
Improving catalyst activity in secondary amine catalysed transformations†
John B.
Brazier
a,
Timothy J. K.
Gibbs
a,
Julian H.
Rowley
b,
Leopold
Samulis
a,
Sze Chak
Yau
a,
Alan R.
Kennedy
b,
James A.
Platts
a and
Nicholas C. O.
Tomkinson
*b
aSchool of Chemistry, Main Building, Cardiff University, Park Place, Cardiff, CF10 3AT, UK
bWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: Nicholas.Tomkinson@strath.ac.uk
First published on 14th October 2014
Abstract
The effect on catalyst performance of altering substituents at the 2-position of the Macmillan imidazolidinone has been examined. Condensation of L-phenylalanine N-methyl amide with acetophenone derivatives results in a series of imidazolidinones whose salts can be used to accelerate the Diels–Alder cycloaddition. Electron withdrawing groups significantly increase the overall rate of cycloaddition without compromise in selectivity. The most effective catalyst was shown to be efficient for a variety of substrates and the applicability of this catalyst to alternative secondary amine catalysed transformations is also discussed.
Introduction
Catalyst design generally relies upon serendipity and extensive high throughput screening which has frequently proved to be effective in asymmetric synthesis. However, pressure to reduce financial and environmental footprints provides inspiration for innovation. The use of predictive models represents the future of asymmetric synthesis whereby bespoke catalysts can be tailored to specific reactions with minimal laboratory screening. Current models, however, frequently fall short of the accuracy required to be effective predictors of both activity and selectivity.1 An alternative approach is to draw inspiration from mechanistic knowledge where a combination of kinetic experiment and intelligent design allows interrogation, reinforcing and underpinning of hypotheses.2Amine catalysis represents an important area of contemporary synthetic chemistry. Since their introduction the fields of LUMO, HOMO and SOMO catalysis have been established as the most significant areas of Organocatalysis.3 This is due, in part, to the wide variety of reactions which have been developed using secondary amines along with the excellent yields and outstanding levels of selectivity which are commonly achieved in these transformations.4,5 The field also follows some of the principles of Green chemistry which has further enhanced applicability, particularly in the industrial environment. Reducing levels of catalyst loading necessary to bring about reaction at an effective rate would further increase this potential.6
Within the field of LUMO catalysis using secondary amines a consistent mechanism has been accepted, the fundamental steps of which are outlined in Fig. 1.7–9 In Step 1 the secondary amine salt 1 condenses with the α,β-unsaturated carbonyl compound 2 to form the reactive iminium ion 3. In Step 2 the diene 4 undergoes cycloaddition with the activated π-system of 3 to form the iminium ion of the product 5. Finally, in Step 3 the iminium ion 5 undergoes hydrolysis/solvolysis to release the product (e.g. acetal 6) and regenerate the secondary amine salt 1.10
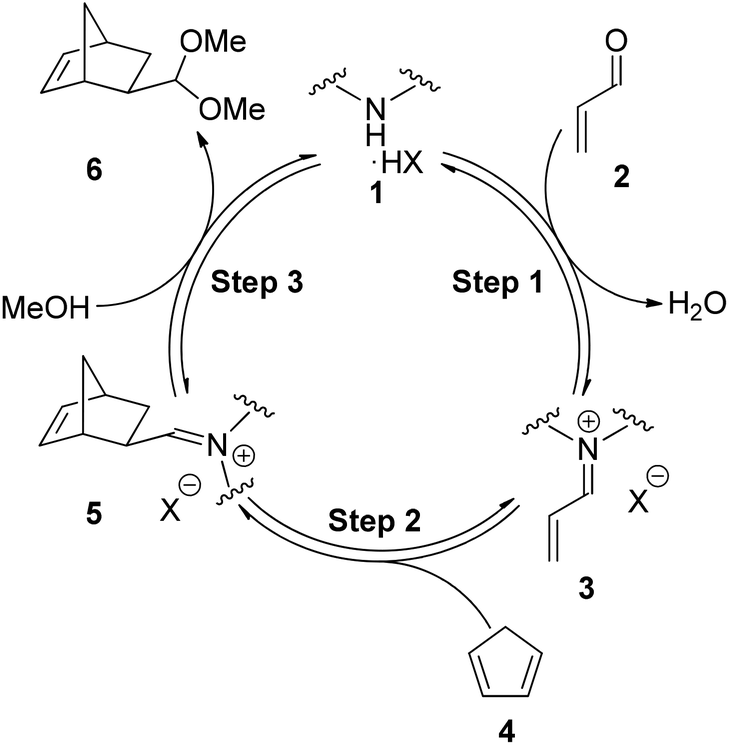 | ||
| Fig. 1 Catalytic cycle for imidazolidinone catalysed Diels–Alder cycloaddition. | ||
For the Diels–Alder cycloaddition reaction, under literature optimised conditions, it has been shown the rate determining step of the catalytic cycle is iminium ion formation (Step 1).11 Iminium ion formation consists of a number of mechanistic steps, whereby distinct equilibria require the amine to have different properties.12Fig. 2 shows a series of productive equilibria from Step 1 leading to a reactive iminium ion 12. Reduced basicity of the amine 7 drives Equilibrium 1 and Equilibrium 4 in the forward direction whereas amine nucleophilicity is required for Equilibrium 3 to proceed forwards. It is well established that the basicity of an amine is intimately linked to its nucleophilicity, therefore, positively influencing Equilibrium 1 and Equilibrium 4 by reducing basicity can negatively impact Equilibrium 3 by reducing nucleophilicity. Therefore in order to influence Step 1 of the catalytic cycle the electronics of the amine must be balanced through appropriate substitution.
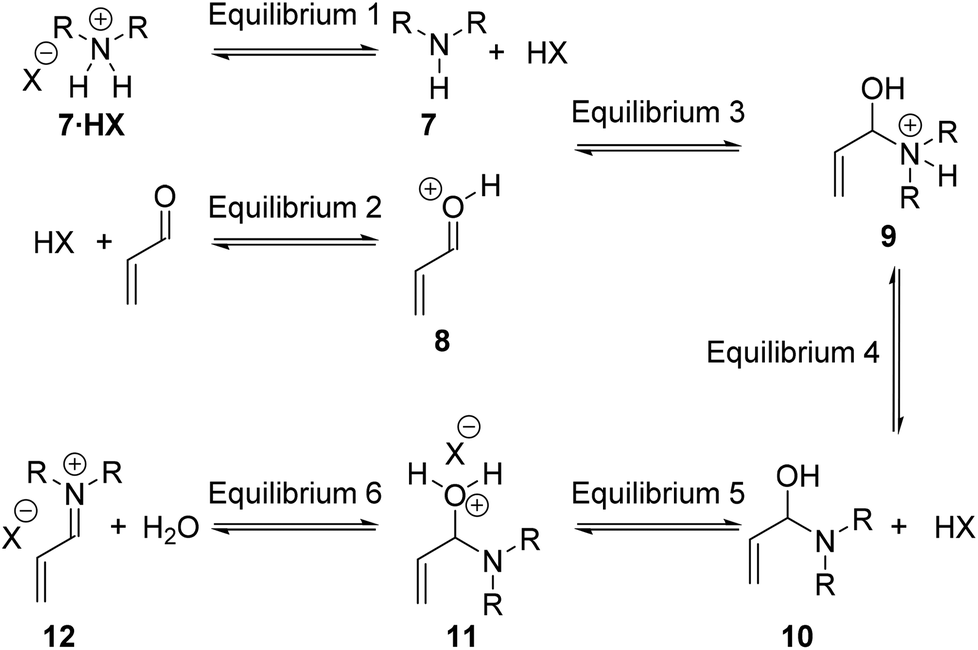 | ||
| Fig. 2 Considerations within Step 1 of the catalytic cycle. | ||
Within the literature there are two principal catalyst scaffolds developed for accelerating the Diels–Alder reaction through iminium ion intermediates (Fig. 3): the imidazolidinones13 and the diarylprolinol ethers;14 the imidazolidinones having a higher level of activity in this reaction.15 We therefore elected to examine the imidazolidinone architecture to discover catalysts with higher levels of activity.16,17 Within this manuscript we prepare a series of imidazolidinone structures based upon our mechanistic understanding of the reaction and explore the reactivity of these catalysts.
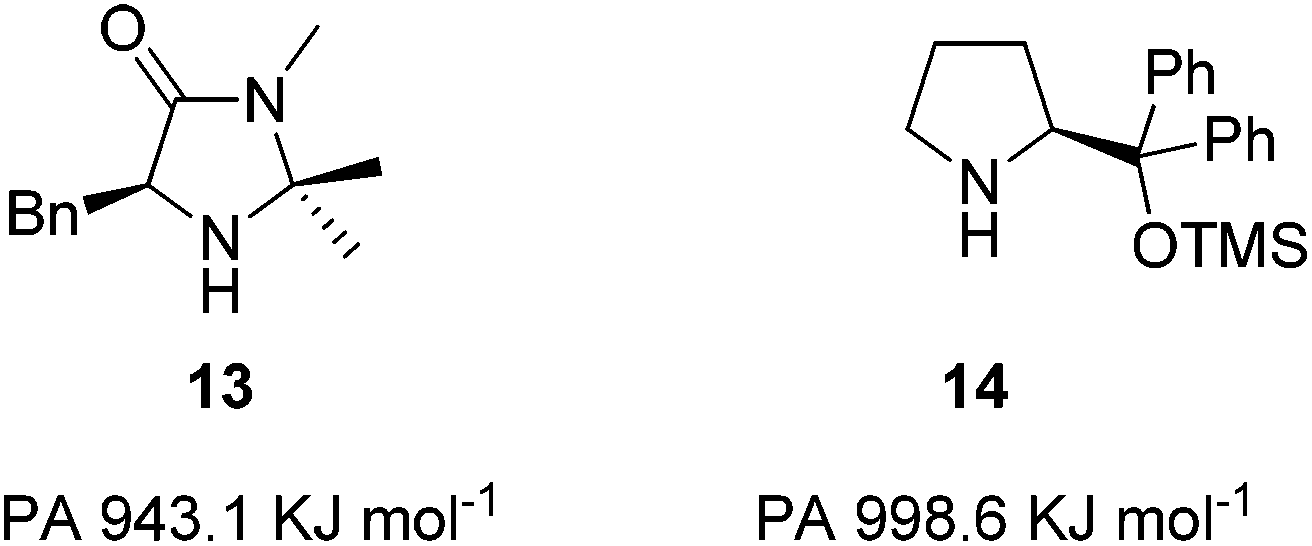 | ||
| Fig. 3 Imidazolidinone and diarylprolinol ether catalysts. | ||
Understanding how substituents on the secondary amine can affect basicity, nucleophilicity, and hence the rate of Step 1 was deemed crucial for influencing reactivity. Modelling studies had previously shown the highest activation barrier within iminium ion formation was Equilibrium 1 where the ammonium salt 7·HX loses a proton (Fig. 2).18 When comparing the imidazolidinone 13 and the diarylprolinol ether 14 it was found proton affinity (PA) was a readily calculable theoretical measure of amine basicity to act as a predictor of catalyst activity.15 The benchmark imidazolidinone 13 has a PA of 943.1 KJ mol−1 whereas the diarylprolinol ether 14, which has a lower level activity, has a PA of 998.6 KJ mol−1 (Fig. 3).15 Therefore we sought to alter the basicity of 13 through substitution and determine the influence on reactivity within a benchmark Diels–Alder reaction, with the expectation that lowering proton affinity would improve reactivity.
The imidazolidinone architecture 15 offers a number of places for developing an understanding of the relationship between structure and catalytic activity (Fig. 4). We elected to examine the substituents R2 and R3 where it was thought ease of synthesis and proximity to the reactive nitrogen would engender the greatest effect on reaction outcome.
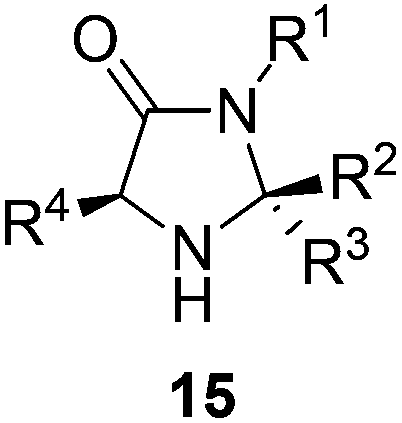 | ||
| Fig. 4 Potential points of substitution in the imidazolidinone architecture. | ||
A second fundamental requirement of the secondary amine catalyst is the ability to control the stereochemical outcome of the transformation. It was crucial for any amine with improved activity over 13 maintained the exceptional levels of asymmetry generally associated with this catalyst.7 Within the reaction of a secondary amine 13 and a α,β-unsaturated carbonyl compound two potential iminium ions can be formed, the E- and the Z-isomers 16 and 17 (Scheme 1). Ratios of 16 and 17 have been shown to be dictated by the steric requirements of the substituents on the imidazolidinone ring.9
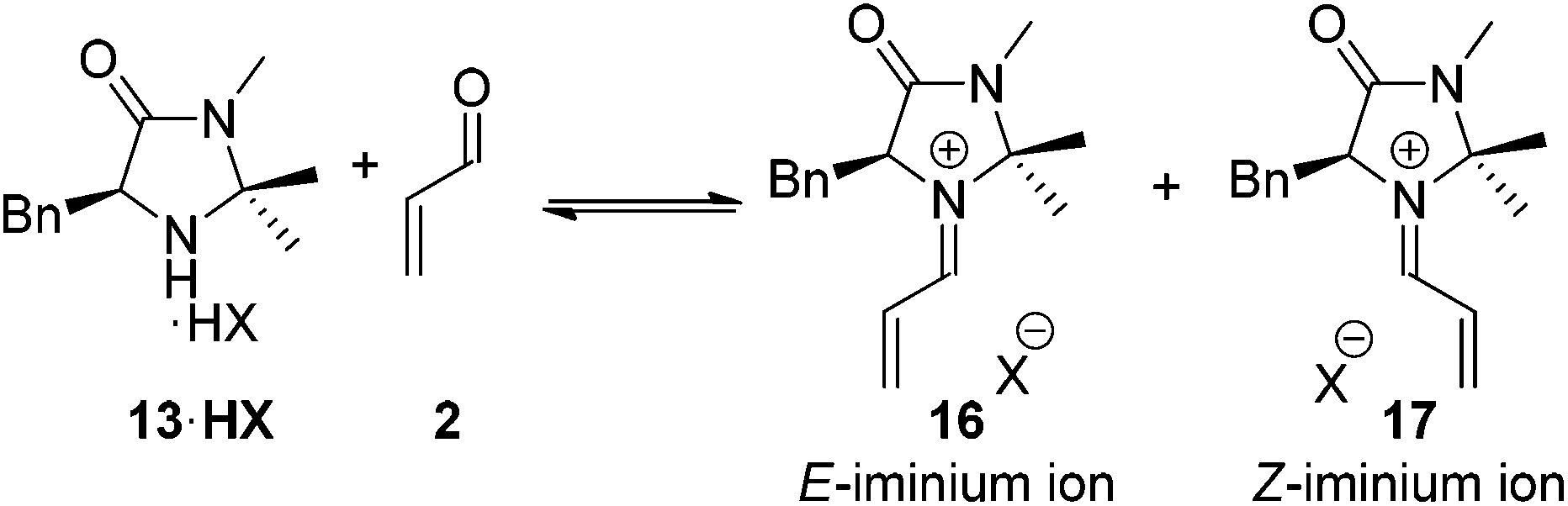 | ||
| Scheme 1 Origins of stereoselectivity in the imidazolidinone catalysed Diels–Alder cycloaddition. | ||
For the imidazolidinone 13, following selective formation of the E-iminium ion 16, the catalyst architecture renders subsequent Diels–Alder cycloaddition asymmetric, with the benzyl arm of the catalyst directing approach of the diene from the lower face of the iminium ion as shown (Scheme 1). For any alternative imidazolidinone structures it is essential that high levels of E/Z iminium control is observed to maintain levels of selectivity.9a
Results and discussion
Based upon the hypothesis that introduction of an electron withdrawing group α- to the reactive nitrogen (R2 and R3 in 15) would reduce basicity and therefore enhance reactivity a series of imidazolidinones 20–24 were prepared through condensation of phenylalanine-N-methyl amide (18) and a substituted acetophenone (Table 1). Reaction of 18 and 19 in toluene under microwave irradiation in the presence of ytterbium triflate gave the corresponding imidazolidinones 20–23 with a variety of substitution on the aromatic ring.19 For preparation of the imidazolidinone 24, derived from 4-nitroacetophenone, it proved necessary to develop alternative conditions to access the catalyst with high levels of ee. Reaction of 18 with the ketone in DMF at 150 °C for 30 minutes in the presence of methane sulfonic acid gave 24 enantiomerically pure (entry 6) (see ESI† for full details of catalyst preparation). Proton affinity (PA) for each catalyst was calculated using Gaussian 09 (B3LYP/6-31+G(d,p)) which showed a clear influence on predicted basicity of the secondary nitrogen (entries 2–6) when compared to the parent structure 13 (entry 1). Introduction of an aromatic ring with an electron donating substituent displayed a raised proton affinity (entry 2, 970 KJ mol−1) when compared to 13 (entry 1, 943 KJ mol−1). Whereas increasing the strength of electron withdrawing group progressively decreased the proton affinity of the secondary amine (entries 3–6), with the lowest value being shown for imidazolidinone 24 (R = 4-NO2C6H4, PA = 924 KJ mol−1).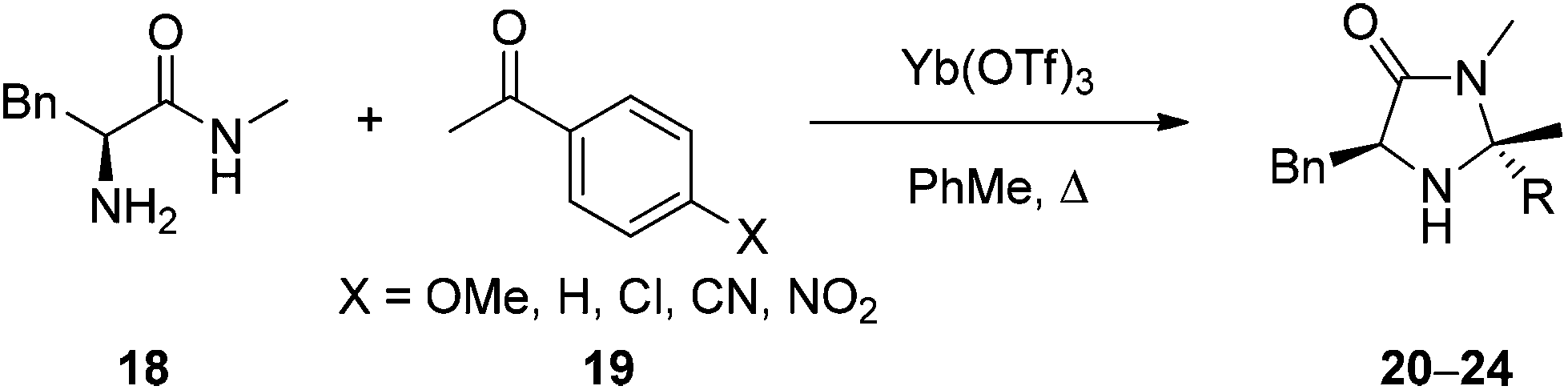
The Diels–Alder cycloaddition of cinnamaldehyde and cyclopentadiene was used to benchmark the acetophenone derived catalysts 20·HCl–24·HCl (Scheme 2).7 Each reaction was performed under literature optimised reaction conditions (3 equiv. cyclopentadiene, MeOH–H2O (19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 25 °C, 5 mol% catalyst), monitoring reaction progress by 1H NMR spectroscopy (see ESI† for full details). As can be seen in Fig. 5, introduction of an aromatic ring greatly increased the rate of the Diels–Alder cycloaddition when compared to the parent system 13·HCl (
:1), 25 °C, 5 mol% catalyst), monitoring reaction progress by 1H NMR spectroscopy (see ESI† for full details). As can be seen in Fig. 5, introduction of an aromatic ring greatly increased the rate of the Diels–Alder cycloaddition when compared to the parent system 13·HCl ( ). This was the case for all acetophenone derived catalysts examined. Subtleties in the electronic substitution of the aromatic ring had less influence on the overall rate than proton affinity predictions had suggested (Table 1), showing a deficiency in this ground state prediction, however, the significantly increased reaction rate observed was exciting and warranted further investigation.
). This was the case for all acetophenone derived catalysts examined. Subtleties in the electronic substitution of the aromatic ring had less influence on the overall rate than proton affinity predictions had suggested (Table 1), showing a deficiency in this ground state prediction, however, the significantly increased reaction rate observed was exciting and warranted further investigation.
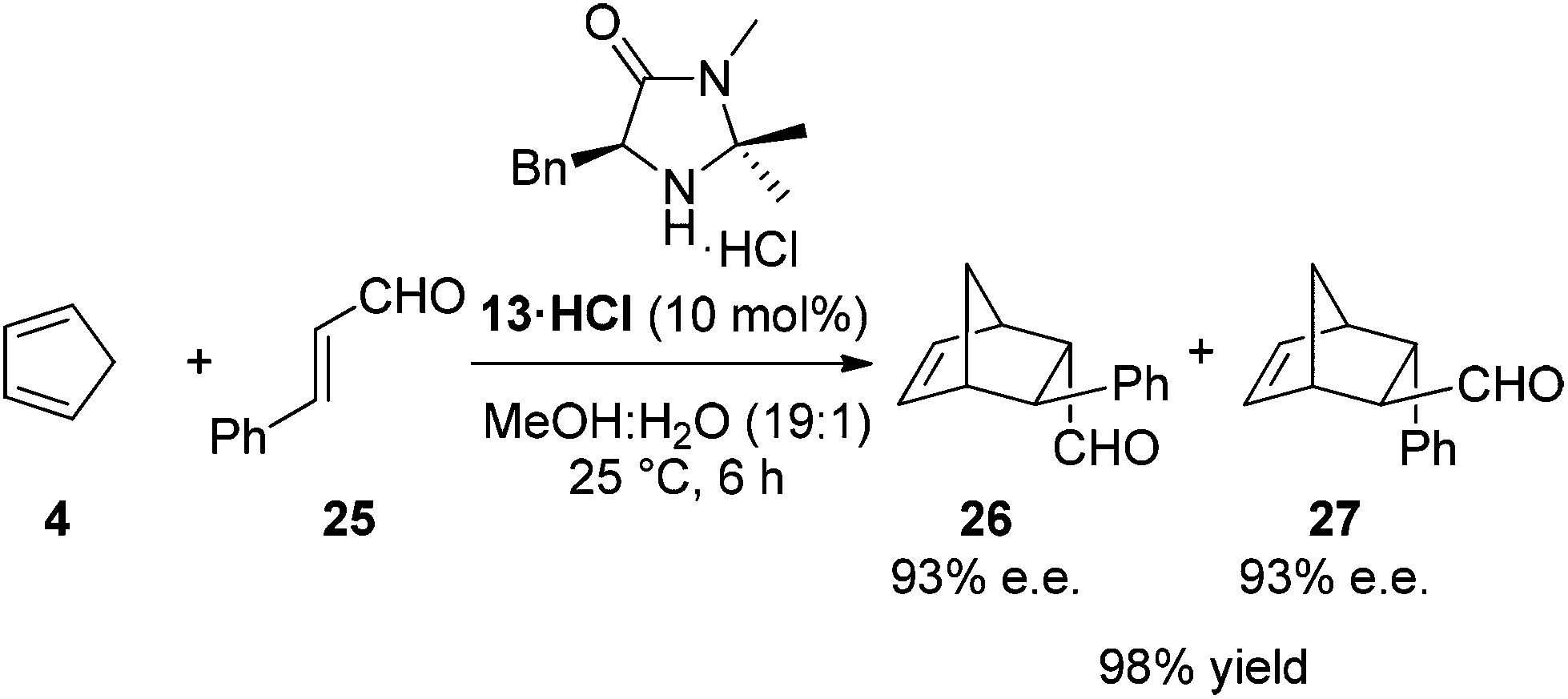 | ||
| Scheme 2 Diels–Alder cycloaddition catalysed by 13·HCl. | ||
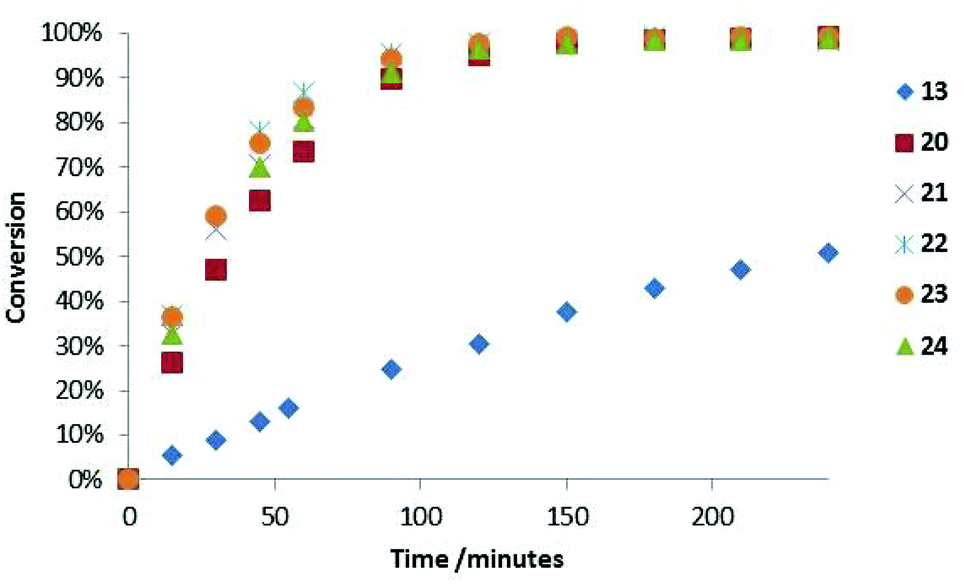 | ||
| Fig. 5 Relative rates of imidazolidinone catalysed Diels–Alder cycloaddition. | ||
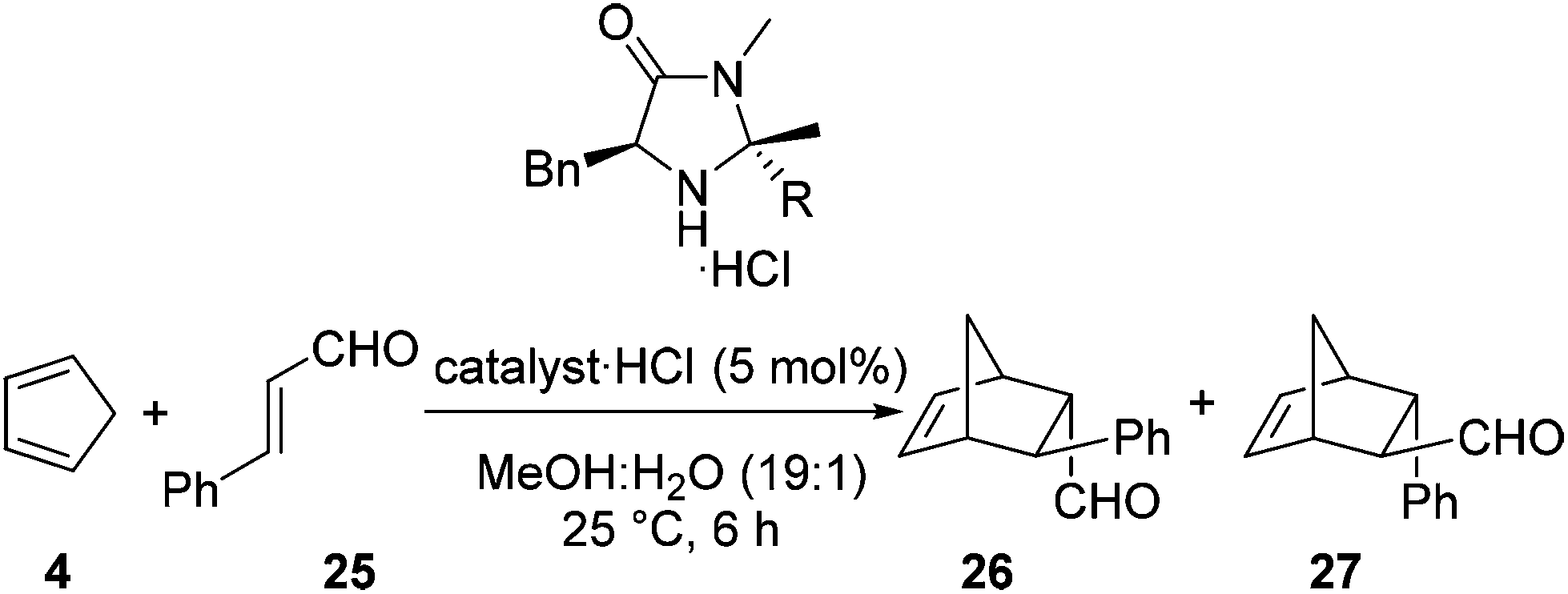
As stated previously, it was deemed essential that alongside an increase in reaction rate we required the high levels of selectivity observed with the imidazolidinone 13·HCl. For each reaction selectivities were determined for the Diels–Alder adducts 26 and 27 (Table 2).20 Based upon these reaction outcomes we selected the imidazolidinone derived from 4-nitroacetophenone 24 (entry 3) for further investigations due to the high levels of enantioselectivity observed for both the endo (95% ee) and exo (87% ee) isomers of the Diels–Alder product.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Yield (%) |
endo:exoa |
endo eeb | exo eeb |
| a Determined by 1H NMR spectroscopy on crude reaction mixture. b Determined by conversion to 2,4-dinitrophenylhydrazine derivative and examination by HPLC. | |||||
| 1 | 13 | 57 | 1:1.32 |
93 | 93 |
| 2 | 20 | 96 | 1:1.20 |
95 | 84 |
| 3 | 24 | 97 | 1:1.05 |
95 | 87 |
| 4 | 21 | 97 | 1.24:1 |
96 | 82 |
| 5 | 22 | 95 | 1:1.18 |
90 | 78 |
| 6 | 23 | 97 | 1.08:1 |
83 | 73 |
To profile the reactivity of the imidazolidinone 24 further we examined a series of alternative dienophiles within the Diels–Alder cycloaddition (Table 3). For cinnamaldehyde derivatives (entries 1–4) high yields and enantioselectivities were maintained for both electron withdrawing and electron donating substituents on the aromatic ring. Reactions with aliphatic substituted substrates proceeded slower than reactions with cinnamaldehyde derivatives, however, they still progressed cleanly and mass balance could be accounted for through recovered starting material. It is believed that parasitic equilibria of the catalyst involving dienamine 3021 formation from the iminium intermediate 29 accounts for this observed loss in reactivity (Scheme 3).
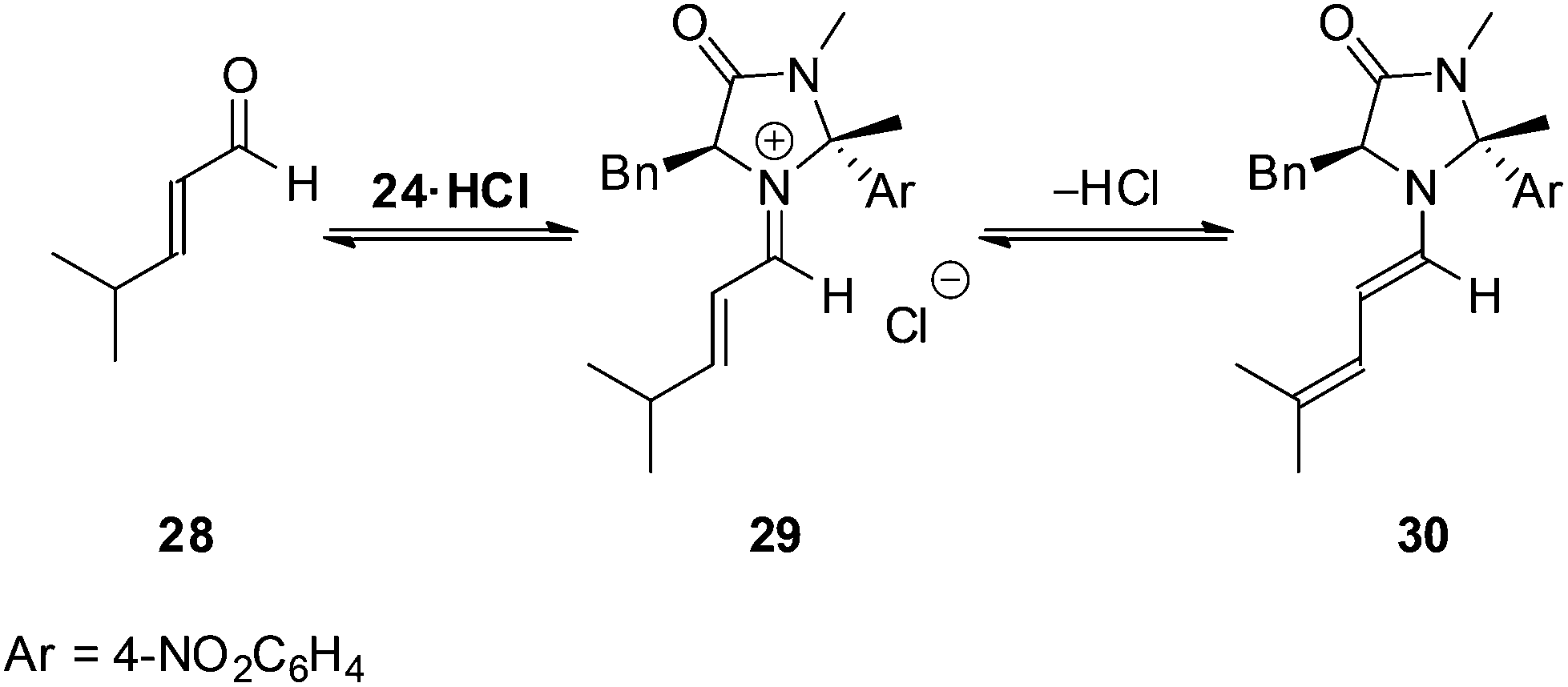 | ||
| Scheme 3 Parasitic dienamine formation. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Yield (%) |
endo:exoa |
endo ee | exo ee |
| a Determined by 1H NMR spectroscopy on crude reaction mixture. b Determined by reduction to corresponding alcohol and examination by HPLC. c Determined by conversion to 2,4-dinitrophenylhydrazine derivative and examination by HPLC. | |||||
| 1 | 4-OMeC6H4 | 95 | 1:1.1 |
96b | 87b |
| 2 | 4-MeC6H4 | 75 | 1:1 |
94b | 86b |
| 3 | 4-ClC6H4 | 95 | 1.1:1 |
96b | 84b |
| 4 | 4-NO2C6H4 | 94 | 1.1:1 |
94b | 86b |
| 5 | n Pr | 72 | 1.2:1 |
81c | 63c |
| 6 | iPr | 18 | 1:1 |
80c | 64c |
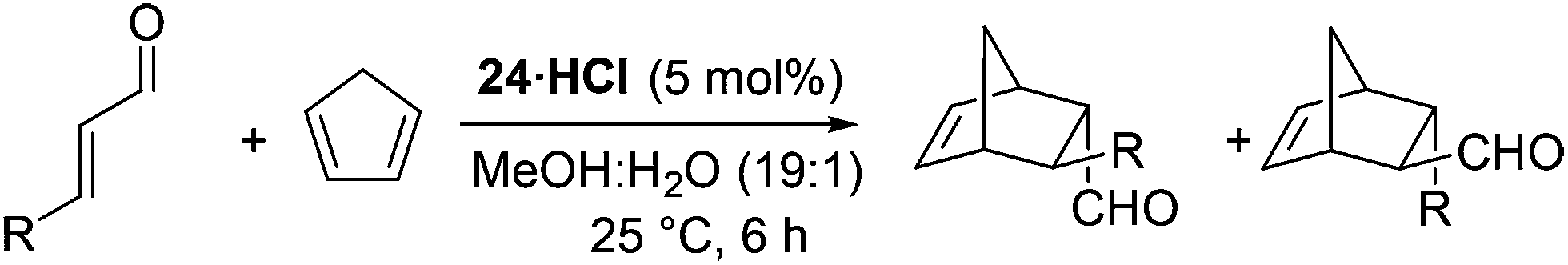
Crystals of an iminium ion suitable for X-ray analysis were obtained through reaction of 24·HPF6 and 4-iodocinnamaldehyde (Fig. 6). A low energy solid-state conformation of the E-iminium ion with the benzyl arm residing over the top of the imidazolidinone ring was observed. Direction of the diene to the bottom face of this iminium ion leads to the stereochemical outcome observed in reactions of 24.22 Consistent with observations on the MacMillan catalyst 13, this suggests use of 24 as a catalyst in conjugate addition reactions would lead to products with low levels of enantioselectivity,23 and increasing the steric requirement of the methyl group on the β-face would be necessary to prepare alternative catalysts to accelerate this class of reaction.
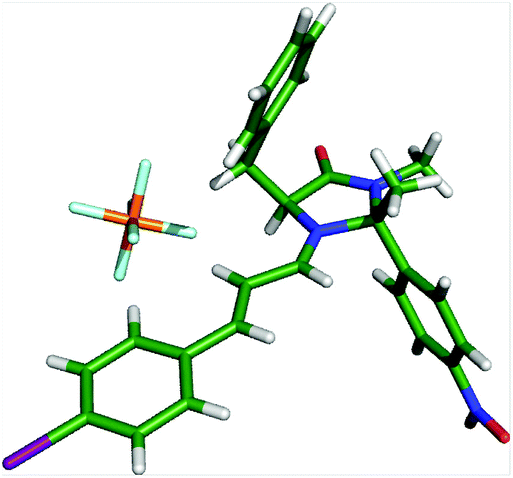 | ||
| Fig. 6 X-ray structure of iminium ion derived from 24 and 4-iodocinnamaldehyde. | ||
The importance of knowing the rate determining step of the catalytic cycle when altering catalyst structure is highlighted by examination of an alternative transformation which proceeds via iminium ion intermediates. For example, Mayr has shown that in the conjugate addition of electron rich heteroaromatics, such as N-methyl pyrrole 31, to an α,β-unsaturated carbonyl compound, C–C bond formation is rate determining.24 From this it could be expected that the 4-nitroacetophenone derived catalyst 24 would not significantly alter the rate of addition of 31 to 25 (Scheme 4).
 | ||
| Scheme 4 Conjugate addition of N-methyl pyrrole to cinnamaldehyde. | ||
The addition of 31 to 25 catalysed by 13 and 24 under literature optimised conditions was monitored by 1H NMR spectroscopy (Fig. 7). Each reaction proceeded smoothly to completion after 6 hours. As can be seen from Fig. 7 each catalyst accelerates the reaction at a similar rate showing that improved activity for 24 will only be observed in transformations where iminium ion formation is rate determining.
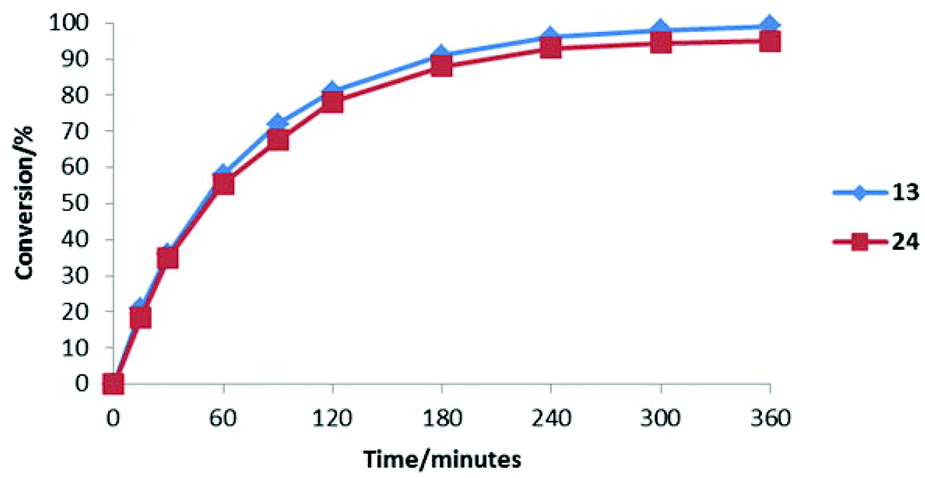 | ||
| Fig. 7 Comparison of the reactivity of 13 and 24 in the conjugate addition of N-methyl pyrrole to cinnamaldehyde. | ||
Conclusions
In summary, we have presented a series of highly reactive imidazolidinone catalysts, which significantly increase the rate of Diels–Alder cycloaddition reactions. A low energy crystal structure of a proposed iminium ion intermediate suggests that salts of 24 induce asymmetry in a similar manner to the MacMillan imidazolidinone 13. The catalysts described would be predicted to accelerate cycloaddition reactions and conjugate addition reactions that proceed through a closed transition state and provide enhanced rates to those observed with 13 where iminium ion formation is the rate determining step of the catalytic cycle. Central to this method of catalyst design is a thorough understanding of reaction mechanism providing an attractive and practical method to improve catalyst activity.Experimental
L-Phenylalanine N-methyl amide 18
L-Phenylalanine ethyl ester hydrochloride (30.0 g, 131 mmol) was stirred in an ethanolic solution of methylamine 33 wt% (150 mL, 1.2 mol) for 72 hours at ambient temperature. The solvent was removed under reduced pressure. The resultant slurry was taken up in saturated sodium carbonate (50 mL) and extracted with chloroform (3 × 50 mL). The combined organic extracts were dried over anhydrous potassium carbonate and the solvent removed under reduced pressure to give a white solid. The residue was recrystallized from ethyl acetate and petroleum ether to give the product as white needles (17.6 g, 75%). mp: 67–69 °C; [α]20D = −89.6 (c = 1, CHCl3); νmax (film)/cm−1 3371, 3345, 3290, 2939, 2875, 1644; 1H NMR (400 MHz, CDCl3) δ 7.47–7.12 (6H, m), 3.54 (1H, dd, J = 9.5, 3.9 Hz), 3.22 (1H, dd, J = 13.7, 3.9 Hz), 2.83 (3H, d, J = 5.0 Hz), 2.60 (1H, dd, J = 13.7, 9.5 Hz), 1.38 (2H, s); 13C NMR (101 MHz, CDCl3) δ 174.8, 138.0, 129.3, 128.7, 126.8, 56.5, 41.0, 25.8; m/z (ES): 179.1 (M + H+).(S)-5-Benzyl-2,2,3-trimethylimidazolidin-4-one 13
L-Phenylalanine-N-methyl amide (2.00 g, 11.2 mmol) was dissolved in acetone (10 mL) and methanol (30 mL). A small crystal of p-toluenesulfonic acid (<1 mg) was added and the mixture was heated under reflux for 18 h. The solvent was removed under reduced pressure and the residue taken up in chloroform (30 mL) and saturated sodium carbonate solution (30 mL). The aqueous layer was extracted with chloroform (2 × 30 mL) and the combined organics were dried over potassium carbonate. The solvent was removed under reduced pressure to give the target compound as a pale yellow oil (2.40 g, quant.). [α]20D = −33.5 (c = 1.3, MeOH); νmax (film)/cm−1 3473, 3329, 3060, 3030, 2975, 2929, 1685; 1H NMR (400 MHz, CDCl3) δ 7.38–7.14 (5H, m), 3.82 (1H, dd, J = 6.5, 4.5 Hz), 3.17 (1H, dd, J = 14.2, 4.5 Hz), 3.03 (1H, dd, J = 14.2, 6.5 Hz), 2.77 (3H, s), 1.28 (3H, s), 1.18 (3H, s); 13C NMR (126 MHz, CDCl3) δ 173.7, 137.6, 129.9, 129.0, 127.2, 75.9, 59.7, 37.7, 27.6, 25.7, 25.6; m/z (APCI): 219.2 (M + H+).General procedure for the preparation of imidazolidinones 20–23
A toluene solution of L-phenylalanine-N-methyl amide (0.6 M), the appropriate substituted acetophenone (0.9 eq.) and ytterbium(III) trifluoromethanesulfonate (0.05 eq.) were heated under reflux for 16–69 h. The mixture was then allowed to cool and diethyl ether (2 vol) was added. The solution was washed with 4 M potassium carbonate (0.3 vol), water (0.3 vol), and brine (0.3 vol). The volatiles were then removed under reduced pressure and the desired products obtained by flash chromatography (ethyl acetate/heptane).General procedure for imidazolidinone salt formation
A solution of the amine in diethyl ether was treated with hydrogen chloride gas for 10 minutes. The precipitate was recovered on a sinter, washed with diethyl ether and petroleum ether, then dried under reduced pressure.General procedure for Diels Alder cycloaddition (Tables 2 and 3)
Imidazolidinone hydrochloride salt (0.1 mmol, 5 mol%) was dissolved in a 19:1 methanol–water mixture (2 mL). The flask was placed into a 25 °C oil bath. The appropriate aldehyde (2.0 mmol) was added and the mixture was stirred for 10 minutes before freshly distilled cyclopentadiene (420 μL, 5.0 mmol) was added in one portion. The reaction mixture was stirred for 6 h and then the volatiles were removed under reduced pressure. The residue was taken up into chloroform (10 mL) and water (10 mL) and the aqueous layer was extracted with chloroform (2 × 10 mL). The combined organics were dried over sodium sulfate and the solvent was removed under reduced pressure. Chloroform (2 mL), water (1 mL) and TFA (1 mL) were added and the biphasic mixture was vigorously stirred for 2 hours. Potassium carbonate (20 mL) was added and the mixture was extracted with chloroform (3 × 10 mL). The combined organics were dried over sodium sulfate and the solvent was removed under reduced pressure. The products were isolated using flash chromatography (10% ethyl acetate in petroleum ether) as oils.
Monitoring Diels–Alder cycloaddition between cinnamaldehyde and cyclopentadiene (Fig. 5)
Secondary amine salt (0.25 mmol, 5 mol%) was dissolved in methanol (4.75 mL) and water (0.25 mL). The mixture was stirred at 25 °C. After 5 minutes, cinnamaldehyde (630 μL, 5 mmol) was added. After another 10 minutes, freshly distilled cyclopentadiene (1020 μL, 12.5 mmol) was added and the reaction timer started. 100 μL aliquots were periodically removed and concentrated under reduced pressure (25 °C, 15 torr, 10 minutes). Water (5 mL) was added and extracted with diethyl ether (3 × 5 mL). The combined organics were concentrated under reduced pressure. Chloroform (2 mL) was added followed by a mixture of TFA–water 1:1 (2 mL). The biphasic mixture was vigorously stirred for 2 h. The reaction was quenched with saturated sodium carbonate solution (5 mL) and extracted with diethyl ether (3 × 5 mL). The combined organics were dried over magnesium sulfate and the solvent was removed under reduced pressure to give a yellow oil. 1H NMR (CDCl3) was used to assess reaction conversion from the CHO resonances: exo 9.93 ppm (1H, d, J = 2.0 Hz, CHO), cinnamaldehyde 9.71 ppm (1H, d, J = 7.7 Hz, CHO) and endo 9.60 ppm (1H, d, J = 2.2 Hz, CHO). The Diels–Alder adducts can be isolated by flash chromatography (20% ethyl acetate in petroleum ether) to give a pale yellow viscous oil.
:1 water and TFA mixture (4 mL). The biphase was vigorously stirred for 2 hours. The reaction was then quenched with saturated sodium carbonate solution and the aqueous layer extracted with diethyl ether (3 × 20 mL). The combined organics were dried over sodium sulfate and the solvent removed under reduced pressure (100 mbar, 25 °C). The product was isolated by flash chromatography (5% diethyl ether in petroleum ether) to give a colourless oil. νmax (ATR)/cm−1: 2957, 2918, 2870, 1717; exo1H NMR (400 MHz, CDCl3) δ 9.77 (1H, d, J = 2.7 Hz, CHO, 6.20 (1H, dd, J = 5.6, 3.1 Hz), 6.13 (1H, dd, J = 5.6, 2.9 Hz), 3.01 (1H, s, J = 1.3 Hz), 2.87 (1H, s), 2.32–2.24 (1H, m), 1.77–1.73 (1H, m), 1.55–1.04 (6H, m), 0.90 (3H, t, J = 7.1 Hz); endo1H NMR (400 MHz, CDCl3) δ 9.36 (1H, d, J = 3.4 Hz), 6.27 (1H, dd, J = 5.7, 3.2 Hz), 6.05 (1H, dd, J = 5.7, 2.8 Hz), 3.11 (1H, s), 2.66 (1H, d, J = 1.5 Hz), 2.37 (1H, dd, J = 7.8, 3.4 Hz), 1.71–1.65 (1H, m), 1.56–1.03 (6H, m), 0.90 (3H, t, J = 7.1 Hz). endo and exo mixture: 13C NMR (101 MHz, CDCl3) δ 205.2, 204.1, 138.9, 136.2, 136.2, 132.9, 60.2, 58.9, 47.4, 47.2, 46.6, 45.2, 42.1, 38.2, 36.6, 21.8, 21.7, 14.3; m/z (CI): 181 (M+ + CH5), 163 (M–H−).
:1 water and TFA mixture (4 mL). The biphase was vigorously stirred for 2 hours. The reaction was then quenched with saturated sodium carbonate solution and the aqueous layer extracted with diethyl ether (3 × 20 mL). The combined organics were dried over sodium sulfate and the solvent removed under reduced pressure (100 mbar, 25 °C). The product was isolated by flash chromatography (5% diethyl ether in petroleum ether) to give a colourless oil. νmax (ATR)/cm−1:2957, 2911, 2895, 2870, 1701; exo1H NMR (400 MHz, CDCl3) δ 9.78 (1H, d, J = 2.6 Hz, CHO), 6.19 (1H, dd, J = 5.6, 3.1 Hz), 6.15 (1H, dd, J = 5.6, 2.8 Hz), 3.04–3.00 (1H, m), 2.96 (1H, s), 1.92–1.84 (2H, m), 1.51–1.40 (2H, m), 1.08–0.97 (1H, m), 0.94 (3H, d, J = 6.2 Hz), 0.84 (3H, d, J = 6.4 Hz); endo1H NMR (400 MHz, CDCl3) δ 9.36 (1H, d, J = 3.4 Hz, CHO), 6.26 (1H, dd, J = 5.7, 3.3 Hz), 6.06 (1H, dd, J = 5.7, 2.8 Hz), 3.11 (1H, s, CH), 2.87–2.83 (1H, m), 2.51–2.47 (1H, m), 1.51–1.39 (1H, m), 1.34–1.29 (1H, m), 1.01 (1H, d, J = 6.5 Hz), 0.91 (1H, d, J = 6.6 Hz); endo–exo mixture 13C NMR (101 MHz, CDCl3) δ 205.4, 204.3, 139.1, 136.4, 135.9, 133.2, 58.8, 58.1, 50.4, 50.2, 47.0, 46.6, 45.3, 45.3, 45.1, 45.1, 32.9, 32.6, 22.1, 22.1, 21.9, 21.6; m/z (CI): 181 (M+ + CH5), 163 (M–H−).
General procedure for monitoring reaction between cinnamaldehyde and N-methyl pyrrole
Acknowledgements
The authors thank EPSRC for financial support and the National Mass Spectrometry Facility, Swansea, U.K., for high-resolution spectra.Notes and references
- K. N. Houk and P. H.-Y. Cheong, Nature, 2008, 455, 309 CrossRef CAS PubMed.
- (a) P. H.-Y. Cheong, C. Y. Legault, J. M. Um, N. ÇelebiÖlçüm and K. N. Houk, Chem. Rev., 2011, 111, 5042 CrossRef CAS PubMed; (b) H. Yang and M. W. Wong, J. Org. Chem., 2011, 76, 7399 CrossRef CAS PubMed; (c) Y. Wang, J. Wang, J. Su, F. Huang, L. Jiao, Y. Liang, D. Yang, S. Zhang, P. A. Wender and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 10060 CrossRef CAS PubMed; (d) S. T. Schneebeli, M. L. Hall, R. Breslow and R. Friesner, J. Am. Chem. Soc., 2009, 131, 3965 CrossRef CAS PubMed; (e) J. M. Brown and R. J. Deeth, Angew. Chem., Int. Ed., 2009, 48, 4476 CrossRef CAS PubMed.
- (a) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed; (b) Asymmetric Organocatalysis Workbench Edition. 2 Volumes, Science of Synthesis, ed. B. List and K. Maruoka, Thieme, Stuttgart, 2012 Search PubMed.
- J. B. Brazier and N. C. O. Tomkinson, Top. Curr. Chem., 2010, 291, 281 CrossRef CAS.
- A. Erkkilae, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef CAS PubMed.
- F. Giacalone, M. Gruttadauria, P. Agrigento and R. Noto, Chem. Soc. Rev., 2012, 41, 2406 RSC.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS.
- (a) G. Evans, T. J. K. Gibbs, R. L. Jenkins, S. J. Coles, M. B. Hursthouse, J. A. Platts and N. C. O. Tomkinson, Angew. Chem., Int. Ed., 2008, 47, 2820 CrossRef CAS PubMed; (b) J. B. Brazier, G. Evans, T. J. K. Gibbs, S. J. Coles, M. B. Hursthouse, J. A. Platts and N. C. O. Tomkinson, Org. Lett., 2009, 11, 133 CrossRef CAS PubMed.
- (a) D. Seebach, R. Gilmour, U. Grošelj, G. Deniau, C. Sparr, M.-O. Ebert, A. K. Beck, L. B. McCusker, D. Šišak and T. Uchimaru, Helv. Chim. Acta, 2010, 93, 603 CrossRef CAS; (b) D. Seebach, U. Grošelj, W. B. Schweizer, S. Grimme and C. Muck-Lichtenfeld, Helv. Chim. Acta, 2010, 93, 1 CrossRef CAS; (c) U. Grošelj, W. B. Schweizer, M.-O. Ebert and D. Seebach, Helv. Chim. Acta, 2009, 92, 1 CrossRef; (d) D. Seebach, U. Grošelj, D. M. Badine, W. B. Schweizer and A. K. Beck, Helv. Chim. Acta, 2008, 91, 1999 CrossRef CAS.
- J. H. Rowley and N. C. O. Tomkinson, In Asymmetric Synthesis II, ed. M. Christmann and S. Bräse, Wiley-VCH, Weinheim, 2012, pp. 29–34 Search PubMed.
- J. B. Brazier, K. M. Jones, J. A. Platts and N. C. O. Tomkinson, Angew. Chem., Int. Ed., 2011, 50, 1613 CrossRef CAS PubMed.
- W. P. Jencks, Catalysis in Chemistry and Enzymology, McGraw Hill Text, New York, 1969 Search PubMed.
- For a review on the use of imidazolidinone catalysts see: G. Lelais and D. W. C. MacMillan, Aldrichimica Acta, 2006, 39, 79 CAS.
- For a review on the use of diarylprolinol ether catalysts see: A. Mielgo and P. Claudio, Chem. – Asian J., 2008, 3, 922 CrossRef CAS PubMed.
- J. B. Brazier, G. P. Hopkins, M. Jirari, S. Mutter, R. Pommereuil, L. Samulis, J. A. Platts and N. C. O. Tomkinson, Tetrahedron Lett., 2011, 52, 2783 CrossRef CAS PubMed.
- (a) J. L. Cavill, J.-E. Peters and N. C. O. Tomkinson, Chem. Commun., 2003, 728 RSC; (b) J. L. Cavill, R. L. Elliott, G. Evans, I. L. Jones, J. A. Platts, A. M. Ruda and N. C. O. Tomkinson, Tetrahedron, 2006, 62, 410 CrossRef CAS PubMed; (c) J. B. Brazier, J. L. Cavill, R. L. Elliott, G. Evans, T. J. K. Gibbs, I. L. Jones, J. A. Platts and N. C. O. Tomkinson, Tetrahedron, 2009, 65, 9961 CrossRef CAS PubMed.
- For studies on alternative secondary amine scaffolds see: (a) E. Gould, T. Lebl, A. M. Z. Slawin, M. Reid, T. Davies and A. D. Smith, Org. Biomol. Chem., 2013, 11, 7877 RSC; (b) I. Suzuki, M. Ando, R. Shimabara, A. Hirata and K. Takeda, Org. Biomol. Chem., 2011, 9, 3033 RSC; (c) Q. Li, W.-Y. Wong, W.-H. Chan and A. W. M. Lee, Adv. Synth. Catal., 2010, 352, 2142–2146 CrossRef CAS; (d) Y. Langlois, A. Petit, P. Rémy, M.-C. Scherrmann and C. Kouklovsky, Tetrahedron Lett., 2008, 49, 5576 CrossRef CAS PubMed; (e) H. He, B. J. Pei, H.-H. Chou, T. Tian, W.-H. Chan and A. W. M. Lee, Org. Lett., 2008, 10, 2421 CrossRef CAS PubMed; (f) M. Lemay, L. Aumand and W. W. Ogilvie, Adv. Synth. Catal., 2007, 349, 441 CrossRef CAS; (g) M. Lemay and W. W. Ogilvie, Org. Lett., 2005, 7, 4141 CrossRef CAS PubMed.
- G. J. S. Evans, K. White, J. A. Platts and N. C. O. Tomkinson, Org. Biomol. Chem., 2006, 4, 2616 CAS.
- L. Samulis and N. C. O. Tomkinson, Tetrahedron, 2011, 67, 4263 CrossRef CAS PubMed.
- A. Hall, L. D. Harris, C. L. Jones and N. C. O. Tomkinson, Tetrahedron Lett., 2003, 44, 111 CrossRef CAS.
- For a review on dienamines in catalysis, see: D. B. Ramachary and Y. V. Reddy, Eur. J. Org. Chem., 2012, 865 CrossRef CAS.
-
endo
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :exo ratios observed for transformations accelerated with imidazolidinones have been rationalised through DFT calculations:
(a) C. Alleman, R. Gordillo, F. R. Clemente, P. H.-Y. Cheong and K. N. Houk, Acc. Chem. Res., 2004, 37, 558 CrossRef PubMed;
(b) R. Gordillo and K. N. Houk, J. Am. Chem. Soc., 2006, 128, 3543 CrossRef CAS PubMed.
:exo ratios observed for transformations accelerated with imidazolidinones have been rationalised through DFT calculations:
(a) C. Alleman, R. Gordillo, F. R. Clemente, P. H.-Y. Cheong and K. N. Houk, Acc. Chem. Res., 2004, 37, 558 CrossRef PubMed;
(b) R. Gordillo and K. N. Houk, J. Am. Chem. Soc., 2006, 128, 3543 CrossRef CAS PubMed. - J. F. Austin and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 1172 CrossRef CAS PubMed.
- S. Lakhdar and H. Mayr, Chem. Commun., 2011, 47, 1866 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C spectra for compounds reported. CCDC 1017471. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob01916d |
| This journal is © The Royal Society of Chemistry 2015 |