
Oxidative activation of dihydropyridine amides to reactive acyl donors†
Erik Daa
Funder
,
Julie B.
Trads
and
Kurt V.
Gothelf
*
Danish National Research Foundation, Center for DNA Nanotechnology, Department of Chemistry and iNANO, Gustav Wieds Vej 14, 8000 Aarhus C, Denmark. E-mail: kvg@chem.au.dk
First published on 31st October 2014
Abstract
Amides of 1,4-dihydropyridine (DHP) are activated by oxidation for acyl transfer to amines, alcohols and thiols. In the reduced form the DHP amide is stable towards reaction with amines at room temperature. However, upon oxidation with DDQ the acyl donor is activated via a proposed pyridinium intermediate. The activated intermediate reacts with various nucleophiles to give amides, esters, and thio-esters in moderate to high yields.
Introduction
The amide bond is among the most common and important functional groups in chemistry and biochemistry. Thus, it is an integral part of molecules such as proteins, peptides, pharmaceuticals, and polymers.1–4 The classical approach to the formation of an amide is to use a reactive acyl derivative or in situ activation of carboxylic acids for the reaction with an amine.5 A plethora of efficient reagents have been developed for this transformation, including the arsenal of carbodiimides for in situ carboxylic acid activation or the generation of activated esters taking advantage of electron-poor alcohols.6–8 Reactive acyl derivatives such as acyl chlorides and N-hydroxysuccinimides can be stored in pure form or in non-nucleophilic solvents, however they are highly sensitive to moisture and nucleophiles.An alternative method to create an amide bond is to take advantage of transamidation strategies. In most cases these take advantage of primary amides using either high temperatures (100 to 160 °C) or added metals as essential activating reagents.9–13 Others have used tertiary amide derivatives, that can chelate metals such as copper, as protecting groups for carboxylic acids.14–18 The strategy resulted in either the free acid or a new ester bond. Also, tertiary and secondary amides, activated by Tf2O and Zn(OTf)2 respectively, have resulted in clever ways of creating intra- and inter-molecular ester bonds from amide bonds.19,20
Herein, we present a new activatable acyl donor which in the “off-state” is stable towards nucleophilic attack by e.g. amines, whereas in the “on-state” it reacts with nucleophiles such as amines, alcohols, and thiols. The activation is orthogonal to most other known methods for amide bond formation as it exploits a metal free oxidation to form a highly reactive acyl transfer intermediate.
Amides are generally much more stable towards nucleophiles than esters.21,22 For this reason, amides are not immediately recognizable as acyl donors. A few examples for the release of quinoline and acridine amides from a solid support have been demonstrated.23–25 Acyl-pyridinium derivatives are known to be highly reactive acyl donors and molecules such as 4-dimethylaminopyridine (DMAP) are commonly used as acyl-transfer catalysts.26,27 Inspired by the redox relay of the pyridinium-1,4-dihydropyridine (DHP) redox couple known as NAD+ and NADH, found in all living systems, we speculated if it was feasible to prepare a stable reduced acyl DHP derivative that could be oxidized to form a reactive acyl pyridinium species.28–30 Thus capturing the inherent reactivity of e.g. an acid chloride in an inactive DHP amide.31,32
Results and discussion
Only a few amides of 1,4-dihydropyridines are described in the literature33 and their use as acyl donors has, to our knowledge, remained unexplored until the current study. Initially, we investigated the amide of an unsubstituted DHP (Fig. 1B).34 This derivative was stable towards hydrolysis and did not react with amines in CDCl3. Oxidation with DDQ activated the DHP for acyl transfer with amines. However, the unsubstituted DHP amide was not stable towards silica gel flash column chromatography. It appears that it was oxidized by oxygen when exposed to silica gel which diminishes the usefulness of the described approach. In order to avoid degradation on silica gel we investigated electron deficient 3,5-diethoxycarbonyl-DHP derivatives which have higher oxidation potentials.35,36 For this reason, an electron deficient 4-phenyl DHP amide was prepared and this proved stable towards purification by silica gel chromatography (Fig. 1B). However, this amide was inert towards oxidation by DDQ and CAN. Further fine-tuning of the properties was achieved with the propionyl 3,5-diethoxycarbonyl-DHP (DDP) amide 1, which had the desired properties of stability and reactivity (Fig. 1B and 1C).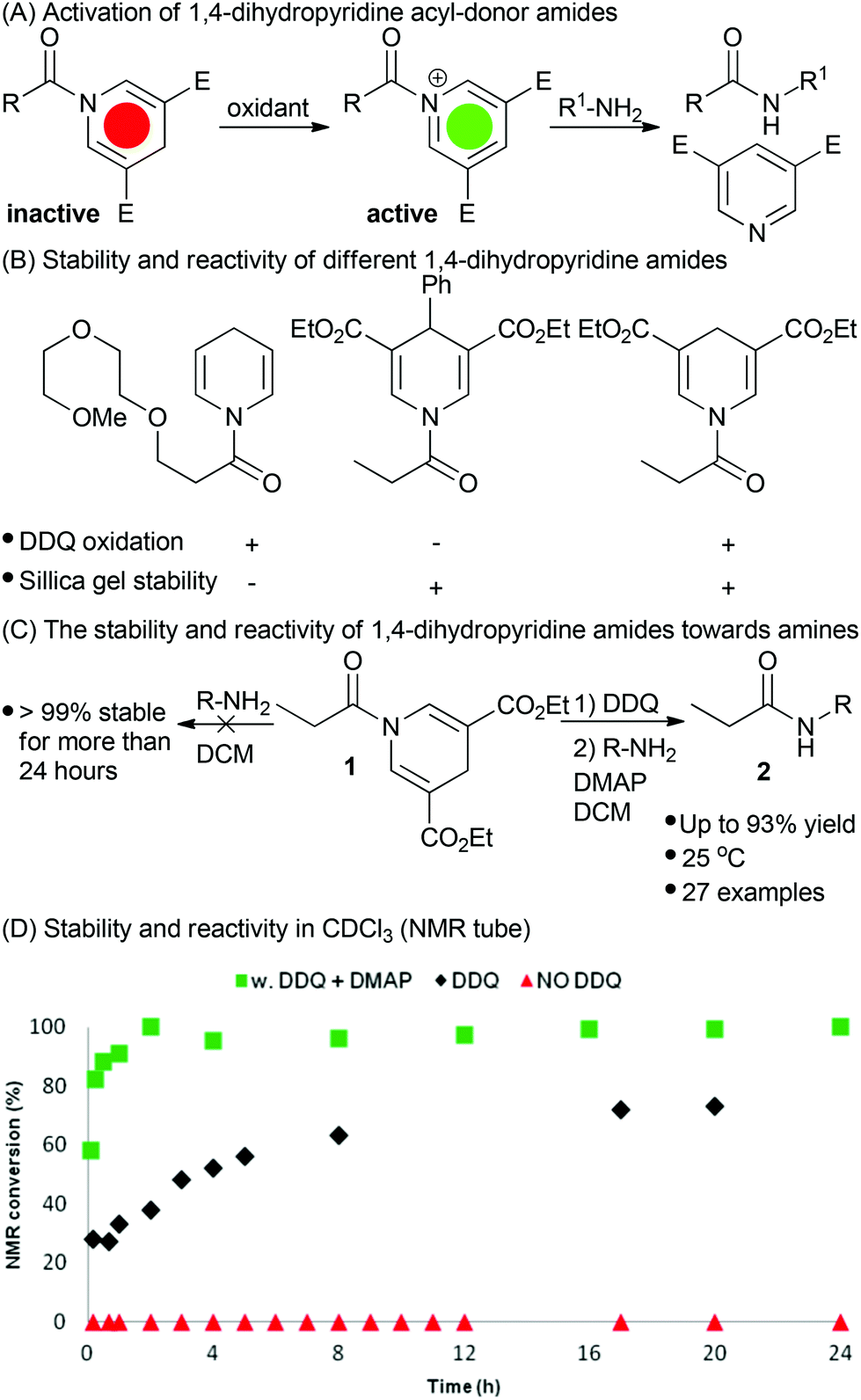 | ||
| Fig. 1 (A) Principle of activation for acyl dihydropyridine donors. By oxidation a reactive pyridinium intermediate is proposed which react with amines to form amides. (B) Three initial DHP-amides investigated for their stability and reactivity towards silica gel purification and DDQ-oxidation. (C) and (D) The stability (without DDQ) and reactivity (with DDQ and additive) of dihydropyridine amide 1 towards amine nucleophiles. | ||
The synthesis of 1 is straight-forward (Scheme 1). It is performed by converting the commercially available, and inexpensive, pyridine-3,5-dicarboxylic acid 3 to the di-ethyl ester 4 by treatment with oxalyl chloride and ethanol. The resulting crude mixture was used directly in the reduction to the 1,4-dihydropyridine 5 using NaCNBH3 in acetic acid.37 Conveniently, the reaction could be performed on 10 g scale and upon the addition of water DHP 5 precipitates as a yellow powder to provide 68% yield over two steps. The DDP-amide was prepared by deprotonating DHP 5 with NaH followed by the addition of propionyl chloride to furnish DDP amide 1 in an isolated yield of 78%. In addition, amide bond formation with 5 could also be achieved by the use of EDC, HOBT, and amine-base albeit in a lower yield of 73%.
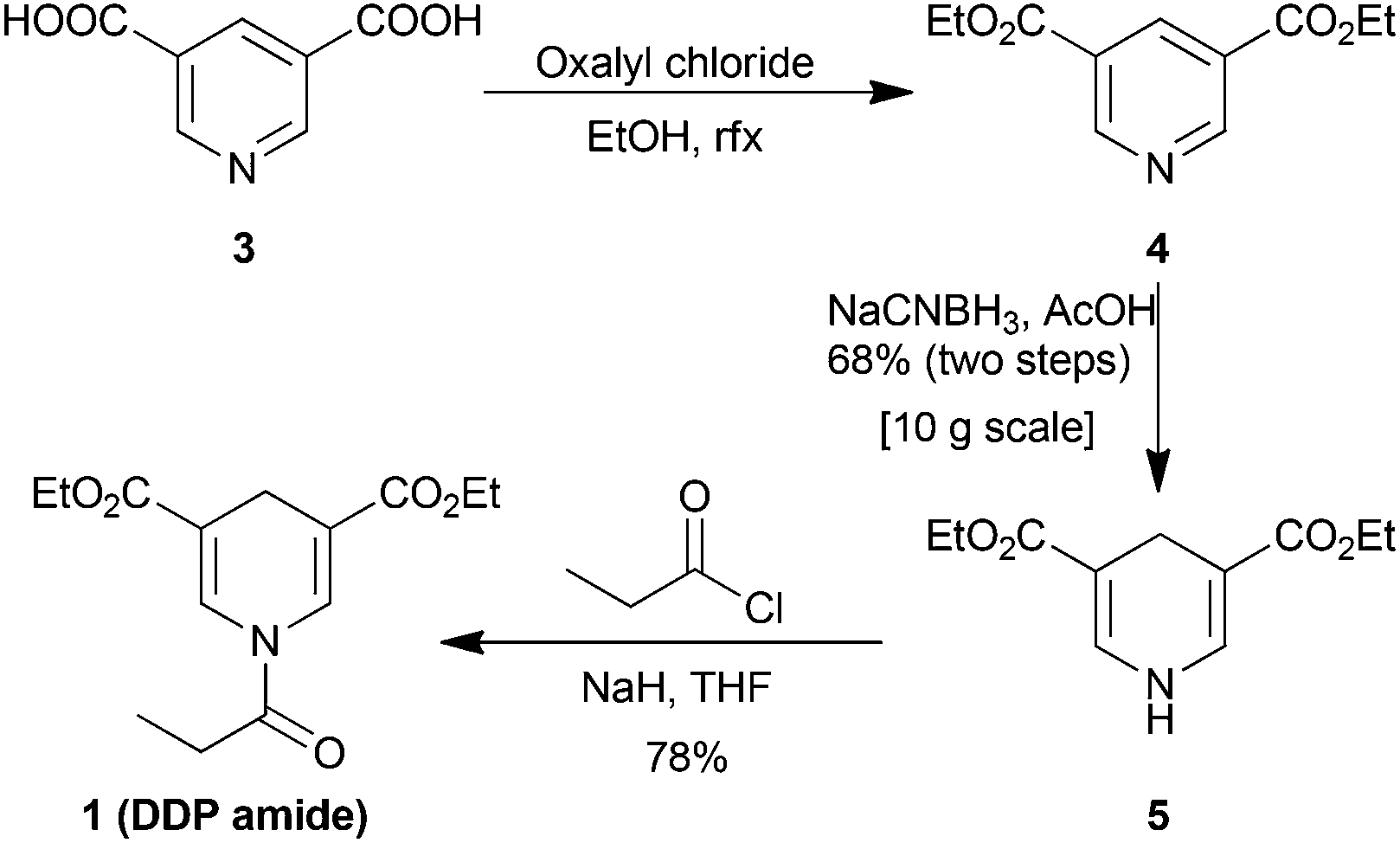 | ||
| Scheme 1 The synthesis of 1,4-dihydropyridine amide 1. | ||
The amide 1 was stable towards silica gel flash column chromatography as well as acidic and basic work-up conditions.
With these preliminary results at hand, we tested the stability and reactivity of the DDP amide 1. Without activation the amide was stable (>99% by 1H-NMR in CDCl3) towards aminolysis with 1 eq. of phenylethylamine over a period of more than 48 h at rt, 40 and 60 °C. With 3 eq. DMAP and 1 eq. amine present at rt the stability in CDCl3 was determined by 1H-NMR to >99% after 24 h and >98% after 48 h. After raising the temperature to 40 and 60 °C and using a prolonged reaction time (48 h) the stability of the amide was determined to be >94% and >93% respectively. In addition, 14 other solvents were investigated in order to explore solvent effects on the amide bond formation and it was observed that the amide was slightly less stable in more polar solvents (see ESI† for specific details). For storage the DDP amide 1 was determined to be completely stable to oxidation in the freezer under argon for a period of more than 4 months.
In the event of DDP amide 1 oxidation using DDQ with subsequent addition of amine, the reaction generates the desired amide 2a (Table 1).38 Initially, the reaction was, however, rather slow providing only 70% conversion after 17 h. After carefully investigating the mechanism using HPLC analysis of the crude-reaction mixture we identified the different intermediates in the reaction. From this we concluded that the proposed acyl pyridinium intermediate, formed after oxidation, is immediately trapped as the mono- or di-dichlorodicyano hydroquinone ester, since these intermediates were identified using HPLC analysis (see ESI†). Such electron-deficient esters are reactive towards acyl transfer, though less active than the acyl pyridinium salt. To increase the rate of the reaction we have investigated the effect of adding various bases and acyl transfer catalysts. The results are presented in Table 1.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Base | Amine (eq.) | Solvent | Time (h) | Conversion (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions for entry 1–10: 1 (0.036 mmol) and DDQ (0.036 mmol) were stirred in 0.6 mL of solvent in a sealed vial (all reactions with isolated yields were performed on 0.18 mmol scale using 3.0 mL of solvent). After 30 minutes phenylethylamine (0.11 mmol) and base (0.11 mmol) were added. Reaction conditions for entry 11–14: 1 (0.18 mmol) and DDQ (0.18 mmol) were stirred in 3 mL DCM in a sealed vial (entry 14 was performed with DDQ (1 eq.) and 1 (1.1 eq.). After 30 minutes phenylethylamine and base were added according to the table. b Conversions were determined by 1H-NMR spectroscopy based on 1. Isolated yields are reported in parenthesis. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | — | 3 | CDCl3 | 17 | 72 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | DIPEA | 3 | CDCl3 | 2 | 47 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 2,6-Lutidine | 3 | CDCl3 | 2 | 65 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Pyridine | 3 | CDCl3 | 2 | 75 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | K2CO3 | 3 | CDCl3 | 2 | 77 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Cs2CO3 | 3 | CDCl3 | 2 | 100 (50) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | DMAP | 3 | CDCl3 | 1 | 100 (86) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | DMAP | 3 | DCM | 1 | 100 (87) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | DMAP | 3 | EtOAc | 26 | (44) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | DMAP | 3 | Dioxane | 26 | (65) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | DMAP (0.2 eq.) | 3 | DCM | 4 | (84) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | DMAP (1 eq.) | 3 | DCM | 1.5 | (94) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | DMAP (1 eq.) | 1 | DCM | 6 | (83) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | DMAP (3 eq.) | 1.1 | DCM | 1 | (90) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
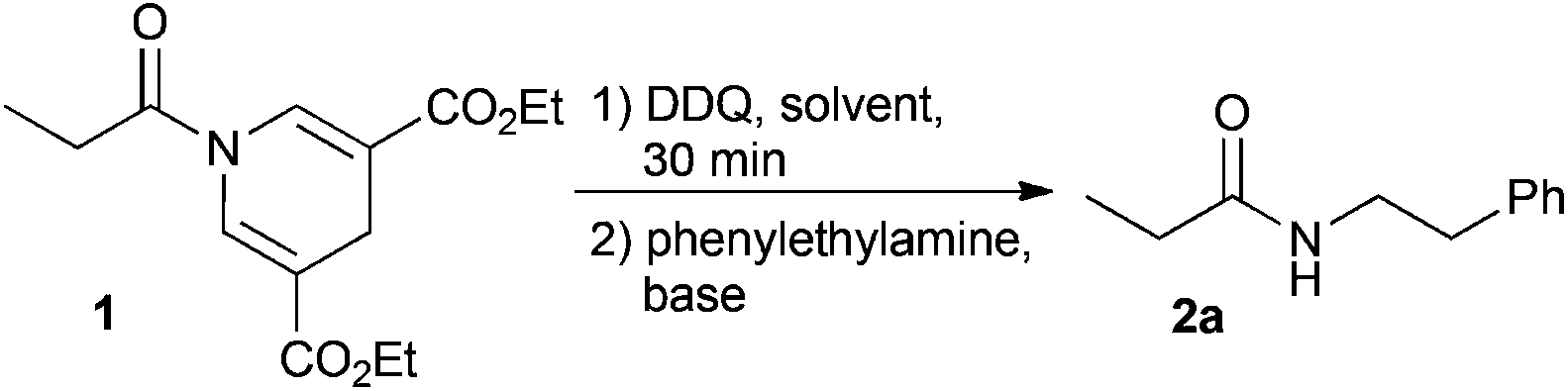
The addition of weak non-nucleophilic bases increased the rate of the reaction to some extent. Addition of DMAP increased the rate of the reaction significantly and complete conversion was observed after only 1 h (this included 30 min oxidation) as shown by 1H-NMR. Also, carbonate bases increased the conversion significantly, however, the isolated yields were inferior to the yields obtained using DMAP in DCM or chloroform. As DCM is a less toxic alternative to chloroform while still preserving the reactivity of the system, it was used in the following optimization with regard to yield. Next the stoichiometry of the amine and DMAP were systematically varied (Table 1). It was observed that the reaction proceeds well using only catalytic amounts of DMAP and 3 eq. of amine, yielding the product in a satisfactory 84% yield after 4 h. However, in order to decrease the equivalents of amine used and increase the rate of the reaction, other conditions were investigated. The optimal conditions were identified using 3 eq. DMAP and 1.1 eq. amine. The optimized reaction conditions gave rise to an isolated yield of 2a in 90% yield after a total reaction time of only 1 h.
To probe the generality of the reaction, different amine nucleophiles were employed in the reaction with 1 (Fig. 2). Primary aliphatic amines furnished good yields using a total reaction time of only 1 h. Electron-rich and electron-poor benzylic amines provided yields from 73 to 86% with reaction times between 1 and 20 h.
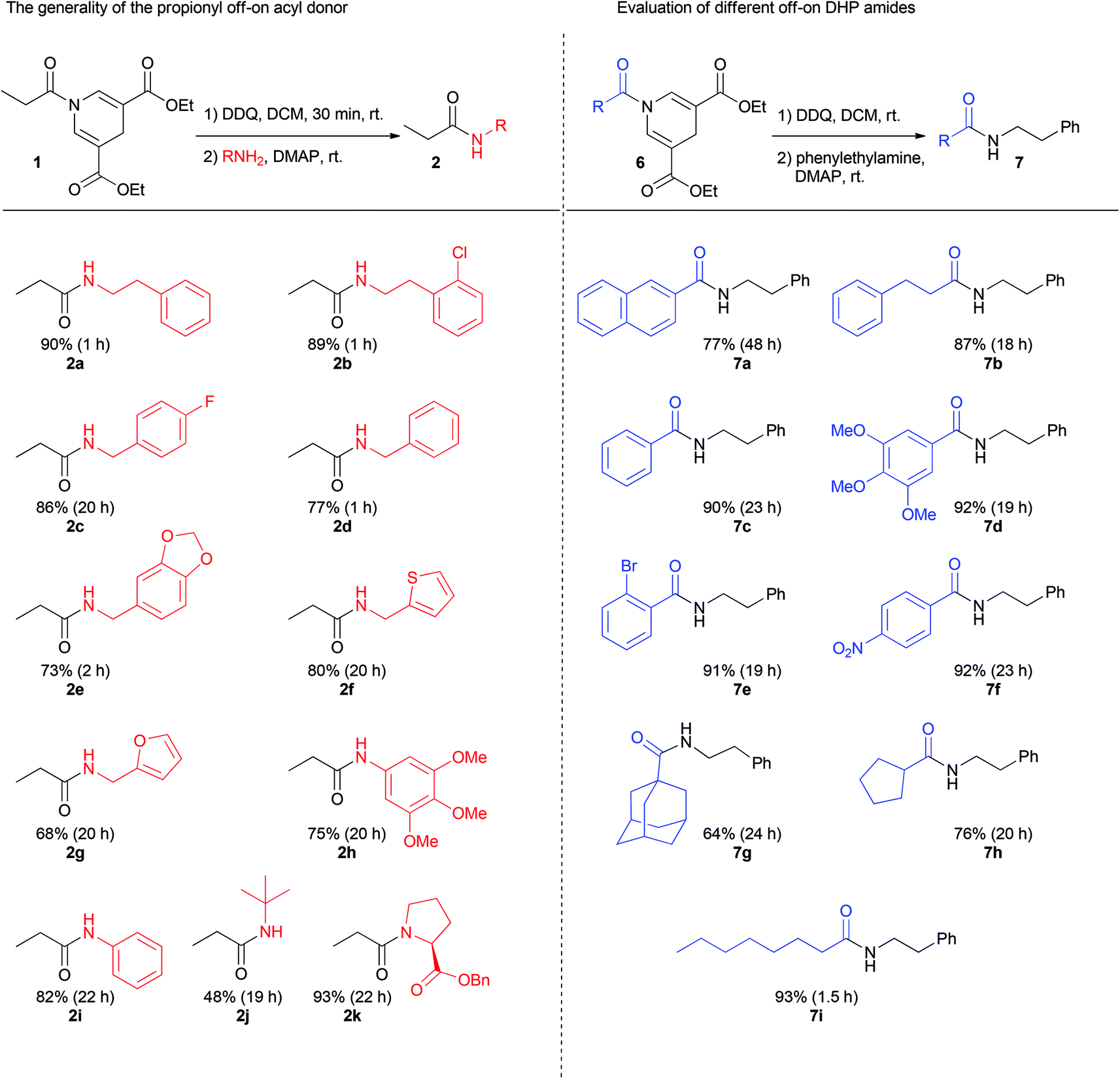 | ||
| Fig. 2 The generality of DDP acyl donor 1 with various amines and different DHP amides. | ||
Heteroaryl methylene amines such as furfuryl amine and 2-thiophenemethylamine also proved reactive giving rise to yields up to 80%.
In order to test the reactivity of the acyl donor's on-state, neutral and electron-rich anilines were investigated. Although slower, these amines proved viable in the amide bond forming reaction providing isolated yields between 75 and 82%.
Furthermore, sterically hindered tert-butyl amine gave rise to 48% yield whereas the secondary amine in benzyl protected proline furnished an excellent yield of 93% after a total reaction time of 22 h.
To expand the generality of the reaction different 1,4-dihydropyridine amides were synthesized and investigated in the reaction with phenylethylamine. After oxidation, electron-rich, neutral, and electron-poor aromatic benzoyl DHP amides provided the desired amide product in excellent yields ranging from 90 to 92%. Primary and secondary aliphatics as well as sterically hindered adamantyl-DHP also provided good yields ranging from 64 to 93%. In addition, naphthalene-DHP amide gave rise to a yield of 77% after a total reaction time of 48 h. The oxidation with DDQ was in most cases complete within 30 minutes. However, in a few cases the oxidation was slower, as in the case with the napthalene-DHP where the oxidation was complete after approximately 24 h.
The most notable characteristic of the reaction is the off-on feature, which prevents aminolysis and hydrolysis without prior activation and permits the reaction only in the presence of an activator. Using this methodology it is possible to have a free acid moiety present during the activation step (Fig. 3A). Thus, only one of the acids has the possibility of being activated and the free acid remains unscathed (94% recovery by flash column chromatography) in the amide bond forming step. It is not possible to obtain such off-on reactivity with conventional amide bond forming reagents without the use of protecting groups. We envision that this conceptually new reactivity can be used in directed chemistry thus enhancing specificity and suppressing unwanted reactions.
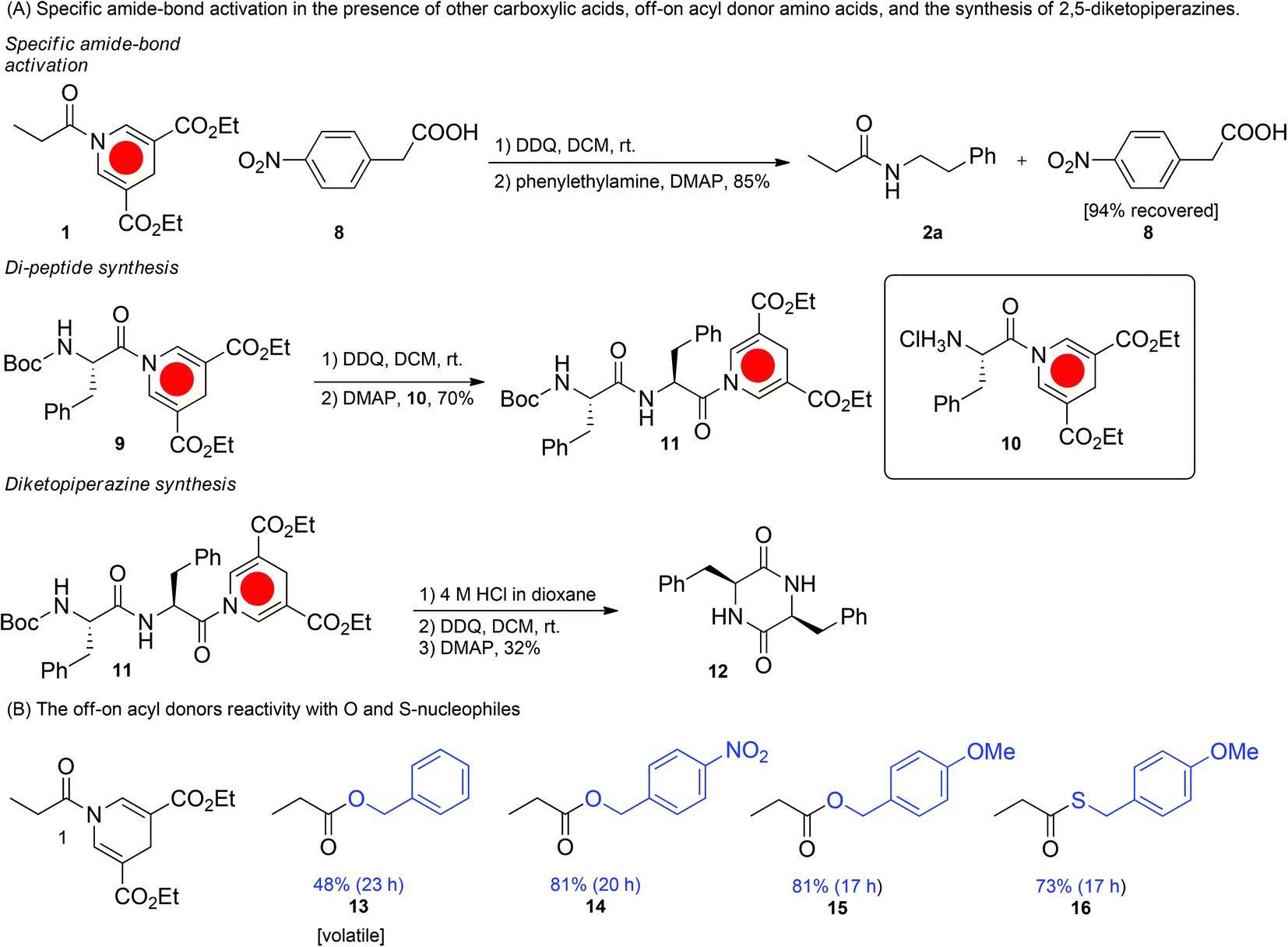 | ||
| Fig. 3 (A) The application of DHP amides in specific amide-bond activation, peptide synthesis, and the intramolecular synthesis of 2,5-diketopiperazines. (B) The reactivity of the DDP amide 1 with oxygen and sulfur nucleophiles. | ||
One of the drawbacks of the reaction is that the amine has to be added after the oxidation step as it will otherwise react with DDQ. This obstacle can however be overcome by using the ammonium salt of the amine during the oxidation step, which prevents unwanted side reactions with DDQ and gives rise to an isolated yield of the desired amide product in 87% (see ESI†).
To explore the applications of the optimized off-on acyl donor system, we first synthesized a Boc-protected phenyl-alanine DHP-amide (Fig. 3A). Secondly, a phenylalanine DHP-amide with a free amine was obtained. We envisioned that this approach would allow for the iterative synthesis of amide bonds. Gratifyingly, the amide bond formation between the two fragments resulted in a phenylalanine DHP di-peptide in an isolated yield of 70%, which can be activated for further couplings. The crude 1H-NMR analysis of the reaction mixture,after workup, indicated a 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 diastereomeric ratio of the dipeptide. Next, we also demonstrated the formation of a cyclic dipeptide by an analogous intramolecular reaction (Fig. 3A).
:15 diastereomeric ratio of the dipeptide. Next, we also demonstrated the formation of a cyclic dipeptide by an analogous intramolecular reaction (Fig. 3A).
Finally, other nucleophiles besides amines were investigated. Employing either alcohols or thiols provided the corresponding esters and thio-esters in yields between 48 and 81% yield. Thus the DHP-amide can function as a useful ester or thio-ester surrogate.39 The off-on switchable DHP-acyl donor is the first example of its kind which provides an off-state that is non-reactive at room temperature and reactive when activated. The advantage of this method is two-fold. It provides shielding of the acyl group in the off-state and thus carboxylic acids located elsewhere in a molecule can undergo reactions selectively. Compared to the free carboxylic acid group it is non-acidic and provides better solubility of the compound in organic solvents. Compared to protected carboxylic acids such as methyl, benzyl, and tert-butyl esters it offers an orthogonal (oxidation) one-pot route to the activated acyl derivative. In terms of yield and reaction rate the DHP-acyl donor method presented here cannot compete with the traditional activation by carbodiimides and other reagents that have been explored and optimized exhaustively. Other drawbacks of the method is that the amine (unless protonated) cannot be present during the oxidation and that 3-fold of the amine is used to give an optimal yield. However, we believe that the DHP-acyl donor method can be further optimized, e.g. by improving the oxidation reagent and optimizing the substituents of the DHP. Furthermore, other methods for activation such as metal-catalyzed activation by oxygen or photochemical activation may become feasible. We are exploring the application of the DHPacyl donor in aqueous media as we believe that the next generation of the method may have potential for bioconjugation.
Conclusions
In summary, we have described the development of an off-on switchable acyl donor in the form of different 1,4-dihydropyridine amides. In the absence of activating reagent (DDQ) the amide is inert towards aminolysis and hydrolysis. In contrast, after activation by DDQ, the DHP-system is activated to a proposed pyridinium intermediate which rapidly is captured by reduced DDQ as either a mono- or di-ester. These then readily reacts with a variety of nitrogen, oxygen, and sulfur nucleophiles. Reaction times can be as short as one hour with yields up to 93% thus demonstrating the reactivity of the on-stage. In addition to these findings the DHP-amides have been utilized in amide couplings in the presence of a free carboxylic acid and for peptide couplings.Experimental section
General methods
For all water and/or oxygen sensitive reactions were used flame dried glassware, which was purged with argon and evacuated a total of three times. All reactions were monitored by thin layer chromatography (TLC) analysis on either Merck® silica gel 60 F254 TLC plates or TLC Silica gel 60 F254 glass plates. The TLC plates were visualized by exposure to UV (254 nm) or by staining with either a basic aq. KMnO4 solution or p-anisaldehyde stain. Flash column chromatography was performed using Merck® silica gel 60 (230–400 mesh). Preparative TLC was carried out using TLC Silica gel 60 F254 glass plates. All solvents used were of HPLC grade quality. For inert reactions the solvents THF and DCM were dried prior to use by the use of a MBRAUN solvent purification system 800. Triethylamine was distilled from CaH2 before use. Unless noted all reagents are commercially available. The reagents were purchased at the highest quality and used without further purification.NMR spectra were recorded at 400 MHz (1H-NMR) and at 100 MHz (13C-NMR) on a Bruker® 400 spectrometer and calibrated to the residual solvent peak. In the interpretation of the 1H-NMR spectra the following abbreviations are used: s, singlet; d, doublet; t, triplet; q, quartet; qui, quintet; sext, sextet; m, multiplet; br, broad. Melting points are uncorrected. High-resolution mass spectrometry (HRMS) was recorded using electrospray ionization. LC-MS measurements were done on a Shimadzu LCMS-2020EV connected to a Shimadzu prominence RP-ULPC system equipped with a YMCbasic 50 × 4.6 mml D. S-3 μm column using a gradient from 20 to 95% MeCN in a solution of 0.1% formic acid in MilliQ purified water.
:1:97 v/v/v). mp (uncorr.) 124.4–132.3 °C (lit.40 122–123 °C). IR (neat) νmax 2995, 2927, 1726 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 5.1 Hz, 2H), 5.76–5.61 (m, 1H), 4.19 (q, J = 7.2 Hz, 4H), 3.26 (s, 2H), 1.27 (t, J = 7.2 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 168.2, 135.0, 105.1, 60.1, 21.5, 14.2. HRMS (ES) m/z [M + H]+ calcd for C11H16NO4: 226.1074. Found: 226.1074.
:1:97 v/v). mp (uncorr.) 107.8–109.3 °C. IR (neat) νmax 2941, 2983, 1720, 1704 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.86 (bs, 2H), 4.23 (q, J = 7.1 Hz, 4H), 3.22 (s, 2H), 2.60 (q, J = 7.3 Hz, 2H), 1.30 (t, J = 7.1 Hz, 6H), 1.22 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.3, 166.6, 146.1, 112.4, 61.0, 27.1, 28.2, 14.5, 8.7. HRMS (ES) m/z [M + H]+ calcd for C14H20NO5: 282.1336. Found: 282.1337.
:1:97. The product was isolated as a light yellow solid (228 mg, 73%).
General procedure A using various amines with DHP amide 1
To a stirred solution of 1,4-dihydropyridine amide 1 (110 mg, 0.393 mmol, 1.1 eq.) in DCM (6 mL) was added DDQ (81 mg, 0.355 mmol, 1 eq.) after which the color changed from light yellow to a milky white color. After 30 minutes the amine nucleophile (0.393 mmol, 1.1 eq.) followed by DMAP (131 mg, 1.07 mmol, 3 eq.) were added. The reaction was monitored by TLC and crude 1H-NMR until completion. Most amines provided full conversion after 0.5 to 1 hours (see individual experiments for exact reaction time) after which the mixture was diluted with EtOAc (20 mL). The mixture was transferred to a separation funnel and washed with 1 M HCl (2 × 10 mL) and saturated NaHCO3 (2 × 10 mL). The remaining organic phase was dried with MgSO4 or Na2SO4, filtered, and evaporated to dryness in vacuo. In most cases the remaining residue consisted exclusively of the amide product and the pyridine 4 (observed by 1H-NMR in CDCl3) which cannot be removed by acidic work-up using 1 M HCl. Increasing the acidity of the aqueous wash to 6 M HCl facilitates the protonation and removal of 4, however the amide products are in many cases not stable towards these conditions or removed from the organic phase.The products were purified by flash column chromatography using silica gel (see details in the following individual experimentals).
:1 EtOAc–pentane). The product was isolated as a white solid (57 mg, 90%). Rf = 0.28 (silica, EtOAc–pentane 50:50 v/v). mp (uncorr.) 58.6–60.6 °C (lit.42 57–57.5 °C). IR (neat) νmax 3273, 3070, 2970, 1643, 1552 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.33–7.26 (m, 2H), 7.24–7.12 (m, 3H), 5.78 (br s, 1H), 3.49 (q, J = 7.0 Hz, 2H), 2.80 (t, J = 7.1 Hz, 2H), 2.14 (q, J = 7.6 Hz, 2H), 1.10 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.5, 139.4, 129.1, 128.9, 126.8, 40.4, 35.5, 29.4, 9.5. HRMS (ES) m/z [M + H]+ calcd for C11H15NO: 178.1226 Found: 178.1230.
The synthesis of 2a was repeated, however in this case the oxidation was performed in the presence of the amine as its hydrochloride salt. The DDP amide 1 and amine hydrochloride was first dissolved in DCM after which DDQ was added. After oxidation for 30 minutes in the presence of the hydrochloride salt of the amine, DMAP was added. Amounts and equivalents of reagents were used as described in the general procedure A. The desired amide 2a was isolated using flash column chromatography using EtOAc–pentane (gradient from 20% EtOAC in pentane to 1:1 EtOAc–pentane). The product was isolated as a white solid (55 mg, 87%). The characterization of 2a matched the data given above.
1H-NMR, 13C-NMR and IR are in accordance with literature values.
:1 EtOAc–pentane). The product was isolated as a white solid (67 mg, 89%). Rf = 0.24 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 50.5–51.4 °C. IR (neat) νmax 3264, 3083, 2939, 1631, 1560 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 8.9 Hz, 1H), 7.23–7.07 (m, 3H), 5.85 (br s, 1H), 3.49 (q, J = 6.9 Hz, 2H), 2.94 (t, J = 7.0 Hz, 2H), 2.15 (q, J = 7.6 Hz, 2H), 1.10 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.0, 136.7, 134.1, 131.0, 129.6, 128.0, 127.0, 39.2, 33.4, 29.7, 9.9. HRMS (ES) m/z [M + H]+ calcd for C11H14ClNO: 212.0837 Found: 212.0840.
:1 EtOAc–pentane). The product was isolated as a white solid (55.5 mg, 86%). Rf = 0.5 (silica, EtOAc–pentane 50:50 v/v). mp (uncorr.) 102.0–102.5 °C.
IR (neat) νmax 3292, 2941, 1641, 1510 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.24–7.13 (m, 2H), 6.96 (t, J = 8.7 Hz, 2H), 6.21 (br s, 1H), 4.33 (d, J = 5.8 Hz, 2H), 2.20 (q, J = 7.6 Hz, 2H), 1.13 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.5, 162.7 (d, JCF = 246), 134.8 (d, JCF = 3.2), 129.8 (d, JCF = 8.0), 115.7 (d, JCF = 21.4), 42.6, 29.4, 9.5. 19F NMR (376 MHz, CDCl3) δ −116.0. HRMS (ES) m/z [M + H]+ calcd for C10H13FNO: 182.0976 Found: 182.0978.
:1 EtOAc–pentane). The product was isolated as white solid with trace amount of red (45 mg, 77%). Rf = 0.14 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 51.2–52.3 °C (lit.44 49–50 °C). IR (neat) νmax 3290, 3031, 2979, 1637, 1540 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.40–7.26 (m, 5H), 5.72 (s, 1H), 4.44 (d, J = 5.7 Hz, 2H), 2.25 (q, J = 7.6 Hz, 2H), 1.18 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.7, 138.5, 128.9, 128.0, 127.7, 43.8, 29.9, 10.0. HRMS (ES) m/z [M + H]+ calcd for C10H13NO: 164.1070 Found: 164.1070. 1H-NMR, 13C-NMR and IR are in accordance with literature values.
:1 EtOAc–pentane). The product was isolated as a white solid (54 mg, 73%). Rf = 0.23 (silica, EtOAc–pentane 50:50 v/v). mp (uncorr.) 99.3–99.7 °C (lit.46 99 °C). IR (neat) νmax 3286, 1636, 1499, 1441 cm−1. 1H NMR (400 MHz, CDCl3) δ 6.82–6.65 (m, 3H), 5.93 (s, 2H), 5.77 (br s, 1H), 4.32 (d, J = 5.6 Hz, 2H), 2.22 (q, J = 7.6 Hz, 2H), 1.16 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.3, 148.3, 147.4, 132.8, 121.3, 108.6, 108.5, 101.2, 43.2, 29.4, 9.5. HRMS (ES) m/z [M + H]+ calcd for C11H14NO3: 208.0968 Found: 208.0969.
:1 EtOAc–pentane). The product was isolated as a taint solid (48.5 mg, 80%). Rf = 0.33 (silica, EtOAc–pentane 50:50 v/v). mp (uncorr.) 45.4–46.5 °C. IR (neat) νmax 3262, 1551, 1633, 696 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.17 (dd, J = 4.6, 1.5 Hz, 1H), 6.99–6.75 (m, 2H), 6.31 (br s, 1H), 4.54 (d, J = 5.7 Hz, 2H), 2.19 (q, J = 7.6 Hz, 2H), 1.12 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.3, 141.7, 127.2, 126.2, 125.4, 38.0, 29.2, 9.4. HRMS (ES) m/z [M + H]+ calcd for C8H12NOS: 170.0634 Found: 170.0634.
:1 EtOAc–pentane). The product was isolated as a light brown oil (37 mg, 68%). Rf = 0.33 (silica, EtOAc–pentane 50:50 v/v). IR (neat) νmax 3285, 1647, 1541 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.37–7.33 (m, 1H), 6.32 (dd, J = 2.9, 1.8 Hz, 1H), 6.23 (d, J = 3.2 Hz, 1H), 5.69 (br s, 1H), 4.44 (d, J = 5.4 Hz, 2H), 2.23 (q, J = 7.6 Hz, 2H), 1.17 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.1, 152.0, 142.7, 110.8, 107.7, 36.4, 29.4, 9.4. HRMS (ES) m/z [M + H]+ calcd for C8H12NO2: 154.0863 Found: 154.0862.
:1 EtOAc–pentane). The product was isolated as a yellow solid (64 mg, 75%). Rf = 0.23 (silica, EtOAc–pentane 50:50 v/v). mp (uncorr.) 120.0–122.0 °C. IR (neat) νmax 3262, 2941, 1610, 1505, 1122 cm−1. 1H NMR (400 MHz, CDCl3) δ 6.84 (s, 2H), 3.82 (s, 6H), 3.80 (s, 3H), 2.37 (q, J = 7.5 Hz, 2H), 1.23 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 172.8, 153.9, 135.0, 134.7, 97.6, 61.0, 56.1, 30.6, 9.3. HRMS (ES) m/z [M + H]+ calcd for C12H18NO4: 240.1230 Found: 240.1234.
:1 EtOAc–pentane). The product was isolated as a white solid (43.5 mg, 82%). Rf = 0.23 (silica, EtOAc–pentane 30:70 v/v). mp (uncorr.) 106.0–107.0 °C (lit.50 106–107 °C). IR (neat) νmax 3196, 2975, 1600, 1495 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.62 (br s, 1H), 7.52 (d, J = 7.9 Hz, 2H), 7.29 (t, J = 7.8 Hz, 2H), 7.08 (t, J = 7.3 Hz, 1H), 2.37 (q, J = 7.6 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.0, 138.5, 129.3, 124.5, 120.3, 30.5, 9.4. HRMS (ES) m/z [M + H]+ calcd for C9H12NO: 150.0913 Found: 150.0915. 1H-NMR, 13C-NMR and IR are in accordance with literature values.
:1 EtOAc–pentane). The product was isolated as a white solid (22 mg, 48%). Rf = 0.47 (silica, EtOAc–pentane 50:50 v/v). mp (uncorr.) 50.5–51.4 °C (lit.51 87–91 °C). IR (neat) νmax 3315, 2967, 2930, 1643, 1542 cm−1. 1H NMR (400 MHz, CDCl3) δ 5.21 (s, 1H), 2.11 (q, J = 7.6 Hz, 2H), 1.34 (s, 9H), 1.11 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.8, 50.9, 30.4, 28.6, 9.5. HRMS (ES) m/z [M + H]+ calcd for C7H16NO: 130.1226 Found: 130.1228. 1H-NMR, 12C-NMR and IR are in accordance with literature values.
:1 EtOAc–pentane). The product was isolated as a clear oil (87 mg, 93%). Rf = 0.23 (silica, EtOAc–pentane 50:50 v/v). IR (neat) νmax 2966, 2933, 2878, 1743, 1647, 1425 cm−1. 1H NMR 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :5 inseparable mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 7.38–7.27 (m, 5H), 5.27–5.00 (m, 2H), 4.53 (dd, J = 8.6, 3.3 Hz, 0.8H), 4.38 (dd, J = 8.5, 2.3 Hz, 0.2H)*, 3.67–3.36 (m, 2H), 2.43–1.75 (m, 6H), 1.13 (t, J = 7.4 Hz, 2.4H), 1.04 (t, J = 7.4 Hz, 0.6H)*. *minor rotamer. 13C-NMR major rotamer: 13C NMR (100 MHz, CDCl3) δ 173.3, 173.0, 136.2, 128.9, 128.5, 128.4, 66.7, 58.7, 46.7, 28.9, 27.4, 24.5, 8.3. 13C NMR minor rotamer: 13C-NMR (100 MHz, CDCl3) δ 173.3, 172.8, 135.7, 129.0, 129.0, 128.7, 67.2, 59.4, 46.2, 31.2, 27.2, 22.3, 8.6. HRMS (ES) m/z [M + H]+ calcd for C15H20NO3: 262.1438 Found: 262.1439. 1H-NMR is in accordance with literature values.
:90 v/v). mp (uncorr.) 31.9–33.0 °C (lit.55 33 °C). IR (neat) νmax 3087, 2994, 2945, 1734, 1323 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.5 Hz, 2H), 7.50 (d, J = 8.5 Hz, 2H), 5.20 (s, 2H), 2.41 (q, J = 7.6 Hz, 2H), 1.16 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.6, 148.2, 144.0, 128.7, 124.1, 64.6, 27.2, 8.7. HRMS (ES) m/z [M + H]+ calcd for C10H12NO4: 210.0761 Found: 210.0762.
:90 v/v). IR (neat) νmax 2977, 2936, 1686, 1510, 1243 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J = 8.5 Hz, 2H), 6.83 (d, J = 8.5 Hz, 2H), 4.08 (s, 2H), 3.78 (s, 3H), 2.58 (q, J = 7.5 Hz, 2H), 1.18 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 200.7, 159.4, 130.4, 130.2, 114.4, 55.3, 37.1, 32.4, 9.3. HRMS (ES) m/z [M + Na]+ calcd for C11H14O2SNa: 233.0607 Found: 233.0607.
:90 v/v). IR (neat) νmax 2981, 2943, 1735, 1172, 696 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.44–7.28 (m, 5H), 5.13 (s, 2H), 2.39 (q, J = 7.6 Hz, 2H), 1.17 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 175.0, 136.6, 129.0, 128.6 (2C), 66.2, 27.4, 8.8. HRMS (ES) m/z [M + Na]+ calcd for C10H12O2Na: 187.0730 Found: 187.0728. 1H-NMR, 12C-NMR and IR are in accordance with literature values.
:95). The product was isolated as a clear oil (55.5 mg, 81%). Rf = 0.59 (silica, EtOAc–pentane 20:80 v/v). IR (neat) νmax 2955, 2837, 1732, 1612, 1514, 1246 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.30 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 5.05 (s, 2H), 3.80 (s, 3H), 2.35 (q, J = 7.6 Hz, 2H), 1.15 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.5, 159.7, 130.1, 128.4, 114.0, 66.0, 55.3, 27.7, 9.2. HRMS (ES) m/z [M + Na]+ calcd for C11H14NaO3: 217.0835 Found: 217.0837. 1H-NMR and IR are in accordance with literature values.
:5 inseparable mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 7.38–7.27 (m, 5H), 5.27–5.00 (m, 2H), 4.53 (dd, J = 8.6, 3.3 Hz, 0.8H), 4.38 (dd, J = 8.5, 2.3 Hz, 0.2H)*, 3.67–3.36 (m, 2H), 2.43–1.75 (m, 6H), 1.13 (t, J = 7.4 Hz, 2.4H), 1.04 (t, J = 7.4 Hz, 0.6H)*. *minor rotamer. 13C-NMR major rotamer: 13C NMR (100 MHz, CDCl3) δ 173.3, 173.0, 136.2, 128.9, 128.5, 128.4, 66.7, 58.7, 46.7, 28.9, 27.4, 24.5, 8.3. 13C NMR minor rotamer: 13C-NMR (100 MHz, CDCl3) δ 173.3, 172.8, 135.7, 129.0, 129.0, 128.7, 67.2, 59.4, 46.2, 31.2, 27.2, 22.3, 8.6. HRMS (ES) m/z [M + H]+ calcd for C15H20NO3: 262.1438 Found: 262.1439. 1H-NMR is in accordance with literature values.
:90 v/v). mp (uncorr.) 31.9–33.0 °C (lit.55 33 °C). IR (neat) νmax 3087, 2994, 2945, 1734, 1323 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.5 Hz, 2H), 7.50 (d, J = 8.5 Hz, 2H), 5.20 (s, 2H), 2.41 (q, J = 7.6 Hz, 2H), 1.16 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.6, 148.2, 144.0, 128.7, 124.1, 64.6, 27.2, 8.7. HRMS (ES) m/z [M + H]+ calcd for C10H12NO4: 210.0761 Found: 210.0762.
:90 v/v). IR (neat) νmax 2977, 2936, 1686, 1510, 1243 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J = 8.5 Hz, 2H), 6.83 (d, J = 8.5 Hz, 2H), 4.08 (s, 2H), 3.78 (s, 3H), 2.58 (q, J = 7.5 Hz, 2H), 1.18 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 200.7, 159.4, 130.4, 130.2, 114.4, 55.3, 37.1, 32.4, 9.3. HRMS (ES) m/z [M + Na]+ calcd for C11H14O2SNa: 233.0607 Found: 233.0607.
:90 v/v). IR (neat) νmax 2981, 2943, 1735, 1172, 696 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.44–7.28 (m, 5H), 5.13 (s, 2H), 2.39 (q, J = 7.6 Hz, 2H), 1.17 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 175.0, 136.6, 129.0, 128.6 (2C), 66.2, 27.4, 8.8. HRMS (ES) m/z [M + Na]+ calcd for C10H12O2Na: 187.0730 Found: 187.0728. 1H-NMR, 12C-NMR and IR are in accordance with literature values.
:95). The product was isolated as a clear oil (55.5 mg, 81%). Rf = 0.59 (silica, EtOAc–pentane 20:80 v/v). IR (neat) νmax 2955, 2837, 1732, 1612, 1514, 1246 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.30 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 5.05 (s, 2H), 3.80 (s, 3H), 2.35 (q, J = 7.6 Hz, 2H), 1.15 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.5, 159.7, 130.1, 128.4, 114.0, 66.0, 55.3, 27.7, 9.2. HRMS (ES) m/z [M + Na]+ calcd for C11H14NaO3: 217.0835 Found: 217.0837. 1H-NMR and IR are in accordance with literature values.
General procedure B for the synthesis of DHP amides
To a 0 °C solution of 1,4-dihydropyridine 5 (1 eq.) in a minimum of dry THF was added NaH (2.2 eq.) from a 60% suspension in mineral oil. After the addition of sodium hydride the color changed from yellow to a very distinct clear light yellow. The mixture was left to stir for 30 minutes after which the acid chloride (1.5 eq.) was added. After reacting overnight the reaction was diluted with water (10 mL) and extracted with EtOAc (3 × 15 mL). The combined organic phases were washed with saturated NaHCO3, dried (Na2SO4), filtered, and evaporated to dryness in vacuo. The residue was purified by flash column chromatography using silica gel.:90 to 20:80) and then a second column with the eluent EtOAc–NEt3–DCM (1:2:97). The product was isolated as a white solid (0.909 g, 61%). Rf = 0.62 (silica, EtOAc–pentane 50:50 v/v). mp (uncorr.) 141.1–142.8 °C. IR (neat) νmax 3116, 2983, 1710, 1620 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.96 (s, 2H), 6.81 (s, 2H), 4.24 (q, J = 7.0 Hz, 4H), 3.93 (s, 3H), 3.89 (s, 6H), 3.31 (s, 2H), 1.30 (t, J = 7.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 167.3, 166.3, 153.4, 141.6, 132.3, 126.6, 111.6, 106.5, 61.0, 60.9, 56.5, 23.0, 14.3. HRMS (ES) m/z [M + H]+ calcd for C21H26NO8: 420.1653 Found: 420.1657.
:99). The product was isolated as yellow oil (0.617 g, 64%). Rf = 0.63 (silica, EtOAc–DCM 5:95 v/v). IR (neat) νmax 2939, 1715, 1681, 1621 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.06 (br. s, 1H), 7.59 (br. s, 1H), 7.30–7.21 (m, 2H), 7.21–7.10 (m, 3H), 4.17 (q, J = 7.1 Hz, 4H), 3.15 (s, 2H), 2.97 (t, J = 7.5 Hz, 2H), 2.82 (t, J = 7.5 Hz, 2H), 1.24 (t, J = 7.1 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 168.8, 166.5, 140.1, 130.6, 128.8, 128.6, 126.7, 111.9, 61.0, 35.5, 30.5, 22.8, 14.5. HRMS (ES) m/z [M + H]+ calcd for C20H24NO5: 358.1649 Found: 358.1652.
:5:94). The solvent was removed using a flow of N2 (g). The product was isolated as a yellow solid (363 mg, 74%). The product is light sensitive and therefore it should be stored in the dark in the freezer. Rf = 0.21 (silica, EtOAc–pentane 20:80). mp (uncorr.) 123.4–125.2 °C. IR (neat) νmax 2984, 1713, 1624, 1231 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.31 (br. s, 1H), 7.69 (d, J = 7.4 Hz, 1H), 7.48 (t, J = 7.4 Hz, 1H), 7.42 (d, J = 7.4 Hz, 1H), 7.38 (t, J = 7.4 Hz, 1H), 7.17 (br. s, 1H), 4.39–4.07 (m, 4H), 3.31 (s, 2H), 1.42–1.14 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 165.9, 154.2, 134.6, 133.4, 132.3, 129.6, 129.1, 128.2, 119.7, 114.0, 111.9, 61.0, 23.2, 14.4. HRMS (ES) m/z [M + H]+ calcd for C18H19BrNO5: 408.0441 Found: 408.0438.
:90 to 20:90) and then a second column with the eluent EtOAc–pentane (10:90). The product was isolated as a yellow solid (238 mg, 58%). Rf = 0.64 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 150.3–150.8 °C. IR (neat) νmax 2986, 1671, 1527, 1343, 1233 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 8.7 Hz, 2H), 7.83 (br. s, 2H), 7.76 (d, J = 8.7 Hz, 2H), 4.24 (q, J = 7.1 Hz, 4H), 3.32 (s, 2H), 1.30 (t, J = 7.1 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 166.0, 165.7, 149.8, 138.0, 131.1, 129.8, 124.3, 113.2, 61.2, 23.2, 14.4. HRMS (ES) m/z [M + H]+ calcd for C18H19N2O7: 375.1187 Found: 375.1187.
:90) and then Et3N–EtOAc–DCM (2:1:97). The product was isolated as a yellow solid (154 mg, 11%). Rf = 0.42 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 139.4–141.3 °C. IR (neat) νmax 2982, 1713, 1621, 1191, 1223 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.12 (s, 1H), 7.97 (s, 1H), 7.95–7.81 (m, 4H), 7.63–7.52 (m, 3H), 4.20 (q, J = 7.1 Hz, 4H), 3.32 (s, 2H), 1.24 (t, J = 7.1 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 167.8, 166.2, 134.9, 132.4, 132.3, 130.0, 129.1, 129.0, 128.9, 128.5, 127.9, 127.3, 124.6, 111.7, 60.8, 23.0, 14.3. HRMS (ES) m/z [M + H]+ calcd for C22H22NO5: 380.1492 Found: 380.1495.
:90). The product was isolated as a yellow solid (0.951 g, 76%). Rf = 0.65 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 118.2–119.7 °C. IR (neat) νmax 3055, 2921, 1782, 1722, 1257 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.97 (br. s, 2H), 4.25 (q, J = 7.2 Hz, 4H), 3.24 (s, 2H), 2.57 (t, J = 7.2 Hz, 2H), 1.76–1.63 (m, 2H), 1.41–1.18 (m, 14H), 0.94–0.82 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 169.5, 166.3, 130.3, 111.3, 60.7, 33.3, 31.6, 28.95, 28.91, 24.3, 22.5, 22.5, 14.2, 14.0. HRMS (ES) m/z [M + H]+ calcd for C19H30NO5: 352.2118 Found: 352.2123.
:90 to 20:80). The product was isolated as yellow solid (0.365 g, 27%). Rf = 0.71 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 124.2–126.6 °C. IR (neat) νmax 2907, 2852, 1723, 1450, 1236 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 2H), 4.24 (q, J = 7.1 Hz, 4H), 3.21 (s, 2H), 2.23–1.99 (m, 9H), 1.85–1.61 (m, 6H), 1.31 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.0, 166.8, 132.7, 110.5, 60.9, 43.3, 39.0, 36.5, 28.2, 22.4, 14.5. HRMS (ES) m/z [M + H]+ calcd for C22H30NO5: 388.2118 Found: 388.2122.
:90). The product was isolated as a yellow solid (0.582 g, 68%). Rf = 0.58 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 42.0–42.7 °C. IR (neat) νmax 2961, 1721, 1702, 1621, 1233, 1189 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.95 (br. s, 2H), 4.30–4.17 (m, 4H), 3.22 (s, 2H), 3.10 (p, J = 7.2 Hz, 1H), 2.00–1.80 (m, 4H), 1.80–1.51 (m, 4H), 1.35–1.21 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 172.9, 166.6, 131.1, 111.3, 60.9, 41.6, 30.1, 26.1, 22.8, 14.4. HRMS (ES) m/z [M + H]+ calcd for C17H24NO5: 322.1649 Found: 322.1652.
General procedure C for the synthesis of amides from various DHP-amides
To a solution of DHP amide (175 μmol, 1.1 eq.) in DCM (3 mL) was added DDQ (1 eq.). The reaction was left to stir until full oxidation by TLC analysis was observed. Hereafter the amine (1.1 eq.) and DMAP (3 eq.) was added. The reaction was followed by 1H NMR. The reaction was diluted with EtOAc (10 mL) and washed with saturated NaHCO3, 1 M HCl, dried (Na2SO4), filtered, and evaporated to dryness in vacuo. The residue was purified by flash column chromatography.:80 to 40:60). The product was isolated as a white solid (46.3 mg, 92%). Rf = 0.32 (silica, EtOAc–pentane 50:50 v/v). mp (uncorr.) 125.3–128.8 °C (lit.58 122–123 °C). IR (neat) νmax 3297, 2938, 1631, 1580, 1537, 1501, 1337, 1233, 1127 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.39–7.30 (m, 2H), 7.30–7.21 (m, 3H), 6.94 (s, 2H), 6.30 (br. s, 1H), 3.88 (s, 3H), 3.86 (s, 6H), 3.70 (q, J = 6.8 Hz, 2H), 2.95 (t, J = 6.8 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 167.3, 153.2, 140.8, 139.0, 130.2, 128.9, 128.8, 126.7, 104.3, 61.0, 56.3, 41.3, 35.7. HRMS (ES) m/z [M + H]+ calcd for C18H22NO4: 316.1543 Found: 316.1545.
:90). The product was isolated as red solid (33.4 mg, 93%). Rf = 0.21 (silica, EtOAc–pentane 20:80 v/v). mp 115.3–116.0 °C (lit.60 115–116 °C). IR (neat) νmax 3341, 3059, 2968, 1637, 1534 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 7.4 Hz, 2H), 7.39 (t, J = 7.4 Hz, 1H), 7.31 (t, J = 7.4 Hz, 2H), 7.24 (t, J = 7.3 Hz, 2H), 7.20–7.10 (m, 3H), 6.17 (br. s, 1H), 3.63 (q, J = 6.7 Hz, 2H), 2.85 (t, J = 6.7 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 167.6, 139.0, 134.8, 131.5, 128.9, 128.8, 128.7, 126.9, 126.7, 41.3, 35.8. HRMS (ES) m/z [M + H]+ calcd for C15H16NO: 266.1226 Found: 266.1667. 1H-NMR, 13C-NMR and IR are in accordance with literature values.
:80). The product was isolated as a white compound with trace amounts of a reddish taint (35.0 mg, 87%). Rf = 0.62 (silica, EtOAc–pentane 50:50 v/v). mp (uncorr.) 94.8–95.4 °C (lit.61 94–95 °C). IR (neat) νmax 3297, 2929, 1635, 1541 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.26–7.16 (m, 4H), 7.16–7.07 (m, 4H), 7.01 (d, J = 7.3 Hz, 2H), 5.33 (br. s, 1H), 3.39 (q, J = 6.7 Hz, 2H), 2.86 (t, J = 7.6 Hz, 2H), 2.65 (t, J = 6.7 Hz, 2H), 2.34 (t, J = 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 172.1, 141.0, 139.0, 128.9, 128.75, 128.65, 128.5, 126.6, 126.4, 40.7, 38.7, 35.8, 31.8. HRMS (ES) m/z [M + H]+ calcd for C17H20NO: 254.1539 Found: 254.1544. 1H-NMR, 13C-NMR and IR are in accordance with literature values.
:90 to 20:80). The product was isolated as white solid (39.8 mg, 92%). Rf = 0.26 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 149.8–150.6 °C (lit.63 149.5–150.5 °C). IR (neat) νmax 3328, 1641, 1541, 1516 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.25 (d, J = 8.6 Hz, 2H), 7.83 (d, J = 8.6 Hz, 2H), 7.34 (t, J = 7.3 Hz, 2H), 7.29–7.21 (m, 3H), 6.23 (br. s, 1H), 3.75 (q, J = 6.7 Hz, 2H), 2.96 (t, J = 6.7 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 165.6, 149.7, 140.3, 138.6, 129.0, 128.9, 128.1, 127.0, 124.0, 41.5, 35.6. HRMS (ES) m/z [M + H]+ calcd for C15H15N2O3: 271.1077 Found: 271.1079. 1H-NMR, 13C-NMR and IR are in accordance with literature values.
:80). The product was isolated as white solid (44.2 mg, 91%). Rf = 0.26 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 120.9–121.1 °C. IR (neat) νmax 3284, 2928, 1640, 1547 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 8.8 Hz, 1H), 7.45 (dd, J = 7.6, 1.7 Hz, 1H), 7.39–7.29 (m, 3H), 7.29–7.19 (m, 4H), 6.06 (br. s, 1H), 3.74 (q, J = 6.9 Hz, 2H), 2.97 (t, J = 6.9 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 167.7, 138.8, 138.0, 133.5, 131.3, 129.6, 129.0, 128.8, 127.6, 126.8, 119.4, 41.3, 35.6. HRMS (ES) m/z [M + H]+ calcd for C15H15BrNO: 304.0332 Found: 304.0330. 1H-NMR is in accordance with literature values.
:90 to 20:80). The product was isolated as white solid (26.4 mg, 76%). Rf = 0.27 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 107.7–108.7 °C. IR (neat) νmax 3293, 2949, 2866, 1635, 1548, 1236 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.31 (t, J = 7.3 Hz, 2H), 7.25–7.21 (m, 1H), 7.21–7.16 (m, 2H), 5.48 (br. s, 1H), 3.51 (q, J = 6.9 Hz, 2H), 2.81 (t, J = 6.9 Hz, 2H), 2.44 (q, J = 7.9 Hz, 1H), 1.85–1.64 (m, 6H), 1.61–1.45 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 176.3, 139.2, 128.9, 128.7, 126.6, 46.0, 40.7, 35.9, 30.5, 26.0. HRMS (ES) m/z [M + H]+ calcd for C14H20NO: 218.1539 Found: 218.1544. 1H-NMR is in accordance with literature values.
:90 to 20:80). The product was isolated as red solid (33.6 mg, 77%). Rf = 0.23 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 138.9–140.1 °C (lit.66 133–134 °C). IR (neat) νmax 3334, 3057, 2936, 1640, 1543, 1305 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 7.86–7.75 (m, 3H), 7.69 (d, J = 8.6 Hz, 1H), 7.54–7.42 (m, 2H), 7.32–7.25 (m, 2H), 7.25–7.14 (m, 3H), 6.23 (br. s, 1H), 3.72 (q, J = 6.7 Hz, 2H), 2.92 (t, J = 6.7 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 167.6, 139.1, 134.8, 132.8, 132.0, 129.04, 129.02, 128.9, 128.6, 127.9, 127.7, 127.4, 126.9, 126.8 123.6, 41.4, 35.9. HRMS (ES) m/z [M + H]+ calcd for C19H18NO: 276.1383 Found: 276.1384. 1H-NMR, 13C-NMR and IR are in accordance with literature values.
:90 to 20:90). The product was isolated as a white solid (36.8 mg, 93%). Rf = 0.17 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 54.9–55.4 °C (lit.43 54–54.5 °C). IR (neat) νmax 3301, 2924, 1636, 1545 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.30 (t, J = 7.3 Hz, 2H), 7.25–7.21 (m, 1H), 7.21–7.14 (m, 2H), 5.57 (br. s, 1H), 3.51 (q, J = 6.8 Hz, 2H), 2.81 (t, J = 6.8 Hz, 2H), 2.11 (t, J = 7.6 Hz, 2H), 1.62–1.53 (m, 2H), 1.31–1.20 (m, 8H), 0.87 (t, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.3, 139.1, 128.9, 128.7, 126.6, 40.6, 36.9, 35.8, 31.8, 29.3, 29.1, 25.9, 22.7, 14.2.
HRMS (ES) m/z [M + H]+ calcd for C16H26NO: 248.2009 Found: 248.2013. 1H-NMR and IR are in accordance with literature values.
:90 to 20:90). The product was isolated as a white solid (28.8 mg, 64%). Rf = 0.29 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 132.2–134.4 °C (lit.68 133 °C). IR (neat) νmax 3317, 2903, 2849, 1627, 1547, 1279 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.31 (t, J = 7.3 Hz, 2H), 7.27–7.14 (m, 3H), 5.59 (br. s, 1H), 3.49 (q, J = 6.9 Hz, 2H), 2.80 (t, J = 6.9 Hz, 2H), 2.01 (s, 3H), 1.88–1.75 (m, 6H), 1.75–1.60 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 178.0, 139.2, 129.0, 128.7, 126.6, 40.7, 40.5, 39.3, 36.6, 35.8, 28.2. HRMS (ES) m/z [M + H]+ calcd for C16H26NO: 248.2009 Found: 248.2012. 1H-NMR is in accordance with literature values.
:80 to 50:50) yielding the N-phenethylpropionamide as a white solid (55.5 mg, 89%).
In order to recover the acid the basic aqueous solution was acidified using 6 M HCl to pH = 1 and extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with brine, dried with Na2SO4, evaporated to dryness in vacuo. The residue was purified by flash column chromatography using (formic acid–DCM 1:99) thus recovering the 2-(4-nitrophenyl)acetic acid as a with solid (66.4 mg, 94%). Rf = 0.33 (silica, formic acid–DCM 1:99 v/v). mp (uncorr.) 153.7–154.3 °C (lit.70 153–155 °C). IR (neat) νmax 2919, 2848, 2641, 1700, 1509, 1339, 1250 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 8.7 Hz, 1H), 3.78 (s, 1H).
13C NMR (100 MHz, CDCl3) δ 175.5, 147.5, 140.5, 130.6, 124.0, 40.6. HRMS (ES) m/z [M − H]− calcd for C8H6NO4: 180.0302 Found: 180.0301. 1H-NMR, 13C-NMR and IR are in accordance with literature values.
:80 v/v). mp (uncorr.) 144.5–147.5 °C. IR (neat) νmax 3325, 2989, 1697, 1678, 1240 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.99 (br s, 1H), 7.47 (br s, 1H), 7.24–7.12 (m, 3H), 7.09 (d, J = 7.2 Hz, 2H), 5.25 (d, J = 8.2 Hz, 1H), 5.11–4.75 (m, 1H), 4.17 (q, J = 7.0 Hz, 4H), 3.08 (br s, 2H), 3.02–2.87 (m, 2H), 1.34 (s, 9H), 1.25 (t, J = 7.1 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 169.8, 166.6 (br s), 155.5, 135.5, 130.3 (br s), 129.8, 129.2, 127.7, 113.6, 112.4, 80.6, 60.9, 52.5, 39.4, 28.1, 22.3, 14.0. 13C NMR (100 MHz, CDCl3) δ 169.8 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 166.6 (br s, (CO)), 155.5 (CO), 135.5 (Ar), 130.3 (br s, (–CHCCO2CH2CH3), 129.8 (Ar), 129.2 (Ar), 127.7 (Ar), 113.6 (CCO2CH2CH3), 112.4 (CCO2CH2CH3), 80.6 (C(CH3)3)., 60.9 (–CO2CH2CH3), 52.5 (–CHCH2Ph), 39.4 (–CHCH2Ph), 28.1 (–CO2CH2CH3), 22.3 (DHP Sp3), 14.0 (C(CH3)3). HRMS (ES) m/z [M + Na]+ calcd for C25H32N2O7Na: 495.2102 Found: 495.2108.
O), 166.6 (br s, (CO)), 155.5 (CO), 135.5 (Ar), 130.3 (br s, (–CHCCO2CH2CH3), 129.8 (Ar), 129.2 (Ar), 127.7 (Ar), 113.6 (CCO2CH2CH3), 112.4 (CCO2CH2CH3), 80.6 (C(CH3)3)., 60.9 (–CO2CH2CH3), 52.5 (–CHCH2Ph), 39.4 (–CHCH2Ph), 28.1 (–CO2CH2CH3), 22.3 (DHP Sp3), 14.0 (C(CH3)3). HRMS (ES) m/z [M + Na]+ calcd for C25H32N2O7Na: 495.2102 Found: 495.2108.
Second, a solution of unprotected 9 (104 mg, 0.218 mmol, 1.1 eq.) in DCM (3 mL) was added DDQ (45 mg, 0.198 mmol, 1 eq.). After 6 hours activated 9 was added to flask A containing 10 followed by DMAP (73 mg, 0.595 mmol, 3 eq.). A clear change in color occurs after the addition of DMAP. The resulting mixture was left to stir overnight after which it was diluted with EtOAc. The resulting mixture was then transferred to a separation-funnel and washed with 1 M HCl (×2) and NaHCO3 (×2). The organic phase was dried (MgSO4), filtered, and evaporated in vacuo. The desired compound was isolated using flash column chromatography (silica) with a gradient from 5% acetone–pentane, 10% acetone–pentane, 20% acetone–pentane, and finally 50% acetone–pentane. The compound was loaded onto the column using Celite.
The product was isolated as a beige solid (86 mg, 70%). Rf = 0.3 (30% EtOAc–pentane). mp (uncorr.) 179.0–180.4 °C. IR (neat) νmax 3348, 3272, 2980, 1720, 1697, 1655, 1231, 1176 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.80 (br s, 1H), 7.27 (br s, 1H), 7.16–6.95 (m, 8H), 6.89 (d, J = 6.6 Hz, 2H), 6.47 (d, J = 7.6 Hz, 1H), 5.16–4.99 (m, 1H), 4.77 (br s, 1H), 4.18 (br s, 1H), 4.08 (q, J = 7.1 Hz, 4H), 3.06–2.90 (m, 2H), 2.90–2.74 (m, 4H), 1.24 (s, 9H), 1.16 (t, J = 6.8 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 171.5, 168.8, 166.8, 155.9, 136.8, 135.1, 130.4, 129.7, 129.7, 129.3, 129.2, 127.9, 127.5, 113.9, 112.5, 80.6, 61.0, 55.8, 51.0, 39.1, 38.1, 28.1, 22.3, 14.1. 13C NMR (100 MHz, CDCl3) δ 171.5, 168.8, 166.8, 155.9, 136.8 (Ar), 135.1 (Ar), 130.4 (–CHCCO2CH2CH3), 129.7 (Ar), 129.7 (Ar), 129.3 (Ar), 129.2 (Ar), 127.9 (Ar), 127.5 (Ar), 113.9 (–CHCCO2CH2CH3), 112.5 (–CHCCO2CH2CH3), 80.6 (–CH(CH3)3), 61.0 (–CO2CH2CH3)., 55.8 (–CHCH2Ph), 51.0 (–CHCH2Ph), 39.1 (–CHCH2Ph), 38.1 (–CHCH2Ph), 28.1 (–CH(CH3)3), 22.3 (DHP Sp3), 14.1 (–CO2CH2CH3). HRMS (ES) m/z [M + Na]+ calcd for C34H41N3O8Na: 642.2786 Found: 642.2790.
:80) as the eluent. The product was isolated as a yellow solid (0.206 g, 14%). Rf = 0.29 (silica, EtOAc–pentane 20:80 v/v). mp (uncorr.) 114.6–117.0 °C (lit.74 117–118 °C). IR (neat) νmax 3314, 2900, 1689, 1667 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.52–7.21 (m, 6H), 7.17 (t, J = 7.3 Hz, 1H), 6.75 (br s, 1H), 4.92 (s, 1H), 4.24–3.88 (m, 4H), 1.25 (t, J = 7.1 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 168.0, 147.5, 134.6, 128.6, 128.3, 126.7, 108.1, 59.9, 37.4, 13.8. HRMS (ES) m/z [M + H]+ calcd for C17H20NO4: 302.1387. Found: 302.1390. 1H-NMR, 13C-NMR and IR are in accordance with literature values.
:80 v/v). mp (uncorr.) 125.0–125.5 °C. IR (neat) νmax 2986, 1697, 1637 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.09 (br. s, 2H), 7.27–7.25 (m, 4H), 7.21–7.15 (m, 1H), 4.89 (s, 1H), 4.20–4.05 (m, 4H), 2.75 (q, J = 7.2 Hz, 2H), 1.30 (t, J = 7.2 Hz, 3H); 1.21 (t, J = 7.1 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 170.7, 166.0, 143.8, 129.4, 128.7, 128.3, 127.1, 115.6, 60.9, 38.7, 27.3, 14.2, 8.7. HRMS (ES) m/z [M + H]+ calcd for C20H24NO5: 358.1649. Found: 358.1653.
Acknowledgements
We thank the Danish National Research Foundation Center for DNA Nanotechnology (DNRF81), The Danish Council for Strategic Research, and The Danish Council for Independent Research for financial support.Notes and references
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC
.
- E. Armelin, L. Franco, A. Rodriguez-Galan and J. Puiggalí, Macromol. Chem. Phys., 2002, 203, 48–58 CrossRef CAS
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed
- J. M. Chalker, Chem. Biol. Drug Des., 2013, 81, 122–135 CAS
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed
- A. Williams and I. T. Ibrahim, Chem. Rev., 1981, 81, 589–636 CrossRef CAS
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS PubMed
- R. M. Lanigan, P. Starkov and T. D. Sheppard, J. Org. Chem., 2013, 78, 4512–4523 CrossRef CAS PubMed
- T. A. Dineen, M. A. Zajac and A. G. Myers, J. Am. Chem. Soc., 2006, 128, 16406–16409 CrossRef CAS PubMed
- M. Zhang, S. Imm, S. Bahn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 3905–3909 CrossRef CAS PubMed
- C. L. Allen, B. N. Atkinson and J. M. J. Williams, Angew. Chem., Int. Ed., 2012, 51, 1383–1386 CrossRef CAS PubMed
- S. N. Rao, D. C. Mohan and S. Adimurthy, Org. Lett., 2013, 15, 1496–1499 CrossRef CAS PubMed
- M. Tamura, T. Tonomura, K.-I. Shimizu and A. Satsuma, Green Chem., 2012, 14, 717–724 RSC
- S. Mundinger, U. Jakob and W. Bannwarth, Chem. – Eur. J., 2014, 20, 1258–1262 CrossRef CAS PubMed
- S. Mundinger, U. Jakob, P. Bichovski and W. Bannwarth, J. Org. Chem., 2012, 77, 8968–8979 CrossRef CAS PubMed
- M. C. Bröhmer, S. Mundiger, S. Bräse and W. Bannwarth, Angew. Chem., Int. Ed., 2011, 50, 6175–6177 CrossRef PubMed
- R. A. Kramer, M. C. Bröhmer, N. V. Forkel and W. Bannwarth, Eur. J. Org. Chem., 2009, 4273–4283 CrossRef CAS
- M. C. Bröhmer and W. Bannwarth, Eur. J. Org. Chem., 2008, 4412–4415 CrossRef
- V. Valerio, D. Petkova, C. Madelaine and N. Maulide, Chem. – Eur. J., 2013, 19, 2606–2610 CrossRef CAS PubMed
- Y. Kita, Y. Nishii, Y. Higuchi and K. Mashima, Angew. Chem., Int. Ed., 2012, 124, 5821–5824 CrossRef
- A. Radzicka and R. Wolfenden, J. Am. Chem. Soc., 1996, 118, 6105–6109 CrossRef CAS
- R. M. Smith and D. E. Hansen, J. Am. Chem. Soc., 1998, 120, 8910–8913 CrossRef CAS
- S. Arseniyadis, A. Wagner and C. Mioskowski, Tetrahedron Lett., 2004, 45, 2251–2253 CrossRef CAS PubMed
- W.-R. Li, N.-M. Hsu, H.-H. Chou, S. T. Lin and Y.-S. Lin, Chem. Commun., 2000, 401–402 RSC
- T. Kamijo, R. Yamamoto, H. Harada and K. Iizuka, Chem. Pharm. Bull., 1982, 30, 4242–4244 CrossRef CAS
- W. Steglich and G. Höfle, Angew. Chem., Int. Ed. Engl., 1969, 8, 981 CrossRef CAS
- U. Ragnarsson and L. Grehn, Acc. Chem. Res., 1998, 31, 494–501 CrossRef CAS
-
J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman and Company, USA, 5th edn, 2003 Search PubMed
- P. Belenky, K. L. Bogan and C. Brenner, Trends Biochem. Sci., 2007, 32, 12–19 CrossRef CAS PubMed
- J.-P. Cheng, Y. Lu, X. Zhu and L. Mu, J. Org. Chem., 1998, 63, 6108–6114 CrossRef CAS PubMed
- A. R. Katritzky, K. Suzuki and Z. Wang, Synlett, 2005, 11, 1656–1665 CrossRef PubMed
- J. E. Tulor, M. D. Jones, J. M. J. Williams and S. D. Bull, J. Org. Chem., 2012, 77, 2808–2818 CrossRef PubMed
- To our knowledge only one example in the literature describes the acylating effect of a 4-substituted 1,4-dihydropyridine amide: J. Bergman, R. Carlsson and S. Misztal, Acta Chem. Scand., 1976, 30, 853–862 CrossRef PubMed
- D. L. Comins and A. H. Abdullah, J. Org. Chem., 1984, 49, 3392–3394 CrossRef CAS
- J. Arguello, L. J. Núnez-Vergara, J. C. Sturm and J. A. Squella, Electrochim. Acta, 2004, 49, 4849–4856 CrossRef CAS PubMed
- Y. P. Stradyn, Y. I. Beilis, Y. R. Uldrikis, G. Y. Dubur, S. E. Sausin and B. S. Chekavichus, Chem. Heterocycl. Compd., 1975, 11, 1299–1303 CrossRef
- E. Booker and U. Eisner, J. Chem. Soc., Perkin Trans. 1, 1975, 929–931 RSC
- A variety of different reagents for oxidation were investigated. In detail these attempts can be seen in the ESI†.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CAS
- P. J. Brignell, U. Eisner and P. G. J. Farrell, Chem. Soc. B, 1966, 1083–1089 RSC
- K. Singh and K. Singh, Tetrahedron, 2009, 65, 10395–10399 CrossRef CAS PubMed
- A. J. Hill and G. E. J. Hall, J. Am. Chem. Soc., 1952, 74, 666–668 CrossRef CAS
- C. L. Allen, A. R. Chhatwal and J. M. J. Williams, Chem. Commun., 2012, 48, 666–668 RSC
- S. Y. Lee, C. Lee and D. Y. J. Oh, Org. Chem., 1999, 64, 7017–7022 CrossRef CAS
- P. Capdevielle, A. Lavigne and M. Maumy, Tetrahedron, 1990, 46, 2635–2844 CrossRef
- P. Borgna, L. Vicarini and C. Calderara, Farmaco, Ed. Sci., 1977, 32, 813–820 CAS
- F. Belin, P. Barthélémy, K. Ruiz, J. M. Lacombe and B. Pucci, Helv. Chim. Acta, 2003, 86, 247–265 CrossRef CAS
- P. S. Mahajan, J. P. Mahajan and S. B. Mhaske, Synth. Commun., 2013, 43, 2508–2516 CrossRef CAS
- H.-J. Pi, J.-D. Dong, N. An, W. Du and W.-P. Deng, Tetrahedron, 2009, 65, 7790–7793 CrossRef CAS PubMed
- S. R. Thopate, S. R. Kote, S. V. Rohokale and N. M. Thorat, J. Chem. Res., 2011, 35, 124–125 CrossRef CAS
- C. A. Goodman, J. B. Eagles, L. Rudahindwa, C. G. Hamaker and S. R. Hitchcock, Synth. Commun., 2013, 43, 2155–2164 CrossRef CAS
- R. G. Kalkhambkar, S. N. Waters and K. K. Laali, Tetrahedron Lett., 2011, 52, 867–871 CrossRef CAS PubMed
- L. Muley, B. Baum, M. Smolinski, M. Freindorf, A. Heine, G. Klebe and D. G. Hangauer, J. Med. Chem., 2010, 53, 2126–2135 CrossRef CAS PubMed
- M. Bolton and J. Walker, J. Chem. Soc. C, 1967, 2095–2096 RSC
- E. Reid, J. Am. Chem. Soc., 1917, 39, 124–136 CrossRef CAS
- I. Dhimitruka and J. SantaLucia, Org. Lett., 2006, 8, 47–50 CrossRef CAS PubMed
- R. Hirose, B. G. Kopek, Z.-H. Wang, R. Yusa and B. W. Baldwin, Tetrahedron, 2003, 9, 1831–1833 CrossRef
- A. Lasslo and W. D. Jordan, J. Org. Chem., 1956, 21, 805–805 CrossRef CAS
- S. A. Rossi, K. W. Shimkin, Q. Xu, L. M. Mori-Quiroz and D. A. Watson, Org. Lett., 2013, 15, 2314–2317 CrossRef CAS PubMed
- B. R. Kim, H. Lee, S. Kang, G. H. Sung, J. Kim, J. K. Park, S. Lee and U. Yoon, Synthesis, 2012, 42–50 CAS
- J.-B. Gualtierotti, X. Schumancher, P. Fontaine, G. Masson, Q. Wang and J. Zhy, Chem. – Eur. J., 2012, 18, 14812–14819 CrossRef CAS PubMed
- V. Prasad, R. R. Kale, B. B. Mishra, D. Kumar and V. K. Tiwari, Org. Lett., 2012, 14, 2936–2939 CrossRef CAS PubMed
- M. Kunishima, C. Kawachi, K. Hioki, K. Terao and S. Tani, Tetrahedron, 2001, 57, 1551–1558 CrossRef CAS
- M. Hellal and G. D. Cuny, Tetrahedron Lett., 2011, 42, 5508–5511 CrossRef PubMed
- M. D. Wendt, T. W. Rockway, A. Geyer, W. McClellan, M. Weitzberg, X. Zhao, R. Mantei, V. L. Nienaber, K. Stewart, V. Klinghofer and V. L. Giranda, J. Med. Chem., 2004, 47, 303–324 CrossRef CAS PubMed
- T. Gonec, P. Bobal, J. Sujan, M. Pesko, J. Guo, K. Kralova, L. Pavlacka, L. Vesely, E. Kreckova, J. Kos, A. Coffey, P. Kollar, A. Imramovsky, L. Placek and J. Jampilek, Molecules, 2012, 17, 613–644 CrossRef CAS PubMed
- T. Ogawa, T. Hikasa, T. Ikegami, N. Ono and H. Suzuki, Chem. Lett., 1993, 22, 815–818 CrossRef
- I. Kim, Y. Park, B. D. Hammock and K. J. Nishi, Med. Chem., 2011, 54, 1752–1761 CrossRef CAS PubMed
- G.-B. Zhou, P.-F. Zhangb and Y.-J. Pan, Tetrahedron, 2005, 61, 5671–5677 CrossRef CAS PubMed
- D. H. Huh, J. S. Jeong, H. B. Lee, H. Ryu and Y. G. Kim, Tetrahedron, 2002, 58, 9925–9932 CrossRef CAS
- J.-M. Wang, G.-Z. Ding, L. Fang, J.-G. Dai, S.-S. Yu, Y.-H. Wang, X.-G. Chen, S.-G. Ma, J. Qu, S. Xu and D. Du, J. Nat. Prod., 2010, 73, 1240–1249 CrossRef CAS PubMed
- K. B. Joshi and S. Verma, Tetrahedron Lett., 2008, 49, 4231–4234 CrossRef CAS PubMed
- N. A. Al-Awadi, M. R. Ibrahim, M. H. Elnagdi, E. John and Y. A. Ibrahim, Beilstein J. Org. Chem., 2012, 8, 441–447 CrossRef CAS PubMed
- B. Loev, M. M. Goodman, K. M. Snader, R. Tedeschi and E. Macko, J. Med. Chem., 1974, 17, 956–965 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Including additional spectroscopic data and solvent effect data. See DOI: 10.1039/c4ob01931h |
| This journal is © The Royal Society of Chemistry 2015 |