
Copper-catalyzed regioselective synthesis of furan via tandem cycloaddition of ketone with an unsaturated carboxylic acid under air†
Monoranjan
Ghosh
,
Subhajit
Mishra
,
Kamarul
Monir
and
Alakananda
Hajra
*
Department of Chemistry, Visva-Bharati University, Santiniketan-731235, India. E-mail: alakananda.hajra@visva-bharati.ac.in
First published on 20th October 2014
Abstract
A catalytic decarboxylative annulation has been developed for the regioselective synthesis of trisubstituted furans by the cycloaddition of ketones with α,β-unsaturated carboxylic acids under ambient air. A library of furan derivatives were obtained in good yields from the readily available substrates in the combination of a catalytic amount of Cu-salt and a stoichiometric amount of water. Water plays a crucial role in this catalytic transformation.
Introduction
Polysubstituted furans represent an important class of five-membered heterocycles ubiquitous in natural products, pharmaceuticals, and agrochemicals as well as act as useful intermediates in organic synthesis.1 The multisubstituted furan is a basic scaffold of various biologically active alkaloids and pharmaceutical drugs, such as dantrolene and (+)-(Z)-deoxypukalide (Fig. 1).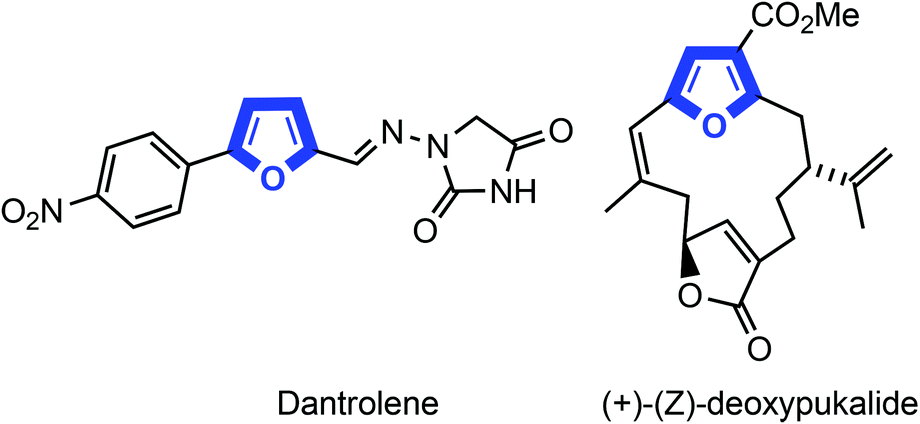 | ||
| Fig. 1 Some biologically active furan containing drugs. | ||
Various methods have been reported for the synthesis of furan derivatives due to their important biological activities and usefulness.2 Classical approaches such as Paal–Knorr synthesis3a,b and Feist–Benary synthesis3c–f have already been established for the assembly of polysubstituted furans from dicarbonyl compounds. In addition, several methodologies have been reported from the prefunctionalized acyclic ketone derivatives such as cyclopropenyl ketones,4a β-acyloxy acetylenic ketones,4b allenyl ketones,4co-alkoxyketones,4d,e α-aryloxyketones4fetc. However, the synthetic potential of these strategies is limited by the requirement of functionalized precursors, which can only be obtained through specialized protocols involving multi-step procedures. As a consequence, the development of a new synthetic route to synthesize furan derivatives from simple and commercially available starting materials is still in demand.
Transition metal-catalyzed decarboxylative coupling has emerged as a powerful tool to form carbon–carbon or carbon–heteroatom bonds starting from carboxylic acids.5 Use of carboxylic acid has several advantages due to its ready availability, easy handling as well as environmentally benign properties. Moreover, it produces nontoxic CO2 as the byproduct. From the point of view of atom and process-economical chemistry, the most concise route with reduced waste and fewer steps is needed.
To the best of our knowledge, no catalytic method has been reported for the synthesis of furans via decarboxylative annulation reaction. Recently, Zhang et al. developed an elegant protocol for the synthesis of furan derivatives through a decarboxylative cyclization between ketones and α,β-unsaturated carboxylic acids.6 However, stoichiometric amounts of two different copper salts are required for this reaction. Catalytic decarboxylative methods are well-known and have been used in various chemical transformations.7 Based on our experience in copper catalysis,8 we envisaged that the synthesis of furans might be possible by the annulation of ketones with α,β-unsaturated carboxylic acids via decarboxylative cycloaddition using a catalytic amount of copper salt in the presence of a suitable oxidant. Generally, oxidants such as PIDA, K2S2O8, TBHP, O2etc. are used for copper-catalyzed oxidative coupling reactions.9 Among them molecular O2 is considered to be the most ideal oxidant due to its high abundance, low cost, and environment friendly nature and therefore shows a great demand in the industrial prospects.10 Moreover, water is produced as the only byproduct using molecular O2 as an oxidant. So, the construction of heterocycles from simple and readily accessible substrates using molecular O2 is compatible with the increasing requirements for green chemistry and an efficient process.
Recently, we have reported the synthesis of arenofuran derivatives by the coupling of phenols/naphthols with nitroalkenes via tandem Michael addition/denitration.11 In continuation of our research interest in furan synthesis as well as in coupling between nitroalkenes with various nucleophiles,12 herein we report an efficient synthetic method for the synthesis of 2,3,5-trisubstituted furans via decarboxylative annulation employing a catalytic amount of copper salt in the presence of a stoichiometric amount of water under ambient air (Scheme 1).
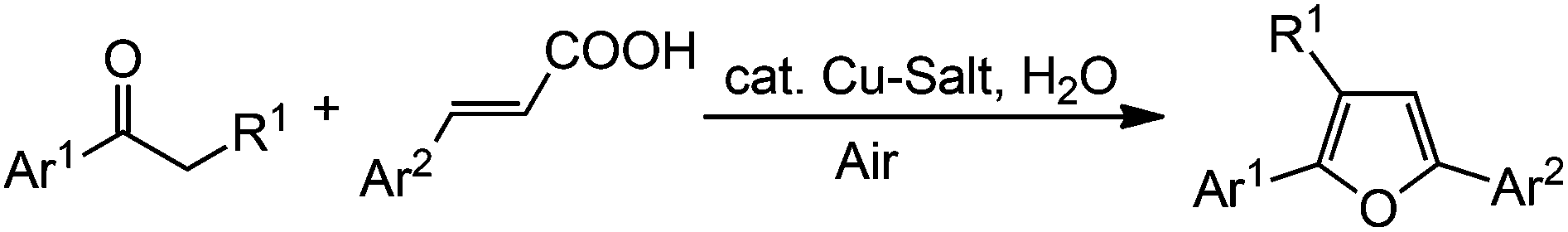 | ||
| Scheme 1 Catalytic decarboxylative synthesis of furans. | ||
Results and discussion
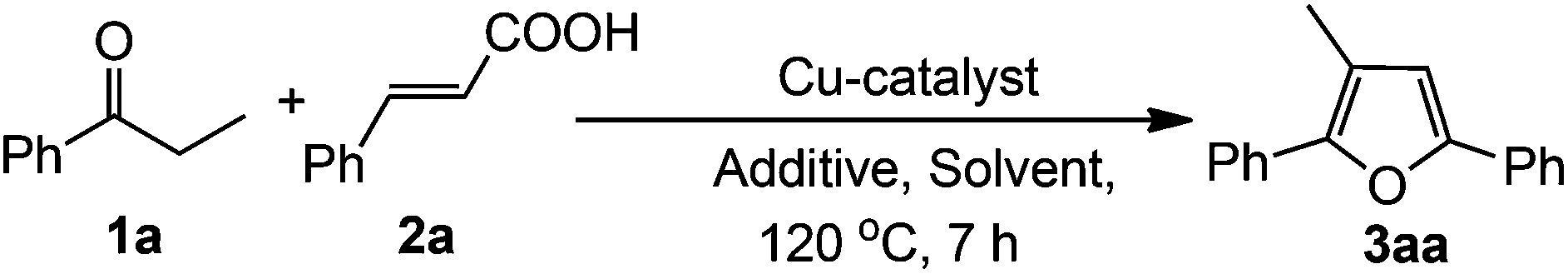
We commenced our study by choosing propiophenone and cinnamic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the model substrates to find out the optimized reaction conditions using various copper salts, additives and solvents. The results are summarized in Table 1. Initially, the reaction was carried out employing 10 mol% Cu(OAc)2·H2O in DMA at 120 °C under an air atmosphere. The reaction did not proceed at all even after 24 h (Table 1, entry 1). On increasing the amount of Cu(OAc)2·H2O (20 mol%), the reaction proceeded in very low conversion (Table 1, entry 2). Encouraged by this result, we turned our attention to check the role of an acid and a base in this transformation. In the presence of various acids (1 equiv.) the desired products were obtained in low to moderate yields (Table 1, entries 3–6). Similar effects were observed in the case of bases (Table 1, entries 7 and 8). On the other hand, in the presence of 1 equiv. of water, the reaction proceeded with 44% yield (Table 1, entry 9). On increasing the amount of H2O (3 equiv.), the desired product was obtained in 67% yield (Table 1, entry 11). The yield of the product was decreased by changing the molar ratios of the reactants (Table 1, entries 12 and 13). Different solvents like NMP, toluene, xylene, DMSO and DMF (Table 1, entries 14–18) were also screened, among them DMA gave the best result compared to others (Table 1, entry 11). Use of other Cu-salts such as Cu(OTf)2, Cu(ClO4)2·6H2O resulted in lower yields (Table 1, entries 19 and 20) whereas CuCl2 and CuBr2 were almost ineffective for this reaction (Table 1, entries 21 and 22). Finally, the optimized reaction conditions were achieved using the combination of propiophenone and cinnamic acid (1:1), 20 mol% Cu(OAc)2·H2O in the presence of 3 equiv. of water at 120 °C for 7 h under ambient air (Table 1, entry 11).
:1) as the model substrates to find out the optimized reaction conditions using various copper salts, additives and solvents. The results are summarized in Table 1. Initially, the reaction was carried out employing 10 mol% Cu(OAc)2·H2O in DMA at 120 °C under an air atmosphere. The reaction did not proceed at all even after 24 h (Table 1, entry 1). On increasing the amount of Cu(OAc)2·H2O (20 mol%), the reaction proceeded in very low conversion (Table 1, entry 2). Encouraged by this result, we turned our attention to check the role of an acid and a base in this transformation. In the presence of various acids (1 equiv.) the desired products were obtained in low to moderate yields (Table 1, entries 3–6). Similar effects were observed in the case of bases (Table 1, entries 7 and 8). On the other hand, in the presence of 1 equiv. of water, the reaction proceeded with 44% yield (Table 1, entry 9). On increasing the amount of H2O (3 equiv.), the desired product was obtained in 67% yield (Table 1, entry 11). The yield of the product was decreased by changing the molar ratios of the reactants (Table 1, entries 12 and 13). Different solvents like NMP, toluene, xylene, DMSO and DMF (Table 1, entries 14–18) were also screened, among them DMA gave the best result compared to others (Table 1, entry 11). Use of other Cu-salts such as Cu(OTf)2, Cu(ClO4)2·6H2O resulted in lower yields (Table 1, entries 19 and 20) whereas CuCl2 and CuBr2 were almost ineffective for this reaction (Table 1, entries 21 and 22). Finally, the optimized reaction conditions were achieved using the combination of propiophenone and cinnamic acid (1:1), 20 mol% Cu(OAc)2·H2O in the presence of 3 equiv. of water at 120 °C for 7 h under ambient air (Table 1, entry 11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additive (equiv.) | Solvent |
1a:2a |
Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (0.5 mmol), solvent: 2 mL, 120 °C, 7 h, air. b Isolated yield. c n.d. = not determined. DMA = N,N-dimethylacetamide. DMF = N,N-dimethylformamide. DMSO = dimethylsulfoxide. NMP = N-methyl-2-pyrrolidone. | |||||
| 1. | Cu(OAc)2·H2O (10) | — | DMA | 1:1 |
n.d.c |
| 2. | Cu(OAc)2·H2O (20) | — | DMA | 1:1 |
5 |
| 3. | Cu(OAc)2·H2O (20) | AcOH (1) | DMA | 1:1 |
33 |
| 4. | Cu(OAc)2·H2O (20) | TFA (1) | DMA | 1:1 |
40 |
| 5. | Cu(OAc)2·H2O (20) | HCl (1) | DMA | 1:1 |
<5 |
| 6. | Cu(OAc)2·H2O (20) | p-TSA (1) | DMA | 1:1 |
8 |
| 7. | Cu(OAc)2·H2O (20) | NaOAc (1) | DMA | 1:1 |
15 |
| 8. | Cu(OAc)2·H2O (20) | KOAc (1) | DMA | 1:1 |
22 |
| 9. | Cu(OAc)2·H2O (20) | Water (1) | DMA | 1:1 |
44 |
| 10. | Cu(OAc)2·H2O (20) | Water (2) | DMA | 1:1 |
55 |
| 11. | Cu(OAc)2·H2O (20) | Water (3) | DMA | 1:1 |
67 |
| 12. | Cu(OAc)2·H2O (20) | Water (3) | DMA | 2:1 |
45 |
| 13. | Cu(OAc)2·H2O (20) | Water (3) | DMA | 1:2 |
20 |
| 14. | Cu(OAc)2·H2O (20) | Water (3) | NMP | 1:1 |
37 |
| 15. | Cu(OAc)2·H2O (20) | Water (3) | Toluene | 1:1 |
22 |
| 16. | Cu(OAc)2·H2O (20) | Water (3) | Xylene | 1:1 |
25 |
| 17. | Cu(OAc)2·H2O (20) | Water (3) | DMSO | 1:1 |
32 |
| 18. | Cu(OAc)2·H2O (20) | Water (3) | DMF | 1:1 |
60 |
| 19. | Cu(OTf)2 (20) | Water (3) | DMA | 1:1 |
43 |
| 20. | Cu(ClO4)2·6H2O (20) | Water (3) | DMA | 1:1 |
45 |
| 21. | CuCl2 (20) | Water (3) | DMA | 1:1 |
n.d. |
| 22. | CuBr2 (20) | Water (3) | DMA | 1:1 |
n.d. |
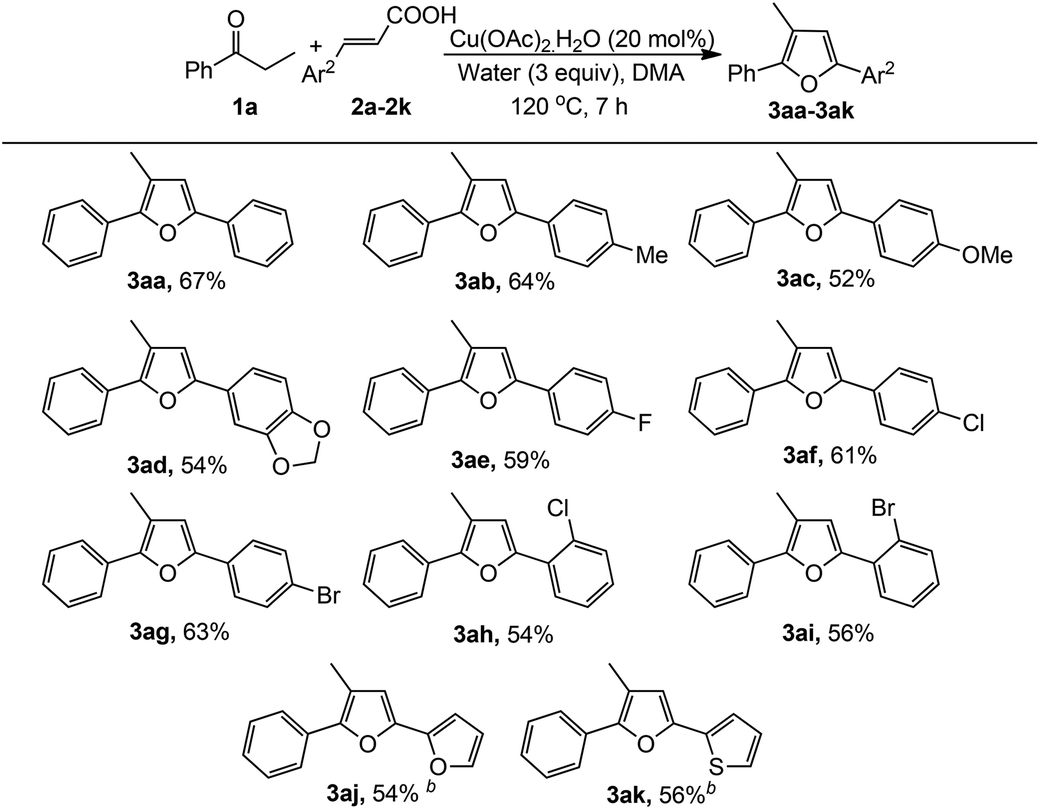
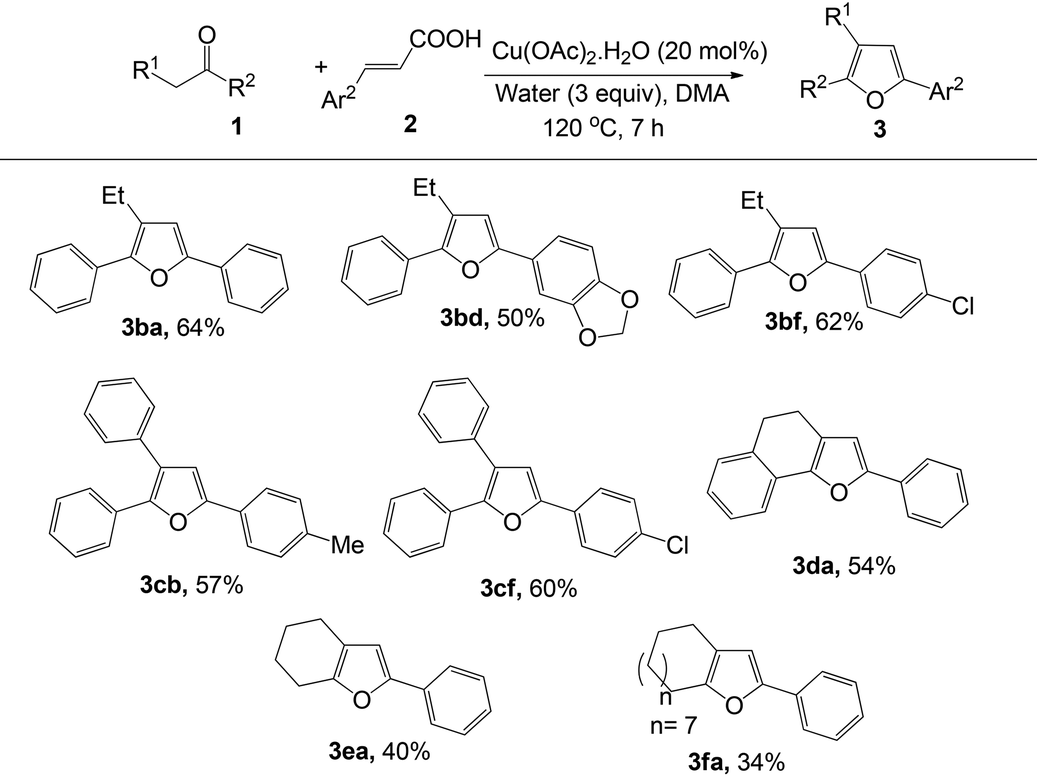
To explore the general applicability, scope and limitation of this method, various cinnamic acids and ketones were investigated. At first, various cinnamic acid derivatives were examined (Table 2). Electron-donating substituents (–Me, –OMe) in cinnamic acid produced the corresponding furans in moderate to good yields (3ab and 3ac). Moreover, halogen substituents in the aryl moiety also produced the furan derivatives with moderate yields (3ae–3ai). Substituents at the different positions in the aryl moiety slightly affected the yields of this reaction. Interestingly, in all cases only one regioisomeric product was obtained. Heteroaryl unsaturated carboxylic acids were also examined and were found to be effective for the synthesis of furan derivatives (3aj and 3ak).
Furthermore, various commercially available ketones were also tested to explore the scope of the reaction (Table 3). Aryl alkyl ketones, such as butyrophenone (3ba, 3bd and 3bf) and 2-phenyl acetophenone (3cb and 3cf) reacted smoothly to afford the desired products. Cyclic ketones such as α-tetralone, cyclohexanone and cyclododecanone also produced the corresponding furan derivatives (3da, 3ea and 3fa) in moderate yields. However, no reaction occurred in the case of acyclic ketones (ethylmethyl ketone and isobutylmethyl ketone) and crotonic acid.
| a Reaction conditions: 1 (0.5 mmol), 2 (0.5 mmol), water (0.27 mL, 3 equiv.), DMA (2 mL), 120 °C, 7 h, air; isolated yields. |
|---|
|
Some controlled experiments were performed to evaluate the mechanism of this reaction (Scheme 2). When the reaction was carried out in the absence of copper, the reaction did not proceed at all (eqn (1)), which signifies that the presence of copper as a catalyst is necessary in this reaction. Only a trace amount of product was detected under an argon atmosphere (eqn (2)), which confirms that molecular O2 (air) is essential for the completion of the catalytic cycle. It is noteworthy to mention that styrene did not produce the desired product under the optimized reaction conditions.6 Moreover, we did not observe the formation of styrene during this reaction. This result indicates that the reaction did not proceed through the formation of a styrene intermediate. Moreover, methyl cinnamate was unreacted under the present reaction conditions (eqn (3)). Furthermore the reaction probably does not follow a radical pathway6,13 as the radical scavengers like quinone and 2,6-di-tert-butyl-4-methylphenol (BHT) (1.5 equiv.) did not inhibit the reaction (eqn (4) and (5)).
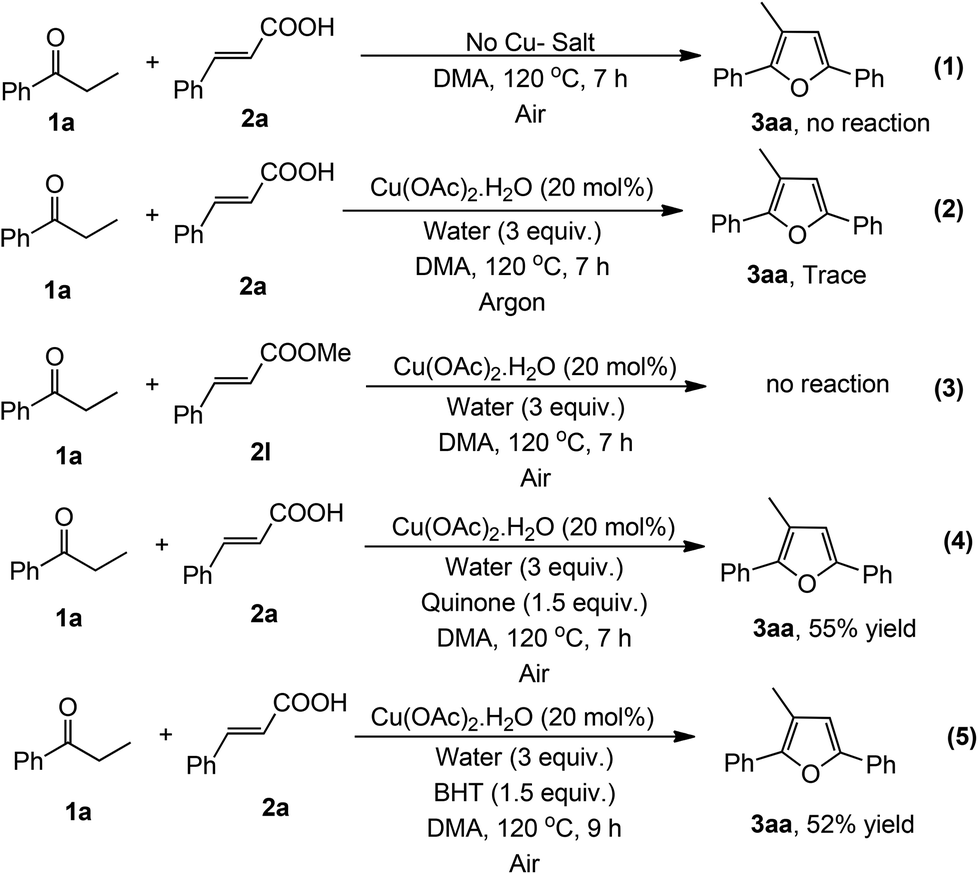 | ||
| Scheme 2 Some controlled experiments. | ||
On the basis of results obtained from these controlled experiments, a probable mechanism of the reaction is depicted in Scheme 3. Nucleophilic addition of the hydroxyl group in enol A, in situ generated from ketones, to cinnamic acids in the presence of Cu(II) salt affords alkyl–Cu(II) intermediate C. The intermediate C reacts with enol to lead the intermediate D which is oxidized to give the C–Cu(III)–C intermediate E.14 The intermediate F is formed from Evia reductive elimination and subsequently oxidative aromatization occurs to convert the furan intermediate G. Finally the intermediate G affords the desired furan derivative via decarboxylation.15 Although the specific role of water is not clear at the present stage, it probably facilitates the formation of enol.
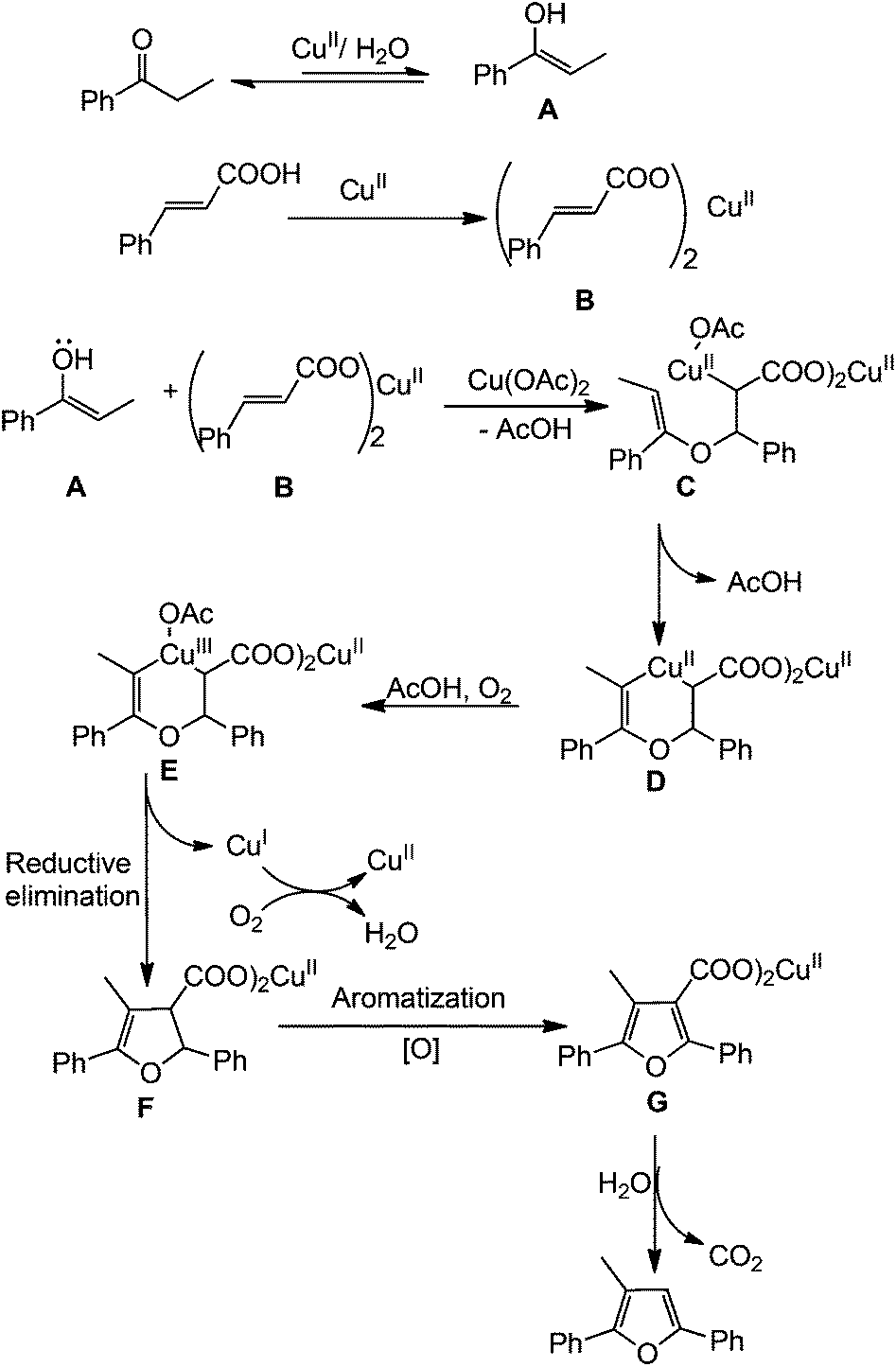 | ||
| Scheme 3 Probable reaction mechanism. | ||
Conclusions
In summary, we have developed a copper-catalyzed regioselective synthesis of 2,3,5-trisubstituted furans from the readily available ketones and α,β-unsaturated carboxylic acids via decarboxylative annulation. Water plays a crucial role in this transformation. Operational simplicity, broad substrates scope, low cost of the catalyst, and use of green oxidant make this protocol a practical one in diversity-oriented synthesis.Experimental section
Typical experimental procedure for the synthesis of 3-methyl-2,5-diphenylfuran product (3aa)
A mixture of propiophenone (1a, 0.5 mmol, 67 mg), cinnamic acid (2a, 0.5 mmol, 74 mg), Cu(OAc)2·H2O (20 mol%, 19 mg) and H2O (0.27 mL, 3 equiv.) in DMA (2 mL) was taken in a reaction vessel and stirred at 120 °C for 7 h under an open atmosphere. After completion of the reaction (TLC), the reaction mixture was cooled to room temperature, and extracted with ethyl acetate. The organic phase was dried over anhydrous Na2SO4. The crude residue was obtained after evaporating the solvent in vacuum and was purified by column chromatography on silica gel (60–120 mesh) using petroleum ether as an eluent to afford the pure product as a white solid (75 mg, 67% yield). Mp 45–46 °C (lit.6 46–47 °C); 1H NMR (400 MHz, CDCl3): δ 7.62 (d, J = 7.6 Hz, 4H), 7.36–7.27 (m, 4H), 7.18–7.14 (m, 2H), 6.51 (s, 1H), 2.23 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 151.7, 148.3, 131.8, 130.9, 128.7, 128.6, 127.3, 126.7, 125.3, 123.7, 118.7, 110.9, 12.2.Acknowledgements
A. H. acknowledges the financial support from CSIR, New Delhi (grant no. 02(0168)/13/EMR-II). We are thankful to DST-FIST and UGC-SAP. M. G. and S. M. thank UGC and K. M. thanks CSIR for their fellowships. Authors thank the reviewers for their valuable suggestions and comments to improve the quality of the paper.Notes and references
- (a) A. R. Katritzky, Comprehensive Heterocyclic Chemistry III, Elsevier, Amsterdam, New York, 1st edn, 2008 Search PubMed; (b) A. F. Pozharskii, A. R. Katritzky and A. T. Soldatenkov, Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry, Biochemistry, and Applications, Wiley, Chichester, West Sussex, 2nd edn, 2011 Search PubMed; (c) X. L. Hou, H. Y. Cheung, T. Y. Hon, P. L. Kwan, T. H. Lo, S. Y. Tong and H. N. C. Wong, Tetrahedron, 1998, 54, 1955 CrossRef CAS; (d) B. H. Lipshutz, Chem. Rev., 1986, 86, 795 CrossRef CAS.
- (a) S. F. Kirsch, Org. Biomol. Chem., 2006, 4, 2076 RSC; (b) M. E. Jung and L. J. Street, J. Am. Chem. Soc., 1984, 106, 8327 CrossRef CAS; (c) C. He, S. Guo, J. Ke, J. Hao, H. Xu, H. Chen and A. Lei, J. Am. Chem. Soc., 2012, 134, 5766 CrossRef CAS PubMed; (d) D. K. Nair, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron Lett., 2012, 53, 3349 CrossRef CAS PubMed; (e) U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 12669 CrossRef CAS PubMed; (f) W.-Y. Huang, Y.-C. Chen and K. Chen, Chem. – Asian J., 2012, 7, 688 CrossRef CAS PubMed; (g) H. Tsuji, C. Mitsui, L. Illies, Y. Sato and E. Nakamura, J. Am. Chem. Soc., 2007, 129, 11902 CrossRef CAS PubMed; (h) F.-Q. Yuan and F.-S. Han, Adv. Synth. Catal., 2013, 355, 537 CAS; (i) S. Anwar, W.-Y. Huang, C.-H. Chen, Y.-S. Cheng and K. Chen, Chem. – Eur. J., 2013, 19, 4344 CrossRef CAS PubMed; (j) H. Tsuji, K. Yamagata, Y. Ueda and E. Nakamura, Synlett, 2011, 1015 CrossRef CAS PubMed; (k) M. R. Kuram, M. Bhanuchandra and A. K. Sahoo, Angew. Chem., Int. Ed., 2013, 52, 4607 CrossRef CAS PubMed.
- (a) V. Amarnath and K. Amarnath, J. Org. Chem., 1995, 60, 301 CrossRef CAS; (b) G. Minetto, L. F. Raveglia, A. Sega and M. Taddei, Eur. J. Org. Chem., 2005, 5277 CrossRef CAS; (c) F. Feist, Chem. Ber., 1902, 35, 1537 CrossRef CAS; (d) E. Benary, Chem. Ber., 1911, 44, 489 CrossRef CAS; (e) G. Mross, E. Holtz and P. Langer, J. Org. Chem., 2006, 71, 8045 CrossRef CAS PubMed; (f) N. Ayyagari, D. Jose, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron Lett., 2011, 52, 258 CrossRef CAS PubMed.
- (a) S. Ma and J. Zhang, J. Am. Chem. Soc., 2003, 125, 12386 CrossRef CAS PubMed; (b) L. K. Sydnes, B. Holmelid, M. Sengee and M. Hanstein, J. Org. Chem., 2009, 74, 3430 CrossRef CAS PubMed; (c) A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2007, 46, 5195 CrossRef CAS PubMed; (d) M. Abdul-Aziz, J. V. Auping and M. A. Meador, J. Org. Chem., 1995, 60, 1303 CrossRef CAS; (e) K. K. Park, H. Lim, S.-H. Kim and D. H. Bae, J. Chem. Soc., Perkin Trans. 1, 2002, 310 RSC; (f) J. Habermann, S. V. Ley and R. Smits, J. Chem. Soc., Perkin Trans. 1, 1999, 2421 RSC.
- (a) N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (b) L. J. Goossen, N. Rodriguez and K. Goossen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef CAS PubMed; (c) S. M. Bonesi and M. Fagnoni, Chem. – Eur. J., 2010, 16, 13572 CrossRef CAS PubMed; (d) L. J. Gooßen and K. Gooßen, Top. Organomet. Chem., 2013, 44, 121 CrossRef; (e) J. Cornella and I. Larrosa, Synthesis, 2012, 653 CAS; (f) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed; (g) O. Baudoin, Angew. Chem., Int. Ed., 2007, 46, 1373 CrossRef CAS PubMed; (h) R. Shang and L. Liu, Sci. China, Ser. B: Chem., 2011, 54, 1670 CrossRef CAS PubMed; (i) J. Moon, M. Jeong, H. Nam, J. Ju, J. H. Moon, H. M. Jung and S. Lee, Org. Lett., 2008, 10, 945 CrossRef CAS PubMed; (j) K. Park and S. Lee, RSC Adv., 2013, 3, 14165 RSC.
- Y. Yang, J. Yao and Y. Zhang, Org. Lett., 2013, 15, 3206 CrossRef CAS PubMed.
- (a) F. Jafarpour, S. Zarei, M. B. A. Olia, N. Jalalimanesh and S. Rahiminejadan, J. Org. Chem., 2013, 78, 2957 CrossRef CAS PubMed; (b) J. Miao and H. Ge, Org. Lett., 2013, 15, 2930 CrossRef CAS PubMed; (c) Z. Li, Z. Cui and Z.-Q. Liu, Org. Lett., 2013, 15, 406 CrossRef CAS PubMed; (d) Y. Yang, C. Xie, Y. Xie and Y. Zhang, Org. Lett., 2012, 14, 957 CrossRef CAS PubMed.
- (a) A. K. Bagdi, M. Rahman, S. Santra, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1741 CrossRef CAS; (b) A. K. Bagdi, A. Majee and A. Hajra, Tetrahedron Lett., 2013, 54, 3892 CrossRef CAS PubMed; (c) A. K. Bagdi, S. Santra, M. Rahman, A. Majee and A. Hajra, RSC Adv., 2013, 3, 24034 RSC; (d) S. Santra, A. K. Bagdi, A. Majee and A. Hajra, RSC Adv., 2013, 3, 24931 RSC.
- (a) H. Li, C. Pan, Y. Cheng and C. Zhu, Tetrahedron Lett., 2013, 54, 6679 CrossRef CAS PubMed; (b) S. Sanjaya and S. Chiba, Org. Lett., 2012, 14, 5342 CrossRef CAS PubMed; (c) S. Seo, M. Slater and M. F. Greaney, Org. Lett., 2012, 14, 2650 CrossRef CAS PubMed; (d) C. W. Cheung and S. L. Buchwald, J. Org. Chem., 2012, 77, 7526 CrossRef CAS PubMed; (e) S. E. Allen, R. R. Walvoord, R. P. Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed.
- (a) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851 CrossRef CAS PubMed; (b) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (c) S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400 CrossRef CAS PubMed; (d) A. Hajra, Y. Wei and N. Yoshikai, Org. Lett., 2012, 14, 5488 CrossRef CAS PubMed.
- D. Kundu, M. Samim, A. Majee and A. Hajra, Chem. – Asian J., 2011, 6, 406 CrossRef CAS PubMed.
- (a) S. Santra, A. K. Bagdi, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1065 CrossRef CAS; (b) S. Mitra, A. K. Bagdi, A. Majee and A. Hajra, Tetrahedron Lett., 2013, 54, 4982 CrossRef CAS PubMed.
- (a) Z. Cui, X. Shang, X. F. Shao and Z. Q. Liu, Chem. Sci., 2012, 3, 2853 RSC; (b) G. A. Russell and P. Ngoviwatchai, J. Org. Chem., 1989, 54, 1836 CrossRef CAS; (c) J. Xiang and P. L. Fuchs, J. Am. Chem. Soc., 1996, 118, 11986 CrossRef CAS.
- A. E. King, L. M. Huffman, A. Casitas, M. Costas, X. Ribas and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 12068 CrossRef CAS PubMed.
- L. J. Goossen, N. Rodriguez, B. Melzer, C. Linder, G. Deng and L. M. Levy, J. Am. Chem. Soc., 2007, 129, 4824 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All 1H and 13C NMR spectra. See DOI: 10.1039/c4ob01320d |
| This journal is © The Royal Society of Chemistry 2015 |