 136, 1842–1855 CrossRef CAS PubMed.
- C. Blaby-Haas and S. Merchant, J. Biol. Chem., 2014, 289, 28129–28136 CrossRef CAS PubMed.
- S. S. Merchant and J. D. Helmann, in Adv. Microbial Physiol., ed. R. K. Poole, 2012, vol. 60, pp. 91–210 Search PubMed.
- C. E. Blaby-Haas and S. S. Merchant, Curr. Opin. Microbiol., 2013, 16, 677–685 CrossRef CAS PubMed.
- M. Huang, M. Parker and J. Stubbe, J. Biol. Chem., 2014, 289, 28104–28111 CrossRef CAS PubMed.
- B. Michalke, s. Halbach and V. Nischwitz, J. Environ. Monit., 2007, 9, 650–656 RSC.
- S. Mounicou, J. Szpunar and R. Lobinski, Chem. Soc. Rev., 2009, 38, 1119–1138 RSC.
- B. Michalke and K. Fernsebner, J. Trace Elem. Med. Biol., 2014, 28, 106–116 CAS.
- A. Cvetkovic, A. L. Menon, M. P. Thorgersen, J. W. Scott, F. L. Poole, F. E. Jenney, W. A. Lancaster, J. L. Praissman, S. Shanmukh, B. J. Vaccaro, S. A. Trauger, E. Kalisiak, J. V. Apon, G. Siuzdak, S. M. Yannone, J. A. Tainer and M. W. W. Adams, Nature, 2010, 466, 779–782 CrossRef CAS PubMed.
- R. L. McNaughton, A. R. Reddi, M. H. S. Clement, A. Sharma, K. Barnese, L. Rosenfeld, E. B. Gralla, J. S. Valentine, V. C. Culotta and B. M. Hoffman, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15335–15339 CrossRef CAS PubMed.
- L. C. Tabares and S. Un, J. Biol. Chem., 2013, 288, 5050–5055 CrossRef CAS PubMed.
- A. Sharma, E. K. Gaidamakova, V. Y. Matrosova, B. Bennett, M. J. Daly and B. M. Hoffman, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5945–5950 CrossRef CAS PubMed.
- J. Park, S. P. McCormick, M. Chakrabarti and P. A. Lindahl, Metallomics, 2013, 5, 656–672 RSC.
- P. A. Lindahl and G. P. Holmes-Hampton, Curr. Opin. Chem. Biol., 2011, 15, 342–346 CrossRef CAS PubMed.
- C. Rivasseau, E. Farhi, A. Atteia, A. Coute, M. Gromova, D. d. G. Saint Cyr, A.-M. Boisson, A.-S. Feret, E. Compagnon and R. Bligny, Energy Environ. Sci., 2013, 6, 1230–1239 CAS.
- S. V. Torti and F. M. Torti, Nat. Rev. Cancer, 2013, 13, 342–355 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |