
Catalytic asymmetric desymmetrization approaches to enantioenriched cyclopentanes
Madhu Sudan
Manna
and
Santanu
Mukherjee
*
Department of Organic Chemistry, Indian Institute of Science, Bangalore 560 012, India. E-mail: sm@orgchem.iisc.ernet.in; Fax: +91-80-2360-0529
First published on 29th September 2014
Abstract
Catalytic asymmetric desymmetrization represents an excellent strategy for accessing highly functionalized chiral building blocks. However, the application of desymmetrization for the synthesis of enantioenriched cyclopentane derivatives remained limited, when compared to chiral cyclohexanes. We have recently developed a desymmetrization protocol for prochiral 2,2-disubstituted cyclopentene-1,3-diones by direct catalytic asymmetric vinylogous nucleophilic addition of deconjugated butenolides. In this perspective, we give an overview of asymmetric desymmetrization reactions leading to enantioenriched cyclopentanes and their derivatives. The focus is kept confined to the diverse nature of reactions used for this purpose. A brief discussion on the potential future directions is also provided.
 Madhu Sudan Manna | Madhu Sudan Manna received his B.Sc. degree (Chemistry Hons.) from Bajkul Milani Mahavidyalaya, West Bengal, in 2008, and his M.Sc. degree (Chemistry) from the Indian Institute of Technology, Guwahati in 2010. He is currently pursuing his Ph.D. at the Indian Institute of Science, Bangalore, under the supervision of Professor Mukherjee. His research interests include application of asymmetric organocatalysis for studying vinylogous nucleophilic reactivity. |
 Santanu Mukherjee | Santanu Mukherjee obtained his B.Sc. degree from Ramakrishna Mission Residential College, Narendrapur (2000) and M.Sc. degree from the Indian Institute of Technology, Kanpur (2002). After earning his Ph.D. (summa cum laude) in 2006 working with Professor Albrecht Berkessel at Universität zu Köln, he worked as a postdoctoral fellow with Professor Benjamin List at Max-Planck Institut für Kohlenforschung in Mülheim an der Ruhr (2006–2008) and with Professor E. J. Corey at Harvard University (2008–2010). Since 2010, he has been an Assistant Professor at the Department of Organic Chemistry at Indian Institute of Science, Bangalore. His research interests revolve around various aspects of asymmetric catalysis. He is a recipient of the Thieme Chemistry Journals Award (2011), the National Academy of Sciences, India (NASI)-Young Scientist Platinum Jubilee Award (2013) and the Indian National Science Academy (INSA) Medal for Young Scientist (2014). He is also an associate of the Indian Academy of Sciences, Bangalore (2012–2015). |
Introduction
Five-membered carbocycles containing one or more stereogenic centers on the ring are a structural element found in a wide variety of natural products and biologically relevant non-natural compounds (Fig. 1).1 As a consequence, various approaches to their synthesis have been investigated.2 [3 + 2]-Cycloaddition,3 Nazarov cyclization,4 Pauson–Khand reaction5 and ring-closing metathesis (RCM)6 are some of the commonly employed methods for the construction of the cyclopentane scaffold. However, while these reactions have been routinely used for accessing racemic cyclopentanes, the corresponding enantioselective variants are less developed. The other approaches to enantioenriched cyclopentanes mostly rely on the enantioselective transformations on the already existing functionalized five-membered carbocyclic backbones. Even though extremely successful for the asymmetric synthesis of a range of chiral cyclopentane derivatives, the limitation of this strategy becomes apparent for cyclopentanes decorated with one or more quaternary stereogenic centers.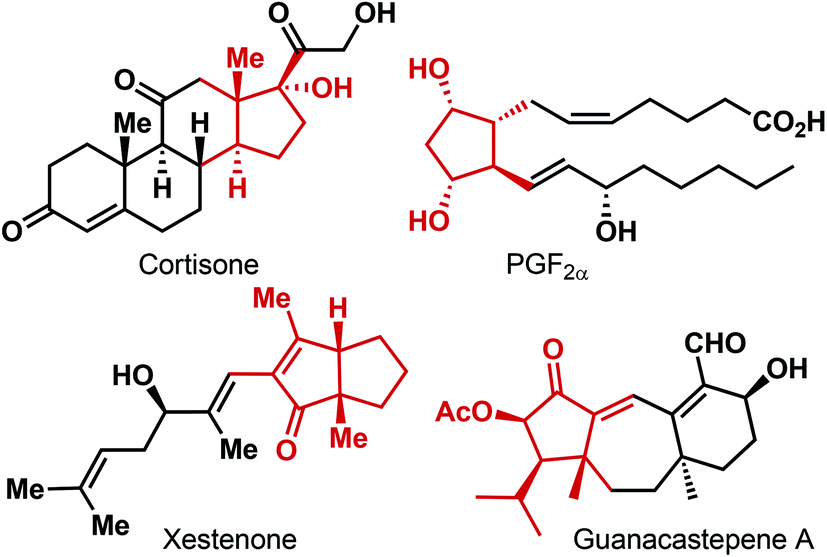 | ||
| Fig. 1 Selected examples of natural products containing chiral five-membered carbocycle moiety. | ||
Desymmetrization of achiral or meso compounds, on the other hand, constitutes a general and powerful strategy for obtaining enantiomerically enriched compounds.7 As opposed to kinetic resolution, desymmetrization holds the intrinsic advantage of delivering the desired product with theoretically 100% chemical yield. Additionally, multiple stereogenic centers can be established simultaneously, some of which can be distant from the reaction site and would be difficult to install by other means. However, application of any catalytic enantioselective transformation to the desymmetrization of prochiral substrates is not a trivial task. Typically a catalytic asymmetric desymmetrization reaction requires a symmetry breaking operation where the chiral catalyst must discriminate between two enantiotopic atoms or groups.8
Due to the relative conformational rigidity, cyclic compounds are often preferred as substrates for desymmetrization reactions and a wide variety of highly functionalized enantioenriched carbocyclic compounds has been synthesized by catalytic asymmetric desymmetrization reactions. In this context, chiral cyclohexane derivatives enjoyed particular attention. In comparison, desymmetrization routes to enantiopure cyclopentanes remained somewhat less explored, despite their widespread occurrence in complex targets (Fig. 1).
Very recently, we have developed a highly efficient desymmetrization protocol for 2,2-disubstituted cyclopentene-1,3-diones via direct vinylogous nucleophilic addition of deconjugated butenolides with the help of a tertiary amine–thiourea bifunctional catalyst.9
This perspective is intended to provide an overview of catalytic asymmetric desymmetrization reactions leading to enantioenriched cyclopentanes and their derivatives. The content of this article is by no means comprehensive, and the discussion is confined to selected earlier developments and concluded with the contribution from our own laboratory.
Catalytic asymmetric desymmetrization routes to chiral cyclopentanes
Catalyst controlled direct nucleophilic ring-opening of achiral or meso epoxides, aziridines and anhydrides is one of the most popular and commonly investigated desymmetrization reactions.10 Numerous catalysts have been developed for these reactions and led to the asymmetric synthesis of a wide variety of chiral building blocks. Quite naturally these reactions provide important and useful routes to highly functionalized chiral cyclopentane derivatives. Nonetheless, these desymmetrization reactions are kept outside the scope of this perspective, since they have previously been reviewed in sufficient detail.The following section will summarize various other types of reactions used for the desymmetrization of achiral or meso compounds en route to enantioenriched cyclopentane derivatives.
One of the earlier examples of such desymmetrization is the classical Hajos–Parrish–Eder–Sauer–Wiechert reaction, a proline-catalyzed enantiogroup-differentiating 6-enolendo aldolization of cyclopentane-1,3-dione 1 (Scheme 1).11 The resulting fused bicyclic compounds (2 or 3) were obtained with high enantioselectivity. Irrespective of the poor mechanistic understanding at the time of its discovery, this reaction found immediate application in steroid and other natural product syntheses, and also bears historical significance as the origin of asymmetric enamine catalysis.
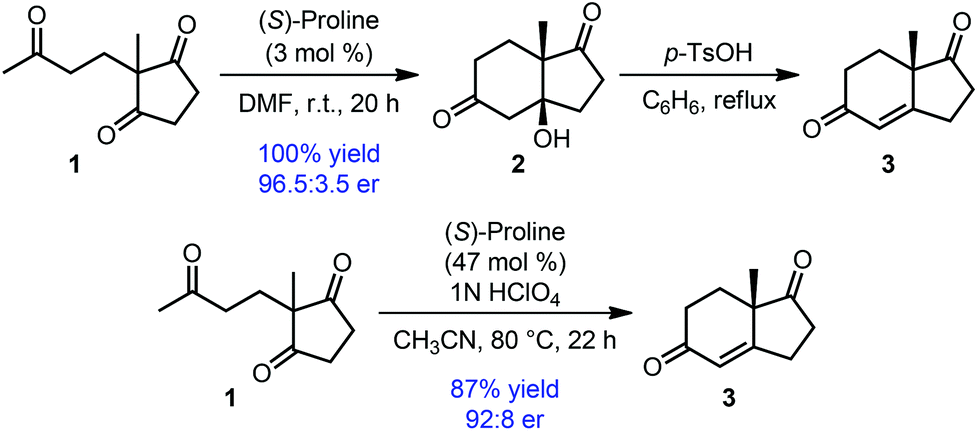 | ||
| Scheme 1 Hajos–Parrish–Eder–Sauer–Wiechert reaction: an enantiogroup-differentiating 6-enolendo aldolization. | ||
Enantioselective alcohol protection served as a popular strategy for the desymmetrization of meso and achiral alcohols.12 Among the most commonly used protecting groups for alcohols, acyl has remained the most developed for this purpose. Either acylation of the free alcohols by means of a transesterification reaction or hydrolysis of the appropriate acyl derivative of the alcohols has been employed, particularly under enzyme catalysis. By jointly using these two processes, in certain cases, both the product enantiomers can be accessed using the same enzyme, provided the enzyme maintains the enantiopreference in both processes. During the enantioselective synthesis of the core structure of the neocarzinostatin chromophore, Hirama et al. prepared both the enantiomers of 4via desymmetrization of meso 5-tert-butyldimethylsilyloxy-2-cyclopentene-1,4-diol 5 and its diacetate derivative 6 using the lipase Amano AK (Scheme 2).13 In both the cases, monoacetates were obtained essentially in enantiopure form in excellent yield.
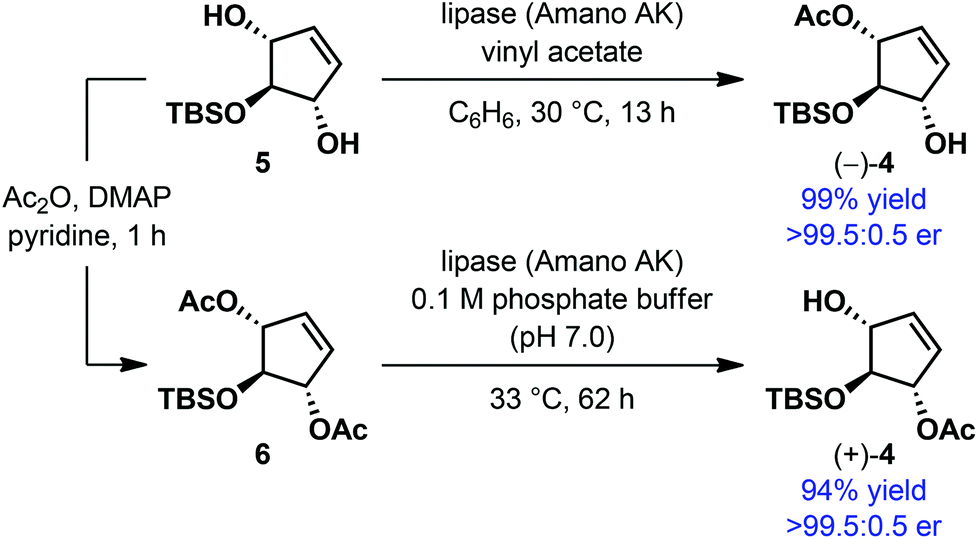 | ||
| Scheme 2 Enzymatic desymmetrization of meso-diol by Hirama et al. | ||
However, the use of acylated building blocks is often avoided in multistep syntheses due to the requirement of rather harsh reaction conditions for their deprotection. In contrast, silyl ethers represent the most commonly used protecting group for alcohols. Selective removal of silyl ethers is possible under mild conditions and does not usually involve the risk of undesired side reactions. Despite such obvious advantages associated with silyl protecting groups and the availability of a number of differently substituted silyl groups, catalytic enantioselective silyl protection of alcohols was rarely studied. In 2006, Snapper, Hoveyda and co-workers developed a simple chiral catalyst 7 for the enantioselective silyl protection of meso-diols (Scheme 3).14 Using this catalyst, cyclopentane-based meso-diols (8–9) were desymmetrized under mild reaction conditions to afford the monosilylated products in high yield with good to excellent enantioselectivities.
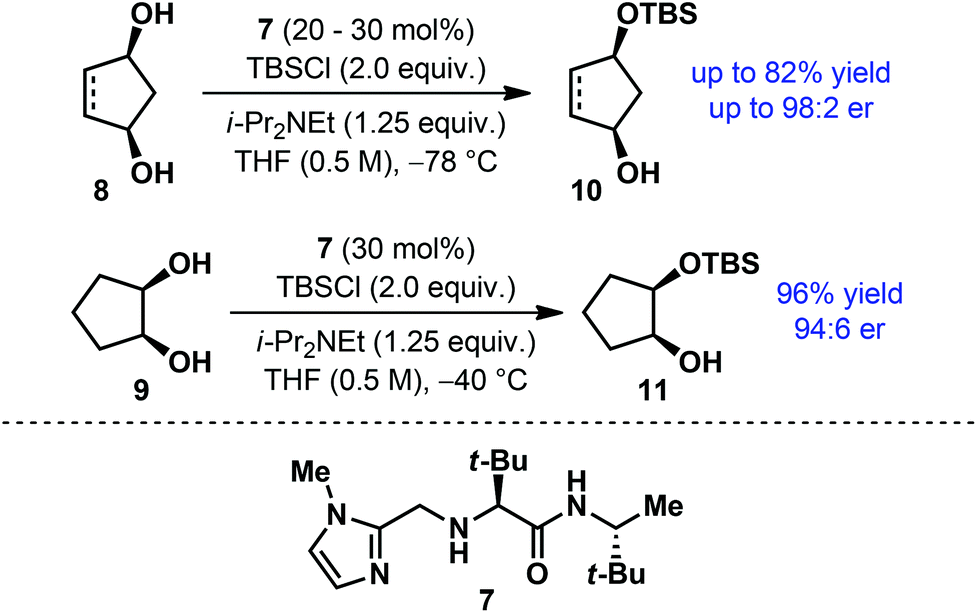 | ||
| Scheme 3 Catalytic enantioselective silylation of meso-diols by Snapper, Hoveyda and co-workers. | ||
Reductive transformations have also been utilized for the desymmetrization of achiral or meso compounds.15 However, the number of such examples is very limited, especially when considering cyclopentane derivatives as the product.
During the conversion of Torgov's estrone synthesis into an enantioselective process, Corey and co-workers developed an elegant reductive desymmetrization of achiral Torgov diketone 12a as the key step using a modified CBS reduction (Scheme 4).16 The resulting product 13a was obtained in high yield and with excellent enantioselectivity when oxazaborolidine 14 was employed as the catalyst. A similar level of selectivity was also observed for a simplified substrate 12b. However, a detailed scope of this desymmetrization protocol has never been explored. It must be mentioned that similar transformation has also been accomplished under enzymatic conditions using Baker's yeast17 and by transfer hydrogenation using a chiral Ru-catalyst.18
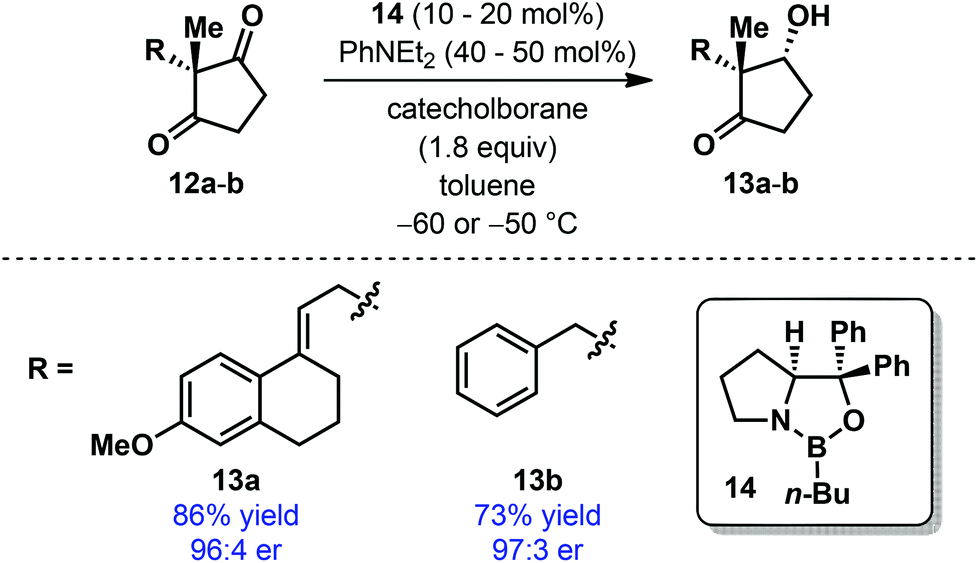 | ||
| Scheme 4 Reductive desymmetrization of cyclopentane-1,3-dione by Corey et al. | ||
Whereas the majority of desymmetrization processes proceeds through bond-forming transformations, desymmetrization through bond-breaking/fragmentation is quite rare. Taking a cue from Corey's brefeldin A synthesis,19 Jørgensen and co-workers developed an asymmetric desymmetrization–fragmentation of meso-cyclopentanone derivatives 15 (Scheme 5).20 Under cinchona alkaloid-derived bifunctional thiourea catalysis (17 or 18), meso epoxycyclopentanones [X = O] and cyclopropane cyclopentanone [X = C(CO2Et)2] underwent desymmetrizing fragmentation to generate synthetically versatile cyclopentenone derivatives 16 in high yield with good to excellent enantioselectivities. However, the scope of this reaction is relatively narrow.
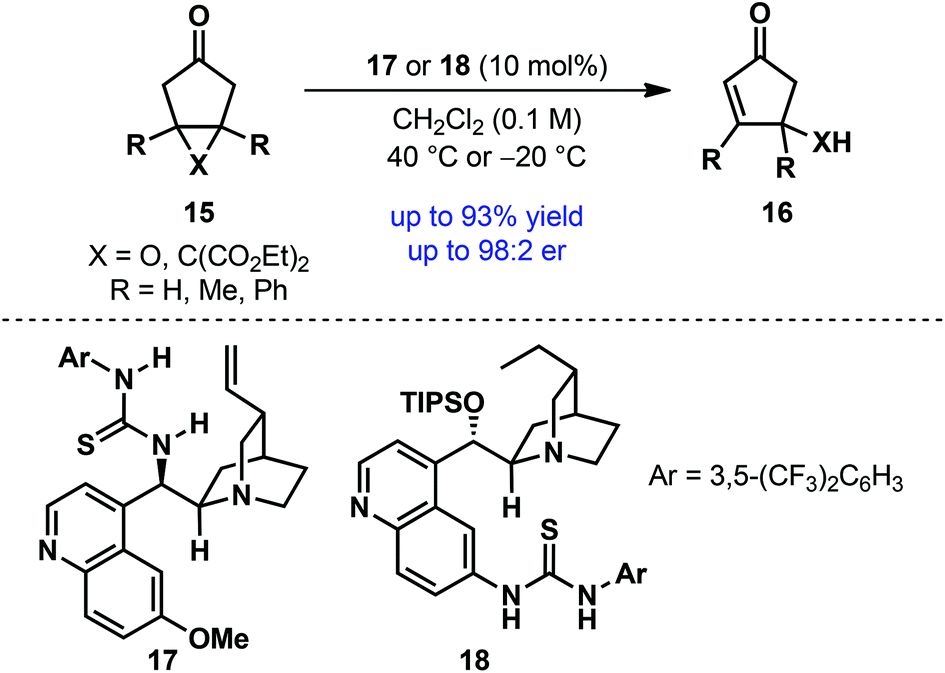 | ||
| Scheme 5 Desymmetrization–fragmentation of fused bicyclic cyclopentanones by Jørgensen et al.20 | ||
The asymmetric desymmetrization reactions described so far are clearly substrate specific and rather narrow in scope, at least as far as the synthesis of enantioenriched cyclopentanes is concerned. In fact, there are only a handful of desymmetrization reactions that display a broad substrate scope for the synthesis of enantioenriched cyclopentanes.
Palladium-catalyzed asymmetric allylic alkylation is a well-established strategy for the synthesis of a wide range of chiral building blocks.21 This strategy has also been exploited for the synthesis of enantioenriched five-membered carbocycles. Desymmetrization of meso disubstituted cyclopentene derivatives proved to be an extremely effective strategy for this purpose (Scheme 6). Initial efforts were directed towards intramolecular allylic alkylation with meso bis-carbamates 19a for the synthesis of fused bicyclic oxazolidin-2-ones 20: the use of Trost's bisphosphine ligand (21) ensured high yield and excellent enantioselectivity (Scheme 6A).22 More recently, a number of reports described the corresponding intermolecular variant with various nucleophiles (Scheme 6B).23 Uniformly high yield and enantioselectivities were encountered while using the Trost ligand 21. The resulting products were often used as substrates for further Pd-catalyzed allylic alkylation reaction with a second nucleophile.23b,c The products of such a tandem reaction were applied by Blechert and co-workers for the enantioselective synthesis of tetraponerines.23b
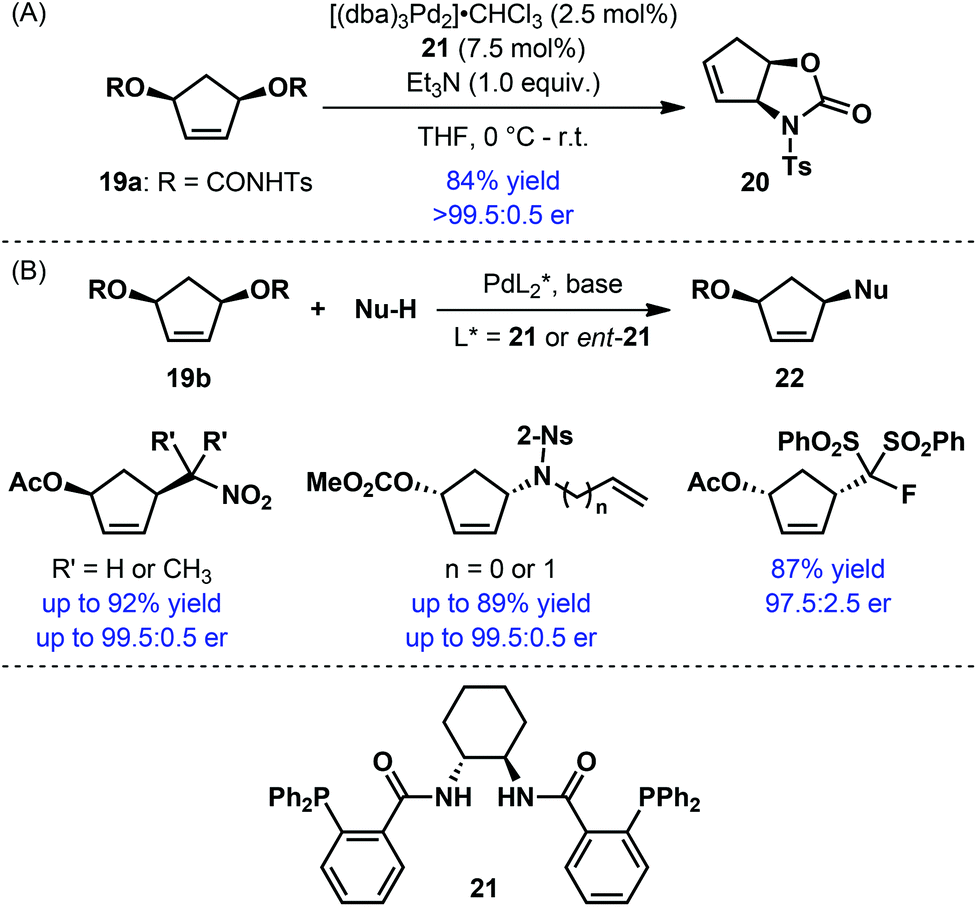 | ||
| Scheme 6 Desymmetrization of meso disubstituted cyclopentene derivatives by Pd-catalyzed asymmetric allylic alkylation. | ||
As mentioned above, a unique advantage of desymmetrization reactions is that stereocenters can be established away from the reaction site. A reaction of this type was reported by Mikami et al. in 2012, when they developed a Cu-catalyzed conjugate addition of dialkylzinc to achiral 2,2-disubstituted cyclopentene-1,3-diones 23.24 Using axially chiral monodentate phosphoramidite 25, a wide range of cyclopentene-1,3-diones 23 were desymmetrized to afford the product (24) containing a remote all-carbon quaternary stereogenic center in high yield with good to excellent diastereo- and enantioselectivity (Scheme 7). Besides the broad substrate scope, a remarkable feature of this protocol is the low catalyst loading; in some cases, an equally high level of efficiency and selectivities was achieved with a catalyst loading as low as 0.5 mol%. The scope of other alkylating agents has also been tested and a similar level of selectivities was obtained with trimethylaluminum.
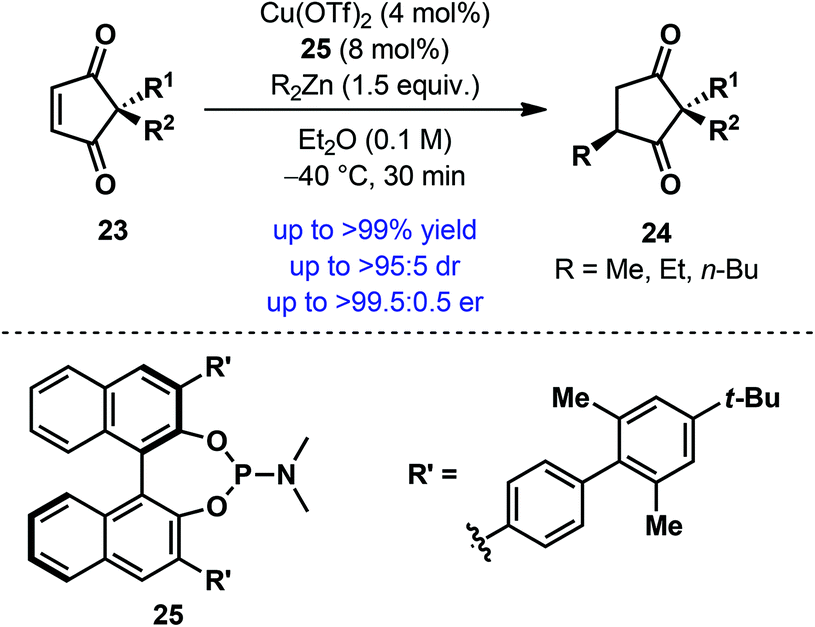 | ||
| Scheme 7 Desymmetrization of cyclopentene-1,3-diones via Cu-catalyzed enantioselective conjugate addition of dialkylzinc by Mikami et al. | ||
In the past couple of years, our laboratory has developed a number of asymmetric direct vinylogous nucleophilic addition reactions of butyrolactam and deconjugated butenolides to various electrophiles.25 Inspired by Mikami's report (Scheme 7)24 and as a continuation of our own work on the conjugate addition to C2v-symmetric maleimides,25c we envisioned an asymmetric vinylogous nucleophilic addition of deconjugated butenolides to prochiral 2,2-disubstituted cyclopentene-1,3-diones. We realized that a successful implementation of this strategy would not only desymmetrize cyclopentene-1,3-diones, but also create multiple stereogenic centers, even outside the cyclopentane scaffold. The latter feature would stand in sharp contrast to the desymmetrization reactions described so far, where the stereocenters are created only on the cyclopentane ring. In addition, given the modular nature of both the reaction partners, we anticipated such a reaction to offer a broad substrate scope. Our choice of the reaction was further motivated by the presence of both butenolide and cyclopentane moieties in some natural products (Fig. 2).26
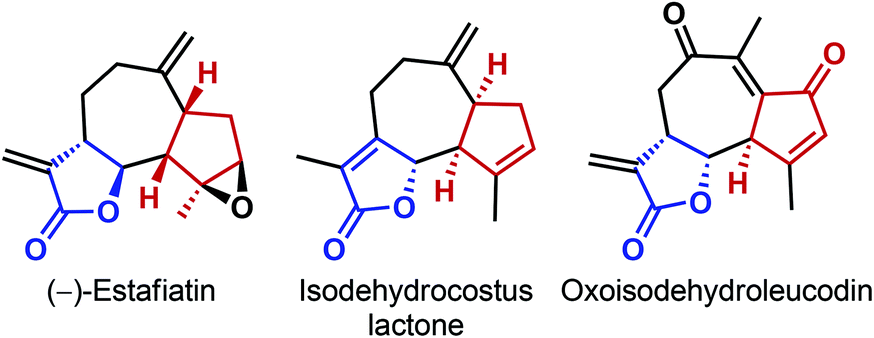 | ||
| Fig. 2 Representative examples of natural products containing both the butenolide and cyclopentane moiety. | ||
A thorough and systematic catalyst optimization established the bifunctional thiourea derivative containing a secondary amide side chain 28a as the optimum catalyst (Scheme 8).9 Using only 10 mol% of the catalyst 28a under mild reaction conditions, we were able to desymmetrize a wide range of prochiral cyclopentene-1,3-diones (23) generally in high yield with outstanding diastereoselectivity and excellent enantioselectivity (Scheme 8). The electrophile scope (23) includes the combination of either methyl or ethyl (R3) with benzylic, allylic, alkyl, aryl and functionalized alkyl substituents (R2) at the quaternary center. A similar level of diastereo- and enantioselectivity was also observed with deconjugated butenolides (26) containing a variety of γ-substituents (R1).
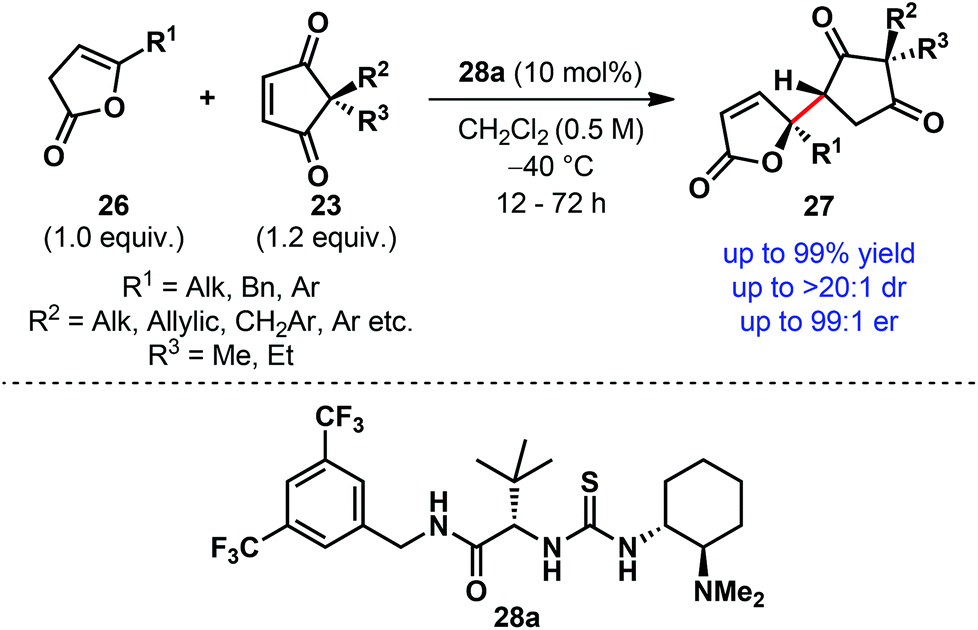 | ||
| Scheme 8 Desymmetrization of cyclopentene-1,3-diones via direct enantioselective vinylogous addition of deconjugated butenolides by Mukherjee et al. | ||
In addition to the broad substrate scope, we have also demonstrated the robustness of our reaction to a large number of potentially competing electrophiles and nucleophiles using the robustness screening protocol devised by Glorius et al.27 Whereas the high level of diastereo- and enantioselectivities was mostly retained in the presence of diverse additives, no desired product formation was observed in the presence of aliphatic amines. Our report represents the first example of the application of this robustness screening protocol to an asymmetric transformation.
A remarkable influence of the catalyst side chain (amide functionality) on the enantioselectivity was observed: methylation led to a drastic drop in enantioselectivity (Scheme 9A).9 Similar effects were also apparent on the catalytic efficiency and diastereoselectivity, albeit to a lesser extent. These observations point towards a dual role played by the amide side chain of the catalyst. According to our current understanding whereas an additional H-bonding from the secondary amide N–H (Scheme 9B) could be held responsible for the enhanced activity of the corresponding catalyst 28a, a methylation-induced conformational switch (with respect to the rotation around C–CO) accounts for the observed difference in enantio- and diastereoselectivity. For the secondary amide side chain (in catalyst 28a), the alignment of all the three NHs in the same direction makes only one face of the activated electrophile available for the nucleophilic attack. On the other hand, in the methylated catalyst (28b), the amide carbonyl orients itself towards thiourea NHs and results in the opening of both the faces of the activated electrophile. To the best of our knowledge, this is the first time such a structure-activity-relationship was observed between the apparently innocuous catalyst side chain and the enantioselectivity of a reaction.
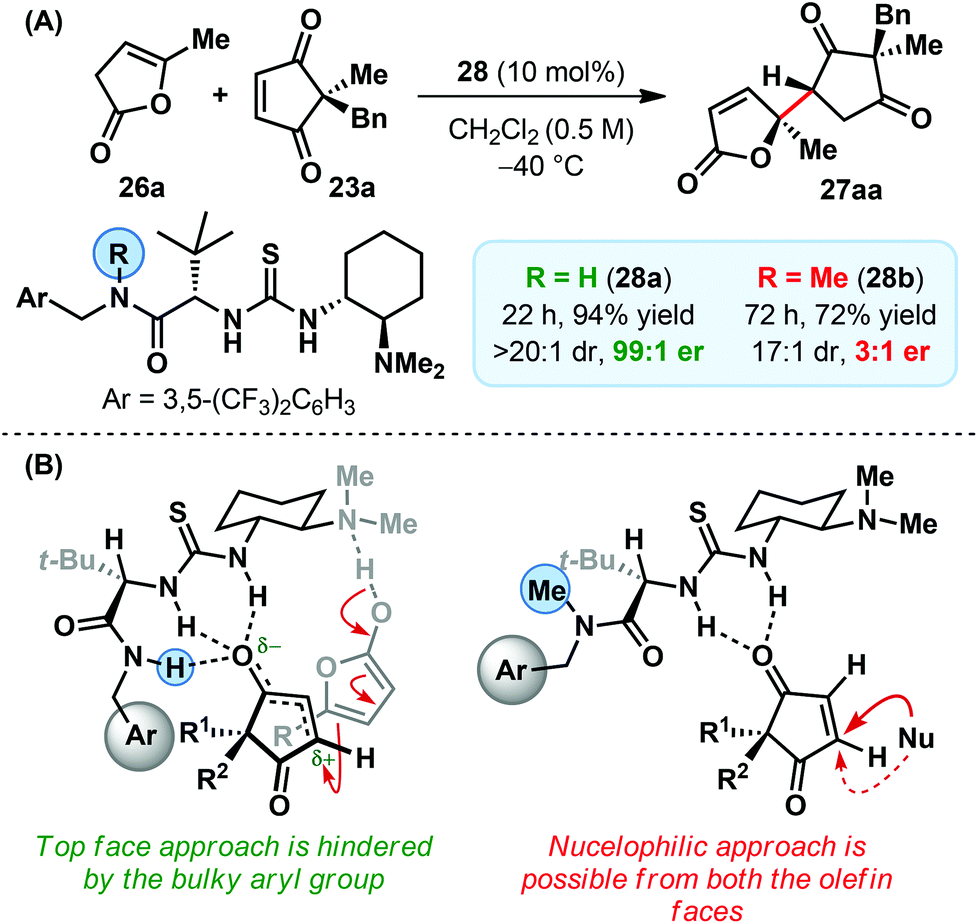 | ||
| Scheme 9 (A) Effect of catalyst methylation on the reaction outcome and (B) mechanistic rationale of the direct vinylogous addition of deconjugated butenolides to cyclopentene-1,3-diones. | ||
Conclusion
Catalytic asymmetric desymmetrization has been and will continue to be an exciting strategy for accessing enantioenriched chiral building blocks. However, application of this strategy for the enantioselective synthesis of chiral five-membered carbocycles has remained rather sporadic, particularly when compared to their six-membered counterparts. As evident from the examples above, a large variety of reactions can be used to desymmetrize meso or prochiral cyclopentane derivatives. In fact, seemingly unspectacular transformations, when used for symmetry breaking, can open up avenues for achieving complex targets. Similarly, shortcomings of well-established catalytic asymmetric methods in desymmetrizing meso or prochiral substrates should continue to be the motivation behind better mechanistic understanding and development of new chiral catalysts. More stimulating advancements along these directions can be expected during the coming years.While the generation of a large number of stereocenters (and hence stereochemical diversity) through desymmetrization can be viewed as an advantage, an ideal desymmetrization should avoid creation of new stereocenters and yet would proceed through the synthetically progressive bond-formation step. The Hajos–Parrish–Eder–Sauer–Wiechert reaction represents probably the most successful example of such a desymmetrization reaction. Its enormous application in the synthesis of steroids and other natural products clearly shows that such reactions could be highly rewarding. However, planning and realization of these types of reactions with a useful level of stereocontrol is extremely challenging. Therefore it is not a surprise that the examples of such reactions are rare. Considering the rapid expansion of asymmetric catalysis in terms of new concepts and catalysts, the emergence of more of this type of transformation can be anticipated in not so distant future.
Overall, going by the appearance of a large number of catalytic asymmetric transformations, it would be surprising if they do not contribute to the findings of more desymmetrization protocols including those delivering enantioenriched cyclopentanes.
Acknowledgements
Generous financial support from the Indian Institute of Science, Bangalore in the form of a start-up grant is gratefully acknowledged. We thank the Department of Science and Technology (DST), the Council of Scientific and Industrial Research (CSIR) and the Department of Atomic Energy (DAE) for funding our research program. M.S.M. thanks CSIR for a doctoral fellowship.Notes and references
- For selected reviews, see: (a) S. Das, S. Chandrasekhar, J. S. Yadav and R. Grée, Chem. Rev., 2007, 107, 3286–3337 CrossRef CAS PubMed; (b) J.F. Biellmann, Chem. Rev., 2003, 103, 2019–2033 CrossRef CAS PubMed; (c) G. Mehta and A. Srikrishna, Chem. Rev., 1997, 97, 671–719 CrossRef CAS PubMed.
- L. F. Silva Jr., Tetrahedron, 2002, 58, 9137–9161 CrossRef.
- B. M. Trost, Angew. Chem., Int. Ed., 1986, 25, 1–20 CrossRef.
- T. Vaidya, R. Eisenberg and A. J. Frontier, ChemCatChem, 2011, 3, 1531–1548 CrossRef CAS.
- (a) J. Habermann, Curr. Org. Chem., 2010, 14, 1139–1152 CrossRef CAS; (b) T. Shibata, Adv. Synth. Catal., 2006, 348, 2328–2336 CrossRef CAS.
- V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2009, 109, 6809–6857 CrossRef CAS PubMed.
- (a) T. Rovis, in New Frontiers in Asymmetric Catalysis, ed. K. Mikami and M. Lautens, Wiley, Hoboken, NJ, 2007, pp. 275–311 Search PubMed; (b) M. C. Willis, J. Chem. Soc., Perkin Trans. 1, 1999, 1765–1784 RSC.
- R. S. Ward, Chem. Soc. Rev., 1990, 19, 1–19 RSC.
- M. S. Manna and S. Mukherjee, Chem. Sci., 2014, 5, 1627–1633 RSC.
- (a) P.-A. Wang, Beilstein J. Org. Chem., 2013, 9, 1677–1695 CrossRef PubMed; (b) M. D. D. de Villegas, J. A. Gálvez, P. Etayo, R. Badorrey and P. López-Ram-de-Víu, Chem. Soc. Rev., 2011, 40, 5564–5587 RSC; (c) M. Pineschi, Eur. J. Org. Chem., 2006, 4979–4988 CrossRef CAS; (d) E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS PubMed.
- (a) Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621 CrossRef CAS; (b) U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496–497 CrossRef CAS.
- Á. Enríquez-García and E. P. Kündig, Chem. Soc. Rev., 2012, 41, 7803–7831 RSC.
- K. Toyama, S. Iguchi, H. Sakazaki, T. Oishi and M. Hirama, Bull. Chem. Soc. Jpn., 2001, 74, 997–1008 CrossRef CAS.
- Y. Zhao, J. Rodrigo, A. H. Hoveyda and M. L. Snapper, Nature, 2006, 443, 67–70 CrossRef CAS PubMed.
- H. Fernández-Pérez, P. Etayo, J. R. Lao, J. L. Núñez-Rico and A. Vidal-Ferran, Chem. Commun., 2013, 49, 10666–10675 RSC.
- Y.-Y. Yeung, R.-J. Chein and E. J. Corey, J. Am. Chem. Soc., 2007, 129, 10346–10347 CrossRef CAS PubMed.
- D. W. Brooks, P. G. Grothaus and W. L. Irwin, J. Org. Chem., 1982, 47, 2820–2821 CrossRef CAS.
- B. Shi, S. Merten, D. K. Y. Wong, J. C. K. Chu, L. L. Liu, S. K. Lam, A. Jäger, W.-T. Wong, P. Chiu and P. Metz, Adv. Synth. Catal., 2009, 351, 3128–3132 CrossRef CAS.
- E. J. Corey and R. H. Wollenberg, Tetrahedron Lett., 1976, 17, 4705–4708 CrossRef.
- G. Dickmeiss, V. D. Sio, J. Udmark, T. B. Poulsen, V. Marcos and K. A. Jørgensen, Angew. Chem., Int. Ed., 2009, 48, 6650–6653 CrossRef CAS PubMed.
- (a) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS PubMed.
- (a) B. M. Trost and D. E. Patterson, J. Org. Chem., 1998, 63, 1339–1341 CrossRef CAS; (b) B. M. Trost, D. L. Van Vranken and C. Bingel, J. Am. Chem. Soc., 1992, 114, 9327–9343 CrossRef CAS.
- (a) T. Fukuzumi, N. Shibata, M. Sugiura, H. Yasui, S. Nakamura and T. Toru, Angew. Chem., Int. Ed., 2006, 45, 4973–4977 CrossRef CAS PubMed; (b) R. Stragies and S. Blechert, J. Am. Chem. Soc., 2000, 122, 9584–9591 CrossRef CAS; (c) B. M. Trost and J.-P. Surivet, Angew. Chem., Int. Ed., 2000, 39, 3122–3124 CrossRef CAS.
- K. Aikawa, T. Okamoto and K. Mikami, J. Am. Chem. Soc., 2012, 134, 10329–10332 CrossRef CAS PubMed.
- (a) V. Kumar and S. Mukherjee, Chem. Commun., 2013, 49, 11203–11205 RSC; (b) V. Kumar, B. Ray, P. Rathi and S. Mukherjee, Synthesis, 2013, 1641–1646 CAS; (c) M. S. Manna and S. Mukherjee, Chem. – Eur. J., 2012, 18, 15277–15282 CrossRef CAS PubMed; (d) A. Ray Choudhury and S. Mukherjee, Org. Biomol. Chem., 2012, 10, 7313–7320 RSC; (e) M. S. Manna, V. Kumar and S. Mukherjee, Chem. Commun., 2012, 48, 5193–5195 RSC.
- (a) M. Ando, K. Ibayashi, N. Minami, T. Nakamura, K. Isogai and H. Yoshimura, J. Nat. Prod., 1994, 57, 433–445 CrossRef CAS; (b) A. E. Greene and M. T. Edgar, J. Org. Chem., 1989, 54, 1468–1470 CrossRef CAS; (c) M. T. Edgar, A. E. Greene and P. Crabbé, J. Org. Chem., 1979, 44, 159–160 CrossRef CAS.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |