
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Therapeutic and analytical applications of arsenic binding to proteins
Beibei
Chen
a,
Qingqing
Liu
a,
Aleksandra
Popowich
b,
Shengwen
Shen
a,
Xiaowen
Yan
a,
Qi
Zhang
a,
Xing-Fang
Li
a,
Michael
Weinfeld
c,
William R.
Cullen
d and
X. Chris
Le
*ab
aDivision of Analytical and Environmental Toxicology, Department of Laboratory Medicine and Pathology, University of Alberta, Edmonton, Canada. E-mail: xc.le@ualberta.ca
bDepartment of Chemistry, University of Alberta, Edmonton, Canada
cExperimental Oncology, Cross Cancer Institute, Edmonton, Canada
dDepartment of Chemistry, University of British Columbia, Vancouver, Canada
First published on 21st October 2014
Abstract
Arsenic binding to proteins plays a pivotal role in the health effects of arsenic. Further knowledge of arsenic binding to proteins will advance the development of bioanalytical techniques and therapeutic drugs. This review summarizes recent work on arsenic-based drugs, imaging of cellular events, capture and purification of arsenic-binding proteins, and biosensing of arsenic. Binding of arsenic to the promyelocytic leukemia fusion oncoprotein (PML-RARα) is a plausible mode of action leading to the successful treatment of acute promyelocytic leukemia (APL). Identification of other oncoproteins critical to other cancers and the development of various arsenicals and targeted delivery systems are promising approaches to the treatment of other types of cancers. Techniques for capture, purification, and identification of arsenic-binding proteins make use of specific binding between trivalent arsenicals and the thiols in proteins. Biarsenical probes, such as FlAsH-EDT2 and ReAsH-EDT2, coupled with tetracysteine tags that are genetically incorporated into the target proteins, are used for site-specific fluorescence labelling and imaging of the target proteins in living cells. These allow protein dynamics and protein–protein interactions to be studied. Arsenic affinity chromatography is useful for purification of thiol-containing proteins, and its combination with mass spectrometry provides a targeted proteomic approach for studying the interactions between arsenicals and proteins in cells. Arsenic biosensors evolved from the knowledge of arsenic resistance and arsenic binding to proteins in bacteria, and have now been developed into analytical techniques that are suitable for the detection of arsenic in the field. Examples in the four areas, arsenic-based drugs, imaging of cellular events, purification of specific proteins, and arsenic biosensors, demonstrate important therapeutic and analytical applications of arsenic protein binding.
 Beibei Chen | Beibei Chen received her BSc degree in Chemistry (2005) and her PhD degree in Analytical Chemistry (2010) with Professor Bin Hu, from Wuhan University (China). She spent a year as a visiting postdoctoral fellow with Professors Xing-Fang Li and X. Chris Le at the University of Alberta. Since 2011 she has been a Lecturer in the College of Chemistry and Molecular Sciences, Wuhan University (China). Her research focuses on the development of new hyphenated techniques, involving mass spectrometry and chromatography, for elemental speciation and metallomics. |
 Qingqing Liu | Qingqing Liu received her BSc degree from Sun Yat-Sen University (China) in 2011. She is currently pursuing her PhD in Medical Sciences (with specialization in Analytical and Environmental Toxicology) at the University of Alberta. Her PhD research is focused on arsenic speciation, arsenic metabolism, and human dietary exposure to arsenic species. |
 Aleksandra Popowich | Aleksandra Popowich is currently pursuing her PhD in Analytical Chemistry at the University of Alberta. She received her BSc in Chemistry from the University of British Columbia in 2012. As a summer student at the University of Calgary in 2011, Aleksandra studied the bioinorganic chemistry of cadmium. Her current research focuses on characterizing the binding between arsenic species and several biologically relevant proteins using mass spectrometry. |
 Xiaowen Yan | Xiaowen Yan received his BSc degree in Chemical Biology (2007) and his PhD degree in Analytical Chemistry (2012) with Professor Qiuquan Wang, from Xiamen University (China). He is currently a postdoctoral fellow with Professor X. Chris Le. His research focuses on the development of new chemical techniques and approaches to study arsenic–protein interactions and on the development of arsenic-based drugs for cancer treatment. |
 Qi Zhang | Qi Zhang is currently a PhD student in the Department of Laboratory Medicine and Pathology, University of Alberta (Canada). She obtained her BSc in Chemistry from Zhejiang University (China) in 2010. Her current research focuses on encapsulation of arsenic species in polymeric micelles and development of targeted delivery of arsenical drugs to cancer cells. |
 X. Chris Le | X. Chris Le is Distinguished University Professor with cross appointments in the departments of Laboratory Medicine and Pathology, Chemistry, and Public Health Sciences. He is Canada Research Chair in Bioanalytical Technology and Environmental Health. He is an elected Fellow of the Royal Society of Canada (Academy of Science). He leads an interdisciplinary team studying human health effects in relation to the environment. His research on metallomics focuses on arsenic binding to proteins, human exposure to arsenic species, and mechanisms of arsenic metabolism and health effects. He is also interested in the development of DNA and protein binding assays for measuring proteins, DNA damage, and molecular interactions. Dr Le is an Editorial Advisory Board member for Analytical Chemistry and Metallomics. |
1. Introduction
The binding of arsenic to proteins plays an important role in various biological processes.1 For example, the well-characterized arsenic resistance system in Escherichia coli R773 involves several proteins that bind to arsenic.2 When arsenic enters the living prokaryotic cells, the resistance system in the cells utilizes several specific proteins, such as ArsA, ArsB, and ArsC (a reductase), to enhance arsenic efflux.3 These proteins act through their cysteine thiolates, which have the ability to bind to arsenic. Fig. 1 presents a typical coordination of arsenic with E. coli ArsA ATPase.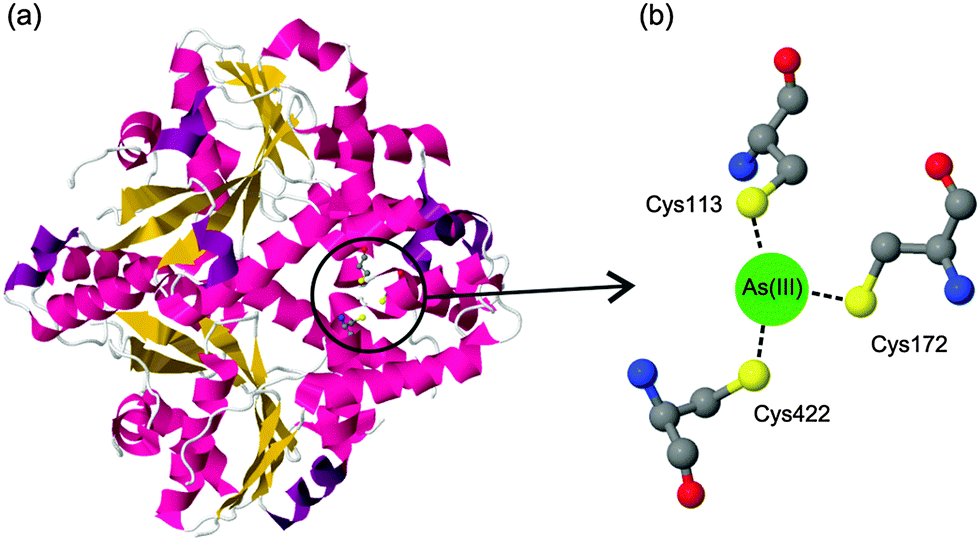 | ||
| Fig. 1 (a) Crystal structure of E. coli ArsA ATPase (PDB: 1f48). (b) Coordination of arsenic to the residues Cys113, Cys172 and Cys422. | ||
The oxidation states of arsenic include −3 in AsH3, 0 in elemental arsenic, +2 in As4S4, +3 in arsenous acid, and +5 in arsenic acid. Although most of the more than 50 arsenic compounds encountered in the environment are known to be pentavalent arsenicals, trivalent arsenicals are the main focus of this review because trivalent arsenicals have a high affinity for thiols. It is the interaction between thiols and the trivalent arsenicals that gives rise to arsenic binding to proteins. The amino acid cysteine is the only source of thiol groups in proteins, and is the main binding site for trivalent arsenicals.4 Arsenite can typically form three-coordinate trigonal–pyramidal complexes with three cysteines in proteins, which can disrupt the activity of certain enzymes. Arsenate usually disrupts cellular processes as a phosphate mimic, but has weak interaction with proteins.
The chemistry of arsenic binding to specific proteins can be utilized for the development of analytical tools and therapeutic treatment. This review discusses these therapeutic and analytical applications of arsenic binding, including cancer chemotherapy, molecular imaging, protein purification, and biosensing. Specific examples include arsenic-based drugs, the imaging of cellular events using arsenic-based dyes, affinity purification of arsenic-binding proteins, and arsenic biosensors.
2. Arsenic-based drugs
Arsenicals have been used therapeutically for more than 2400 years.5,6 Ancient Chinese alchemists prepared elixirs with the minerals orpiment (As4S6) and realgar (As4S4), as ingredients which were prescribed for various imbalances in soul, health, and discipline.7 A number of traditional Chinese medicines contain these minerals, and several formulations are still in use today.8 Hippocrates (460–375 B.C.) used orpiment and realgar to treat tumors and cancerous ulcers.9 Medicinal uses of arsenic reached their peak in the eighteenth and nineteenth centuries, but arsenic was used mainly externally until the development of Fowler's solution, a solution of 1% potassium arsenite (KAsO2) in alcohol and water, which became one of the principal therapeutic drugs of that time, and was prescribed for epilepsy, asthma, eczema, Hodgkin's disease, leukemia, anemia, rheumatism, and psoriasis.10 In the early 1900s, the first organic arsenical used therapeutically, Salvarsan (arsphenamine), was introduced to treat syphilis and African trypanosomiasis (sleeping sickness). The structure of Salvarsan was identified by Ronimus et al. using electrospray ionization mass spectrometry (ESI-MS).11 Salvarsan is actually a mixture of cyclic (3-amino-4-hydroxylphenylarsine)n species, with n = 3 and n = 5. It quickly became the most widely prescribed drug in the world. Melarsaprol, a related trivalent organoarsenical developed in 1949, is still being used for the treatment of sleeping sickness.12 The structures of some medicinal arsenic compounds are illustrated in Table 1.| Arsenical | Structure |
|---|---|
| Arsenic trioxide, arsenous acid |
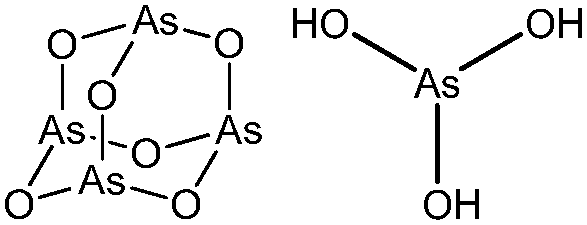
|
| As4S4 |
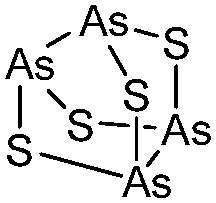
|
| Arsphenamine (Salvarsan) |

|
| Melarsaprol (Mel B) |
|
| Melarsen oxide |

|
| 4-(N-(S-Penicillaminylacetyl)amino)phenylarsonous acid (PENAO) |
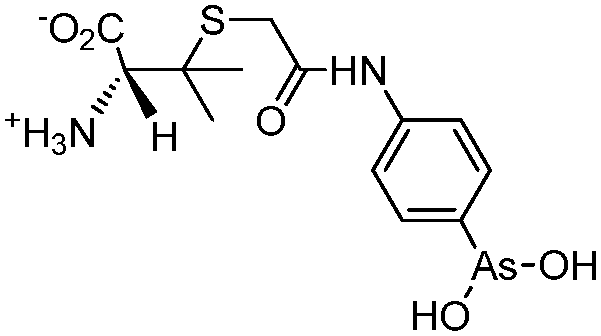
|
| 4-(N-Glutathionylacetyl)aminophenylarsonous acid (GSAO) |
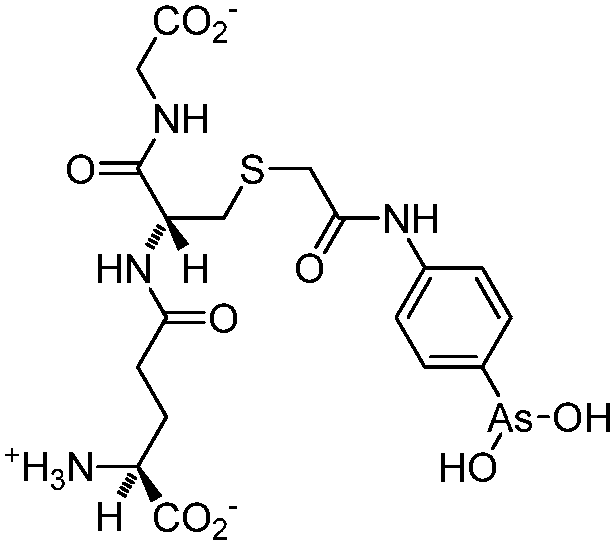
|
| S-Dimethylarsinoglutathione (ZIO-101, Darinaparsin, SGLU-1) |
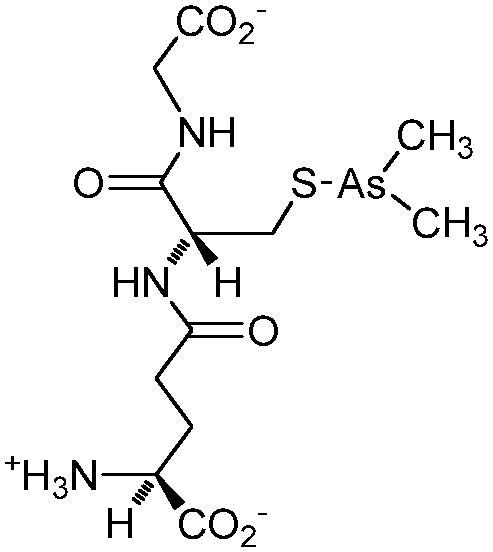
|
| S-Dimethylarsinothiosuccinic acid (MER1) |

|
| Dipropyl-S-glycerol arsenic (GMZ27) |
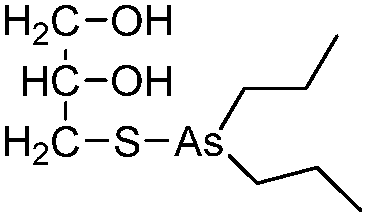
|
| Arsenolipids and arsenosugars |
|
| Glycol-lipid-arsenical |
|
| Arsenicin A |
|
| Phenylarsonous acid (PAO) |

|
In the 1970s Chinese researchers found that dissolved arsenic trioxide (As2O3), which forms arsenous acid [As(OH)3] in aqueous solution, was a relatively safe and effective treatment for relapsed and refractory acute promyelocytic leukemia (APL).6,10,13,14 The success of As2O3 (intravenous injection of its aqueous solution) in APL treatment led to its approval9 as a first-line treatment for APL by the United States Food and Drug Administration in September 2000. Cytotoxicity studies showed that As2O3 treatment has dual effects on APL cells. Low concentration of arsenic (0.25–0.50 μM) triggers differentiation of APL cells, while a high concentration of arsenic (1–2 μM) induces apoptosis (programmed cell death) of APL cells.
Recently, the direct binding of arsenic to cysteine residues in zinc fingers located in the promyolocytic leukemia fusion protein (PML-RARα) was demonstrated to be one of the plausible modes of action leading to APL remission.15,16 The arsenic binding to the PML moiety of PML-RARα oncogenic protein induces a protein conformational change. This conformational change triggers the oligomerization of PML-RARα, which in turn enhances the attachment of Small Ubiquitin-like Modifier proteins to PML-RARα oligomers (SUMOylation) and ubiquitylation, and subsequently results in the degradation of the PML-RARα oncoprotein (Fig. 2).17
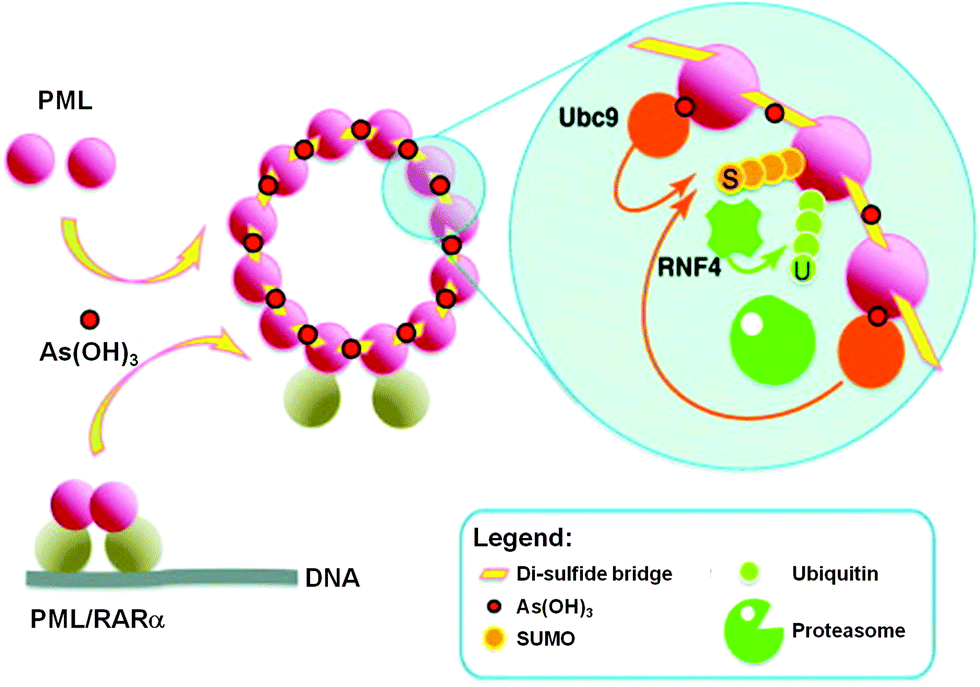 | ||
| Fig. 2 Trivalent arsenical, e.g., As(OH)3, crosslinks cysteine residues in zinc fingers in PML and PML/RARα to induce their degradation through a SUMO-triggered RNF4/ubiquitin-mediated pathway.17 | ||
A more generic version of the mechanism of cell death through the inhibition of thioredoxin reductase (TrxR) by arsenic has been proposed.18 The thioredoxin (Trx) system, especially TrxR, has been suggested as a new target for anticancer drug development because TrxR and Trx are overexpressed in many aggressive tumors, which is likely a consequence of the constant requirement of DNA and protein synthesis. Accumulating evidence suggests that TrxR is essential for tumor cell growth in vivo.19 Both the N-terminal dithiols and the C-terminal selenothiol of TrxR may participate in the reaction with the arsenic compound.18 A low basal level of cellular glutathione and a high expression level of aquaglyceroporin 9, a membrane transporter that mediates uptake of arsenite, make the cells uniquely sensitive to arsenic.10
The clinical utility of As2O3 has been expanded to other types of malignancies (reviewed by Dilda and Hogg10), but is limited by either toxicity at higher doses, leading to peripheral neuropathies, liver failure and cardiac toxicity, or by bioavailability of the arsenic compounds for targeting solid tumors.
Drug delivery systems incorporating nanomaterials are expected to increase drug efficacy and reduce the adverse side effects through targeted delivery of the drug to tumor areas. Inspired by the clinical success of liposomal doxorubicin, O'Halloran and coworkers devised a lipid encapsulation approach.20–23 Arsenite was loaded into liposomes that were preloaded with transitional metal ions (e.g. Ni+, Pb+, Fe+, etc.). Arsenic formed co-precipitates with these transition metal ions, which allows for triggered release of arsenic at low pH and prolonged arsenic circulation in blood. A folate-mediated intracellular delivery system was later introduced to increase targeted arsenic delivery into cancer cells.24 Other nano-carriers such as polymeric vesicles25 and polymeric micelles26 were also developed for targeted delivery of arsenic species. The polymeric vesicles are composed of double layers of amphiphilic polymers, which have a similar structure to liposomes. The polymeric micelles contain a single layer of amphiphilic polymers and the micelle core is hydrophobic. The polymer was functionalized with a thiol-containing pendent group to increase the loading of hydrophilic arsenous acid and slow down the arsenic release. The thiol groups in the micelle core provide binding sites for arsenic, which allows for increased arsenic loading. The release of arsenic is triggered by glutathione (GSH) via a competing reaction to form As–S bonds. Arsenolipid-containing liposomes were also prepared and demonstrated antiparasitic activity in vitro.27 Some glycol-lipid-arsenicals were synthesized based on p-aminophenylarsine oxide (PAPAO) by attaching a lipoamino acid to a sugar, and they were shown to have differential anti-proliferative effects on MCF-7 human breast cancer cells.28 The conjugation with lipoamino acids could protect the trivalent arsenical from oxidation and polymerization and enhance membrane permeability by increasing its lipophilicity. However, the arsenolipids by themselves were judged to be inadequate for therapeutic applications.
As4S4 has been tested to treat APL because of the ease of oral administration and its assumed similar mode of action as As2O3. Lu et al. reported a 6 year pilot study on As4S4 for APL treatment.29 One hundred and twenty-nine patients were involved, among whom 19 were newly diagnosed, 7 were first relapsed, and 103 had hematologic complete remission (HCR). In the HCR group, 44 patients were PML-RARα positive. With a dosage of 50 mg kg−1 of body weight per day, 14 patients in the newly diagnosed group, 5 patients in the first relapsed group, and 35 patients in the HCR group with PML-RARα expression achieved cytogenetic and molecular complete remission (CR). Another As4S4 containing formulation named Realgar-Indigo Naturalis Formula (RIF), which contains realgar, Indigo naturalis, Radix salvia miltiorrhizae (Chinese sage), and Radix pseudostellariae (root of Tai Zi Shen), was shown to be active in APL treatment. The efficacy of RIF for the treatment of APL was comparable to intravenous administration of As2O3 combined with all-trans retinoic acid (ATRA), according to a multicenter phase III clinical trial.30 As4S4 has a low bioavailability (∼4%), because of its insolubility in water.8 The absorption and cytotoxicity were increased31 when the realgar was ground to nano sizes, typically with diameter of 100 to 200 nm. An animal model study demonstrated the enhanced absorption of realgar nanoparticles.32 More than 50% of orally administered arsenic in the form of realgar nanoparticles was recovered from urine during the first 48 hours post administration to rats, which doubled the recovery when coarse realgar was administered. Realgar nanoparticles can also be administered transdermally.33 Tumor areas in rats were found to have the highest arsenic concentration, followed by liver, kidney, lung, small intestine, and heart.
Several other organoarsenicals such as dipropyl-S-glycerol arsenic (GMZ27) and S-dimethylarsino-thiosuccinic acid (MER1) are much less toxic than As2O3 yet have comparable or higher anti-leukemic activity.34,35 Arsenicin A, a naturally occurring adamantine-type tetraarsenical,36 was recently shown to be 20-fold more potent than As2O3 for the induction of proliferation arrest and cell death in APL cells and it can bind to dithiols.37
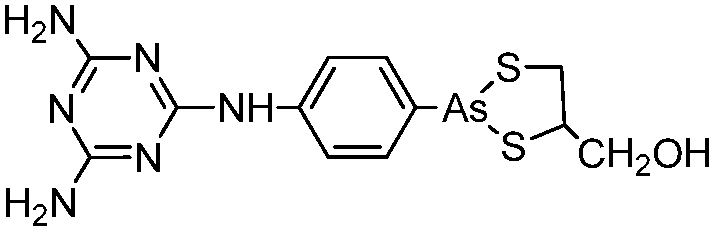
Some organoarsenicals are currently undergoing clinical trials. Those involving dimethylarsinic acid and melarsoprol have been well reviewed by Dilda and Hogg.10 Here we update studies with 4-(N-glutathionylacetyl)aminophenylarsonous acid (GSAO) and S-dimethylarsinoglutathione (ZIO-101, Darinaparsin, SGLU-1) (Chemical structures are illustrated in Table 1).
Phenylarsine oxide (PAO) was precluded from application in clinical cancer therapy because of its high toxicity in vivo and its lack of selectivity for cancerous versus normal cells, in part due to its lipophilicity.38 GSAO, a hydrophilic derivative of PAO, was developed for clinical use. GSAO is a mitochondrial poison that selectively perturbs angiogenic endothelial cells in vitro and in vivo. It inactivates the mitochondrial inner membrane transporter, adenine nucleotide translocase (ANT), by cross-linking the matrix facing thiols Cys160 and Cys257.39 The high selectivity of GSAO for proliferating endothelial cells is a consequence of the higher mitochondrial calcium levels (a several-fold increase over non-proliferating cells) in these cells. ANT is a calcium receptor that undergoes a calcium-induced conformational change. GSAO can bind to ANT when the calcium concentration is high, but binds only minimally in the absence of calcium ions.39 Interestingly, a study addressing the transportation question of GSAO through the endothelial cell membrane revealed that 4-(N-(S-cysteinylglycylacetyl)amino)phenylarsonous acid (GCAO), the metabolite of GSAO cleavage by γ-glutamyltranspeptidase (γGT) on the cell membrane, is transported across the plasma membrane by the organic anion-transporting polypeptide family, instead of by GSAO itself.40 GCAO is further cleaved by dipeptidases in the cytosol, forming 4-(N-(S-cysteinylacetyl)amino)phenylarsonous acid (CAO). CAO reacts with its mitochondria target, ANT, and inactivates the functions of ANT, which further increases the concentration of reactive oxygen species (ROS), arrests proliferation, and induces apoptosis. GSAO is a pro-drug that is dependent on cell surface processing by γGT in order to function. A phase I clinical trial indicated that GSAO with daily 1 hour infusions, across 9 dose levels (1.3–44.0 mg m−2), was well tolerated by patients with advanced solid tumors.41
P-(N-(S-Penicillaminylacetyl)amino)phenylarsonous acid PEANO, a cysteine analogue of GSAO, bypasses the cell membrane processing and accumulates in cells 85 times faster than GSAO, which translates to a 44-fold increase in antiproliferative activity in vitro and 20-fold better antitumor efficacy in vivo.42
ZIO-101, a conjugate of glutathione and dimethylarsinous acid, is a more potent pro-apoptotic agent than As2O3.10 ZIO-101 was well-tolerated in phase I/II clinical trials with both oral and intravenous administration.43 It has lower cardiac toxicity and hepatotoxicity than As2O3. Its activity is probably associated with the hydrolysis of the arsenic–glutathione bond, and the reaction of the resulting dimethylarsinous acid with a protein thiol. The glutathione moiety of ZIO-101 mimics GSAO in the translocation of the arsenicals into cells.44 The hydrolysis of ZIO-101 requires γGT processing. While GCAO is hydrolysed by intracellular dipeptidases, ZIO-101 takes a step further and is hydrolysed to S-dimethylarsinocysteine (DMAC) on the cell membrane. DMAC is transported through the cysteine transporter system.44 However, the possibility that some DMAC is also formed inside the cells cannot be excluded. The existence of the putative hydrolysis intermediate, S-dimethylarsinocysteinylgluamic acid, in the cell cytosol, has yet to be verified.
3. Imaging of cellular events
Biarsenical probes, fluorescein arsenical hairpin binder-ethanedithiol (FlAsH-EDT2) and resorufin arsenical helix binder-EDT2 (ReAsH-EDT2) (Fig. 3a), coupled with tetracysteine tags (CCXXCC, where “C” is cysteine and “X” is any amino acid except cysteine) that are genetically incorporated into the targeted proteins, are used for the site-specific fluorescent labelling and imaging of recombinant targeted proteins in living cells. These fluorescent probes were pioneered by Tsien and coworkers,45,46 expanded upon by other researchers, and reviewed by Goncalves47 and Soh.48 The tetracysteine tags can be genetically fused to either the N- or C-terminus of the protein or to an existing helix within the protein, as long as the tag is placed in a location accessible by the probe. The membrane-permeable biarsenical probes have a high affinity (Kd 0.01–1 nM) and specificity for the optimized tetracysteine motifs in recombinant proteins.49 These biarsenical probes are initially nonfluorescent, but become fluorescent upon the replacement of EDT by the tetracysteine peptide. These probes can be used for sensitive imaging without a washing step.45,46 The tetracysteine–biarsenical system has opened up many application opportunities, providing a tool kit for dynamic protein imaging,50 assessing protein–protein interactions,51 analyzing organelle events,52 visualizing metallic ions,53,54 and detection of bacteria55 (see Fig. 4 for one of the applications).50 Other reviews of the tetracysteine–biarsenical system are available.56–59 Research topics employing this system are still evolving.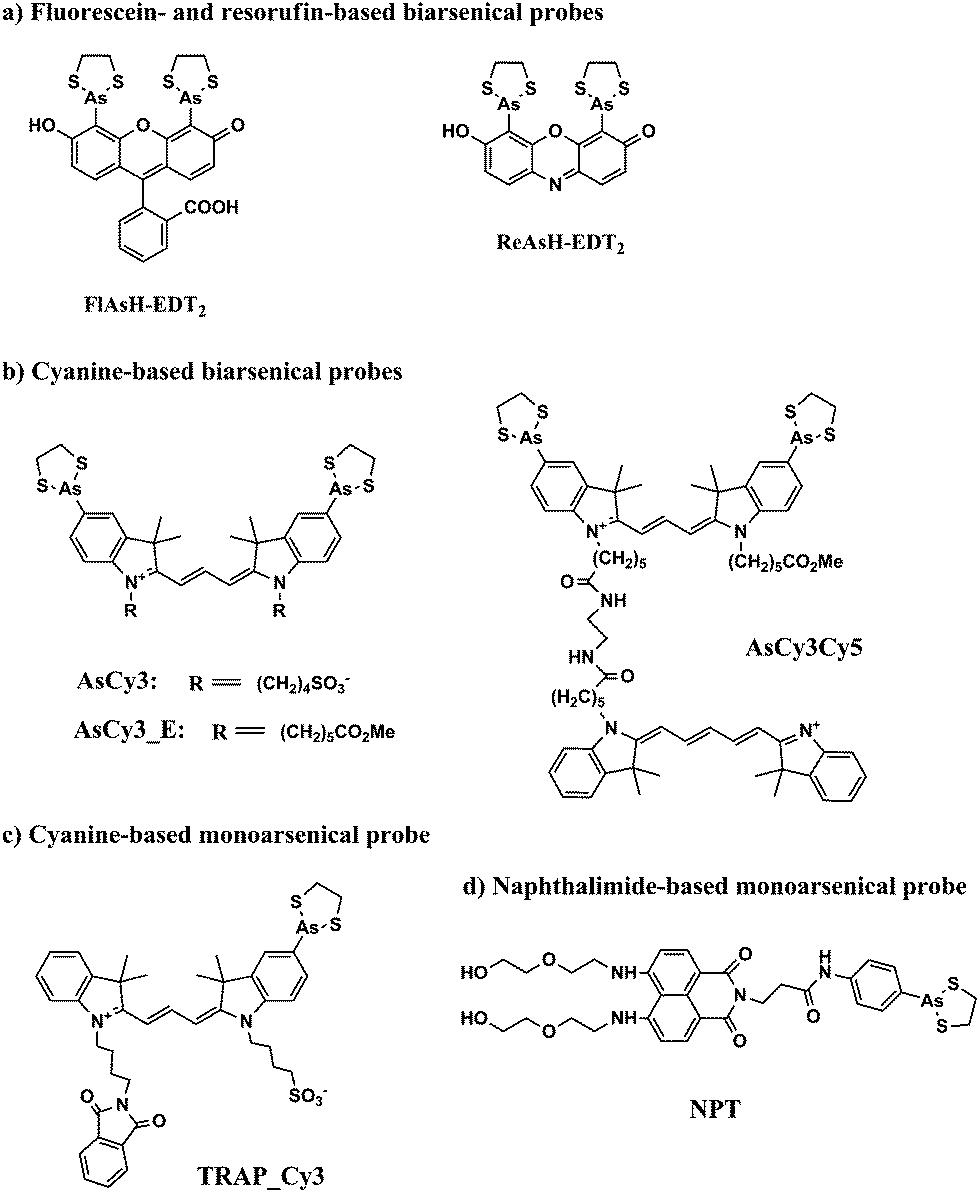 | ||
| Fig. 3 Chemical structures of some typical arsenical probes. | ||
 | ||
| Fig. 4 FlAsH-EDT2 (green) and ReAsH-EDT2 (red) can be used to sequentially stain the old and newly synthesized connexin43 protein that has been genetically fused with the tetracysteine tag (CCRECC) (Cx43-TC). The newly synthesized Cx43-TC, which is stained with red ReAsH, is located in both ends of the junctional plaque. The old Cx43-TC, which is stained with green FlAsH, is located in the central region of junctional plaque (adapted from ref. 50). | ||
FlAsH-EDT2 (green) and ReAsH-EDT2 (red) have different fluorescent emissions, and both bind tightly to the same tetracysteine tag (CCXXCC) due to the conserved distance (∼6 Å) between the two arsenic atoms in their structure. These properties permit the sequential usage of FlAsH-EDT2 and ReAsH-EDT2 to temporally label old and newly synthesized proteins (connexin43) that have been genetically fused with the tetracysteine tag (CCRECC) (Fig. 4).50 This dual-color labeling and imaging strategy enabled Gaietta et al. to monitor the trafficking of connexin43 inside living cells.50 However, it would be difficult to simultaneously label different proteins of interest with FlAsH-EDT2 and ReAsH-EDT2, because the targeted proteins carry the same CCXXCC tag, which cannot be distinguished simultaneously by these probes.
A red Cy3-based biarsenical fluorescent probe (AsCy3) (Fig. 3b), with a longer distance (∼14.5 Å) between the two arsenic atoms, could circumvent this limitation.60 The CCKAEAACC peptide, containing two pairs of vicinal cysteines spaced by five amino acids, was designed as the complementary binding tag. FlAsH-EDT2 and AsCy3 can preferentially bind to their corresponding peptide tags, and the fluorescent spectrum of FlAsH (λem = 528 nm) overlaps partially with the absorption spectrum of AsCy3 (λex = 560 nm). These features enable the FlAsH and AsCy3 probes to act as donor and acceptor for a fluorescent resonance energy transfer (FRET) assay. Through the optimization of the N-alkyl chain and arsenic capping reagent on the AsCy3 probe, a cell-permeable biarsenical cyanine probe (AsCy3_E) (Fig. 3b) has been recently synthesized and successfully applied to the labelling of tagged cellular proteins, for the imaging of live cells and measurements of protein dynamics.61
Another biarsenical-labeled cyanine probe, AsCy3Cy5 (Fig. 3b), was synthesized and utilized to successfully image the 42 kDa α-subunit of RNA polymerase that was genetically fused with the tetracysteine tag (CCKAEAACC).62 The Cy5 probe in AsCy3Cy5 is photoswitchable, i.e., its fluorescence can be turned off when the resonance π-electron cloud is disrupted by the addition of a thiol to its polymethine bridge, but the fluorescence can be switched on again by irradiation with UV light that can photo-remove the attached thiol from the polymethine bridge.63,64 For traditional fluorescence imaging, the spatial resolution of an image is limited by the diffraction of the fluorescence simultaneously emitted from different probes. The photoswitchable Cy5 can be utilized to break the diffraction limit by sequentially activating and localizing a fraction of the dye-labeled targets, and reconstructing the overall images with super-resolution.65 This photoswitchable fluorescent probe (AsCy3Cy5) was therefore developed to enable super-resolution imaging of a tagged protein within a supramolecular complex. Further improvements in state-of-the-art imaging techniques and new fluorescent probes of different colors will facilitate the development of more versatile imaging approaches that are based on arsenic–thiol binding. This methodology will be useful for the analysis of more complicated cellular events. Although great advances have been made by biarsenical fluorescent probes, the requirement of genetically incorporating the tetracysteine tag into the recombinant targeted protein prohibits the analysis of clinical samples from patients for disease diagnosis and restricts their use to basic biological research using cultured cells and animals.
Proximal cysteines are able to undergo reversible conversion between reduced cysteines and oxidized disulfides through highly dynamic cellular redox changes and trivalent arsenic has a high affinity for reduced proximal cysteines. To take advantage of this property, monoarsenical fluorescent probes based on cyanine dye (TRAP_Cy3)66 (Fig. 3c) or naphthalimide (NPE)67,68 (Fig. 3d) have been developed to visualize proximal cysteines in cytosolic proteins so as to trace the cellular redox status corresponding to the environmental conditions. However, since the fluorescence of the monoarsenical dyes is relatively unaffected by its binding to cysteines, intensive washing steps are required to remove the excess unbound dyes for the imaging of labeled cysteines.
GSAO can be tagged with a fluorescent or a radioisotopic reporter through the amine of the γ-glutamyl residue (see Table 1 for its structure), and used to image cell death in cultured Jurkat A3 cells and in murine tumors ex vivo and in situ.69 GSAO is retained in the cytosol predominantly by its covalent reaction with vicinal cysteines in the 90 kDa heat shock protein (Hsp90), which is the most abundant protein chaperone in mammalian cells and whose expression is enhanced by chemotherapy.69 GSAO is unreactive until it reaches the cytosolic proteins because of the low level of thiols in the extracellular milieu. The natural barrier of most cell membranes to GSH prevents GSAO from penetrating the intact plasma membrane of healthy or early-stage apoptotic/necrotic cells.70 Therefore, GSAO shows high selectivity to mid- to late-stage apoptotic/necrotic cells, whose membrane integrity is compromised.71,72 Furthermore, the unbound GSAO can be quickly eliminated from the body by the kidneys, within 3 hours of administration by tail vein injection.71,72 Noninvasive imaging of treatment-related tumor cell death69 and cell death in traumatic brain injury72 were achieved using GSAO conjugated with the near-infrared fluorescent probe AF750 (GSAO-AF750). It can be expected that the nature of the reversible binding of trivalent arsenic to protein thiols, combined with the existing and emerging arsenical fluorescent probes, will play an increasingly important role in fundamental biological studies, clinical diagnosis, and disease treatment.
4. Purification of specific proteins
Hannestad et al.73 first developed affinity chromatography, based on the interaction of trivalent arsenic with thiols, for the separation of mono- and di-thiols. Several research groups have modified this method in order to study interactions between arsenic and proteins. Arsenic-based affinity chromatography has also been employed to purify proteins containing dithiols. Such applications have been well summarized in our previous review.1 Here, we will focus our discussion on two topics, (1) the purification of proteins fused to thioredoxin or tagged with selenoprotein (Sel) or tetracysteinepeptides, by means of arsenic–thiol binding (listed in Table 2), and (2) the combination of arsenic affinity chromatography with tandem mass spectrometry as a proteomic approach to study the interaction of arsenicals with proteins in cells.| Tag | Binding motif | Chromatographic matrix | Elution method | Protein | Ref. |
|---|---|---|---|---|---|
| Thioredoxin | –CGPC– | PAO resin | 0.5 M β-ME | Chlamydomonas outer arm dynein light chains DLC14 and DLC16 | 93 |
| 0.5 M β-ME | Sea urchin outer arm dynein intermediate chain IC1 | 94 | |||
| 5–1000 mM β-ME | Functional connexin 32 domains | 96 | |||
| 100 mM β-ME | Human glutamate decarboxylase 65 (GAD65) | 98 | |||
| 1–250 mM β-ME | Xenopus interphotoreceptor retinoid-binding protein module (X4IRBP) | 99 | |||
| ? mM β-ME | Euglena α-tubulin | 97 | |||
| n.a. | Envelope (E) protein of the West Nile virus | 95 | |||
| 1–100 mM β-ME | Full-length Xenopus IRBP and its individual modules | 102 | |||
| Sel | –GCUG– | PAO resin | 10 mM DMPS | Rat thioredoxin reductase TrxR1 | 104, 106 and 107 |
| Up to 100 mM DMPS | Sec-containing domestic cat allergen Feld 1 | 105 | |||
| Tetracysteine | –CCXXCC– | FlAsH resin | 50 mM DTT or 5 mM DMPS | Kinesin | 89 |
| 10 mM DMPS | Calmodulin | 46 | |||
| 50 mM DMPS | RNA polymerase A | 118 | |||
| SplAsH resin | 10 mM DMPS | Green fluorescent protein (GFP) | 121 | ||
4.1. Arsenic affinity chromatography
Several methods are available for the purification of thiol-containing proteins including the use of immobilized heavy metal affinity resins, thiol–sulfide exchange resins, and chelated zinc affinity resins. Unlike arsenic affinity chromatography, none of these methods permit the separation of mono- and dithiol-containing molecules.74 Heavy metal affinity media interacts similarly with both mono- and dithiols.73 Thiol–sulfide exchange media does not bind to dithiols.74 Zinc affinity media cannot differentiate proteins containing histidines from proteins containing dithiols.75 Trivalent arsenicals such as PAO (Table 1) with at least two binding sites for thiols, bind strongly to proteins containing two thiols in close proximity to one another, forming stable dithioarsenicals. The affinity resins are composed of beads modified by hydrophobic trivalent arsenicals. Fig. 5 depicts the interaction between a typical PAO-modified affinity resin and a dithiol protein. PAO-modified resins have a higher affinity for dithiol proteins than monothiol proteins.76–78 This difference in binding affinity between trivalent arsenicals and mono- and dithiols allows for the separation of mono- and dithiol containing proteins by using arsenic affinity chromatography.79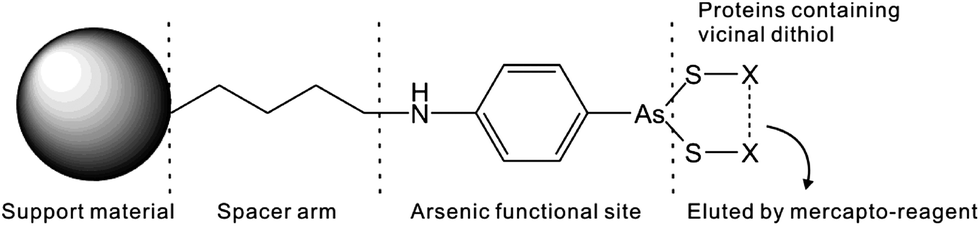 | ||
| Fig. 5 The basis of capturing arsenic-binding proteins with a PAO-based affinity resin. | ||
In the presence of high concentration of low molecular weight dithiols, the binding between arsenic and thiol-containing proteins becomes reversible and the competition can contribute to the elution of thiol-containing proteins from arsenic affinity resins. β-mercaptoethanol, 2,3-dimercapto-1-propanesulfonic acid (DMPS), or dithiolthreitol (DTT) are used for the elution of thiol-containing proteins bound to an arsenic affinity resin. Both gradient and step elutions are possible by increasing the concentration of dithiol in the elution buffer. These techniques allow for the separation of weakly bound proteins from more strongly bound proteins.74 Furthermore, proteins containing dithiols can be separated on the basis of the strength of their interaction with the resin. The strength of protein interaction with an arsenic affinity resin depends on the close proximity of cysteines to each another in the protein.73,79
A summary of arsenic affinity resins is listed in Table 3. There are several varieties of PAO affinity resins including Sepharose,73,74,80–84 Affigel,78,85 Eupergit C beads,86 carboxymethyl (CM)-Bio-Gel supports,87 and Glycidyl methacrylate grafted macroporous polysulfone membranes20 coupled to one of the following arsenic functional groups: p-aminophenylarsine oxide (PAPAO),73,74,78,84–88p-[(bromoacetyl)-amino]phenylarsenoxide (BrAcNPAsO),80 Cymelarsan,81 or p-arsanilic acid (PAPA).82,83,88 An atom spacer is frequently included between the support and the arsenic moiety. The biarsenical fluorescein dye FlAsH can also be incorporated into an affinity resin that binds to proteins containing the sequence CCXXCC in an α-helix.43 FlAsH affinity chromatography employs milder elution conditions than the other arsenic affinity resins, allowing for the fully intact and active protein to be purified.89
| Support material | Functional arsenicals | Spacer arm | Note | Ref. |
|---|---|---|---|---|
| CNBr-activated Sepharose 4B | p-Aminophenylarsine oxide (PAPAO) | n.a. | 50 g CNBr-activated Sepharose 4B was reacted with 122 mg PAPAO in 10% aqueous dimethylsulfoxide (DMSO) for 2 h | 73 |
| Affigel 10 | PAPAO | 10 atom | 73.2 mg PAPAO was dissolved in 4 mL of 0.2 M β-ME in DMSO, and reacted with 25 mL of Affigel 10 for 24 h | 78 |
| Carboxymethyl (CM) Bio-Gel A | PAPAO | 2 carbon | 25 mL CM Bio-Gel A was first reacted with 150 mL of 150 mM N,N′-dicyclohexylcarbodiimide and N-hydroxysuccinimide in DMSO, and then reacted with 92 mg PAPAO in 115 mL DMSO for 3 h | 87 |
| CNBr-activated Sepharose 4B-CL | PAPAO | 6 carbon | 30 g CNBr-activated Sepharose 4B-CL was modified with 6-aminohexanoic acid, and then coupled to 85 mg PAPAO in 4 mL DMSO with the addition of water-soluble carbodiimide | 74 |
| Activated CH-Sepharose 4B | p-[(Bromoacetyl)-amino] phenylarsenoxide (BrAcNPAsO) | n.a. | 2 mM BrAcNPAsO was first reacted with 1 mM β-mercaptoethylamine, and then reacted with activated CH-Sepharose 4B with the ratio of 1–2 μmol BrAcNPAsO per mL resin | 80 |
| Activated CH-Sepharose 4B | Cymelarsan | 6 carbon | 5 mmol Cymelarsan in 5 mL NaCl/phosphate buffer was reacted with activated CH-Sepharose 4B for 2 h | 81 |
| CNBr-activated Sepharose 4B | p-Arsanilic acid (PAPA) | n.a. | 3.4 g CNBr-activated Sepharose 4B was reacted with PAPA (1 mg mL−1) in 0.1 M NaHCO3 buffer for 2 h | 82 |
| Eupergit C | PAPAO | 3 atom | 0.5 g Eupergit C was reacted with 0.1 g PAPAO in 25% DMSO for 24 h | 86 |
| Glycidyl methacrylate (GMA) grafted macroporous polysulfone membranes (PSf) | PAPA, PAPAO, p-aminophenylarsine hydrochloride (PAPACl) | n.a. | GMA-grafted PSf was soaked in a 0.15 M 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMSO–water solution of PAPA, PAPAO or PAPACl for 10 h, and then activated in 0.2 M ME for 3 h. (A reduction step was done in the case of PAPA) :1 DMSO–water solution of PAPA, PAPAO or PAPACl for 10 h, and then activated in 0.2 M ME for 3 h. (A reduction step was done in the case of PAPA) |
88 |
| NHS-activated Sepharose gel | PAPA | n.a. | 60 mM APA was reacted with Sepharose gel (molar ratio was 15) in phosphate buffer overnight to prepare As(V)-immobilized Sepharose. As(V)-immobilized Sepharose was suspended in GSH solution at 15 mg mL−1 overnight to form As diglutathione-immobilized Sepharose. As diglutathione-immobilized Sepharose is stable in 25 mM GSH solution, and can be hydrolyzed to produce As(III)-immobilized Sepharose | 83 |
| Affi-Gel 10 | PAPAO | 10 atom | 32.8 mg PAPAO in 10 methanol was incubated with 4 mL of 50% Affi-Gel 10 in isopropanol | 85 |
| CNBr-activated Sepharose 4B | PAPAO | 6 carbon | 3 g CNBr-activated Sepharose 4B was first reacted with 6 mL of 0.25 M 6-aminohexanoic acid in water containing 0.1 M NaHCO3 overnight, and then coupled with 55 mg PAPAO by cross-linking reagents. | 84 |
4.2. Thioredoxin fusion proteins
E. coli thioredoxin was introduced in 1993 by LaVallie et al.90–92 as a fusion partner of recombinant proteins. Fusion proteins were typically purified by ion exchange chromatography. Patel-King et al. were the first to use PAO affinity chromatography for purifying thioredoxins,93,94 and by using sequence analysis, they demonstrated that the 14- and 16 kDa outer arm dynein light chains (DLC14 and DLC16) from Chlamydomonas flagella were novel members of the thioredoxin superfamily. As a fusion protein with maltose-binding protein (MBP) at the C-terminus or as a fusion protein cleaved from MBP with Factor Xa, both DLCs have high affinity for PAO affinity resins. The immobilized PAO, which binds protein vicinal thiols, has a high affinity for the CysGlyProCys active site of thioredoxin. The strong binding of PAO to the two cysteines in the CGPC region of thioredoxin has been demonstrated from mass spectrometry analyses of interactions between arsenicals and thioredoxin.95 PAO affinity chromatography has been widely used for thioredoxin fusion proteins.96–99In 1996, Lu et al.100 constructed mutant forms of thioredoxin that presented a patch of histidine residues on the surface of the molecule (termed ‘histidine patch’ thioredoxin), which made possible a convenient, generic, and specific affinity purification of thioredoxin fusion proteins, by using metal chelating affinity chromatography (IMAC). To facilitate purification by PAO resins, Wang et al.101 expressed a recombinant envelope (E) protein of the West Nile virus fused to thioredoxin at the N-terminus and a polyhistidine tag at the C-terminus. Recently, Gonzalez-Fernandez et al.102 expressed the first full-length Xenopus interphotoreceptor retinoid-binding protein (IRBP) and its individual modules using either regular thioredoxin or ‘histidine patch’ thioredoxin as fusion partners. The purification was achieved by a combination of ammonium sulfate precipitation, ion exchange chromatography, and affinity chromatography, based on either arsenic or Ni2+, depending on whether the thioredoxin was wild-type or mutant. Thioredoxin expression vectors and PAO affinity resins were commercialized by Invitrogen under the brand names of pTrxFus™ (for wild type thioredoxin), pThiolHis™ (for histidine patch thioredoxin), and Thiobond™. It has been demonstrated that binding to PAO does not induce alterations in the structure of Trx.103
4.3. Sel-tagged proteins
Arnér and coworkers104,105 applied PAO affinity chromatography to purify recombinant rat TrxR1, which is a selenoprotein containing a C-terminal tetrapeptide motif –Gly–Cys–Sec–Gly–COOH, where Sec is selenocysteine (U in one-letter code). They reasoned that the selenothiol motif would have a high affinity toward PAO, because As–Se is more stable than As–S with respect to hydrolysis. This high affinity proved to be true: in contrast to other vicinal dithiol-containing proteins, selenothiol-containing TrxR was not eluted from the resin with either 1 M β-mercaptoethanol or 10 mM DTT. A stronger PAO chelating agent, DMPS, was required to elute the selenothiol-containing protein.106 Inspired by this success Rengby et al. developed a Sel-tag technique,104 and found that the same selenocysteine-containing redox active C-terminal motif naturally in mammalian TrxR1 could be tapped as a fusion tag for recombinant non-selenoproteins expressed in E. coli. The Sel-tag could be used subsequently as a protein tag for PAO Sepharose-based single-step purification of the recombinant protein; selenocysteine-targeting protein labeling with electrophilic fluorescent probes; or radiolabeling with either γ-emitting 75Se or short-lived positron emitters such as 11C suitable for Positron Emission Tomography (PET) imaging.104,107–111 These novel biotechnological applications are based on the unique chemical properties of selenocysteine.112–114 The application of the Sel-tag, however, has a few prerequisites for the protein of interest. Firstly, it must be expressed in E. coli. Secondly, the function or activity of the protein must not rely on its C-terminus since the Sel-tag can only be introduced at the C-terminus of the protein. Finally, the protein must tolerate reduction with DTT or other equivalent reductants because the Se–S bond formed between the selenocysteine and cysteine residues within the Sel-tag has to be reduced to serve as the basis for purification on PAO affinity resins. By use of the genetic tailoring methodology developed by Arnér et al.112 that enables direct Sec-insertion at a pre-defined UGA codon, several proteins have successfully been Sel-tagged and purified by PAO affinity chromatography.112,115,1164.4. Tetracysteine-tagged proteins
Thorn et al.89 were the first to purify tetracysteine-tagged proteins by immobilizing the biarsenical FlAsH. To prepare the FlAsH affinity matrix, they began with aminofluorescein, coupled it to β-alanine, and then converted it to β-alanyl FlAsH, after mercuration and transmetallation. The β-alanyl FlAsH was then conjugated with N-hydroxysuccinmide (NHS) functionalized agarose beads. A fully active tetracysteine-tagged kinesin was purified from crude E. coli lysates using FlAsH affinity chromatography. A similar strategy was also utilized by Adams et al.46 to purify tetracysteine-tagged calmodulin. They immobilized FlAsH in a different manner, by coupling the N-hydroxysuccinimide ester of carboxyFlAsH to an amino-modified agarose support. The tetracysteine tagged proteins obtained by FlAsH affinity chromatography were shown to be generally purer than the corresponding polyhistidine tagged ones obtained with IMAC. The latter are often contaminated with Ni2+ or Co2+ and other histidine-rich intrinsic proteins. Compared with IMAC, which requires high concentrations of imidazole or pH changes for elution, the elution conditions for FlAsH affinity chromatography are mild. Bound proteins are typically eluted by millimolar concentrations of DTT or DMPS, making FlAsH affinity chromatography ideal for recovering fully active proteins and for the purification of intact protein complexes. FlAsH resin was prepared later by Mayer et al.,117,118 to study protein–protein interactions. They used FlAsH resin to pull-down protein complexes and then identified the complexes using mass spectrometry. They synthesized FlAsH–ethylenediamine starting from carboxyfluorescein and coupled it to N-hydroxysuccinimide-activated glass beads. By using a tetracysteine-tagged RNA polymerase A as a bait protein, they not only isolated the Shewanella oneidensis RNA polymerase core enzyme but also identified many other transient interactors. Recently, a tetracysteine tag was introduced by Giannone et al.119,120 into their dual-tagging system for studying mammalian protein–protein interactions. The tetracysteine tag serves not only as a “handle” for purifying protein complexes when combined with FlAsH resins, but also can be used to detect bait protein subcellular localization and expression as described in Section 3. In addition, the putative interacting partners can be confirmed by co-localization and even the downstream purification progress can be monitored directly by SDS-PAGE. In addition to FlAsH, non-fluorescent, photochemically stable analogues have also been explored as affinity ligands for the purification of tetracysteine-tagged proteins.1214.5. Arsenic-binding proteins in cells
The binding of arsenic to cellular proteins affords a plausible explanation for how arsenic causes detrimental health effects because the binding of arsenic to a protein can change the protein's conformation and inhibit its function. Identifying these proteins can guide the development of arsenic-based drugs to target these proteins. Our group has developed a combination of arsenical affinity chromatography and tandem mass spectrometry to identify arsenic-binding proteins in the cellular proteome.86 We identified fifty proteins in the nuclear fraction and 24 proteins in the membrane/organelle fraction of A549 human lung cancer cells. These proteins captured by arsenic affinity chromatography were proven to have at least one cysteine available for arsenic-binding. Most of these were identified as arsenic-binding proteins for the first time and a number were identified in the presence of a large excess of non-specific proteins, indicating that the coupling of arsenical affinity chromatography and tandem mass spectrometry is a promising technique for the specific study of arsenic-binding proteins in the proteome.Joshi et al.122 isolated arsenic hypertolerant bacterial cells, Bacillus sp. Strain DJ-1, from the common industrial effluent treatment plant in Vapi, India, and studied arsenic binding proteins in this new strain. By using PAO affinity chromatography and mass spectrometry, they found that the DNA protection during starvation (DPS) protein in the cytoplasm has a high binding affinity for trivalent arsenicals. The induced DPS protein protects the cellular DNA from arsenic-triggered stress, and the binding of arsenite with DPS protein may be responsible for the hypertolerance and high arsenic accumulation by Bacillus sp. Strain DJ-1. Mizumura et al.83 investigated the binding of hepatic cytosolic proteins to pentavalent, trivalent, and glutathione-conjugated trivalent arsenicals by using three different arsenic-bound Sepharoses (As(V)-, As(III)-, and As(III) diglutathione-immobilized Sepharose). The results show that no protein bound to the As(V)-immobilized Sepharose, indicating that cytosolic proteins in HepG2 liver cells do not bind to As(V). Matrix-assisted laser desorption/ionization tandem mass spectrometry (MALDI-MS/MS) enabled the identification of the binding of four proteins to glutathione conjugated trivalent arsenic: peroxiredoxin 2 (thioredoxin peroxidase), cytosolic inorganic pyrophosphatase, phosphoglycerate kinase, and KM-102-derived reductase-like factor (thioredoxin reductase). The proteins that specifically bound to trivalent arsenic include protein disulfide isomerase-related protein 5 (thioredoxin domain) and peroxiredoxin 1/enhancer protein (peroxiredoxin family). These results demonstrate that the hepatic cytoplasm proteins that bind to hydroxylated As(III) are not the same as those that bind to glutathione conjugated As(III). Chang et al.123 identified arsenic-binding proteins in the arsenite-resistant SA47 and arsenite-sensitive CHOA cells by using a PAO-agarose matrix combined with proteomic techniques to better characterize the interaction of protein–cysteines with arsenite. Nineteen proteins, functionally categorized into three groups (metabolic, stress and developmental processes, according to the gene ontology information), were found to be differentially expressed, either in CHOA or in SA7 cells. The number of cysteine residues ranged widely from zero to nine among the identified arsenic-binding proteins. Recombinant reticulocalbin-3 precursor (RCN3, without cysteine residues), heat shock protein beta-1 (HSP27, with one cysteine residue), peroxiredoxin 6 (Prdx6, with one cysteine residue), and galectin-1 (GAL1, with six cysteine residues) produced in E. coli were re-applied onto the PAO-agarose matrix to verify their arsenite-binding capacity. Recombinant RCN3, HSP27 and GAL1 were retained, but Prdx6 was not retained by the PAO-agarose matrix. These results indicate that cysteine residues may play a crucial role in relation to arsenic binding in some proteins such as HSP27 and GAL1, while this interaction might be mediated by other factors in case of RCN3 and Prdx6.
Arsenical affinity chromatography combined with tandem mass spectrometry provides an efficient proteomic approach to extensively study the interaction of arsenicals with proteins in cells, but it requires cell lysis prior to protein binding on the arsenic affinity column. An alternative to the in situ capture of arsenic-binding proteins in cells is to use an arsenic–biotin conjugate. Biotinylated phenylarsonous acids were first designed as bifunctional reagents to study spatially close thiols.124 The arsenical group can bind to spaced thiols forming a ring structure, and biotin can bind to avidin allowing for the detection of the reagent-macromolecular complex. Based on this concept, Donoghue et al.125 synthesized biotin conjugated GSAO to identify cell-surface proteins that contain closely spaced thiols. Ten and twelve distinct proteins in the surface of the endothelial and fibrosarcoma cells, respectively, were found to incorporate GSAO-B. To capture arsenic-binding proteins in situ in MCF-7 human breast cancer cells, Zhang et al.126 prepared PAPAO–biotin conjugate to treat with cells. After the treated cells are lysed, the proteins binding to the PAPAO–biotin conjugate can be isolated with streptavidin resin. Fifty arsenic-binding proteins were identified using MALDI-MS. These proteins can be classified into three main categories: metabolic enzymes, structural proteins, and stress response proteins. They further confirmed β-tubulin and PKM2 as arsenic-binding proteins, and found that arsenic binding inhibited tubulin polymerization but did not impair PKM2 activity. Recently, our group designed and synthesized an azide-labeled arsenical, p-azidophenylarsenoxide (PAzPAO), as a novel arsenical “bait” to capture cellular arsenic-binding proteins in living cells, using copper-free click chemistry.127 This approach allowed for the identification of arsenic-binding proteins in A549 cells using a shotgun proteomics strategy.
5. Arsenic biosensors
Over the last decade, there has been increased interest in the development of sensors for the field detection of arsenic128 in response to the worldwide arsenic problem. Abiotic sensors involving colourimetry and electrochemistry are well established.128 Silver and coworkers129 developed bacterial biosensors for arsenic detection where the ars operon encoding an arsenic resistance system was utilized; these arsenic biosensors have been further improved by other researchers.130–132The first report133 on a genetically modified bioluminescent bacterial sensor for the detection of naphthalene appeared in 1990, and the basic construction principle has remained essentially unchanged ever since. Briefly, a genetically modified bacterial sensor is analogous to a natural regulatory circuit, consisting of a transcription regulator, promoter, and operator in a microorganism (usually E. coli). The regulatory circuit is artificially fused with a promoterless reporter gene, encoding an easily measurable protein.134 Induction of the promoter by the transcription regulator interacting with a chemical target leads to the expression of the reporter gene, yielding an output signal that can be detected, calibrated and interpreted.134 In the case of arsenic, ArsAB, a highly effective arsenite efflux pump, together with an arsenate reductase ArsC, comprises the bacterial defense system against arsenite and arsenate. This defense system provides the platform for arsenic detection. ArsR, an arsenite sensing protein in this defense system, transcriptionally represses the expression of these arsenic defense genes (the operator), including itself. In the absence of arsenite, ArsR binds to a specific DNA sequence named ParsR overlapping with the binding site for RNA polymerase (the promoter), thereby repressing the arsenic defense genes from being transcribed (Fig. 6a). In the presence of arsenite, the affinity of ArsR to the promoter decreases, allowing transcription to occur (Fig. 6b).128 ArsR has been most commonly used to construct bacterial biosensors for arsenic. Arsenate can be detected at lower sensitivity compared to arsenite probably because arsenate is required to be reduced to arsenite by the ArsC before being effluxed.135
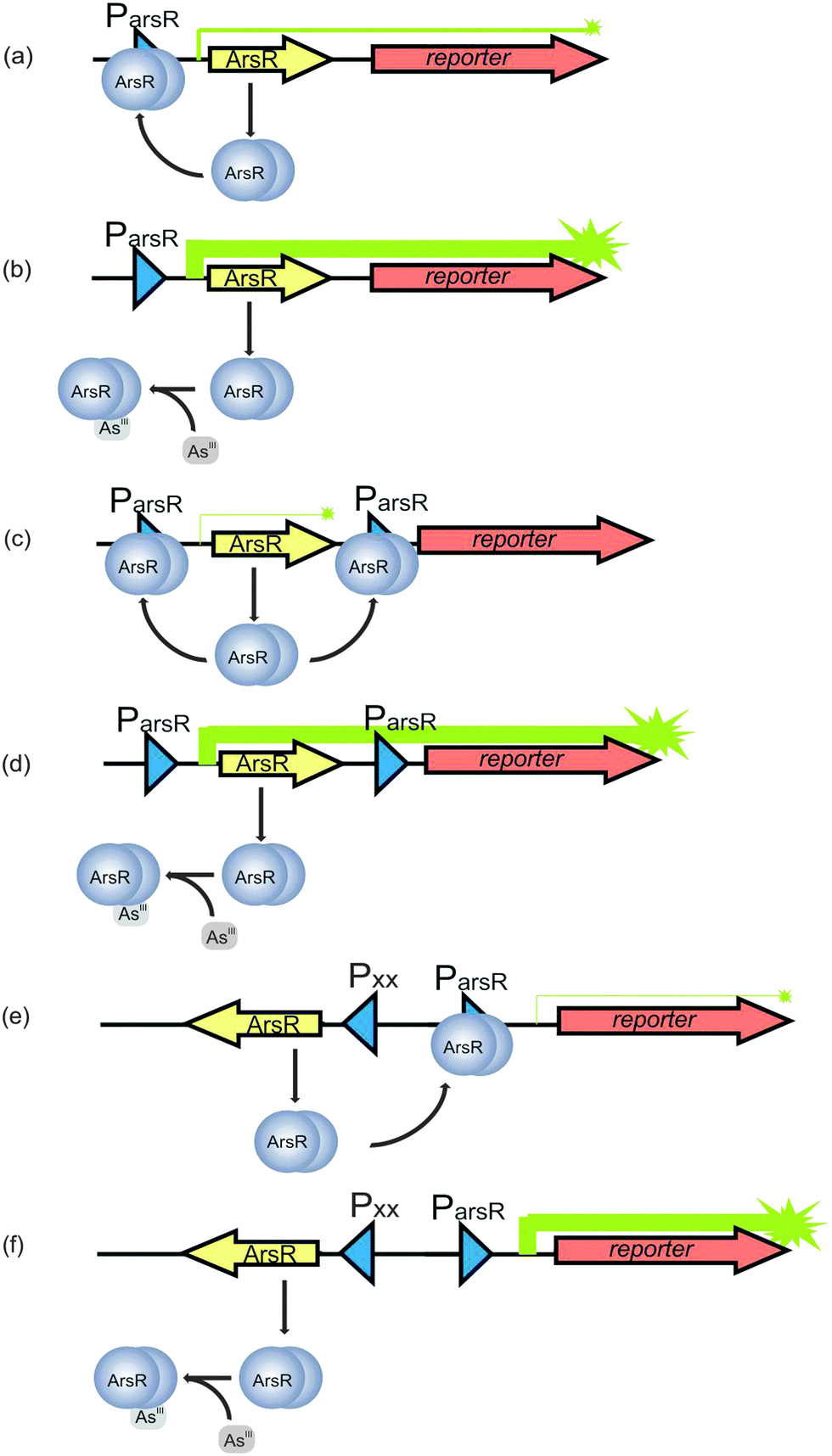 | ||
| Fig. 6 Principle of the arsenic biosensors based on ArsR. The arsR gene controls expression of the reporter gene (symbolized by arrows). (a) In the absence of As(III), transcription is repressed by a basal level of ArsR synthesis via binding of ArsR (symbolized as dimer protein) to the operator site of the arsR promoter (Pars). Some background expression of the reporter protein occurs. (b) In the presence of As(III), ArsR losses its affinity for Pars. Repression is relieved and expression of ArsR itself and the reporter protein is very high. (c) A secondary ArsR promoter is inserted downstream of arsR to reduce background expression of the reporter gene. In the absence of As(III), only the arsR mRNA is produced as background. ArsR binds to both ArsR promoters and prevent RNA polymerase from reading the reporter gene. (d) When the bacteria encounter As(III), both the defense system and the reporter protein synthesis are de-repressed. (e) In the uncoupled circuit, expression of arsR is controlled by an arsenite independent promoter, and the expression of reporter gene is controlled by Pars. In the absent of As(III), the expression of reporter gene is still repressed by ArsR and thus background is reduced. (f) In the presence of As(III), the expression of reporter gene is de-repressed (modified from Stocker et al.132 and Merulla et al.138). | ||
The repression of transcription due to the binding of ArsR to ParsR, however, is incomplete and low levels of ArsR are always present.136 It is accepted that a basal level of ArsR expression is required for the system to function properly. However, the constitutively low levels of gene expression in the absence of arsenic require the optimization of the biosensor to reduce the background expression of the reporter proteins in order to achieve a satisfying detection limit. This is typically done by introducing a second promoter for ArsR.132 Therefore, in the developed arsenic sensor, a second copy of the operator-promoter sequence for ArsR is constructed (Fig. 6c) by linking the arsR, the ParsR upstream and downstream of arsR and the reporter gene together. The assembly is then introduced into bacteria by transfection.128 The most likely explanation of how this approach reduces the background is that the downstream ParsR might block the RNA polymerase even when the transcription starts from the upstream ParsR in the absence of arsenite. When the bacteria encounter arsenic, both the defense system and the reporter protein synthesis are de-repressed (Fig. 6d).128 Aside from insertion of an extra ParsR to insulate the arsR based gene circuitry, other approaches, such as an uncoupled ArsR gene circuitry137,138 and gene oscillator,139 are also used to reduce background expression of the reporter gene. In the uncoupled circuit configuration, expression of arsR is controlled by an arsenite independent promoter, which is uncoupled from the expression of reporter gene controlled by ParsR, while the expression of reporter gene is still under the control of ArsR (Fig. 6e and f). In the other approach, the gene period oscillator measures the arsenite-dependent de-repression of the system not by the absolute expression level of the reporter gene but by the duration of the oscillation period. De-repression signal is recorded from thousands of oscillating colonies of bacterial cells, which smooth out the intercellular variability in circuit behavior and decrease the cellular environmental noise. Another advantage the oscillator offers is that the frequency measurement is less sensitive than the intensity measurement to instrumental factors in optical detection.139
Two other promoters as the alternatives to ParsR for inducing arsR operon are arcA found in Aspergillus niger140 and UFO1 found in Saccharomyces cerevisiae.141 These two promoters are summarized in a previous review.142
Generally, the optical technique is the most widely used signal readout in the microbial biosensors. In most cases, the reporter gene used is one of the following: lacZ, encoding β-galactosidase, which acts on the chromogenic substrate X-gal to generate a blue color; luxAB or luxCDABE, encoding bacterial luciferase, or luc, encoding firefly luciferase, which generates a luminescent signal; or gfp or variants, encoding fluorescent proteins.143–145 The most recently found reporter genes are phiYFP in Phialidium sp.,146 encoding a yellow florescent protein and crtI in Rhodopseudomonas palustris, encoding the carotenoid-metabolizing protein CrtIBS whose color changes from yellow to red when encountering arsenite.147 The applications of these reporter genes have been well summarized in another review.142 Another colorimetric biosensor not utilizing a reporter gene is based on Prussian Blue (PB). In this biosensor, arsenite inhibits the electron transfer from E. coli to ferricyanide during E. coli respiration and thus represses the production of ferrocyanide which subsequently reacts with ferric ions to produce PB.148
In addition to the optical techniques, a pH based biosensor was developed, making use of the induced expression of β-galactosidase (from the fusion gene arsR-lacZ) in E. coli in the presence of arsenic. The formation of β-galactosidase caused a change of pH that could be measured using a simple pH indicator.149
MtrB is essential for metal reduction by Shewanella oneidensis. Webster et al. fused mtrB with arsR to drive the expression of mtrB by the arsenic inducible promoter. This bioelectrochemical system produced a direct electric signal that could be remotely monitored and achieved a detection limit of 40 μM arsenite. The remote controlling property makes the device suitable for field tests without on-site technicians.150
A number of bacteria-free biosensors have been developed based on the binding of arsenic to proteins. These proteins are generally immobilized on the surface of a solid support and the signal of arsenic is determined by electrochemical techniques. Silvia et al. immobilized acetylcholinesterase on screen-printed electrodes and determined arsenite concentration by monitoring the inhibited acetylcholinesterase enzyme activity. The detection limit was 1.1 × 10−8 M.151 Cytochrome C was also used to develop a biosensor since the activity of cytochrome C can be inhibited by arsenite. The detection limit for this biosensor depends on which kind of detection methods is employed (e.g., cyclic voltammetry, square wave voltammetry and electrochemical impedance spectroscopy), but usually it is in the range of 8–22 μM.152 In another approach,153 arsenite oxidase was deposited on the multiwalled carbon nanotube modified glassy carbon electrode and its enzymatic activity against arsenite was exploited to build arsenic biosensors. Liu et al.154 constructed a sensitivity-enhanced 3D localized surface plasmon resonance spectroscopy (LSPR) system by combining a highly sorptive polymer with gold nanoparticles. They then immobilized the ArsA ATPase (the catalytic subunit of the ArsAB arsenite efflux pump and one of the five proteins encoded by the ars operon) to the polymer and used the LSPR to test the affinity of ArsA ATPase to arsenite. This technique was envisioned to provide a strategy to sense and study the kinetics of arsenic with other biomolecules. The amperometric detection methods showing specificity to arsenate were built based on the inhibitory effect of arsenate on acid phosphatase activity towards the hydrolysis of phenyl phosphate155 or 2-phospho-L-ascorbic acid,156 which provided detection limits of 2 nM and 0.11 μM, respectively. Finally, immobilized L-cysteine was used as a redox substrate for arsenate and its oxidation was used to measure arsenate concentration with a detection limit of 1–30 μg L−1.157 Both bacterial and bacteria-free biosensors for arsenic detection have been reviewed.142,158,159
Although most of the biosensors were being developed for inorganic arsenicals, biosensors for organoarsenicals are also in demand because of the environmental and health risks of organoarsenicals. In the regular ArsR system, trimethylarsenoxide was detected at 10% relative response of arsenite.135 A biosensor that specifically detects trivalent methylated arsenicals and aromatic arsenicals was developed by Rosen et al.160 By using AfArsR in Acidithiobacillus ferrooxidans that had a different expression level from the commonly used ArsR in E. coli plasmid R773, researchers could achieve selectivity for phenyl As(III) > methyl As(III) > inorganic As(III).
Typically, the bacteria-based arsenic biosensors require several hours of incubation of the sample with the bacteria to achieve reliable results. For example, Buffi et al.161 reported the time needed to reproducibly detect a concentration of 50 μg L−1 arsenite was up to 120 minutes and up to 3 hours for 10 μg L−1 arsenite. Despite the current technical hurdles facing the field application of bacterial biosensors in general (especially the long incubation time), arsenic biosensors hold some promise for increased application in the field. The biosensors that have been developed feature two designs: bacteria in a liquid phase or freeze-dried bacteria immobilized on a solid chip. The latter are more easily-managed and friendly for field study. In a large-scale comparative field trial, an E. coli-based ArsR-LuxAB biosensor was applied to samples from nearly 200 groundwater wells in Vietnam and this biosensor performed as well as regular chemical analysis.162 In another example, a field test kit based on living, lyophilized bacterial bioreporters emitting bioluminescence was used in six villages in Bangladesh.163 Arsenic in rice was measured using an E. coli ArsR-LuxAB reporter assay after enzymatic pre-treatment of rice powder slurry.134 Using ArsB-LuxAB bacteria sensors, Cai et al.164 detected arsenic near sites harboring chromated copper arsenate (CCA)-treated wood. Truffer et al. has developed portable agarose-encapsulated bacteria chips to detect arsenic in drinking water with detection limits of 0.10 μg L−1 and 0.5 μg L−1 in 100 and 80 minutes, respectively, which is very sensitive.165 Recent work published by Jouanneau et al. shows that it is possible to use a biosensor with immobilized freeze-dried bacteria over a long period (10 days) with a reproducibility of 3% standard error.166
One unique feature associated with bacterial bioassays for arsenic is that they could be species specific. The field arsenic tests at this stage detect only inorganic arsenic species that are more bioaccessible, providing useful information for determining bioremediation or ecotoxicological-safety endpoints.167 Selective detection of bioaccessible arsenic species, and inexpensive on-site application could be advantages that influence the development of arsenic biosensors for field studies, provided that reaction times can be shortened.
6. Conclusions and perspectives
The success of arsenic treatment of human APL cancer has revived research on arsenic-based drugs, and arsenic has been tested for the treatment of solid tumors in the liver, breasts, ovaries, etc. Because of the affinity of arsenic for sulfur and the existence of thiol-containing proteins in living organisms, arsenic can bind to a number of these proteins although the binding strength varies. A knowledge gap on identifying specific arsenic-binding proteins and the consequences of this binding slows down the development of arsenic therapy for various cancers. Although structure modification of both inorganic and organic arsenicals has been attempted to endow arsenic with selectivity for specific cells or certain organelles, the advances achieved thus far are very limited. The implementation of nanomaterials for arsenic delivery confronts the challenge of cell uptake and efflux. In principle, the targeted delivery of arsenic carried by nano-materials can be achieved by proper design of the carrier. Current research on arsenic encapsulation using liposomes and polymeric nanoparticles indicates promising targeted delivery of arsenicals to several solid tumor cell lines.Diverse arsenical fluorescence probes have been created and shown high application potential. The biarsenical probes, with different colors or with different distances between the two arsenic atoms, enable the temporal and spatial labeling and imaging of multiple target proteins that are genetically fused with the tetracysteine tags. Various fundamental biological processes have been revealed by the development of imaging strategies using the biarsenical probes. For the monoarsenical dyes, the high binding affinity of trivalent arsenicals to thiols compared to oxidized disulfide, make them the natural probes to explore the oxidative states of cysteines, so as to trace the cellular redox status. When arsenical probes are modified with functional biomolecule (e.g., GSH in GSAO), they exhibit excellent selectivity and metabolic kinetics in living organism for clinical diagnosis. Further integrating arsenical probes with new fluorescent dyes and novel imaging techniques will facilitate the development of more versatile imaging approaches for fundamental research, biological analysis, and clinical diagnosis.
Arsenic affinity chromatography is an efficient technique for the purification of arsenic-binding proteins. PAO-based resins have a high affinity for proteins containing a C(X)nC motif, where n is from 2 to 6. FlAsH or ReAsH have a high affinity for proteins containing the tetracysteine sequence CCXXCC. The discovery of proteins with specific binding to arsenic via arsenic affinity chromatography can improve our understanding on the interaction of arsenic and proteins. The identified specific arsenic-binding proteins can also serve as new options for arsenic applications, e.g., imaging, sensing, and targeted delivery. Therefore, techniques for selective capture and identification of arsenic-binding proteins will continue to advance research on arsenicals and their applications.
Arsenic biosensors integrate knowledge of arsenic binding to proteins with sensing technology. Biosensors with high sensitivity and selectively for arsenic detection have potential in areas including but not limited to environmental monitoring and public health protection. Important improvements have been made to reduce the background signal and enhance the sensitivity for detecting arsenic. The design originating from the arsenic resistance system in bacteria has now been developed to an analytical device that is suitable for field work. Thus arsenic biosensors have been shown to be effective in detecting arsenic in water at concentrations relevant to the World Health Organization drinking water guideline (10 μg L−1). Improvements are needed to increase the reproducibility, decrease the reaction time, and make the biosensors portable and storable. Although currently most of the arsenic biosensors require online electrochemical or optical instrument to read the signal, a remote-control device could be invented to facilitate ease of use for both trained technicians and general users. Confronting the arsenic problem worldwide, arsenic biosensors hold an untapped promise in rapid and routine monitoring of arsenic.
All four areas of applications described above are based on our understanding of arsenic interaction with proteins. We can expect even more when advances in technology are combined with a deeper understanding of these processes.
Acknowledgements
We thank the Canadian Institutes of Health Research, the Natural Sciences and Engineering Research Council of Canada, the Canada Research Chairs Program, Alberta Innovates, and Alberta Health for their support. We thank the Government of the Province of Alberta for a Queen Elizabeth II Graduate Scholarship (to A.P.) and the China Scholarship Council for a Doctoral Scholarship (to Q.L.) and a Visiting Postdoctoral Fellowship (to B.C.).References
- S. W. Shen, X. F. Li, W. R. Cullen, M. Weinfeld and X. C. Le, Chem. Rev., 2013, 113, 7769–7792 CrossRef CAS PubMed.
- R. W. Hedges and S. Baumberg, J. Bacteriol., 1973, 115, 459–460 CAS.
- B. P. Rosen, Trends Microbiol., 1999, 7, 207–212 CrossRef CAS.
- M. Lu, H. Wang, Z. Wang, X. Li and X. C. Le, J. Proteome Res., 2008, 7, 3080–3090 CrossRef CAS PubMed.
- D. Doyle, Br. J. Haematol., 2009, 145, 309–317 CrossRef CAS PubMed.
- S. Waxman and K. C. Anderson, Oncologist, 2001, 6, 3–10 CrossRef CAS PubMed.
- M. Sarquis, J. Chem. Educ., 1979, 56, 815–818 CrossRef CAS.
- J. Liu, Y. F. Lu, Q. Wu, R. A. Goyer and M. P. Waalkes, J. Pharmacol. Exp. Ther., 2008, 326, 363–368 CrossRef CAS PubMed.
- W. R. Cullen, Is arsenic an aphrodisiac? The sociochemistry of an element, Royal Society of Chemistry, 2008 Search PubMed.
- P. J. Dilda and P. J. Hogg, Cancer Treat. Rev., 2007, 33, 542–564 CrossRef CAS PubMed.
- N. C. Lloyd, H. W. Morgan, B. K. Nicholson and R. S. Ronimus, Angew. Chem., Int. Ed., 2005, 44, 941–944 CrossRef CAS PubMed.
- K. J. Williams, J. R. Soc. Med., 2009, 102, 343–348 CrossRef CAS PubMed.
- H. Sun, L. Ma, X. Hu and T. Zhang, Chin. J. Integr. Tradit. West. Med., 1992, 12, 170–171 Search PubMed.
- Z. X. Shen, G. Q. Chen, J. H. Ni, X. S. Li, S. M. Xiong, Q. Y. Qiu, J. Zhu, W. Tang, G. L. Sun, K. Q. Yang, Y. Chen, L. Zhou, Z. W. Fang, Y. T. Wang, J. Ma, P. Zhang, T. D. Zhang, S. J. Chen, Z. Chen and Z. Y. Wang, Blood, 1997, 89, 3354–3360 CAS.
- X. W. Zhang, X. J. Yan, Z. R. Zhou, F. F. Yang, Z. Y. Wu, H. B. Sun, W. X. Liang, A. X. Song, V. Lallemand-Breitenbach, M. Jeanne, Q. Y. Zhang, H. Y. Yang, Q. H. Huang, G. B. Zhou, J. H. Tong, Y. Zhang, J. H. Wu, H. Y. Hu, H. de The, S. J. Chen and Z. Chen, Science, 2010, 328, 240–243 CrossRef CAS PubMed.
- M. Jeanne, V. Lallemand-Breitenbach, O. Ferhi, M. Koken, M. Le Bras, S. Duffort, L. Peres, C. Berthier, H. Soilihi, B. Raught and H. de The, Cancer Cell, 2010, 18, 88–98 CrossRef CAS PubMed.
- V. Lallemand-Breitenbach, J. Zhu, Z. Chen and H. de The, Trends Mol. Med., 2012, 18, 36–42 CrossRef CAS PubMed.
- J. Lu, E. H. Chew and A. Holmgren, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12288–12293 CrossRef CAS PubMed.
- S. Gromer, S. Urig and K. Becker, Med. Res. Rev., 2004, 24, 40–89 CrossRef CAS PubMed.
- H. Chen, S. Pazicni, N. L. Krett, R. W. Ahn, J. E. Penner-Hahn, S. T. Rosen and T. V. O'Halloran, Angew. Chem., Int. Ed., 2009, 48, 9295–9299 CrossRef CAS PubMed.
- H. M. Chen, R. C. MacDonald, S. Y. Li, N. L. Krett, S. T. Rosen and T. V. O'Halloran, J. Am. Chem. Soc., 2006, 128, 13348–13349 CrossRef CAS PubMed.
- S.-M. Lee, O.-S. Lee, T. V. O'Halloran, G. C. Schatz and S. T. Nguyen, ACS Nano, 2011, 5, 3961–3969 CrossRef CAS PubMed.
- N. D. Winter, R. K. J. Murphy, T. V. O'Halloran and G. C. Schatz, J. Liposome Res., 2011, 21, 106–115 CrossRef CAS PubMed.
- H. Chen, R. Ahn, J. Van den Bossche, D. H. Thompson and T. V. O'Halloran, Mol. Cancer Ther., 2009, 8, 1955–1963 CrossRef CAS PubMed.
- C. Qian, Y. Wang, Y. Chen, L. Zeng, Q. Zhang, X. Shuai and K. Huang, Biomaterials, 2013, 34, 6175–6184 CrossRef CAS PubMed.
- Q. Zhang, M. R. Vakili, X.-F. Li, A. Lavasanifar and X. C. Le, Biomaterials, 2014, 35, 7088–7100 CrossRef CAS PubMed.
- D. G. Fatouros, P. V. Ioannou and S. G. Antimisiaris, J. Nanosci. Nanotechnol., 2006, 6, 2618–2637 CrossRef CAS PubMed.
- N. Wimmer, J. A. Robinson, N. Gopisetty-Venkata, S. J. Roberts-Thomson, G. R. Monteith and I. Toth, Med. Chem. (Sharjah, United Arab Emirates), 2006, 2, 79–87 CrossRef CAS.
- D. P. Lu, J. Y. Qiu, B. Jiang, Q. Wang, K. Y. Liu, Y. R. Liu and S. S. Chen, Blood, 2002, 99, 3136–3143 CrossRef CAS PubMed.
- H.-H. Zhu, D.-P. Wu, J. Jin, J.-Y. Li, J. Ma, J.-X. Wang, H. Jiang, S.-J. Chen and X.-J. Huang, J. Clin. Oncol., 2013, 31, 4215–4221 CrossRef CAS PubMed.
- W. Z. Zhao, X. Lu, Y. Yuan, C. S. Liu, B. C. Yang, H. Hong, G. Y. Wang and F. Y. Zeng, Int. J. Nanomed., 2011, 6, 1569–1577 CrossRef CAS PubMed.
- J. Z. Wu and P. C. Ho, Eur. J. Pharm. Sci., 2006, 29, 35–44 CrossRef CAS PubMed.
- Q. H. Zhao, Y. Zhang, Y. Liu, H. L. Wang, Y. Y. Shen, W. J. Yang and L. P. Wen, Med. Oncol., 2010, 27, 203–212 CrossRef CAS PubMed.
- X. Cheng, A. Quintas-Cardama, M. Golemovic, R. Zingaro, M.-Z. Gao, E. J. Freirech, M. Andreeff, H. M. Kantarjian and S. Verstovsek, Anticancer Res., 2012, 32, 2871–2880 CAS.
- M. Golemovic, A. Quintas-Cardama, T. Manshouri, N. Orsolic, H. Duzkale, M. Johansen, E. J. Freireich, H. Kantarjian, R. A. Zingaro and S. Verstovsek, Invest. New Drugs, 2010, 28, 402–412 CrossRef CAS PubMed.
- I. Mancini, G. Guella, M. Frostin, E. Hnawia, D. Laurent, C. Debitus and F. Pietra, Chem. – Eur. J., 2006, 12, 8989–8994 CrossRef CAS PubMed.
- D. Lu, M. L. Coote, J. Ho, N. L. Kilah, C.-Y. Lin, G. Salem, M. L. Weir, A. C. Willis and S. B. Wild, Organometallics, 2012, 31, 1808–1816 CrossRef CAS.
- S. J. Ralph, Met.-Based Drugs, 2008, 2008, 260146 Search PubMed.
- A. S. Don, O. Kisker, P. Dilda, N. Donoghue, X. Y. Zhao, S. Decollogne, B. Creighton, E. Flynn, J. Folkman and P. J. Hogg, Cancer Cell, 2003, 3, 497–509 CrossRef CAS.
- P. J. Dilda, E. E. Ramsay, A. Corti, A. Pompella and P. J. Hogg, J. Biol. Chem., 2008, 283, 35428–35434 CrossRef CAS PubMed.
- L. Horsley, J. Cummings, M. Middleton, T. Ward, A. Backen, A. Clamp, M. Dawson, H. Farmer, N. Fisher, G. Halbert, S. Halford, A. Harris, J. Hasan, P. Hogg, G. Kumaran, R. Little, G. J. M. Parker, P. Potter, M. Saunders, C. Roberts, D. Shaw, N. Smith, J. Smythe, A. Taylor, H. Turner, Y. Watson, C. Dive, G. C. Jayson and Cancer Research UK Drug Development Office Phase I Clinical Trial, Cancer Chemother. Pharmacol., 2013, 72, 1343–1352 CrossRef CAS PubMed.
- P. J. Dilda, S. Decollogne, L. Weerakoon, M. D. Norris, M. Haber, J. D. Allen and P. J. Hogg, J. Med. Chem., 2009, 52, 6209–6216 CrossRef CAS PubMed.
- K. K. Mann, B. Wallner, I. S. Lossos and W. H. Miller, Expert Opin. Invest. Drugs, 2009, 18, 1727–1734 CrossRef CAS PubMed.
- N. Garnier, G. G. J. Redstone, M. S. Dahabieh, J. N. Nichol, S. V. del Rincon, Y. X. Gu, D. S. Bohle, Y. Sun, D. S. Conklin, K. K. Mann and W. H. Miller, Mol. Pharmacol., 2014, 85, 576–585 CrossRef PubMed.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef CAS.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063–6076 CrossRef CAS PubMed.
- M. S. T. Goncalves, Chem. Rev., 2008, 109, 190–212 CrossRef PubMed.
- N. Soh, Sensors, 2008, 8, 1004–1024 CrossRef CAS PubMed.
- B. R. Martin, B. N. Giepmans, S. R. Adams and R. Y. Tsien, Nat. Biotechnol., 2005, 23, 1308–1314 CrossRef CAS PubMed.
- G. Gaietta, T. J. Deerinck, S. R. Adams, J. Bouwer, O. Tour, D. W. Laird, G. E. Sosinsky, R. Y. Tsien and M. H. Ellisman, Science, 2002, 296, 503–507 CrossRef CAS PubMed.
- C. Hoffmann, G. Gaietta, M. Bunemann, S. R. Adams, S. Oberdorff-Maass, B. Behr, J. P. Vilardaga, R. Y. Tsien, M. H. Ellisman and M. J. Lohse, Nat. Methods, 2005, 2, 171–176 CrossRef CAS PubMed.
- G. M. Gaietta, B. N. G. Giepmans, T. J. Deerinck, W. B. Smith, L. Ngan, J. Llopis, S. R. Adams, R. Y. Tsien and M. H. Ellisman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17777–17782 CrossRef CAS PubMed.
- O. Tour, S. R. Adams, R. A. Kerr, R. M. Meijer, T. J. Sejnowski, R. W. Tsien and R. Y. Tsien, Nat. Chem. Biol., 2007, 3, 423–431 CrossRef CAS PubMed.
- T. Fujii, Y. Shindo, K. Hotta, D. Citterio, S. Nishiyama, K. Suzuki and K. Oka, J. Am. Chem. Soc., 2014, 136, 2374–2381 CrossRef CAS PubMed.
- L. Wu, T. Huang, L. Yang, J. Pan, S. Zhu and X. Yan, Angew. Chem., Int. Ed., 2011, 50, 5873–5877 CrossRef CAS PubMed.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS PubMed.
- A. Pomorski and A. Krężel, ChemBioChem, 2011, 12, 1152–1167 CrossRef CAS PubMed.
- R. A. Scheck and A. Schepartz, Acc. Chem. Res., 2011, 44, 654–665 CrossRef CAS PubMed.
- C. Spagnuolo, M. Joselevich, F. C. Leskow and E. A. Jares-Erijman, in Advanced Fluorescence Reporters in Chemistry and Biology III: Applications in Sensing and Imaging, ed. A. P. Demchenko, Springer-Verlag, Berlin, 2011, vol. 10, pp. 263–295 Search PubMed.
- H. Cao, Y. Xiong, T. Wang, B. Chen, T. C. Squier and M. U. Mayer, J. Am. Chem. Soc., 2007, 129, 8672–8673 CrossRef CAS PubMed.
- N. Fu, Y. Xiong and T. C. Squier, Bioconjugate Chem., 2013, 24, 251–259 CrossRef CAS PubMed.
- N. Fu, Y. Xiong and T. C. Squier, J. Am. Chem. Soc., 2012, 134, 18530–18533 CrossRef CAS PubMed.
- M. Bates, T. R. Blosser and X. Zhuang, Phys. Rev. Lett., 2005, 94, 108101 CrossRef.
- G. T. Dempsey, M. Bates, W. E. Kowtoniuk, D. R. Liu, R. Y. Tsien and X. Zhuang, J. Am. Chem. Soc., 2009, 131, 18192–18193 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS PubMed.
- N. Fu, D. Su, J. R. Cort, B. Chen, Y. Xiong, W. J. Qian, A. E. Konopka, D. J. Bigelow and T. C. Squier, J. Am. Chem. Soc., 2013, 135, 3567–3575 CrossRef CAS PubMed.
- C. Huang, Q. Yin, W. Zhu, Y. Yang, X. Wang, X. Qian and Y. Xu, Angew. Chem., Int. Ed., 2011, 50, 7551–7556 CrossRef CAS PubMed.
- C. Huang, Q. Yin, J. Meng, W. Zhu, Y. Yang, X. Qian and Y. Xu, Chem. – Eur. J., 2013, 19, 7739–7747 CrossRef CAS PubMed.
- D. Park, A. S. Don, T. Massamiri, A. Karwa, B. Warner, J. MacDonald, C. Hemenway, A. Naik, K. T. Kuan, P. J. Dilda, J. W. Wong, K. Camphausen, L. Chinen, M. Dyszlewski and P. J. Hogg, J. Am. Chem. Soc., 2011, 133, 2832–2835 CrossRef CAS PubMed.
- E. J. Levy, M. E. Anderson and A. Meister, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 9171–9175 CrossRef CAS.
- D. Park, B. W. Xie, E. R. Van Beek, V. Blankevoort, I. Que, C. W. Löwik and P. J. Hogg, Mol. Pharmacol., 2013, 10, 3882–3891 CAS.
- B. W. Xie, D. Park, E. R. Van Beek, V. Blankevoort, Y. Orabi, I. Que, E. L. Kaijzel, A. Chan, P. J. Hogg and C. W. Löwik, Cell Death Dis., 2013, 4, e473, DOI:10.1038/cddis.2012.207.
- U. Hannestad, P. Lundqvist and B. Sörbo, Anal. Biochem., 1983, 126, 200–204 CrossRef.
- E. Kalef, P. G. Walfish and C. Gitler, Anal. Biochem., 1993, 212, 325–334 CrossRef CAS PubMed.
- T. T. Yip, Y. Nakagawa and J. Porath, Anal. Biochem., 1989, 183, 159–171 CrossRef CAS.
- J. L. Webb, Enzyme and Metabolic Inhivitors, Academic Press, New York, 1966, vol. 3, pp. 595–793 Search PubMed.
- E. Adams, D. Jeter, A. W. Cordes and J. W. Kolis, Inorg. Chem., 1990, 29, 1500–1503 CrossRef CAS.
- R. D. Hoffman and M. D. Lane, J. Biol. Chem., 1992, 267, 14005–14011 CAS.
- C. Gitler, B. Zarmi and E. Kalef, Anal. Biochem., 1997, 252, 48–55 CrossRef CAS PubMed.
- E. S. Berleth, E. M. Kasperek, S. P. Grill, J. A. Braunscheidel, L. A. Graziani and C. M. Pickart, J. Biol. Chem., 1992, 267, 16403–16411 CAS.
- H. Denise1, C. Giroud, M. P. Barrett and T. Baltz, Eur. J. Biochem., 1999, 259, 339–346 CrossRef.
- R. N. Huang, H. Y. Yeh, S. C. Cheng, L. P. Chow and T. C. Lee, J. Chromatogr. B: Biomed. Sci. Appl., 2000, 740, 109–116 CrossRef CAS.
- A. Mizumura, T. Watanabe, Y. Kobayashi and S. Hirano, Toxicol. Appl. Pharmacol., 2010, 242, 119–125 CrossRef CAS PubMed.
- Y. Y. Chang, T. C. Kuo, C. H. Hsu, D. R. Hou, Y. H. Kao and R. N. Huang, Arch. Toxicol., 2012, 86, 911–922 CrossRef CAS PubMed.
- T. D. Foley, C. M. Stredny, T. M. Coppa and M. A. Gubbiotti, Neurochem. Res., 2010, 35, 306–314 CrossRef CAS PubMed.
- H. Yan, N. Wang, M. Weinfeld, W. R. Cullen and X. C. Le, Anal. Chem., 2009, 81, 4144–4152 CrossRef CAS PubMed.
- G. Zhou, M. Jauhiainen, K. Stevenson and P. Dolphin, J. Chromatogr. B: Biomed. Sci. Appl., 1991, 568, 69–83 CrossRef CAS.
- M. L. Carbajal, S. L. S. Espinoza, S. N. Valdez, E. Poskus, E. E. Smolko and M. Grasselli, React. Funct. Polym., 2009, 69, 816–820 CrossRef CAS PubMed.
- K. S. Thorn, N. Naber, M. Matuska, R. D. Vale and R. Cooke, Protein Sci., 2000, 9, 213–217 CrossRef CAS PubMed.
- E. R. LaVallie and J. M. McCoy, Curr. Opin. Biotechnol., 1995, 6, 501–506 CrossRef CAS.
- E. R. LaVallie, E. A. DiBlasio, S. Kovacic, K. L. Grant, P. F. Schendel and J. M. McCoy, Nat. Biotechnol., 1993, 11, 187–193 CrossRef CAS PubMed.
- E. R. LaVallie, E. A. DiBlasio-Smith, L. A. Collins-Racie, Z. Lu and J. M. McCoy, Methods Mol. Biol., 2003, 205, 119–140 CAS.
- R. S. Patel-King, S. E. Benashski, A. Harrison and S. M. King, J. Biol. Chem., 1996, 271, 6283–6291 CrossRef CAS PubMed.
- K. Ogawa, H. Takai, A. Ogiwara, E. Yokota, T. Shimizu, K. Inaba and H. Mohri, Mol. Biol. Cell, 1996, 7, 1895–1907 CrossRef CAS.
- Z. Wang, H. Zhang, X. Li and X. C. Le, Rapid Commun. Mass Spectrom., 2007, 21, 3658–3666 CrossRef CAS PubMed.
- E. T. Mambetisaeva, P. E. M. Martin and W. H. Evans, Protein Expression Purif., 1997, 11, 26–34 CrossRef CAS PubMed.
- A. C. da Silva, S. U. L. Liu and G. B. Bouck, J. Eukaryotic Microbiol., 1999, 46, 95–104 CrossRef CAS PubMed.
- M. L. Papouchado, S. N. Valdez, D. Ghiringhelli, E. Poskus and M. R. Ermácora, Eur. J. Biochem., 1997, 246, 350–359 CAS.
- C. Baer, J. Retief, E. Van Niel, M. Braiman and F. Gonzalez-Fernandez, Exp. Eye Res., 1998, 66, 249–262 CrossRef CAS PubMed.
- Z. Lu, E. A. DiBlasio-Smith, K. L. Grant, N. W. Warne, E. R. LaVallie, L. A. Collins-Racie, M. T. Follettie, M. J. Williamson and J. M. McCoy, J. Biol. Chem., 1996, 271, 5059–5065 CrossRef CAS PubMed.
- T. Wang, J. F. Anderson, L. A. Magnarelli, S. J. Wong, R. A. Koski and E. Fikrig, J. Immunol., 2001, 167, 5273–5277 CrossRef CAS.
- F. Gonzalez-Fernandez, C. A. Baer and D. Ghosh, BMC Biochem., 2007, 8, 15–34 CrossRef PubMed.
- A. L. Femia, C. F. Temprana, J. Santos, M. L. Carbajal, M. S. Amor, M. Grasselli and S. V. Alonso, Protein J., 2012, 31, 656–666 CrossRef CAS PubMed.
- O. Rengby, L. Johansson, L. A. Carlson, E. Serini, A. Vlamis-Gardikas, P. Karsnas and E. S. J. Arner, Appl. Environ. Microbiol., 2004, 70, 5159–5167 CrossRef CAS PubMed.
- Q. Cheng, S. Stone-Elander and E. S. J. Arnér, Nat. Protoc., 2006, 1, 604–613 CrossRef CAS PubMed.
- O. Rengby, Q. Cheng, M. Vahter, H. Jörnvall and E. S. J. Arnér, Free Radical Biol. Med., 2009, 46, 893–904 CrossRef CAS PubMed.
- L. Johansson, C. Chen, J. O. Thorell, A. Fredriksson, S. Stone-Elander, G. Gafvelin and E. S. J. Arnér, Nat. Methods, 2004, 1, 61–66 CrossRef CAS PubMed.
- Q. Cheng, L. Lu, J. Grafström, M. H. Olofsson, J. O. Thorell, E. Samén, K. Johansson, H. S. Ahlzén, S. Linder and E. S. J. Arnér, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 2562–2573 CrossRef CAS PubMed.
- Q. Cheng, L. Lu, J. Grafström, M. H. Olofsson, J. O. Thorell, E. Samén, K. Johansson, H. S. Ahlzén, S. Stone-Elander and S. Linder, PLoS One, 2012, 8, e42151 Search PubMed.
- A. Chopra, In: Molecular Imaging and Contrast Agent Database (MICAD) [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2004–2013, available from: http://www.ncbi.nlm.nih.gov/books/NBK109731/.
- H. Wållberg, J. Grafström, Q. Cheng, L. Lu, H. S. M. Ahlzén, E. Samén, J. O. Thorell, K. Johansson, F. Dunås and M. H. Olofsson, J. Nucl. Med., 2012, 53, 1446–1453 CrossRef PubMed.
- L. Johansson, G. Gafvelin and E. S. J. Arnér, Biochim. Biophys. Acta, Gen. Subj., 2005, 1726, 1–13 CrossRef CAS PubMed.
- L. Johansson and E. S. J. Arnér, in Selenium: Its molecular biology and role in human health, ed. D. L. Hatfield, M. J. Berry and V. N. Gladyshev, Springer, 2006, vol. 20, pp. 221–230 Search PubMed.
- Q. Cheng, PhD thesis, Karolinska Institutet, 2010.
- Q. Cheng, L. Johansson, J. O. Thorell, A. Fredriksson, E. Samén, S. Stone-Elander and E. S. J. Arnér, ChemBioChem, 2006, 7, 1976–1981 CrossRef CAS PubMed.
- L. Johansson, L. Svensson, U. Bergström, G. Jacobsson-Ekman, E. S. J. Arnér, M. van Hage, A. Bucht and G. Gafvelin, FEBS J., 2005, 272, 3449–3460 CrossRef CAS PubMed.
- M. U. Mayer, L. Shi and T. C. Squier, Mol. BioSyst., 2005, 1, 53–56 RSC.
- S. Verma, Y. Xiong, M. U. Mayer and T. C. Squier, Biochemistry, 2007, 46, 3023–3035 CrossRef CAS PubMed.
- R. J. Giannone, W. H. McDonald, G. B. Hurst, Y. Huang, J. Wu, Y. Liu and Y. Wang, BioTechniques, 2007, 43, 296–302 CrossRef CAS PubMed.
- R. J. Giannone, Y. Liu and Y. Wang, Methods Mol. Biol., 2009, 544, 421–438 CAS.
- L. Q. Ying and B. Branchaud, Bioconjugate Chem., 2011, 22, 987–992 CrossRef CAS PubMed.
- D. N. Joshi, S. J. S. Flora and K. Kalia, J. Hazard. Mater., 2009, 166, 1500–1505 CrossRef CAS PubMed.
- Y. Y. Chang, T. C. Kuo, C. H. Hsu, D. R. Hou, Y. H. Kao and R. N. Huang, Arch. Toxicol., 2012, 86, 911–922 CrossRef CAS PubMed.
- R. Moaddel, A. Sharma, T. Huseni, G. S. Jones Jr., R. N. Hanson and R. H. Loring, Bioconjugate Chem., 1999, 10, 629–637 CrossRef CAS PubMed.
- N. Donoghue, P. T. Yam, X. M. Jiang and P. J. Hogg, Protein Sci., 2000, 9, 2436–2445 CrossRef CAS PubMed.
- X. Zhang, F. Yang, J. Y. Shim, K. L. Kirk, D. E. Anderson and X. Chen, Cancer Lett., 2007, 255, 95–106 CrossRef CAS PubMed.
- X. W. Yan, J. H. Li, Q. Q. Liu, H. Y. Peng, A. Popowich, Z. X. Wang, X. F. Li and X. C. Le, presented at the 97th Canadian Chemistry Conference and Exhibition, Vancouver, Canada, June 1–4, 2014, abstract #00864.
- E. Diesel, M. Schreiber and J. R. van der Meer, Anal. Bioanal. Chem., 2009, 394, 687–693 CrossRef CAS PubMed.
- P. Corbisier, G. Ji, G. Nuyts, M. Mergeay and S. Silver, FEMS Microbiol. Lett., 1993, 110, 231–238 CrossRef CAS PubMed.
- S. Daunert, G. Barrett, J. Feliciano, R. Shetty, S. Shrestha and W. Smith-Spencer, Chem. Rev., 2000, 100, 2705–2738 CrossRef CAS PubMed.
- A. Date, P. Pasini and S. Daunert, Anal. Chem., 2007, 79, 9391–9397 CrossRef CAS PubMed.
- J. Stocker, D. Balluch, M. Gsell, H. Harms, J. Feliciano, S. Daunert, K. A. Malik and J. R. Van der Meer, Environ. Sci. Technol., 2003, 37, 4743–4750 CrossRef CAS.
- J. M. H. King, P. M. Digrazia, B. Applegate, R. Burlage, J. Sanseverino, P. Dunbar, F. Larimer and G. S. Sayler, Science, 1990, 249, 778–781 CAS.
- H. Harms, M. C. Wells and J. R. van der Meer, Appl. Microbiol. Biotechnol., 2006, 70, 273–280 CrossRef CAS PubMed.
- B. Baumann and J. R. van der Meer, J. Agric. Food Chem., 2007, 55, 2115–2120 CrossRef CAS PubMed.
- J. R. van der Meer, Swiss Federal Institute for Environmental Science and Technology (EAWAG), 2003, 11, 12–14 Search PubMed.
- C. Tani, K. Inoue, Y. Tani, M. Harun-ur-Rashid, N. Azuma, S. Ueda, K. Yoshida and I. Maeda, J. Biosci. Bioeng., 2009, 108, 414–420 CrossRef CAS PubMed.
- D. Merulla, V. Hatzimanikatis and J. R. van der Meer, Microb. Biotechnol., 2013, 6, 503–514 CrossRef PubMed.
- A. Prindle, P. Samayoa, I. Razinkov, T. Danino, L. S. Tsimring and J. Hasty, Nature, 2012, 481, 39–44 CrossRef CAS PubMed.
- S. Choe, F. N. Gravelat, Q. Al Abdallah, M. J. Lee, B. F. Gibbs and D. C. Sheppard, Appl. Environ. Microbiol., 2012, 78, 3855–3863 CrossRef CAS PubMed.
- A. Bakhrat, E. Eltzov, Y. Finkelstein, R. S. Marks and D. Raveh, Cell Biol. Toxicol., 2011, 27, 227–236 CrossRef CAS PubMed.
- D. Merulla, N. Buffi, S. Beggah, F. Truffer, M. Geiser, P. Renaud and J. R. van der Meer, Curr. Opin. Biotechnol., 2013, 24, 534–541 CrossRef CAS PubMed.
- K. Yagi, Appl. Microbiol. Biotechnol., 2007, 73, 1251–1258 CrossRef CAS PubMed.
- N. Joshi, X. Wang, L. Montgomery, A. Elfick and C. E. French, Desalination, 2009, 248, 517–523 CrossRef CAS PubMed.
- D. N. Joshi, J. S. Patel, S. J. S. Flora and K. Kalia, Environ. Health Prev. Med., 2008, 13, 257–263 CrossRef CAS PubMed.
- Q. Hu, L. Li, Y. Wang, W. Zhao, H. Qi and G. Zhuang, J. Environ. Sci., 2010, 22, 1469–1474 CrossRef CAS.
- H. Fujimoto, M. Wakabayashi, H. Yamashiro, I. Maeda, K. Isoda, M. Kondoh, M. Kawase, H. Miyasaka and K. Yagi, Appl. Microbiol. Biotechnol., 2006, 73, 332–338 CrossRef CAS PubMed.
- J. Zhai, D. Yong, J. Li and S. Dong, Analyst, 2013, 138, 702–707 RSC.
- K. de Mora, N. Joshi, B. L. Balint, F. B. Ward, A. Elfick and C. E. French, Anal. Bioanal. Chem., 2011, 400, 1031–1039 CrossRef CAS PubMed.
- D. P. Webster, M. A. TerAvest, D. F. R. Doud, A. Chakravorty, E. C. Holmes, C. M. Radens, S. Sureka, J. A. Gralnick and L. T. Angenent, Biosens. Bioelectron., 2014, 7, 320–324 CrossRef PubMed.
- S. Sanllorente-Mendez, O. Dominguez-Renedo and M. Julia Arcos-Martinez, Sensors, 2010, 10, 2119–2128 CrossRef CAS PubMed.
- X. Fuku, F. Iftikar, E. Hess, E. Iwuoha and P. Baker, Anal. Chim. Acta, 2012, 730, 49–59 CrossRef CAS PubMed.
- K. B. Male, S. Hrapovic, J. M. Santini and J. H. T. Luong, Anal. Chem., 2007, 79, 7831–7837 CrossRef CAS PubMed.
- C. Liu, V. Balsamo, D. Sun, M. Naja, X. Wang, B. Rosen and C. Li, Biosens. Bioelectron., 2012, 38, 19–26 CrossRef CAS PubMed.
- S. Cosnier, C. Mousty, X. Cui, X. Yang and S. Dong, Anal. Chem., 2006, 78, 4985–4989 CrossRef CAS PubMed.
- S. Sanllorente-Mendez, O. Dominguez-Renedo and M. J. Arcos-Martinez, Talanta, 2012, 93, 301–306 CrossRef CAS PubMed.
- P. Sarkar, S. Banerjee, D. Bhattacharyay and A. P. F. Turner, Ecotoxicol. Environ. Saf., 2010, 73, 1495–1501 CrossRef CAS PubMed.
- H. Strosnide, US EPA, Office of Solid Waste and Emergency Response, Tech. Rep. 2003, 23.
- S. Kohler, S. Belkin and R. D. Schmid, Fresenius' J. Anal. Chem., 2003, 366, 769–779 Search PubMed.
- J. Chen, S. Sun, C. Li, Y. Zhu and B. P. Rosen, Environ. Sci. Technol., 2014, 48, 1141–1147 CrossRef CAS PubMed.
- N. Buffi, D. Merulla, J. Beutier, F. Barbaud, S. Beggah, H. van Lintel, P. Renaud and J. R. van der Meer, Lab Chip, 2011, 11, 2369–2377 RSC.
- P. T. K. Trang, M. Berg, P. H. Viet, N. Van Mui and J. R. Van Der Meer, Environ. Sci. Technol., 2005, 39, 7625–7630 CrossRef CAS.
- K. Siegfried, C. Endes, A. F. M. K. Bhuiyan, A. Kuppardt, J. Mattusch, J. R. van der Meer, A. Chatzinotas and H. Harms, Environ. Sci. Technol., 2012, 46, 3281–3287 CrossRef CAS PubMed.
- J. Cai and M. S. DuBow, Biodegradation, 1997, 8, 105–111 CrossRef CAS.
- F. Truffer, N. Buffi, D. Merulla, S. Beggah, H. van Lintel, P. Renaud, J. R. van der Meer and M. Geiser, Rev. Sci. Instrum., 2014, 85, 015120 CrossRef PubMed.
- S. Jouanneau, M. J. Durand and G. Thouand, Environ. Sci. Technol., 2012, 46, 11979–11987 CrossRef CAS PubMed.
- J. R. van der Meer and S. Belkin, Nat. Rev. Microbiol., 2010, 8, 511–522 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |