
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the pH-triggered self-assembly of dendritic peptide amphiphiles using fluorinated side chains†
Ralph
Appel
ab,
Sebastian
Tacke
c,
Jürgen
Klingauf
c and
Pol
Besenius
*ab
aOrganic Chemistry Institute, Westfälische Wilhelms-Universität Münster, Corrensstrasse 40, 48149 Münster, Germany. E-mail: p.besenius@uni-muenster.de; Tel: +49 251 83 63928
bCenter for Nanotechnology (CeNTech), Heisenbergstrasse 11, 48149 Münster, Germany
cDepartment of Cellular Biophysics, Institute of Medical Physics and Biophysics, Westfälische Wilhelms-Universität Münster, Robert-Koch-Strasse 31, 48149 Münster, Germany
First published on 13th November 2014
Abstract
We report the synthesis of a series of anionic dendritic peptide amphiphiles of increasing hydrophobic character. By establishing state diagrams we describe their pH and ionic strength triggered self-assembly into supramolecular nanorods in water and highlight the impact of hydrophobic shielding in the supramolecular polymerisation process. Via the incorporation of fluorinated peptide side chains the pH-triggered monomer to polymer transition at physiological ionic strength is shifted from pH 5.0 to pH 7.4. We thereby show that compensating attractive non-covalent interactions and hydrophobic effects with repulsive electrostatic forces, a concept we refer to as frustrated growth, is a sensitive tool in order to manipulate one-dimensional supramolecular polymerisation processes in water.
Introduction
Fabrication of nanoscale materials using supramolecular non-covalent interactions provides access to many exciting material properties. Compared to traditional covalent synthetic approaches, the self-assembly of molecular building blocks to supramolecular polymeric morphologies and nanostructures leads to dynamic materials,1–6 that are responsive towards optical,7–9 mechanical10,11 or biological12 stimuli. Some of the attractive features for supramolecular materials that arise thereof are self-healing properties,10,13 controlled release of cargo,8,14–16 or emergent behaviour like self-replication.11,17,18 In the area of aqueous responsive supramolecular systems, peptide materials such as nanofibres,19–24 nanotubes11,25 and artificial β-barrels26 have attracted much interest. Particularly aliphatic β-sheet encoded peptide amphiphiles, functionalised with hydrophobic alkyl tails and charged hydrophilic head groups, as developed by Stupp and coworkers,19 are prominent building blocks for the fabrication of bioactive gels and supports for tissue engineering and regenerative medicine.21,27 Aromatic peptide amphiphiles have shown growing interest as well, as shown by the groups of Ulijn and Adams,28–32 yielding enzyme or pH-responsive hydrogels.33,34We have recently focused on developing strategies to manipulate one-dimensional (1D) supramolecular polymerisation processes in water. For that purpose we used the concept of frustrated growth that relies on compensating attractive non-covalent interactions with repulsive steric and electrostatic forces. The supramolecular synthons we have used in an earlier study consisted of dendritic peptide amphiphiles, based on a small benzene-1,3,5-carboxamide branching agent. C3-symmetrical hydrophobic cores are indeed known to direct the supramolecular polymerisation into strictly 1D morphologies with negligible secondary aggregation.35–44 We have reported the synthesis of dendritic nonaphenylalanines which were linked to peripheral carboxylic acid Newkome dendrons45,46via a 3-(hydroxypropylthio)propionic acid type linker. Similarly to the examples of van Esch,35 Hartgerink,47 Goldberger48 and others49–52 we were able to study the pH-dependent self-assembly.44 Very recently we have then expanded the core of the branching agent, using 3,4-ethylenedioxythiophene (EDOT) extended 1,3,5-substituted benzene: using dendritic hexaphenylalanine we realised that the loss of non-covalent interactions due to the omission of one phenylalanine per side arm in the supramolecular monomer, was compensated by the larger π-system of the hydrophobic core.53
We hereby aim to systematically study the effect of increasing the hydrophobic shielding of dendritic peptide amphiphiles, and the impact on the pH and ionic strength triggered self-assembly into supramolecular nanorods (Fig. 1). By establishing state diagrams for three different molecules of varying hydrophobic character, we show that the supramolecular polymerisation is fully switched off if the apolar hexyl linker between the dendritic nonaphenylalanines and peripheral carboxylic acid Newkome dendrons is omitted. If the hydrophobic character of the peptide core is increased further by introducing trifluoromethyl groups via thiol–ene click chemistry in O-allyl derivatised tyrosine groups, the thermodynamic driving force for polymerisation is increased and the pH-triggered monomer to polymer transition at physiological ionic strength is shifted from pH 5.0 to pH 7.4. Note that the fluorinated analogues of hydrophobic aliphatic amino acids have been introduced in peptide structures in the past:54–62 it was for example shown that the thermodynamic stability of coiled-coil motifs was increased due to an increased hydrophobic core;63,64 very recently it has been shown that an increased fluorine content increases the hydrophobic character of peptides, decreases their α-helix propensity and increases the formation rates of β-sheet-rich amyloids.65
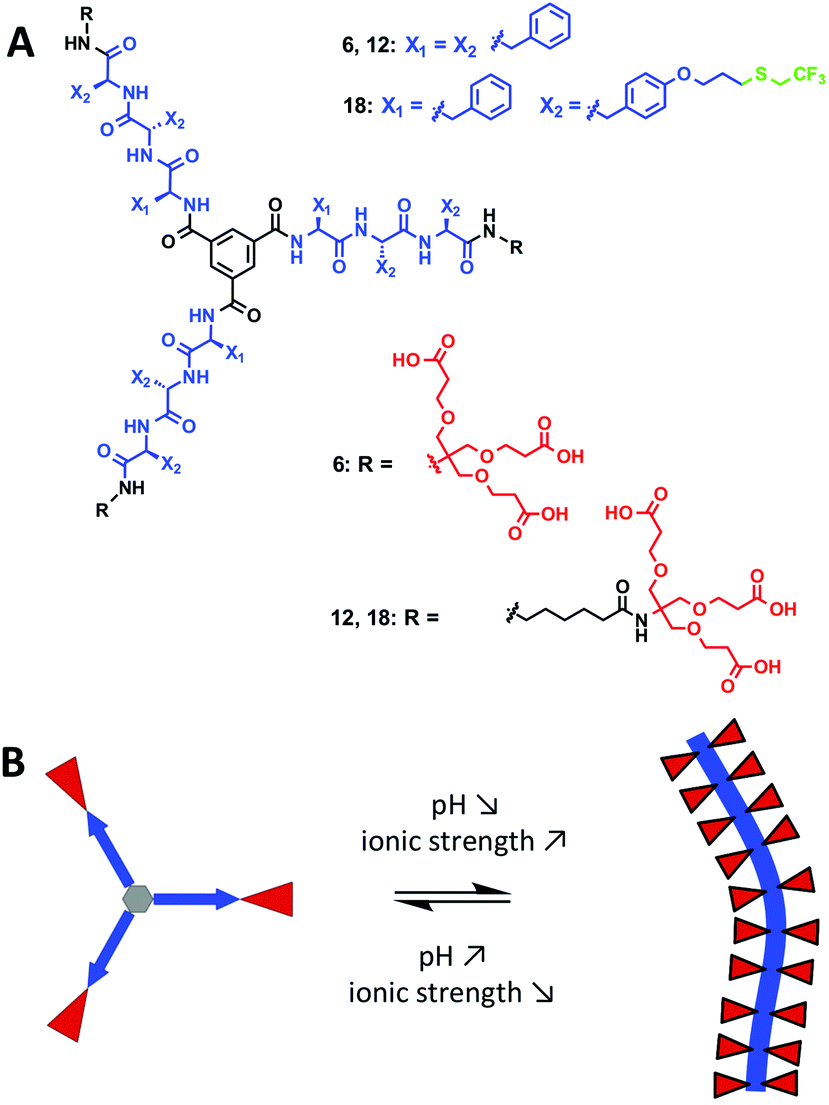 | ||
| Fig. 1 (A) Chemical structures of the three dendritic peptide amphiphiles 6, 12 and 18. (B) The schematic representation of their pH and ionic strength triggered self-assembly into one-dimensional (1D) nanorods. | ||
We are particularly interested in well-defined and stimuli-responsive nanomaterials66–74 that are designed to undergo morphological transitions triggered by specific changes in a physiological environment. These hold much promise for applications in biomedical imaging and therapy.75 We are planning to develop methodologies whereby the acidity in the microenvironment of a tumour interior or inflamed tissue induces the triggered self-assembly of small molecules into larger nanorods. The additional benefit of fluorinated building blocks is their potential use in 19F-MRI,76–78 which would allow to quantify the selective tumour retention and accumulation in cancer diagnosis and therapy.79
Results and discussion
Synthetic routes
The synthesis of the dendritic peptide amphiphiles 6 and 12 followed a convergent approach (Scheme 1A; full details can be found in the ESI†). First, the Cbz-protected tri-L-phenylalanine 2 was synthesized using conventional solid-phase peptide synthesis (SPPS). Using standard peptide coupling reagents, the tert-butyl protected Newkome dendron461, or 6-aminohexanoic acid extended dendron 8 were coupled to 2 in order to obtain the Cbz-protected dendronised triphenylalanines 3 and 9. After hydrogenation using Pd/C as catalyst the amine derivatives 4 and 10 were coupled to 1,3,5-benzenetricarbonyl trichloride to obtain the branched self-assembly units 5 and 11. Finally after acidic deprotection of 5 and 11, the dendritic nona-phenylalanine peptide amphiphile 6 was obtained after four steps in a moderate non-optimised overall yield of 13%, and 12 after four steps in a non-optimised overall yield of 22% (Scheme 1A). In 6 the hydrophobic core and hydrophilic dendron are directly connected, while 12 contains an apolar hexyl linker between the same dendritic nonaphenylalanine core and the peripheral carboxylic acid Newkome dendron.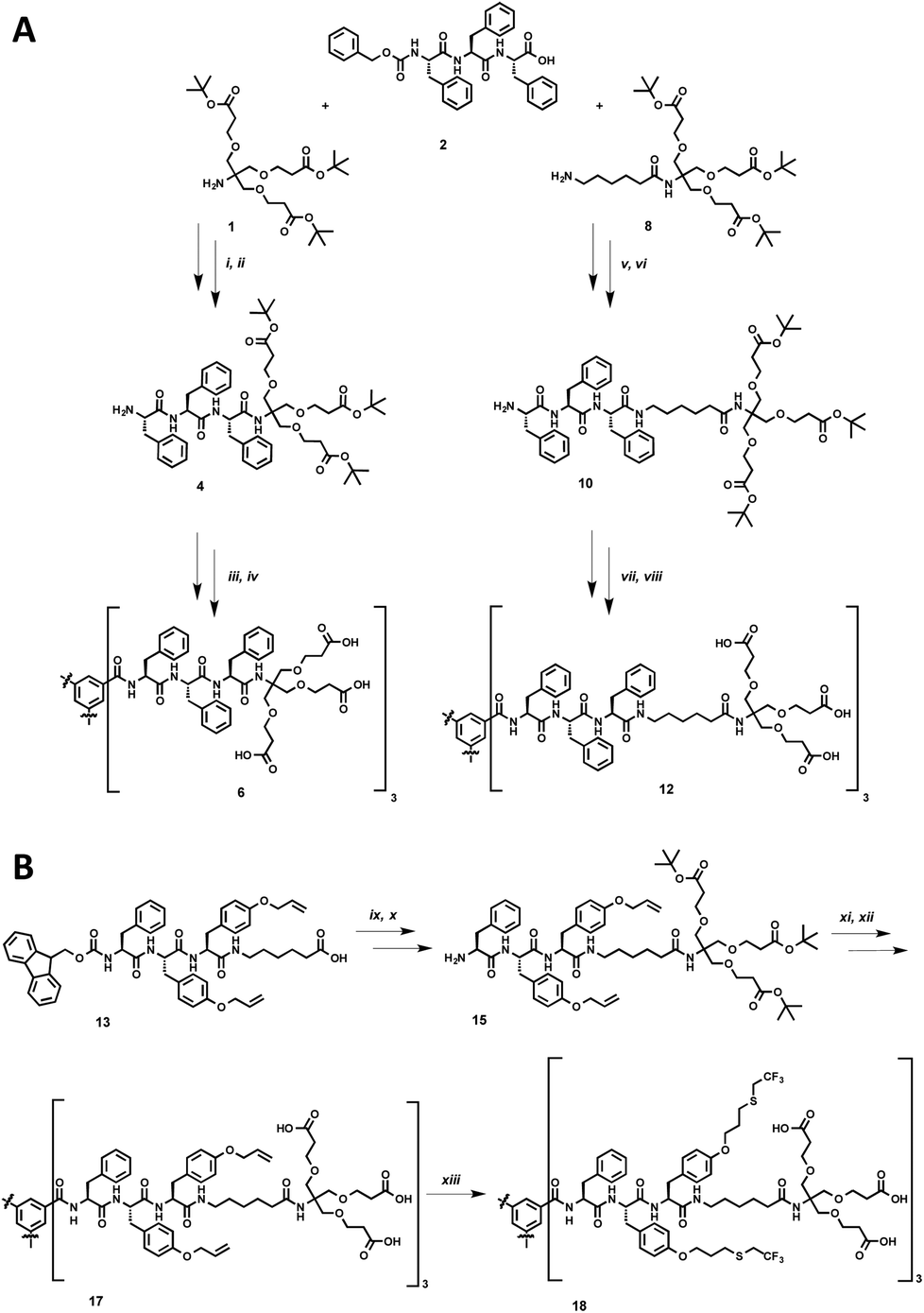 | ||
Scheme 1 Synthetic schemes for the preparation of the dendritic peptide amphiphiles 6, 12 and 18: (A) (i) PyBOP, DIPEA (64%); (ii) H2, Pd/C (91%); (iii) benzene-1,3,5-tricarbonyl trichloride, DIPEA (50%); (iv) TFA–DCM, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (44%); (v) PyBOP, DIPEA (81%); (vi) H2, Pd/C (89%); (vii) benzene-1,3,5-tricarbonyl trichloride, DIPEA (63%); (viii) TFA–DCM, 1:1 (48%); (B) (ix) PyBOP, DIPEA (71%); (x) piperidine, CH3CN (44%); (xi) benzene-1,3,5-tricarbonyl trichloride, DIPEA (50%); (xii) TFA–DCM, 1:1 (93%), (xiii) trifluoroethanethiol, DMPA, THF/H2O, hν (38%). :1 (44%); (v) PyBOP, DIPEA (81%); (vi) H2, Pd/C (89%); (vii) benzene-1,3,5-tricarbonyl trichloride, DIPEA (63%); (viii) TFA–DCM, 1:1 (48%); (B) (ix) PyBOP, DIPEA (71%); (x) piperidine, CH3CN (44%); (xi) benzene-1,3,5-tricarbonyl trichloride, DIPEA (50%); (xii) TFA–DCM, 1:1 (93%), (xiii) trifluoroethanethiol, DMPA, THF/H2O, hν (38%). | ||
In order to create a dendritic core that is more hydrophobic than the one in 12, we incorporated a L-phenylalanine-di-(O-allyl-L-tyrosine) into the same convergent synthesis as described before (Scheme 1B) including an apolar hexyl linker between the dendritic nonapeptide core and the peripheral carboxylic acid Newkome dendron. The allyl ether functional groups are very useful since they can easily be modified via thiol–ene type click chemistry,44,80,81 using 2,2,2-trifluoroethanethiol to incorporate fluorinated side chains into the hydrophobic peptide block. Note that we had to install a phenylalanine amino acid on the N-terminus rather than a protected tyrosine, since we encountered very low yields when coupling tri-(O-allyl-tyrosine) derivatives to the benzenetricarbonyl trichloride core, most likely due to steric issues. Using conventional Fmoc-SPPS chemistry we prepared Fmoc-phenylalanine-di-(O-allyl-L-tyrosine). In four similar steps as described for compound 12 the amphiphiles 17 was successfully obtained. After UV-initiated thiol–ene click chemistry using 2,2-dimethoxy-2-phenylacetophenone (DMPA) in a THF–water mixture 1:1 we isolated the target fluorinated dendritic peptide amphiphile 18, after five steps in a non-optimised overall yield of 6% (Scheme 1B).
Spectroscopic investigations into the pH and ionic strength triggered self-assembly
The dendritic peptide amphiphiles 6, 12 and 18 equipped with nine carboxylic acids groups turned out to be highly soluble in aqueous buffers.First, we investigated the self-assembly of dendritic peptide amphiphile 6 in phosphate buffer using circular dichroism (CD) spectroscopy. At pH 9.0 in 10 mM phosphate buffer, the CD spectra suggest that no aggregation takes place (Fig. 2A and S1†). This agrees with observations we have made before:44 in slightly basic pH and at low ionic strength repulsive Coulombic interactions from the charged dendritic carboxylate groups in the hydrophilic shell of the peptides prevent supramolecular polymerisation into large macromolecular structures. Surprisingly we were not able to screen these charge-charge repulsive interactions by increasing the ionic strength up to 1 M NaCl at pH 9.0 (Fig. 2A) or by acidifying the buffer to pH 6 (Fig. 2B). Since the CD spectra are identical in pH 9.0, 7.4 and 6.0, both at low and high ionic strength (Fig. S1†) it thereby becomes apparent that an apolar spacer between the hydrogen bonding hydrophobic core and hydrophilic shell is critical for self-assembly.
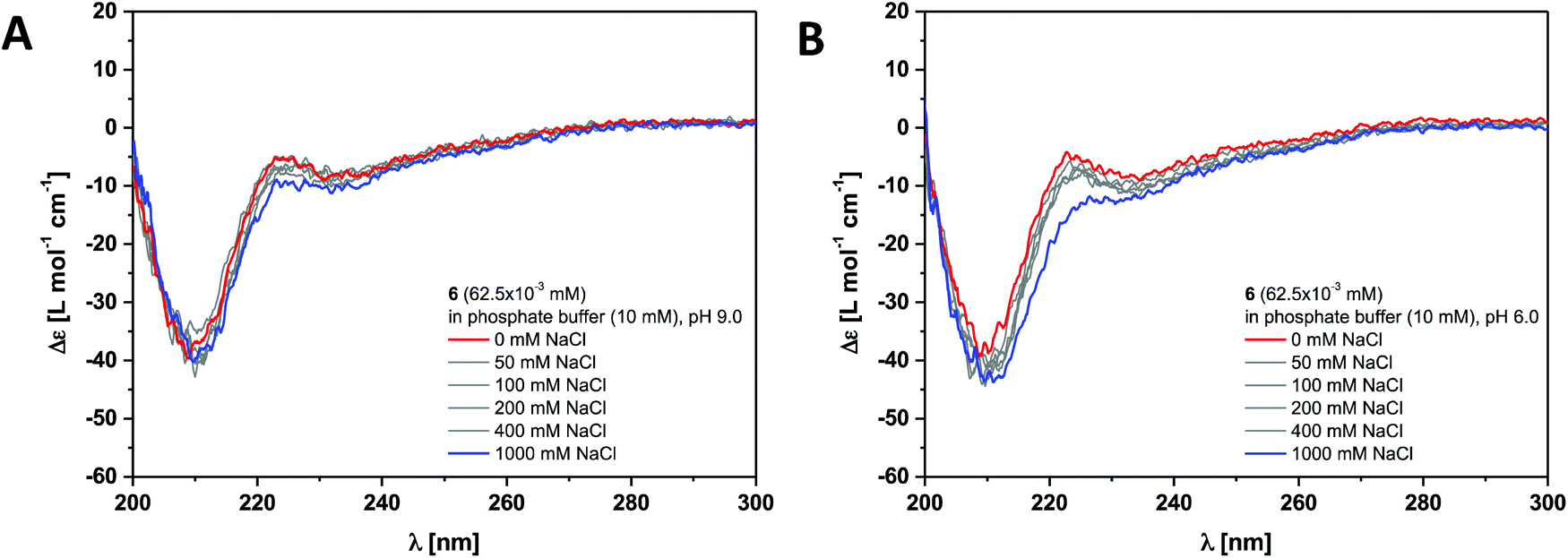 | ||
| Fig. 2 Ionic strength dependent CD spectra for dendritic peptide amphiphile 6 (62.5 × 10−3 mM, 293 K) in 10 mM phosphate buffer at pH 9.0 (A) and pH 6.0 (B). | ||
This was verified by investigating the supramolecular polymerisation of the dendritic peptide amphiphile 12 in full detail. At neutral pH 7.4, in 10 mM phosphate buffer and 0 M NaCl, a CD spectrum with at weak negative band at 210 nm is observed, that is very similar to the one obtained for compound 6 (Fig. 3 and S2†). At pH 7.4, the addition of NaCl leads the appearance of a positive band at λ = 203 nm, to a shift of the negative band from λ = 210 nm to 220 nm and a significant increase in the intensity. This is a good indication for the self-assembly into ordered chiral supramolecular polymers. By electrostatic screening of the negative charges in the periphery of the supramolecular nanorods, the polymerisation becomes more favourable.44,53 Similarly after lowering the pH from 7.4 to pH 5.0 the same transitions in the CD spectra are observed, which indicates the formation of supramolecular polymers. These findings suggest that at physiological pH the self-assembly of the polyanionic dendritic peptide 12 is dictated by a mechanism of frustrated growth: attractive supramolecular interactions, hydrogen-bonding and hydrophobic effects, that drive the monomers to polymerise are opposed by repulsive Coulomb interactions which disfavour self-assembly.35,37,39,44,53
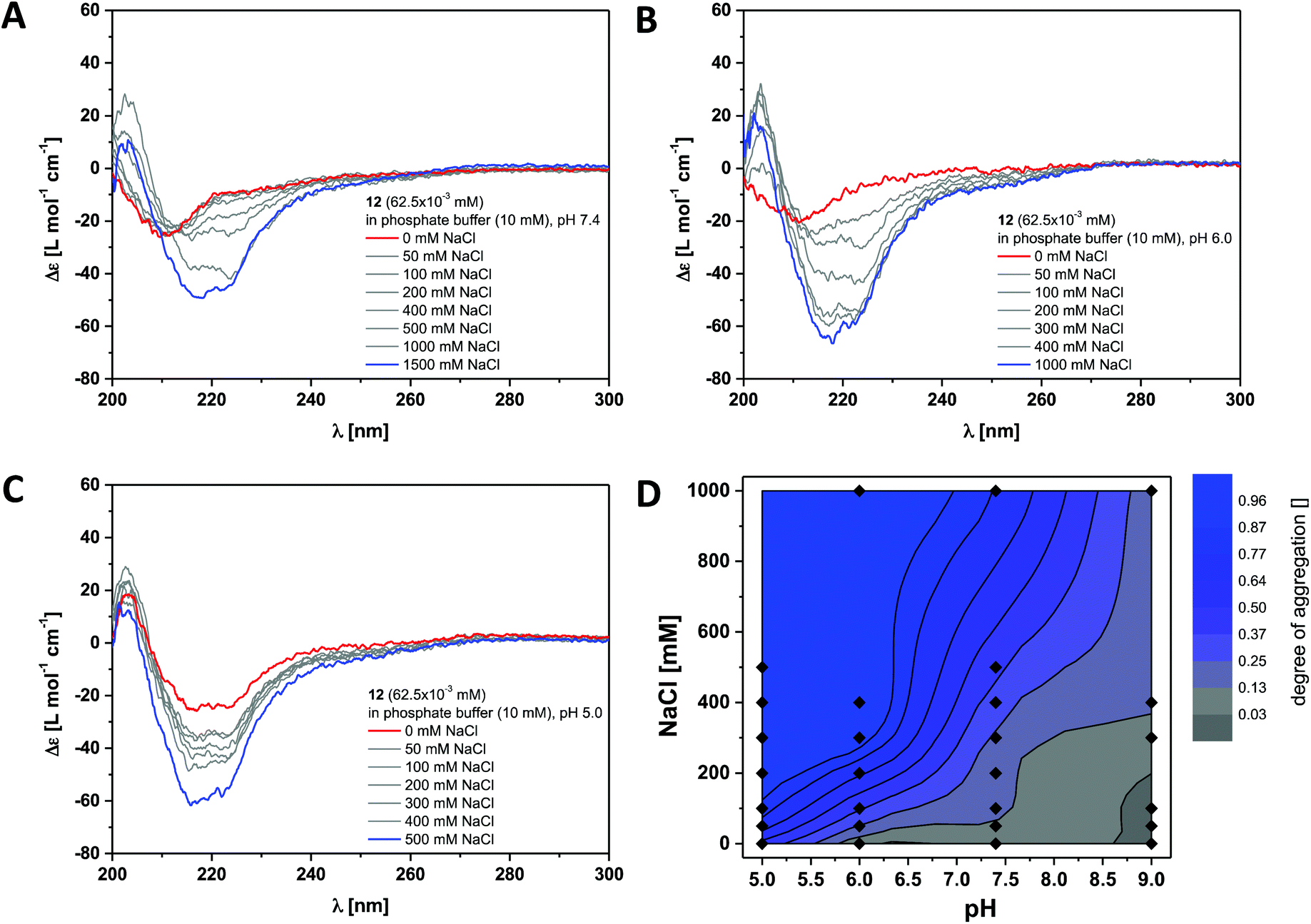 | ||
| Fig. 3 Ionic strength dependent CD spectra for dendritic peptide amphiphile 12 (62.5 × 10−3 mM, 293 K) in 10 mM phosphate buffer at pH 7.4 (A), pH 6.0 (B) and pH 5.0 (C); (D) state diagram depicting the dimensionless degree of aggregation (0 referring to the molecularly dissolved state and 1 to a fully polymerised system, monitored at the CD band of λ = 220 nm) of 12 (62.5 × 10−3 mM), in 10 mM phosphate buffer at 293 K, as a function of the pH and the concentration of added NaCl.‡ | ||
The multistimuli-responsive self-assembly of these peptidic supramolecular polymers can be most conveniently visualised in an ionic strength and a pH dependent contour plot, which we refer to as state diagram‡ (Fig. 3D). It can clearly be observed that at 100 mM NaCl the transition from a disassembled state at pH 7.4 to a fully polymerised state at pH 5.0 is much sharper than the transition at pH 7.4, from 0 to 1 M NaCl. Screening of the repulsive interactions by protonation of the dendritic carboxylic acids occurs at a pH value between pH 5.0 and pH 6.0, which is at least one unit higher than expected for a common organic carboxylic acid in water. The pKa for a Newkome-type dendritic carboxylic acid was reported to be about 4.82 We have previously observed a similar shift in the apparent pKa value for amphiphilic dendritic carboxylic acids which we attributed due to be driven by the aggregation process;44 this is a well-known phenomenon for polymeric weak acids and self-assembled fatty acids.83–85
Finally we investigated the self-assembly of the most hydrophobic compound out of the small series of dendritic peptide amphiphiles 6, 12 and 18. CD bands for compound 18 become red-shifted compared to 6 and 12, due to the presence of the functionalised O-allyl-tyrosine side chains (Fig. 4 and S3†). At pH 9.0 and 0 M NaCl we expect the carboxylic acids to fully deprotonated and the dendritic amphiphiles to be molecularly dissolved. The CD spectra reveal a negative band at λ = 210 nm and a positive band at λ = 225 nm. Upon lowering the pH from pH 9.0 to pH 7.4, pH 6.0 and pH 5.0, or by increasing the ionic strength, the observed bands shift significantly: a strong positive band at λ = 205 nm appears, the negative band shifts from λ = 210 nm to λ = 220 nm, and the positive one from λ = 225 nm to λ = 230 nm. Both CD bands increase in intensity, while another additional weakly negative band at λ = 245 nm appears. The pH and ionic strength dependent contour plot shows that the supramolecular polymerisation for the fluorinated monomer 18 is clearly shifted towards higher pH values and lower ionic strength compared to dendritic peptide amphiphile 12 (Fig. 3D and 4D). The pH-triggered monomer to polymer transition at physiological ionic strength is shifted from pH 5.0 (for monomer 12) to pH 7.4 (for monomer 18). The state diagram thereby highlights the importance of hydrophobic effects in the self-assembly of amphiphilic dendritic carboxylic acids. The previously discussed apparent pKa shifts (the intrinsic pKa of the dendritic carboxylic acid is about 4)82 are increased by another two units in the case of monomer 18 compared to 12: due to the higher thermodynamic driving force for supramolecular polymerisation the carboxylic acids in the hydrophilic shell become weaker.
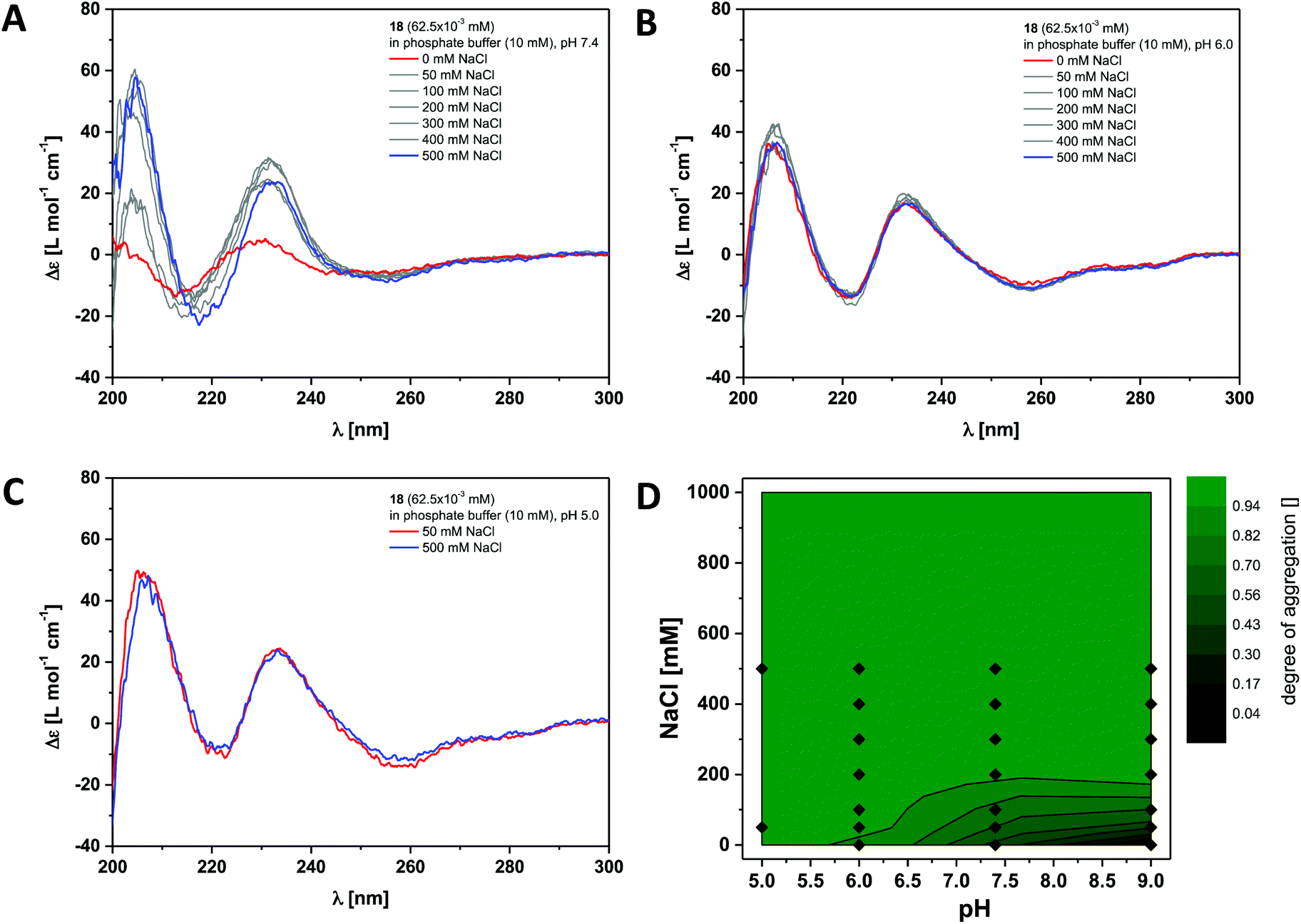 | ||
| Fig. 4 Ionic strength dependent CD spectra for dendritic peptide amphiphile 18 (62.5 × 10−3 mM, 293 K) in 10 mM phosphate buffer at pH 7.4 (A), pH 6.0 (B) and pH 5.0 (C); (D) state diagram depicting the dimensionless degree of aggregation (0 referring to the molecularly dissolved state and 1 to a fully polymerised system, monitored at the CD band of λ = 230 nm) of 18 (62.5 × 10−3 mM), in 10 mM phosphate buffer at 293 K, as a function of the pH and the concentration of added NaCl.‡ | ||
These findings are in good agreement with pKa sifts observed in the self-assembly of linear peptide amphiphiles: the Goldberger lab has reported amphiphiles based on a palmic acid tail coupled to a XAAAEEEE oligopeptide.48 Due to the electrostatic repulsion of the deprotonated glutamic acids (E) at neutral pH, these molecules are present as isolated monomers at low concentration. By acidifying the solution self-assembly into nanofibers occurs, driven by the formation of intermolecular β-sheets. The balance of repulsive to attractive interactions can be manipulated by varying the hydrophobic amino acid (X) embedded in the β-sheet forming region: if the driving force for β-sheet formation in the oligoalanine block XAAA is enhanced via the incorporation of a more hydrophobic amino acid ((tyrosine (Y), valine (V), phenylalanine (F), or isoleucine (I), the propensity for β-sheet formation follows the trend I > F > V > Y), the triggered self-assembly is shifted from pH 6.0 to pH 6.6; the apparent pKa of the oligoglutamic acid EEEE block is therefore shifted from their intrinsic pKa of 4.7–4.9 by up to two units.
Similarly, weakening of the basic character of protonated lysine and histidine side chains (apparent pKa of 7 and 4.5 respectively), as well as a weakening of the acidic character of aspartic acid side chains (apparent pKa of 6) has been observed by Matile and coworkers in the β-sheet directed self-assembly of p-octiphenyl β-barrel pores embedded in lipid bilayers.86 Due to the spacial proximity and electrostatic repulsion of the multiply charged bases or acids in these synthetic pores, the intrinsic pKa of the base/acid is shifted, an observation which was summarized in the intermediate internal charge repulsion (ICR) model.86,87 Indeed, the delicate balance of attractive and repulsive features in these barrel-stave supramolecules is not too dissimilar to the frustrated assembly term we have used to describe the supramolecular 1D nanorod formation: lack of ICR has been thought to account for ‘implosion’, low ICR for contraction, high ICR for expansion, and excess ICR for ‘explosion’ of the β-barrels. Furthermore contributions of the surrounding bilayer have to be taken into account: the external membrane pressure (EMP) model is supposed to protect the overcharged barrels from exploding and to promote the implosion of undercharged barrels.86,87 This effect has important consequences for the catalytic activity of the synthetic pores.87
In summary, we have been able to build state diagrams, which are well known for self-assembled natural spherical or filamentous viruses,88–92 in order to investigate pH and ionic strength dependent self-assembly of dendritic peptide amphiphiles into supramolecular polymers in water. We have highlighted the impact of hydrophobic shielding in the supramolecular polymerisation process: via the incorporation of fluorinated peptide side chains the pH-triggered monomer to polymer transition at physiological ionic strength is shifted from pH 5.0 to pH 7.4.
Morphological characterisation of the supramolecular nanorods via electron microscopy
We performed conventional negative stain transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) experiments in order to investigate the morphology of the supramolecular polymers based on monomers 12 and 18. Negative stain TEM images were recorded after depositing a 1 mg mL−1 concentrated clear solution of dendritic peptide 12 in 10 mM phosphate buffer at pH 7.4 and 500 mM NaCl on carbon film coated EM grids (Fig. 5A). Note that after deposition, the EM grids were washed with an aqueous 2% w/v uranyl acetate solution for 15 s to remove the inorganic salts and in order to fixate the organic peptidic material,93 before blotting excess liquid with filter paper. The TEM micrographs clearly show the presence of nanorods with a diameter of approximately 5 nm in diameter and a contour length distribution ranging from about 150 to 200 nm (Fig. 5A). The morphology thereby justifies the terminology of 1D nanorod-like supramolecular polymers. STEM experiments were furthermore performed using the same solution of 12 to investigate the influence of the staining agent and washing steps on the morphology of the nanorods. After omitting the washing and staining procedures, STEM micrographs reveal the presence of nanorods, albeit with a much higher density on the carbon film coated TEM grid (Fig. S4†).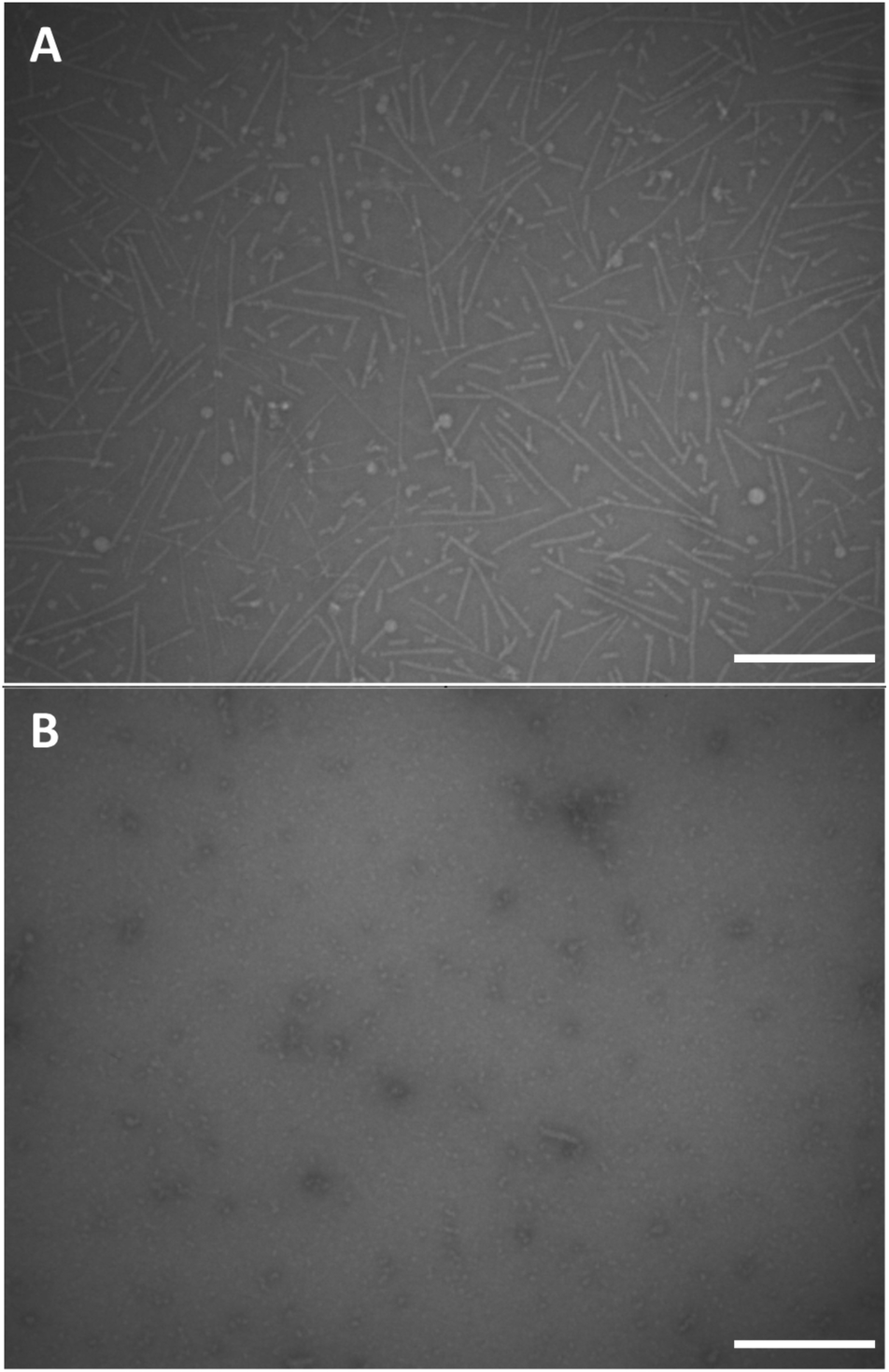 | ||
| Fig. 5 TEM images of the negative stained dendritic peptide amphiphile 12, deposited on a carbon coated EM grid from a 1 mg mL−1 clear solution of 12 in 10 mM phosphate buffer, pH 7.4 and 500 mM NaCl (A) and 0 mM NaCl (B) (negative staining was performed using 2% w/v uranyl acetate), the scale bars represent 200 nm. | ||
In contrast, conventional negative stain TEM experiments of the monomer solution of 12 at pH 7.4 and 0 M NaCl only shows small nanostructures of around 10 nm in size, which are likely monomers or very small oligomers (Fig. 5B). Screening of the repulsive interactions in the anionic amphiphilic building blocks leads to a monomer–polymer transition at higher ionic strength, as expected based on our previous findings.44 Note that ionic strength dependent lengths profiles have also been reported in worm-like micellar solutions of charged surfactants.94–98 These results are therefore in full agreement with the detailed pH and ionic strength titrations probed via CD spectroscopy.
Finally we were also able to visualise the pH-triggered formation of supramolecular polymers from fluorinated amphiphilic monomer 18. At pH 5.0 and 0 mM NaCl STEM images show the presence of nanorods (Fig. 6). At this pH, the protonation of the dendritic carboxylic acid side chains is likely to favour bundling of the nanorods into thin fibres. By increasing the ionic strength at pH 5.0 from 0 mM NaCl to 500 mM NaCl, the tendency for bundling becomes more pronounced and larger structures were observed in these samples (Fig. S5†). In summary, negative stain TEM and STEM experiments corroborate the CD investigations and the pH and ionic strength triggered formation of one-dimensional supramolecular polymers, with a nanorod-like morphology.
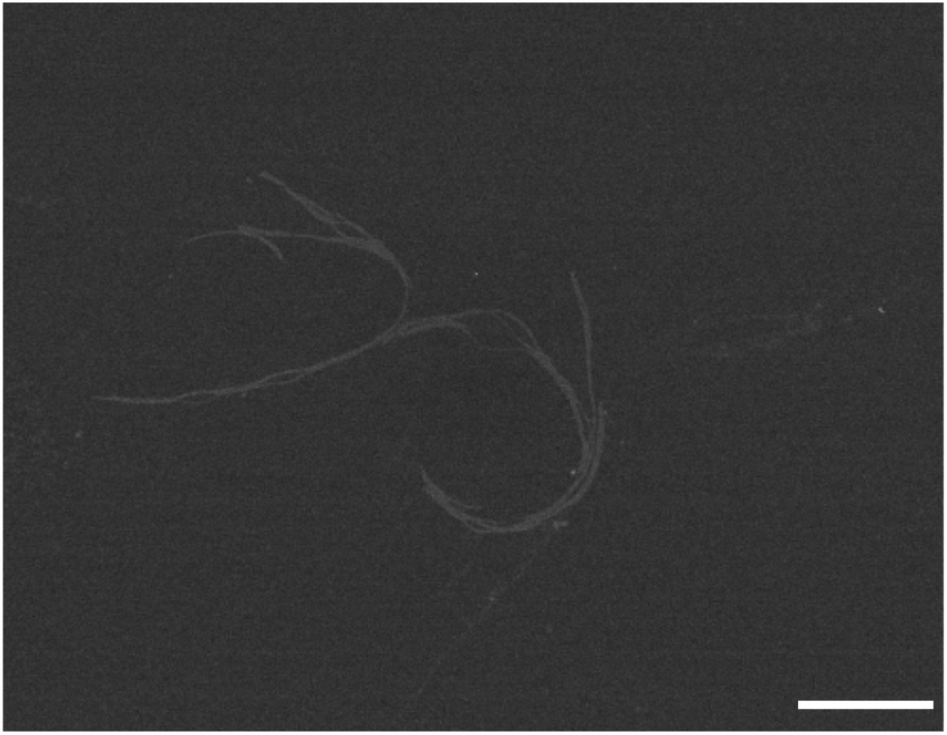 | ||
| Fig. 6 STEM image of the dendritic peptide amphiphile 18, deposited on an EM grid covered with an amorphous carbon film from a 1 mg mL−1 clear solution of 18 in 10 mM phosphate buffer, pH 5.0 and 0 mM NaCl; the scale bar represents 500 nm. | ||
Experimental
Unless stated otherwise, all reagents and chemicals were obtained from commercial sources at the highest purity available and used without further purification. Water was demineralised prior to use. Some solvents were dried using the following drying agents: dichloromethane over sodium hydride, tetrahydrofurane over sodium and benzophenone. Purification via preparative flash column chromatography was carried out using silica gel with an average grain size of 15–40 μm (MERCK). Technical grade solvents that were used as a mobile phase were distilled before use. Analysis of the collected fractions was performed via TLC on silica coated aluminum sheets (60 F254, MERCK). Size exclusion chromatography was carried out using Sephadex™ LH-20 beads (GE Healthcare Bio-Sciences, Uppsala) as stationary phase and distilled methanol as mobile phase or Sephadex™ G-25 beads (GE Healthcare Bio-Sciences, Uppsala) as stationary phase and demineralised water as mobile phase.The NMR-spectra were recorded on the spectrometers AV 300, ARX 300 and AV 400 (BRUKER). All measurements were carried out in deuterated solvents. The chemical shift (δ) is recorded in parts per million (ppm) and relative to the residual solvent protons.99 Mass spectra were recorded on the electro-spray ionisation spectrometers (ESI) Micro Tof (BRUKER) and Orbi-Trap LTQ-XL (THERMO SCIENTIFIC). Molecules of a high molecular weight were analysed using matrix assisted laser desorption ionization time of flight (MALDI-TOF) spectrometry using an Autoflex Speed (BRUKER). CD spectra were recorded on a JASCO J-815.
For transmission electron microscopy (TEM) investigations, standard 200 mesh EM grids, covered with a carbon coated plastic foil (Pioloform), were utilized. To render the support film more hydrophilic, the EM grids were plasma cleaned for 30–45 seconds. For sedimentation, 2–3 μL sample material was left 1 min and afterwards the residual material was blotted. Thereafter, the grids were washed once with 2–3 μL 2% w/v uranyl acetate (15 s). Followed by a second blotting step, the sample material was then stained by uranyl acetate for 45 s, blotted and left for air drying. The samples were examined on a Philips CM10 (FEI, Eindhoven, the Netherlands) operated at 80 kV. Electron micrographs were recorded by a side-mounted CCD camera (IDS, Obersulm, Germany) utilizing acquisition software written in LabView.
For scanning transmission electron microscopy (STEM) investigations, EM high-resolution grids were utilised. Here, a holey carbon film is covered with a 2–4 nm thick amorphous carbon film, which acts as a support for the sample material. As in the case for TEM investigations, the grids were plasma cleaned prior to the sample preparation. Since no staining was performed for STEM investigations the preparation protocol was reduced to the following steps: 2–3 μL sample material was left for sedimentation (1 min). Hereafter, grids were either washed briefly with buffer or blotted directly and left for air drying. The samples were examined in a high-resolution in-lens scanning electron microscope (S-5000, HITACHI, Japan), equipped with a homemade annular dark-field (ADF) detector. ADF images were taken at 30 kV acceleration voltage.
Full experimental procedures, materials synthesis and characterisation for compounds 1–18 can be found in the ESI.†
(5) 4 (159 mg; 0.169 mmol; 3.0 eq.) and 1,3,5-benzenetricarbonyl trichloride (15 mg; 0.056 mmol; 1.0 eq.) were dissolved under argon in dry DMF (5 mL) and DIPEA (109 mg; 0.845 mmol; 15.0 eq.) were then added. The reaction mixture was stirred at room temperature for 20 h, after 2 h PyBOP (179 mg; 0.344 mmol; 7.0 eq.) was added to react hydrolysed acid chloride with remaining amine 10. The organic solvent was removed under reduced pressure. The residue was purified via precipitation in water and isolated via centrifugation. The residue was purified again via SEC in methanol. Rf (DCM–EtOAc 1:1 + 20 vol% MeOH, SiO2) = 0.90. Yield: 83 mg (0.028 mmol, 50%) C165H222N12O39, light brownish solid. 1H NMR (300 MHz, DMSO-d6): δ [ppm] = 8.70–8.54 (m, 3H, NH), 8.43–7.97 (m, 9H, NH and CHaroBTA), 7.60–6.82 (m, 48H, CHaroPhe and NH), 4.80–4.49 (m, 9H, CHα), 3.67–3.44 (m, 36H, CH2OCH2), 2.90–2.58 (m, 18H, CH2Phe), 2.47–2.33 (m, 18H, OCH2CH2), 1.37 (s, 81H, CH3tBu). MALDI-MS (positive mode) (DHB [EtOAc]): m/z calcd for [M + Na]+ 3020.57; found: 3020.71 [M + Na]+.
(6) 5 (80 mg; 0.027 mol) was stirred three times in 3.0 mL of a mixture of TFA and DCM (1:1) for 1 h. The organic solvent was removed in each step under reduced pressure. Finally the residue was taken up in water and freeze-dried over night. Yield: 29 mg (0.012 mmol, 44%) C129H150N12O39, white solid. 1H NMR (300 MHz, DMSO-d6): δ [ppm] = 12.18 (brs, 9H, COOH), 8.69–8.52 (m, 3H, NH), 8.39–7.97 (m, 9H, NH and CHaroBTA), 7.59–6.82 (m, 48H, CHaroPhe and NH), 4.78–4.51 (m, 9H, CHα), 3.75–3.41 (m, 36H, CH2OCH2), 3.05–2.64 (m, 18H, CH2Phe), 2.47–2.33 (m, 18H, OCH2CH2). MALDI-MS (positive mode) (DHB [H2O/ACN]): m/z calcd for [M + Na]+ 2515.00; found: 2515.00 [M + Na]+.
(11) 10 (171.5 mg; 0.162 mmol; 3.3 eq.) and 1,3,5-benzenetricarbonyl trichloride (13.0 mg; 0.049 mmol; 1.0 eq.) were dissolved under argon in dry DMF (5 mL) and DIPEA (82 mg; 0.64 mmol; 4.0 eq.) were then added. The reaction mixture was stirred at room temperature for 18 h. The organic solvent was removed under reduced pressure. The residue was purified via precipitation from THF into a mixture of n-pentan and diethylether (1:1) and then isolated via centrifugation. The residue was purified via flash chromatography over SiO2 (DCM–EtOAc–MeOH 1:1:1), Rf (DCM–EtOAc–MeOH 1:1:1, SiO2) = 0.50. Yield: 105 mg (0.031 mmol, 63%) C183H255N15O42, light brownish solid. 1H NMR (300 MHz, DMSO-d6): δ [ppm] = 8.72–8.61 (m, 3H, NH), 8.41 (d, J = 8.5 Hz, 3H, NH), 8.25–7.99 (m, 9H, CHaroBTA and NH), 7.80 (t, J = 5.6 Hz, 3H, NH), 7.33–6.87 (m, 48H, CHaroPhe and NH), 4.82–4.38 (m, 9H, CHα), 3.59 (m, 36H, CH2OCH2), 3.08–2.67 (m, 24H, CH2Phe and NHCH2Ahx), 2.36 (t, J = 6.3 Hz, 18H, OCH2CH2), 2.01 (t, J = 7.0 Hz, 6H, CH2CONAhx), 1.50–1.28 (m, 99H, CH3tBu and CH2Ahx). MALDI-MS (positive mode) (DHB [EtOAc]): m/z calcd for [M + Na]+ 3357.82; found: 3357.86 [M + Na]+.
(12) 11 (105 mg; 0.031 mol) was stirred three times in 2.5 mL of a mixture of TFA and DCM (1:1) for 1 h. The organic solvent was removed after each step under reduced pressure. Finally the residue was taken up in water and freeze-dried over night. Yield: 42 mg (0.015 mmol, 48%) C147H183N15O42, white solid. 1H NMR (300 MHz, DMSO-d6): δ [ppm] = 12.14 (brs, 9H, COOH), 8.73–8.58 (m, 3H, NH), 8.49–8.32 (m, 3H, NH), 8.15 (s, 3H, CHaroBTA), 8.10–7.96 (m, 3H, NH), 7.84–7.62 (m, 3H, NH), 7.33–6.87 (m, 48H, HaroPhe and NH), 4.83–4.37 (m, 9H, CHα), 3.53 (m, 36H, CH2OCH2), 3.08–2.67 (m, 24H, CH2Phe and NHCH2Ahx), 2.40 (t, J = 6.3 Hz, 18H, OCH2CH2), 2.01 (t, J = 7.0 Hz, 6H, CH2CONAhx), 1.46–1.11 (m, 18H, CH2Ahx). ESI-MS (positive mode): m/z calcd for [M + 2Na]2+ 1438.6231; found: 1438.6249 [M + 2Na]2+.
(16) 15 (89 mg; 0.07 mmol; 3.3 eq.) and 1,3,5-benzenetricarbonyl trichloride (6 mg; 0.02 mmol; 1.0 eq.) were dissolved in 3 mL DMF (SPPS grade) and treated with DIPEA (89 mg; 0.69 mmol; 30.0 eq.). The solution was stirred at room temperature for 45 h. The solvent was removed under reduced pressure. The residue was purification via precipitation from water and washing subsequently with methanol to remove excess 15. Yield: 38 mg (0.01 mmol, 50%), C201H279N15O48, white solid. 1H NMR (300 MHz, DMSO-d6): δ [ppm] = 8.77–8.60 (m, 3H, NH), 8.26–8.08 (m, 9H, NH and CHaroBTA), 7.84–7.69 (m, 3H, NH), 7.26 (d, J = 7.4 Hz, 6H, CHaroTyr), 7.18–7.05 (m, 21H, CHaroPhe and Tyr), 6.88 (s, 1H, s, 3H, NH), 6.79 (d, J = 8.5 Hz, 3H, CHaroTyr), 6.68 (d, J = 8.4 Hz, 3H, CHaroTyr), 5.92 (ddt, J = 17.1, 10.4, 5.1 Hz, 6H, OCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.39–5.04 (m, 12H, OCH2CHCH2), 4.81–4.65 (m, 3H, CHα), 4.53–4.28 (m, 18H, CHα and OCH2CHCH2), 3.57–3.45 (m, 36H, CH2OCH2), 3.08–2.58 (m, 24H, CH2Phe and Tyr and NHCH2Ahx), 2.36 (t, J = 6.1 Hz, 18H, OCH2CH2), 2.00 (t, J = 7.5 Hz, 6H, CH2CONAhx), 1.37 (m, 81H, CH3tBu), 1.30–1.04 (m, 18H, CH2Ahx). MALDI-MS (positive mode) (DHB [H2O/ACN]): m/z calcd for [M + Na]+ 3695.98; found: 3696.16 [M + Na]+.
CH2), 5.39–5.04 (m, 12H, OCH2CHCH2), 4.81–4.65 (m, 3H, CHα), 4.53–4.28 (m, 18H, CHα and OCH2CHCH2), 3.57–3.45 (m, 36H, CH2OCH2), 3.08–2.58 (m, 24H, CH2Phe and Tyr and NHCH2Ahx), 2.36 (t, J = 6.1 Hz, 18H, OCH2CH2), 2.00 (t, J = 7.5 Hz, 6H, CH2CONAhx), 1.37 (m, 81H, CH3tBu), 1.30–1.04 (m, 18H, CH2Ahx). MALDI-MS (positive mode) (DHB [H2O/ACN]): m/z calcd for [M + Na]+ 3695.98; found: 3696.16 [M + Na]+.
(17) 16 (38 mg; 0.01 mmol) was stirred twice in 2.0 mL of a mixture of TFA and DCM (1:1) for 45 min. The organic solvent was removed after each step under reduced pressure. Yield: 30 mg (0.009 mmol, 93%), C165H207N15O48, white solid. 1H NMR (300 MHz, DMSO-d6): δ [ppm] = 8.68–8.58 (m, 3H, NH), 8.27–8.08 (m, 9H, NH and CHaroBTA), 7.80–7.71 (m, 3H, NH), 7.27 (d, J = 6.9 Hz, 6H, CHaroTyr), 7.21–7.06 (m, 21H, CHaroTyr and Phe), 6.92 (s, 3H, NH), 6.81 (d, J = 8.7 Hz, 6H, CHaroTyr), 6.70 (d, J = 8.6 Hz, 6H, CHaroTyr), 5.93 (ddt, J = 17.2, 10.4, 5.2 Hz, 6H, CH2CHCH2), 5.37–5.06 (m, 12H, OCH2CHCH2), 4.81–4.71 (m, 3H, CHα), 4.55–4.27 (m, 21H, CHα and CH2CHCH2), 3.59–3.49 (m, 36H, CH2OCH2), 3.09–2.68 (m, 24H, CH2Tyr and Phe and NHCH2Ahx), 2.41 (t, J = 6.3 Hz, 18H, OCH2CH2), 2.02 (t, J = 7.4 Hz, 6H, CH2CONAhx), 1.46–1.10 (m, 18H, CH2Ahx). MALDI-MS (positive mode) (DHB [EtOAc]): m/z calcd for [M + Na]+ 3191.5172; found: 3191.534 [M + Na]+.
(18) 17 (29.5 mg; 0.0093 mmol; 1.0 eq.) was dissolved in a mixture of 6 mL THF and water (1:1). 1 mg 2,2-dimethoxy-2-phenylacetophenone (DMPA) and trifluoroethanethiol (0.093 mmol, 10.8 mg 10.0 eq.) were added and the mixture was stirred for 18 h under irradiation with UV light (λ = 365 nm and 405 nm). The solvent was removed under reduced pressure and the residue was washed with diethylether. After dissolving the residue in basic water it was purified via SEC with water as a mobile phase. Yield: 13.5 mg (0.0035 mmol, 38%), C177H225F18N15O48S6, white solid. 1H NMR (300 MHz, DMSO-d6): δ [ppm] = 8.71–8.61 (m, 3H, NH), 8.41–8.06 (m, 9H, NH and CHaroBTA), 7.89–7.76 (m, 3H, NH), 7.27 (d, J = 7.4 Hz, 6H, CHaroTyr), 7.21–7.06 (m, 21H, CHaroPhe and Tyr), 6.93 (s, 3H, NH), 6.84–6.64 (m, 12H, CHaroTyr), 4.80–4.66 (m, 3H, CHα), 4.56–4.32 (m, 6H, CHα), 3.95–3.80 (m, 12H, ArOCH2), 3.60–3.50 (m, 36H, CH2OCH2), 3.0–2.7 (m, 36H, CH2Tyr and Phe, NHCH2Ahx and CH2CF3), 2.40 (t, J = 6.3 Hz, 18H, OCH2CH2), 2.02 (t, J = 6.7 Hz, 6H, CH2CONAhx), 1.48–1.34 (m, 6H, CH2Ahx), 1.30–1.23 (m, 6H, CH2Ahx), 1.18–1.09 (m, 6H, CH2Ahx). 19F NMR (282 MHz, DMSO-d6): δ [ppm] = −65.01 (t, J = 11.0 Hz, 9F), −65.20 (t, J = 10.8 Hz, 9F).
Conclusions
In conclusion, we disclose the synthesis of a series of three anionic dendritic peptide amphiphiles. We systematically study the effect of increasing the hydrophobic shielding of dendritic peptide amphiphiles, and the impact on the pH and ionic strength triggered self-assembly. By establishing state diagrams we show that the supramolecular polymerisation is switched off if the apolar hexyl spacer between the dendritic nonaphenylalanines and peripheral carboxylic acid Newkome dendrons is omitted in compound 6, compared to molecule 12. If the hydrophobic character of the peptide core is increased further by introducing trifluoromethyl groups via straightforward thiol–ene click chemistry in the O-allyl derivatised tyrosine groups of 18, the thermodynamic driving force for self-assembly is increased and the pH-triggered monomer to polymer transition at physiological ionic strength is shifted from pH 5.0 to pH 7.4. Using TEM experiments we have characterised the 1D supramolecular polymers as well-defined nanorods with lengths distributions ranging from around 100 nm to 500 nm, depending on the exact pH, ionic strength and hydrophobicity of the peptide backbone. We thereby show that by compensating attractive non-covalent interactions and solvophobic effects with repulsive electrostatic forces, a concept we refer to as frustrated growth, is a sensitive tool in order to manipulate one-dimensional supramolecular polymerisation processes in water.Acknowledgements
We thank U. Malkus, A. Ricker and H. Nüsse (Institute of Medical Physics and Biophysics, Münster) for performing TEM experiments, H. Frisch and B. Kemper for critical reading of the manuscript, and B. J. Ravoo for his support. P.B. acknowledges the ‘Fonds der Chemischen Industrie’ for a Liebig Fellowship, the ‘Nordrhein-Westfälische Akademie der Wissenschaften und der Künste’ for a Fellowship via the ‘Junges Kolleg’, Marie-Curie Actions FP7 (Career Integration Grant, SupraBioMat, PCIG10-GA-2011-303872) and COST Action CM1005 (Supramolecular Chemistry in Water).Notes and references
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151 RSC.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2010, 10, 14 CrossRef PubMed.
- F. B. L. Cougnon and J. K. M. Sanders, Acc. Chem. Res., 2011, 45, 2211 CrossRef PubMed.
- E. Krieg and B. Rybtchinski, Chem. – Eur. J., 2011, 17, 9016 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- T. Fenske, H.-G. Korth, A. Mohr and C. Schmuck, Chem. – Eur. J., 2012, 18, 738 CrossRef CAS PubMed.
- T. Muraoka, C.-Y. Koh, H. Cui and S. I. Stupp, Angew. Chem., Int. Ed., 2009, 48, 5946 CrossRef CAS PubMed.
- S. K. M. Nalluri, J. Voskuhl, J. B. Bultema, E. J. Boekema and B. J. Ravoo, Angew. Chem., Int. Ed., 2011, 50, 9747 CrossRef CAS PubMed.
- T. Hirose, F. Helmich and E. W. Meijer, Angew. Chem., Int. Ed., 2012, 52, 304 CrossRef PubMed.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS PubMed.
- J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J.-P. Peyralans and S. Otto, Science, 2010, 327, 1502 CrossRef CAS PubMed.
- R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19 CrossRef CAS PubMed.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051 CrossRef CAS PubMed.
- R. Klajn, M. A. Olson, P. J. Wesson, L. Fang, A. Coskun, A. Trabolsi, S. Soh, J. F. Stoddart and B. A. Grzybowski, Nat. Chem., 2009, 1, 733 CrossRef CAS PubMed.
- C. Kim, S. S. Agasti, Z. Zhu, L. Isaacs and V. M. Rotello, Nat. Chem., 2010, 2, 962 CrossRef CAS PubMed.
- A. Samanta, M. C. A. Stuart and B. J. Ravoo, J. Am. Chem. Soc., 2012, 134, 19909 CrossRef CAS PubMed.
- T. Tjivikua, P. Ballester and J. Rebek, J. Am. Chem. Soc., 1990, 112, 1249 CrossRef CAS.
- D. H. Lee, J. R. Granja, J. A. Martinez, K. Severin and M. R. Ghadiri, Nature, 1996, 382, 525 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684 CrossRef CAS PubMed.
- S. Zhang, Biotechnol. Adv., 2002, 20, 321 CrossRef CAS PubMed.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352 CrossRef CAS PubMed.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664 RSC.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877 RSC.
- X. Yan, Y. Su, J. Li, J. Früh and H. Möhwald, Angew. Chem., Int. Ed., 2011, 50, 11186 CrossRef CAS PubMed.
- M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301 CrossRef CAS PubMed.
- N. Sakai, J. Mareda and S. Matile, Acc. Chem. Res., 2008, 41, 1354 CrossRef CAS PubMed.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2012, 48, 26 RSC.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37 CrossRef CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856 RSC.
- E. K. Johnson, D. J. Adams and P. J. Cameron, J. Am. Chem. Soc., 2010, 132, 5130 CrossRef CAS PubMed.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O'Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070 CrossRef CAS PubMed.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. v. Esch, S. Santabarbara, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089 CrossRef CAS PubMed.
- K. J. C. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. van Esch, Angew. Chem., Int. Ed., 2004, 43, 1663 CrossRef CAS PubMed.
- M. K. Müller and L. Brunsveld, Angew. Chem., Int. Ed., 2009, 48, 2921 CrossRef PubMed.
- P. Besenius, G. Portale, P. H. H. Bomans, H. M. Janssen, A. R. A. Palmans and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888 CrossRef CAS PubMed.
- M. K. Müller, K. Petkau and L. Brunsveld, Chem. Commun., 2011, 47, 310 RSC.
- P. Besenius, K. P. van den Hout, H. M. H. G. Albers, F. A. de Greef Tom, L. L. C. Olijve, T. M. Hermans, B. F. M. de Waal, P. H. H. Bomans, N. A. J. M. Sommerdijk, G. Portale, A. R. A. Palmans, M. H. P. van Genderen, J. A. J. M. Vekemans and E. W. Meijer, Chem. – Eur. J., 2011, 17, 5193 CrossRef CAS PubMed.
- P. Besenius, Y. Goedegebure, M. Driesse, M. Koay, P. H. H. Bomans, A. R. A. Palmans, P. Y. W. Dankers and E. W. Meijer, Soft Matter, 2011, 7, 7980 RSC.
- C. Schaefer, I. K. Voets, A. R. A. Palmans, E. W. Meijer, P. van der Schoot and P. Besenius, ACS Macro Lett., 2012, 1, 830 CrossRef CAS.
- J. Leenders, L. Albertazzi, T. Mes, M. Koenigs, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963 RSC.
- H. Frisch, J. P. Unsleber, D. Lüdeker, M. Peterlechner, G. Brunklaus, M. Waller and P. Besenius, Angew. Chem., Int. Ed., 2013, 52, 10097 CrossRef CAS PubMed.
- M. von Gröning, I. de Feijter, M. C. A. Stuart, I. K. Voets and P. Besenius, J. Mater. Chem. B, 2013, 1, 2008 RSC.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275 CrossRef CAS PubMed.
- G. R. Newkome and C. Shreiner, Chem. Rev., 2010, 110, 6338 CrossRef CAS PubMed.
- H. Dong, S. E. Paramonov, L. Aulisa, E. L. Bakota and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 12468 CrossRef CAS PubMed.
- A. Ghosh, M. Haverick, K. Stump, X. Yang, M. F. Tweedle and J. E. Goldberger, J. Am. Chem. Soc., 2012, 134, 3647 CrossRef CAS PubMed.
- F. Versluis, I. Tomatsu, S. Kehr, C. Fregonese, A. W. J. W. Tepper, M. C. A. Stuart, B. J. Ravoo, R. I. Koning and A. Kros, J. Am. Chem. Soc., 2009, 131, 13186 CrossRef CAS PubMed.
- F. Versluis, H. R. Marsden and A. Kros, Chem. Soc. Rev., 2010, 39, 3434 RSC.
- D. M. Wood, B. W. Greenland, A. L. Acton, F. Rodríguez-Llansola, C. A. Murray, C. J. Cardin, J. F. Miravet, B. Escuder, I. W. Hamley and W. Hayes, Chem. – Eur. J., 2012, 18, 2692 CrossRef CAS PubMed.
- R. C. T. Howe, A. P. Smalley, A. P. M. Guttenplan, M. W. R. Doggett, M. D. Eddleston, J.-C. Tan and G. O. Lloyd, Chem. Commun., 2013, 49, 4268 RSC.
- P. Ahlers, H. Frisch, D. Spitzer, Z. Vobecka, F. Vilela and P. Besenius, Chem. – Asian J., 2014, 9, 2052 CrossRef CAS PubMed.
- B. Bilgiçer, A. Fichera and K. Kumar, J. Am. Chem. Soc., 2001, 123, 4393 CrossRef.
- J.-C. Horng and D. P. Raleigh, J. Am. Chem. Soc., 2003, 125, 9286 CrossRef CAS PubMed.
- H.-Y. Lee, K.-H. Lee, H. M. Al-Hashimi and E. N. G. Marsh, J. Am. Chem. Soc., 2005, 128, 337 CrossRef PubMed.
- B. Anil, S. Sato, J.-H. Cho and D. P. Raleigh, J. Mol. Biol., 2005, 354, 693 CrossRef CAS PubMed.
- C. Jäckel, M. Salwiczek and B. Koksch, Angew. Chem., Int. Ed., 2006, 45, 4198 CrossRef PubMed.
- H.-P. Chiu, Y. Suzuki, D. Gullickson, R. Ahmad, B. Kokona, R. Fairman and R. P. Cheng, J. Am. Chem. Soc., 2006, 128, 15556 CrossRef CAS PubMed.
- M. Salwiczek and B. Koksch, ChemBioChem, 2009, 10, 2867 CrossRef CAS PubMed.
- M. A. Molski, J. L. Goodman, C. J. Craig, H. Meng, K. Kumar and A. Schepartz, J. Am. Chem. Soc., 2010, 132, 3658 CrossRef CAS PubMed.
- E. K. Nyakatura, O. Reimann, T. Vagt, M. Salwiczek and B. Koksch, RSC Adv., 2013, 3, 6319 RSC.
- B. C. Buer, B. J. Levin and E. N. G. Marsh, J. Am. Chem. Soc., 2012, 134, 13027 CrossRef CAS PubMed.
- B. C. Buer, J. L. Meagher, J. A. Stuckey and E. N. G. Marsh, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4810 CrossRef CAS PubMed.
- U. I. M. Gerling, M. Salwiczek, C. D. Cadicamo, H. Erdbrink, C. Czekelius, S. L. Grage, P. Wadhwani, A. S. Ulrich, M. Behrends, G. Haufe and B. Koksch, Chem. Sci., 2014, 5, 819 RSC.
- A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546 CrossRef CAS PubMed.
- S. R. Diegelmann, J. M. Gorham and J. D. Tovar, J. Am. Chem. Soc., 2008, 130, 13840 CrossRef CAS PubMed.
- J. Baram, E. Shirman, N. Ben-Shitrit, A. Ustinov, H. Weissman, I. Pinkas, S. G. Wolf and B. Rybtchinski, J. Am. Chem. Soc., 2008, 130, 14966 CrossRef CAS PubMed.
- F. Tian, D. Jiao, F. Biedermann and O. A. Scherman, Nat. Commun., 2012, 3, 1207 CrossRef PubMed.
- Z. Huang, S.-K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Science, 2012, 337, 1521 CrossRef CAS PubMed.
- J. del Barrio, P. N. Horton, D. Lairez, G. O. Lloyd, C. Toprakcioglu and O. A. Scherman, J. Am. Chem. Soc., 2013, 135, 11760 CrossRef CAS PubMed.
- S. Biswas, K. Kinbara, T. Niwa, H. Taguchi, N. Ishii, S. Watanabe, K. Miyata, K. Kataoka and T. Aida, Nat. Chem., 2013, 5, 613 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, A. Hashidzume and A. Harada, Angew. Chem., Int. Ed., 2013, 52, 5731 CrossRef CAS PubMed.
- M. M. C. Bastings, S. Koudstaal, R. E. Kieltyka, Y. Nakano, A. C. H. Pape, D. A. M. Feyen, F. J. van Slochteren, P. A. Doevendans, J. P. G. Sluijter, E. W. Meijer, S. A. J. Chamuleau and P. Y. W. Dankers, Adv. Healthcare Mater., 2013, 3, 70 CrossRef PubMed.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822 RSC.
- Z.-X. Jiang, X. Liu, E.-K. Jeong and Y. B. Yu, Angew. Chem., Int. Ed., 2009, 48, 4755 CrossRef CAS PubMed.
- J. Ruiz-Cabello, B. P. Barnett, P. A. Bottomley and J. W. M. Bulte, NMR Biomed., 2011, 24, 114 CrossRef CAS PubMed.
- J.-X. Yu, R. R. Hallac, S. Chiguru and R. P. Mason, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 70, 25 CrossRef CAS PubMed.
- C. C. Lee, J. A. MacKay, J. M. J. Fréchet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 CrossRef CAS PubMed.
- M. I. Montañez, L. M. Campos, P. Antoni, Y. Hed, M. V. Walter, B. T. Krull, A. Khan, A. Hult, C. J. Hawker and M. Malkoch, Macromolecules, 2010, 43, 6004 CrossRef.
- T. J. Cho, R. A. Zangmeister, R. I. MacCuspie, A. K. Patri and V. A. Hackley, Chem. Mater., 2011, 23, 2665 CrossRef CAS PubMed.
- J. R. Kanicky and D. O. Shah, Langmuir, 2003, 19, 2034 CrossRef CAS.
- A. V. Dobrynin and M. Rubinstein, Prog. Polym. Sci., 2005, 30, 1049 CrossRef CAS.
- A. V. Dobrynin, Curr. Opin. Colloid Interface Sci., 2008, 13, 376 CrossRef CAS.
- B. Baumeister, A. Som, G. Das, N. Sakai, F. Vilbois, D. Gerard, S. P. Shahi and S. Matile, Helv. Chim. Acta, 2002, 85, 2740 CrossRef CAS.
- A. Som and S. Matile, Chem. Biodiversity, 2005, 2, 717 CAS.
- K. W. Adolph and P. J. G. Butler, J. Mol. Biol., 1974, 88, 327 CrossRef CAS PubMed.
- A. Klug, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 531 CrossRef CAS PubMed.
- W. K. Kegel and P. van der Schoot, Biophys. J., 2004, 86, 3905 CrossRef CAS PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581 CrossRef CAS PubMed.
- A. Siber, A. L. Bozic and R. Podgornik, Phys. Chem. Chem. Phys., 2012, 14, 3746 RSC.
- M. Ohi, Y. Li, Y. Cheng and T. Walz, Biol. Proced. Online, 2004, 6, 23 CrossRef CAS PubMed.
- T. Imae and S. Ikeda, Colloid Polym. Sci., 1987, 265, 1090 CAS.
- F. C. MacKintosh, S. A. Safran and P. A. Pincus, Europhys. Lett., 1990, 12, 697 CrossRef.
- S. A. Safran, P. A. Pincus, M. E. Cates and F. C. MacKintosh, J. Phys., 1990, 51, 503 CAS.
- T. Odijk, Biophys. Chem., 1991, 41, 23 CrossRef CAS PubMed.
- S. May and M. A. Ben-Shaul, J. Phys. Chem. B, 2001, 105, 630 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, materials synthesis and characterisation, additional CD data, negative stain TEM and STEM micrographs. See DOI: 10.1039/c4ob02185a |
| ‡ The state diagrams were plotted from experimental pH and ionic strength dependent data points. The contour plot was generated using the 2D Interpolation/Extrapolation function in OriginPro 9.1. The boundaries are approximately correct for a monomer concentration of 62.5 × 10−6 M at 293 K. They are likely to shift, favouring aggregation, when the monomer concentration is increased. |
| This journal is © The Royal Society of Chemistry 2015 |