
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion binding and transport properties of cyclic 2,6-bis(1,2,3-triazol-1-yl)pyridines†
Tamara
Merckx
a,
Cally J. E.
Haynes
b,
Louise E.
Karagiannidis
b,
Harriet J.
Clarke
b,
Katie
Holder
b,
Alexandra
Kelly
b,
Graham J.
Tizzard
b,
Simon J.
Coles
 b,
Peter
Verwilst
a,
Philip A.
Gale
*b and
Wim
Dehaen
*a
b,
Peter
Verwilst
a,
Philip A.
Gale
*b and
Wim
Dehaen
*a
aMolecular Design and Synthesis, Department of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: wim.dehaen@chem.kuleuven.be; Tel: +32 16 32 74 39
bChemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: philip.gale@soton.ac.uk; Tel: +44 (0)23 8059 3332
First published on 1st December 2014
Abstract
A series of cyclic 2,6-bis-(1,2,3-triazolyl)-pyridine anion receptors with thiourea functionalities were synthesized by click reaction of 2,6-diazidopyridine with protected propargylamine followed by condensation of a bisthiocyanate derivative with a series of diamines. Their chloride binding affinities as well as their transport properties in POPC bilayers were examined. These receptors were found to function as anion carriers, which can mediate both Cl−/NO3− antiport and H+/Cl− symport, and the transport activity of these hosts were dominated by their lipophilicity.
The development of synthetic transmembrane transporters is a rapidly expanding area of supramolecular chemistry.1–4 The transport of inorganic anions, such as chloride, across cell lipid bilayers is vital to a number of important biological processes. Malfunctioning of certain anion transport mechanisms can lead to serious diseases,5 most notably cystic fibrosis.6–8
Recently, there has been an increased interest in CH hydrogen bond donors.9,10 In particular, 1,2,3-triazoles proved to be interesting hydrogen bond donor groups for anion complexation11 due to their large polarity (dipole moment ∼5D), with the positive end of the dipole situated at the CH group.12 The practical utility of the 1,2,3-triazoles in anion receptor chemistry is further enhanced by the fact that they are readily accessible via the copper catalyzed Huisgen condensation or so-called ‘Click reaction’.13,14
In our studies, we have previously reported the synthesis and anion binding properties of a series of 2,6-bis-(1,2,3-triazolyl)-pyridine receptors, like 1 and 2 (Chart 1)15 with compound 2 showing a promising chloride binding affinity. Prompted by these findings, we investigated the binding and transport properties of some cyclic derivatives of this compound 2 and compared these results to the properties of the acyclic system.
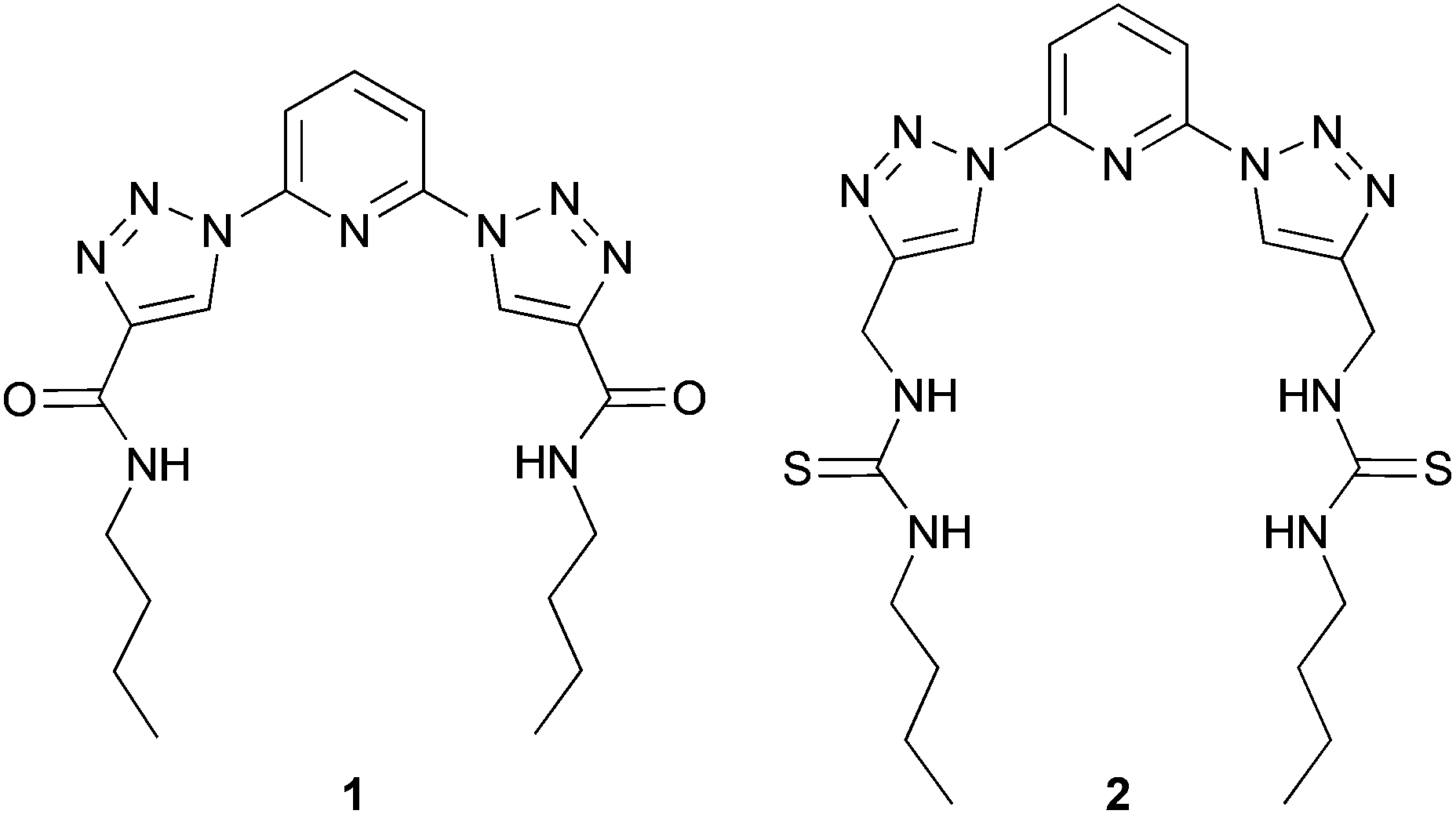 | ||
| Chart 1 Previously reported 2,6-bis-(1,2,3-triazolyl)-pyridine receptors. | ||
Results and discussion
The cyclic receptors were synthesized starting from 2,6-difluoropyridine. In the first step of the synthesis, 2,6-diazidopyridine 3 was synthesized by treatment of 2,6-difluoropyridine with NaN3 in DMF. Molecule 4 was then obtained as previously reported by a click reaction between 2,6-diazidopyridine 3 and N-Boc-propargylamine.15 In a next step, the diisothiocyanate compound 5 was synthesized by a deprotection of the Boc-groups16 of compound 4, followed by treatment of the obtained bisamine intermediate with thiophosgene, yielding the desired product 5. Finally, the macrocyclic receptors 6a–f were obtained in fair to good yield (18–65%) by combination of 5 with a number of α,ω-diaminoalkyl molecules with different chain lengths under high dilution conditions (Scheme 1).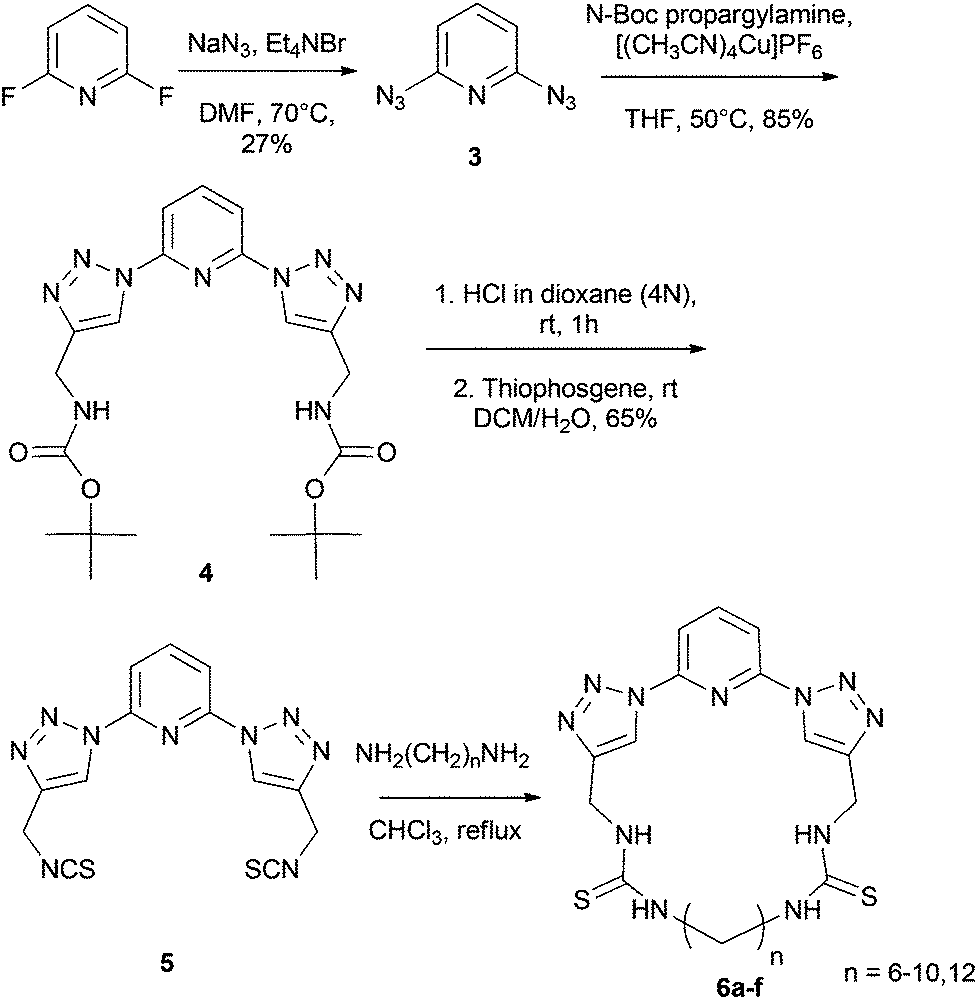 | ||
| Scheme 1 Synthesis of the cyclic hosts 6a–f. | ||
Solution binding studies
The chloride binding affinities of the obtained cyclic hosts 6a–f were determined by 1H NMR titrations with tetra-n-butylammonium chloride in DMSO-d6/0.5% H2O. The data was fitted to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model as confirmed by Job plot analysis (Fig. S1 in the ESI†) and the anion binding constants were calculated with HypNMR.17 Moderate binding affinities were obtained for all the hosts (Table 1), the highest binding constants were obtained for 6b and 6c, with log(Ka) of respectively 1.75 ± 0.02 and 1.76 ± 0.02. For comparison, the binding affinity log(Ka) of the open-chain receptor 215 was now also measured in DMSO-d6/0.5% H2O and found to be 1.20, much lower than the 3.4 reported when the less competitive acetonitrile was the solvent. We could unfortunately not use acetonitrile for studies of compounds 6 due to the low solubility.
:1 binding model as confirmed by Job plot analysis (Fig. S1 in the ESI†) and the anion binding constants were calculated with HypNMR.17 Moderate binding affinities were obtained for all the hosts (Table 1), the highest binding constants were obtained for 6b and 6c, with log(Ka) of respectively 1.75 ± 0.02 and 1.76 ± 0.02. For comparison, the binding affinity log(Ka) of the open-chain receptor 215 was now also measured in DMSO-d6/0.5% H2O and found to be 1.20, much lower than the 3.4 reported when the less competitive acetonitrile was the solvent. We could unfortunately not use acetonitrile for studies of compounds 6 due to the low solubility.
| Nr | n | Yield | log(Ka) |
|---|---|---|---|
| 6a | 6 | 30% | 1.57 ± 0.01 |
| 6b | 7 | 44% | 1.75 ± 0.02 |
| 6c | 8 | 65% | 1.76 ± 0.02 |
| 6d | 9 | 62% | 1.68 ± 0.02 |
| 6e | 10 | 18% | 1.57 ± 0.01 |
| 6f | 12 | 54% | 1.49 ± 0.03 |
| 2 | — | — | 1.20 ± 0.01 |
Crystallography‡
The structures of the complexes of compound 6a with the chloride salts of tetraethylammonium, 1-ethylpyridinium and 3-ethyl-1-methyl imidazolium were obtained by single crystal X-ray diffraction methods.18 The crystals were obtained by slow evaporation from DMSO in the presence of excess chloride salt.Crystals of the tetraethylammonium chloride complex of compound 6a (Fig. 1) were obtained by this method. The macrocyclic receptor was found to be present in two conformations within the crystal (approx. 73:27 occupancy) with the alkyl linker chain adopting a marginally different position in each case. In both conformations the chloride anion is bound in the centre of the macrocycle via six hydrogen bonds (N5⋯Cl1 3.260(1) Å; N6⋯Cl1 3.271(2) Å; N7⋯Cl1 3.382(1) Å; N8⋯Cl1 3.307(1) Å; C6⋯Cl1 3.533(1) Å; C19⋯Cl1 3.535(1) Å).
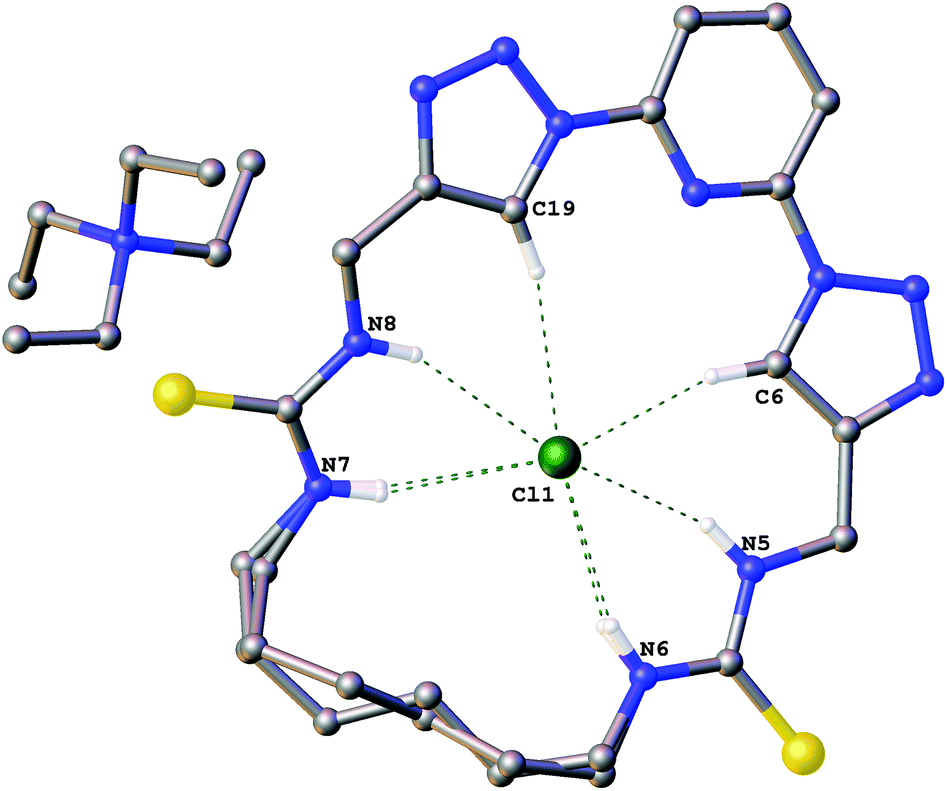 | ||
| Fig. 1 The crystal structure of 6a·(TEA+)·(Cl−). Non-acidic hydrogen atoms have been omitted for clarity and hydrogen bonds are shown as dashed lines. | ||
Crystals of the 1-ethylpyridinium chloride complex of compound 6a (Fig. 2) were also obtained as a hemihydrate. This structure contains two crystallographically independent molecules of 6a in a 2 + 2 arrangement via hydrogen bonding to two bridging chloride anions. One chloride anion bridges via two hydrogen bonds from each macrocycle (N5⋯Cl11 3.263(4) Å; N6⋯Cl11 3.263(4) Å; N107⋯Cl11 3.565(4) Å; N108⋯Cl11 3.167(4) Å). The other, along with the water molecule is disordered over two positions (approx. 62:38 occupancy) so that in the major component the chloride anion and water molecule each bridge via one hydrogen bond from each macrocycle (N7⋯Cl1A 3.453(5) Å; N105⋯Cl1A 3.289(5) Å; N8⋯O1A 3.230(9) Å; N106⋯O1A 2.939(7) Å) and in the minor component the chloride anion bridges via two hydrogen bonds from each macrocycle (N7⋯Cl1B 3.331(7) Å; N8⋯Cl1B 3.056(8) Å; N105⋯Cl1B 3.122(6) Å; N106⋯Cl1B 3.566(7) Å) with no involvement of the water molecule.
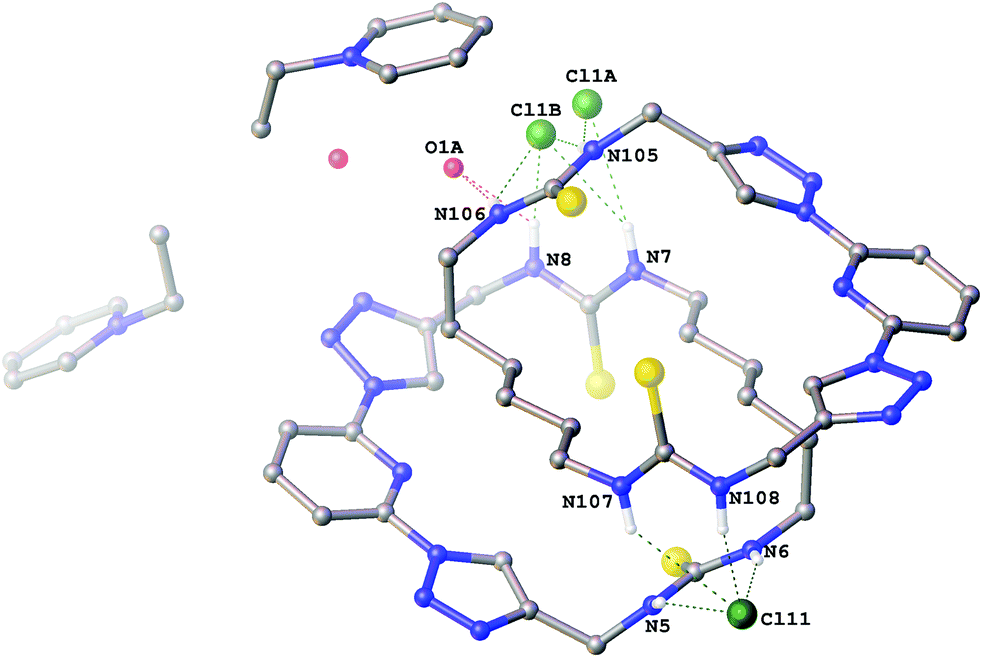 | ||
| Fig. 2 The crystal structure of 2[6a·(C7H10N+)·(Cl−)]·H2O Non-acidic hydrogen atoms have been omitted for clarity and hydrogen bonds are shown as dashed lines. | ||
Interestingly the crystals of the 3-ethyl-1-methylimidazolium chloride complex of compound 6a (Fig. 3) give a structure which combines features from both the structures previously described. It is a Z′ = 2 structure with two crystallographically independent molecules of 6a which each adopt two conformations (approx. 88:12 occupancy). In one of the macrocycles one of the thioureas exhibits minor positional disorder, whereas in the other the disorder is more extensive involving both thioureas and the alkyl linker. In each conformation of both macrocycles a chloride anion is bound in the centre of the macrocycle via six hydrogen bonds (N5⋯Cl1 3.384(3) Å; N6⋯Cl1 3.317(3) Å; N7⋯Cl1 3.339(4) Å; N8⋯Cl1 3.320(4) Å; N7A⋯Cl1 3.31(2) Å; N8A⋯Cl1 3.50(3) Å; C6⋯Cl1 3.708(3) Å; C19⋯Cl1 3.638(4) Å and N105⋯Cl11 3.366(3) Å; N106⋯Cl11 3.327(5) Å; N107⋯Cl11 3.318(3) Å; N206⋯Cl11 3.45(4) Å; N207⋯Cl11 3.40(2) Å; N108⋯Cl11 3.348(3) Å; C106⋯Cl11 3.653(3) Å; C119⋯Cl11 3.660(4) Å).
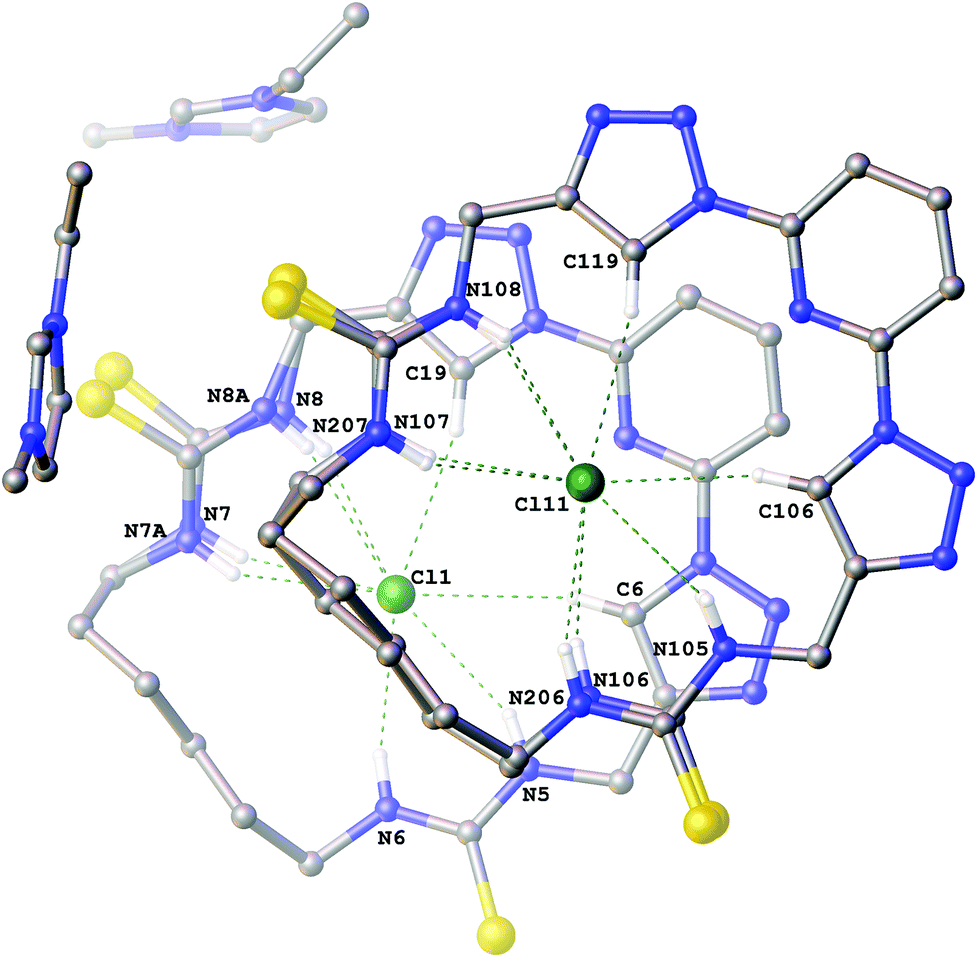 | ||
| Fig. 3 The crystal structure of 2[6a·(C6H11N2+)·(Cl−)]. Non-acidic hydrogen atoms have been omitted for clarity and hydrogen bonds are shown as dashed lines. | ||
Transport studies
The chloride transport activity of the receptors was initially assessed using an ion selective electrode (ISE) assay.19 We prepared a sample of unilamellar POPC vesicles loaded with 489 mM NaCl buffered to pH 7.2 with 5 mM sodium phosphate salts. The vesicles were suspended in 489 mM NaNO3 buffered to pH 7.2 with 5 mM sodium phosphate salts. A sample of the receptor (2 mol% w.r.t. lipid) was added in DMSO and the resulting chloride efflux was monitored using a chloride selective electrode. After 300 s the vesicles were lysed by the addition of detergent and the final electrode reading used to calibrate 100% chloride efflux. The results are shown below in Fig. 4. All of the thiourea containing receptors were shown to mediate chloride efflux from vesicles under these conditions. Compound 6b (C7 macrocycle) seemed to be the most efficient transporter, mediating the efflux of 81% of the encapsulated chloride after 270 s. Compound 2, which is an acyclic amide analogue of 6b showed no activity, highlighting the importance of the thiourea functionality.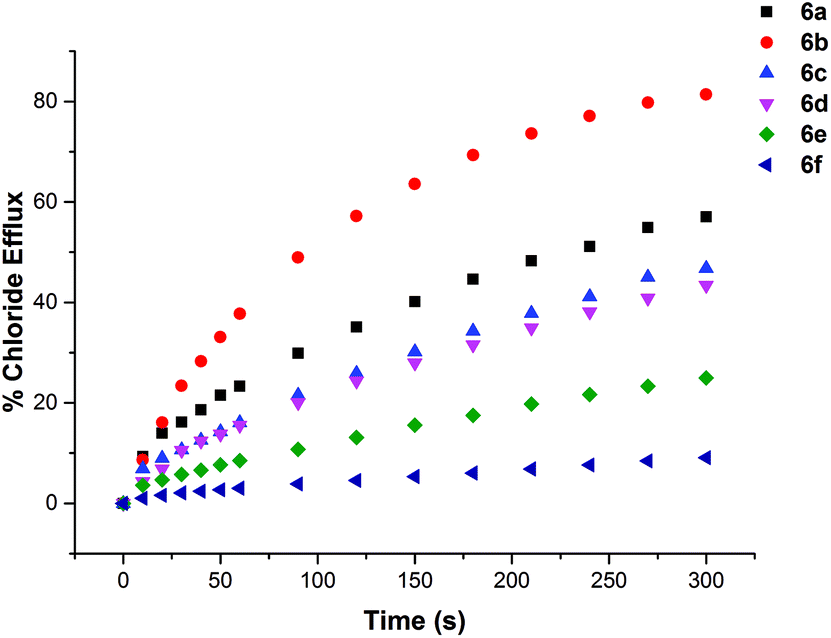 | ||
| Fig. 4 Chloride efflux promoted by thioureas 6a–f (2% molar carrier to lipid) from unilamellar POPC vesicles loaded with 489 mM NaCl buffered to pH 7.2 with 5 mM sodium phosphate salts. The vesicles were dispersed in 489 mM NaNO3 buffered to pH 7.2 with 5 mM sodium phosphate salts. At the end of the experiment, detergent was added to lyse the vesicles and calibrate the ISE to 100% chloride efflux. Each point represents the average of three trials. | ||
We performed a Hill analysis20 for chloride transport under these conditions for all of the active transporters. This enabled us to determine the EC50 for each receptor – the concentration of receptor required to mediate 50% chloride efflux after 270 s. This is a measure of transport activity, with the most active compounds having the lowest EC50 value. The results are summarized in Table 2. This analysis also yielded values for the Hill coefficient (n), which has been interpreted as an indication of the stoichiometry of the transport process. All of the receptors tested in this study had a Hill coefficient <2, which provides support for their mode of transport being via a mobile carrier mechanism rather than aggregation into membrane spanning channels as such channel formation would require a large number of receptor molecules.
| Compound | EC50 Cl−/NO3− | n |
|---|---|---|
| 6a (C6) | 2.7 | 1.2 |
| 6b (C7) | 0.7 | 1.0 |
| 6c (C8) | 3.0 | 1.1 |
| 6d (C9) | 4.4 | 0.7 |
| 6e (C10) | 10.0 | 0.7 |
| 6f (C12) | 48.0* | 0.7 |
The Hill coefficients from the Hill analyses indicated that these receptors were most likely to function as mobile carriers rather than channels. In order to gain further proof for this mechanism, we prepared a sample of vesicle composed of POPC–cholesterol (7:3). The vesicles contained NaCl and were suspended in NaNO3 (both solutions buffered to pH 7.2 with 5 mM sodium phosphate salts). The addition of cholesterol to a bilayer is reported to reduce its fluidity-therefore the action of a mobile carrier, which is diffusion controlled, should be reduced.21 Correspondingly, the addition of 2 mol% of the receptors was found to mediate a reduced level of chloride efflux compared to experiments performed using vesicles composed of pure POPC lipid, thus indicating a mobile carrier mechanism is in effect.
During the Hill analysis, it was often observed that addition of high loadings of receptor did not result in increased chloride efflux, and in some cases, a precipitate was also observed. This indicates a loss of activity as a result of solubility issues.
The most efficient transporter in each experiment is compound 6b (C7 macrocycle). It has previously been observed that there may be an optimum lipophilicity for anion transport, as the transport process requires a balance between aqueous solubility (in order for the transporter to reach the lipid bilayer), and lipophilicity (as the receptor must efficiently partition with the bilayer to mediate anion transport).22 Without conducting a much larger quantitative structure activity relationship study, which is outside the scope of the current project, it is not possible to systematically determine which molecular parameters should be optimised to maximise the rate of transport.23 However based on previous findings22 it is reasonable to assume that the optimum lipophilicity observed for 6b results from compounds having shorter chain lengths not being lipophilic enough to efficiently partition into the bilayer, and above this chain length the compounds become increasingly incompatible with delivery through the aqueous phase, resulting in precipitation – i.e. this is essentially a dependence on a (logP)2 term. The trend in transport activity is summarized in Fig. 5.
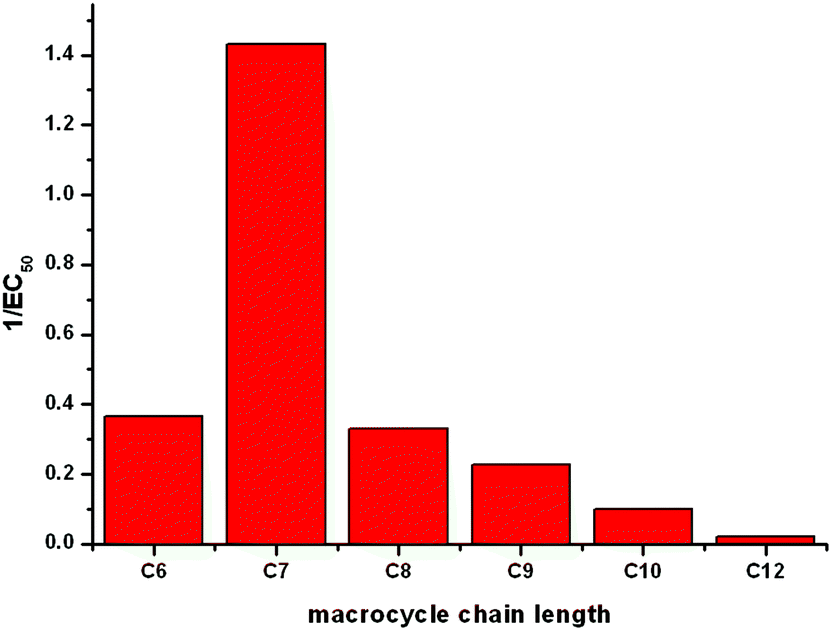 | ||
| Fig. 5 A representation of the relationship between the size of the macrocycle of receptors 6a–f and their Cl−/NO3− antiport activity (1/EC50 as determined by a Hill plot analysis). | ||
Symport vs. antiport?
In order to investigate if the observed chloride transport was by a Cl−/NO3− antiport mechanism (as has been commonly observed for thiourea based transporters) or by a co-transport mechanism, we prepared a sample of vesicles containing 450 mM NaCl buffered to pH 7.2 with 20 mM sodium phosphate salts. The vesicles were suspended in 162 mM Na2SO4 buffered to pH 7.2 with 20 mM sodium phosphate salts. If a transporter functions solely by an anion antiport mechanism, it is expected that no transport will be observed if the extra-vesicular anion is sulfate, as the high dehydration penalty for this anion prevents its transport in the vast majority of cases. However, on addition of a sample of the receptors (2 mol% w.r.t. lipid), significant chloride efflux was observed. We repeated these experiments but added a spike of NaHCO3 to the extravesicular solution after 2 minutes – this was found not to increase the observed chloride efflux, indicating that these receptors do not mediate Cl−/HCO3− antiport. The results for compound 6b are shown below in Fig. 6, and similar results were observed for all thiourea compounds tested.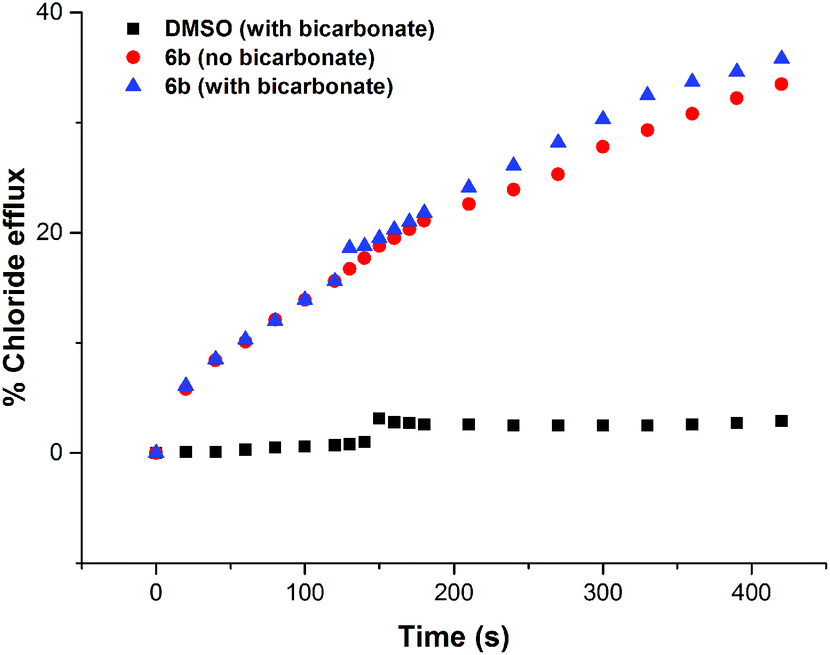 | ||
| Fig. 6 Chloride efflux promoted by compound 6b (2% molar carrier to lipid) from unilamellar POPC vesicles loaded with 450 mM NaCl buffered to pH 7.2 with 20 mM sodium phosphate salts. The vesicles were dispersed in 162 mM Na2SO4 buffered to pH 7.2 with 20 mM sodium phosphate salts. At t = 120 s, a solution of NaHCO3 was added to give a 40 mM external concentration (blue markers) or the experiment was allowed to continue without the addition of NaHCO3 (red markers). At the end of the experiment, detergent was added to lyse the vesicles and calibrate the ISE to 100% chloride efflux. Each point represents the average of three trials. | ||
The chloride efflux in the absence of a readily transported external anion initially indicated that some sort of co-transport process was occurring. We decided to further investigate this effect using the receptors that were found to mediate significant (>15%) chloride efflux after 390 s (6b, 6c, 6a and 6d). All salt solutions referenced in this section contain 20 mM phosphate buffer at pH 7.2 unless otherwise stated.
We considered the possibility of a M+/Cl− co-transport mechanism. We prepared a sample of vesicles encapsulating 450 mM CsCl and suspended them in 162 mM Na2SO4.24 We then monitored chloride efflux mediated by the receptors (2 mol%) using a chloride ISE, and compared the data to that collected using vesicles containing NaCl. The results are summarized below in Fig. 7. We found that there was no significant difference in the behaviour of these compounds when the encapsulated cation was changed from Na+ to Cs+, indicating the nature of the encapsulated cation does not affect the chloride transport properties of the receptors and hence that a M+/Cl− co-transport mechanism is not possible.
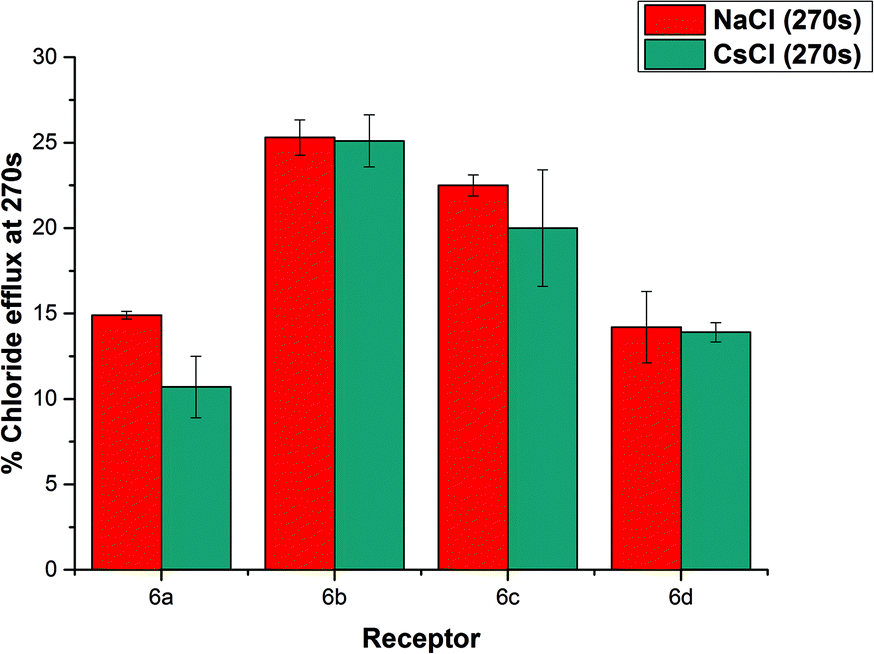 | ||
| Fig. 7 Chloride efflux promoted by 6a–6d (2% molar carrier to lipid) after 270 s from unilamellar POPC vesicles loaded with 450 mM CsCl buffered to pH 7.2 with 20 mM sodium phosphate salts. The vesicles were dispersed in 162 mM Na2SO4 buffered to pH 7.2 with 20 mM sodium phosphate salts. At the end of the experiment, detergent was added to lyse the vesicles and calibrate the ISE to 100% chloride efflux. Each point represents the average of three trials. | ||
We then considered the possibility that these receptors could mediate a Cl−/SO42− antiport mechanism. It is usually assumed that SO42− cannot be transported by synthetic ionophores as it is strongly hydrated. However, we have recently seen examples of extremely potent anion transporters that we believe to mediate this transport mechanism.25 We prepared a sample of vesicles with 100 mM NaCl inside and outside the vesicles, containing the halide sensitive fluorescent probe lucigenin (2 mM). In this experiment, there is no chloride gradient to drive chloride transport. We monitored the fluorescence intensity of the lucigenin on addition of (a) a spike of either NaNO3, NaCl or Na2SO4 at 10 s followed by (b) the addition of the receptor in DMSO (2 mol%) at 40 s (Fig. 8). An increase to the fluorescence intensity of the lucigenin after the addition of the receptor is representative of chloride efflux in the presence of the anion that has been added into the system (an antiport process). We also performed control experiment, in which we added a spike of Na2SO4 followed by DMSO. The results for compound 6b are shown in Fig. 8. In the experiments using NaNO3, all of these receptors were able to mediate chloride transport, confirming that a Cl−/NO3− antiport mechanism is possible. However, no chloride transport was detected in the presence of external SO42−. This indicates that these receptors cannot mediate a Cl−/SO42− antiport mechanism.
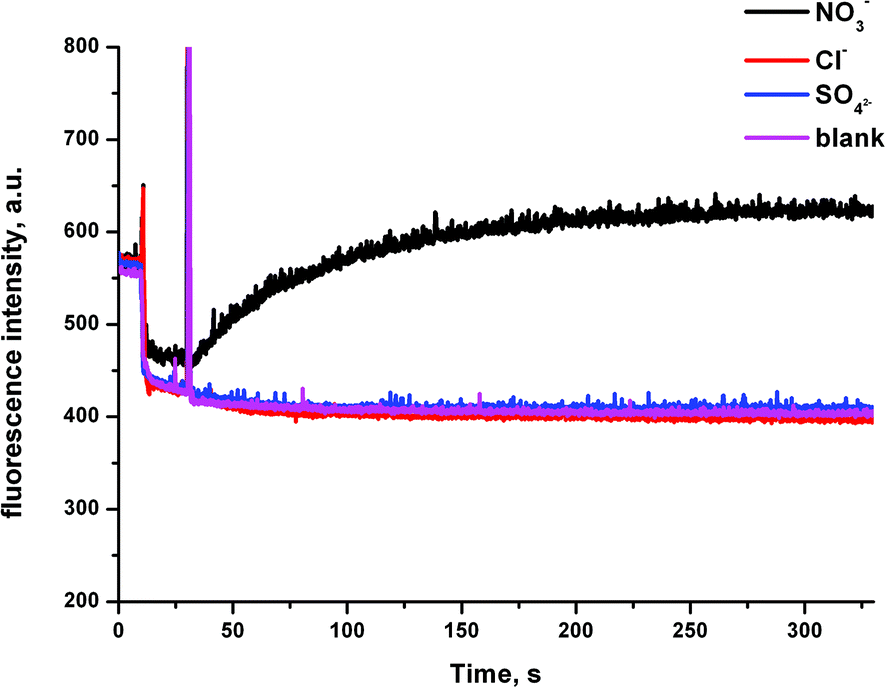 | ||
| Fig. 8 Unilamellar POPC vesicles were loaded with 100 mM NaCl and 2 mM lucigenin buffered to pH 7.2 with 20 mM sodium phosphate salts and dispersed in a 100 mM NaCl solution (buffered to pH 7.2). At t = 10 s, a solution of the appropriate anion was added (final concentration of 40 mM NaNO3, 40 mM Na2SO4 or 40 mM NaCl). At t = 40 s, a DMSO solution of the compound 6b was added. At the end of the experiment (340 s), detergent was added to lyse the vesicles. The blank measurement refers to the addition of Na2SO4, followed by the addition of DMSO. Each point represents the average of three trials. | ||
We then investigated if the chloride efflux was due to a H+/Cl− co-transport mechanism.26 We prepared a sample of vesicles containing 489 mM NaCl buffered to pH 7.2 with 5 mM sodium phosphate salts and the pH sensitive fluorescent probe HPTS (1 mM). The vesicles were suspended in 162 mM Na2SO4 buffered to pH 7.2 with 5 mM sodium phosphate salts and the experiment was initiated by adding a sample of the receptor (2 mol%) in DMSO at 30 s. We followed the ratio of the intensity of the peaks at 510 nm with excitation at 460 nm (basic form of HPTS) and 403 nm (acidic form of HPTS). We then used a previously obtained calibration26 to calculate the corresponding change in internal pH mediated by the receptors. The results for receptors 6a–6d are shown in Fig. 9, and indicate that addition of these receptors causes an increase in intravesicular pH consistent with the efflux of H+/Cl−.
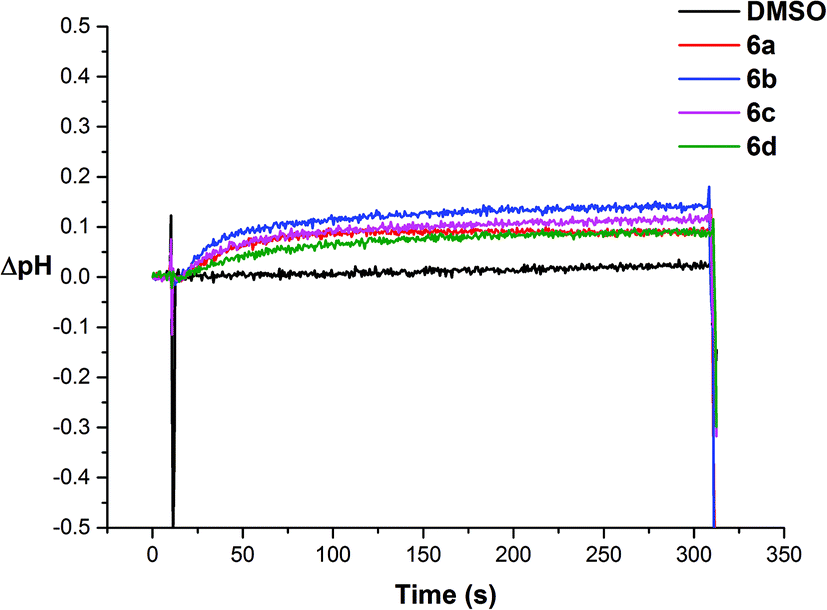 | ||
| Fig. 9 Intravesicular pH change promoted by 6a–6d (2% molar carrier to lipid) from unilamellar POPC vesicles loaded with 1 mM HPTS and 489 mM NaCl, buffered to pH 7.2 with 5 mM sodium phosphate salts. The vesicles were dispersed in 162 mM Na2SO4 buffered to pH 7.2 with 5 mM sodium phosphate salts. At t = 10 s, a DMSO solution of the putative transporters was added to start the experiment. At the end of the experiment (t = 310 s), detergent was added to lyse the vesicles. Each point represents the average of three trials. | ||
Conclusions
Macrocyclic anion receptors 6a–f of different ring sizes, containing thiourea and 2,6-bistriazolepyridinium groups could be prepared by condensation of bisisothiocyanate and bisamine precursors. The binding constants with tetra-n-butylammonium chloride in the relatively competitive solvent DMSO-d6/0.5% H2O are modest but higher than for the open chain analogue. In conclusion, these receptors were found to function as anion carriers which can mediate both a Cl−/NO3− antiport and (to a lesser extent) H+/Cl− symport. The most efficient carrier for Cl−/NO3− antiport was 6b, the structure of which provided the optimum balance between lipophilicity and hydrophilicity/ solubility.Experimental section
General experimental methods
NMR spectra were acquired on commercial Bruker instruments (300 and 400 MHz), and chemical shifts (δ) are reported in parts per million (ppm) referenced to tetramethylsilane (1H) or the internal (NMR) solvent signals (13C). Exact mass measurements were acquired in the EI (at a resolution of 10000) or ESI (at a resolution of 15000) mode. IR spectra were recorded on an FT-IR spectrometer with a universal sampling module. Melting points were determined by using a Reichert Thermovar apparatus and were not corrected. For column chromatography, 70–230 mesh silica 60 was used as the stationary phase. Chemicals received from commercial sources were used without further purification. Tetra-n-butylammonium salts were dried overnight under vacuum at 40 °C prior to use.
2,6-Diazidopyridine (3)
A mixture of 2,6-difluoropyridine (0.3 ml, 3.3 mmol), NaN3 (0.641 g, 9.86 mmol) and tetra-n-butylammonium bromide (0.424 g, 1.32 mmol) in DMF (5 ml) was stirred at 60 °C for 48 hours. The crude product was purified by column chromatography (SiO2, EtOAc) to afford 3 (0.145 g, 27%). 1H NMR (300 MHz, CDCl3, ppm) δ: 7.55 (t, 1H, J = 7.8 Hz), 6.55 (d, 2H, J = 7.8 Hz); 13C NMR (75 MHz, CHCl3, ppm) δ: 153.4, 140.9, 109.8; Tm: 73–75 °C; FT-IR (cm−1) 2109, 1562.Di-tert-butyl((1,1′-(pyridine-2,6-diyl)bis(1H-1,2,3-triazole-4,1-diyl))bis(methylene)) dicarbamate (4)
To a solution of 2,6-diazidopyridine 3 (0.120 g, 0.74 mmol) in dry THF (5 ml) was added N-Boc-propargylamine (0.18 ml, 2 mmol), [(CH3CN)4Cu]PF6 (0.056 g, 0.15 mmol) and 0.03 ml Et3N and the reaction mixture was stirred at 50 °C for 3 hours. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (SiO2, EtOAc) to afford 4 (0.306 g, 87%). 1H NMR (300 MHz, DMSO-d6, ppm) δ: 8.94 (s, 2H), 8.38 (t, 1H, J = 8.3 Hz, 8.21 (d, 2H, J = 7.9 Hz,), 7.45 (br s, 2H), 4.32 (d, 4H, J = 5.6 Hz), 1.40 (s, 18H); 13C NMR (75 MHz, DMSO-d6, ppm) δ: 155.6, 147.3, 146.7, 143.8, 120.2, 112.9, 78.05, 35.5, 28.2; HRMS: (ESI+) calculated mass for C21H29N9O4Na: 494.2235 g mol−1 [M + Na]+, found: 494.2240 g mol−1; Tm: 165–166 °C; FT-IR (cm−1): 3407, 1687, 1519.2,6-Bis[4-(isothiocyanatomethyl)-1H-1,2,3-triazol-1-yl]pyridine (5)
A suspension of 4 (1.233 g, 2.6 mmol) in 4 N HCl in dioxane (7 ml) was stirred for 2 hours at room temperature. The solvent was removed under reduced pressure and the obtained product was redissolved in 12 ml DCM. Subsequently, thiophosgene (0.65 ml, 6.8 mmol) and 12 ml saturated KHCO3 solution were added and the reaction mixture was stirred at room temperature for 16 hours. The product was extracted with DCM (3 × 50 ml), the combined organic layers were dried over Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by column chromatography (SiO2, DCM + 1% MeOH) to afford 5 (0.603 g, 65%). 1H NMR (300 MHz, CDCl3, ppm) δ: 8.64 (s, 2H), 8.30 (d, 2H, J = 7.4 Hz), 8.24 (t, 1H, J = 7.4 Hz), 4.99 (s, 4H); 13C NMR (75 MHz, CHCl3, ppm) δ: 147.7, 143.0, 142.9, 135.6, 119.5, 113.6, 31.0; HRMS: (EI) calculated mass for C13H9N9S2: 355.04062 g mol−1 [M]+, found: 355.04224 g mol−1; Tm: 175–177 °C; FT-IR (cm−1): 2068,14792,3,4,7,9,16,18,21,22,23,28-Undecaazatetracyclo[22.3.1.12,5.120,23]triaconta-1(28),3,5(30),20(29),21,24,26-heptaene-8,17-dithione (6a)
2,3,4,7,9,17,19,22,23,24,29-Undecaazatetracyclo[23.3.1.12,5.121,24]hentriaconta-1(29),3,5(31),21(30),22,25,27-heptaene-8,18-dithione (6b)
Synthesis according to general procedure 1: 0.119 g 5 (0.34 mmol), 0.044 g 1,7-diaminoheptane (0.34 mmol). 0.071 g (44% yield) of receptor 6b was obtained.1H NMR (300 MHz, DMSO-d6, ppm) δ: 8.97 (s, 2H), 8.40 (t, 1H, J = 8 Hz), 8.24 (d, 2H, J = 8 Hz), 7.82 (br s, 2H), 7.56 (br s, 2H), 4.88 (s, 4H), 3.43 (m, 4H), 1.54 (m, 4H), 1.34 (m, 4H), 1.24 (m, 2H); 13C NMR (75 MHz, DMSO-d6, ppm) δ: 182, 147.4, 146.9, 143.9, 119.9, 113.2, 43.7, 43.6, 28.2, 25.9; MS (ESI+): m/z 508 [M + Na]+; Tm: 274–275 °C (decomposition upon melting); FT-IR (cm−1): 3239, 3065, 2927, 2856, 1608, 1544.
2,3,4,7,9,18,20,23,24,25,30-Undecaazatetracyclo[24.3.1.12,5.122,25]dotriaconta-1(30),3,5(32),22(31),23,26,28-heptaene-8,19-dithione (6c)
Synthesis according to general procedure 1: 0.100 g 5 (0.28 mmol), 0.041 g 1,8-diaminooctane (0.28 mmol). 0.092 g (65% yield) of receptor 6c was obtained.1H NMR (300 MHz, DMSO-d6, ppm) δ: 8.90 (s, 2H), 8.40 (t, 1H, J = 8 Hz), 8.23 (d, 2H, J = 8 Hz), 7.84 (br s, 2H), 7.58 (br s, 2H), 4.84 (s, 4H), 3.39 (m, 4H), 1.52 (m, 4H), 1.32 (m, 6H), 1.23 (m, 2H); 13C NMR (75 MHz, DMSO-d6, ppm) δ: 183, 147.3, 146.7, 143.8, 120.1, 113.3, 43.7, 43.6, 28.0, 25.7; MS (ESI+): m/z 822 [M + Na]+; Tm: 267–268 °C (decomposition upon melting); FT-IR (cm−1): 3250, 3065, 2924, 2851, 1607, 1556
2,3,4,7,9,19,21,24,25,26,31-Undecaazatetracyclo[25.3.1.12,5.123,26]tritriaconta-1(31),3,5(33),23(32),24,27,29-heptaene-8,20-dithione (6d)
Synthesis according to general procedure 1: 0.100 g 5 (0.28 mmol), 0.045 g 1,9-diaminononane (0.28 mmol). 0.090 g (62% yield) of receptor 6d was obtained.1H NMR (300 MHz, DMSO-d6, ppm) δ: 9.03 (s, 2H), 8.41 (t, 1H, J = 8 Hz), 8.24 (d, 2H, J = 8 Hz), 7.83 (br s, 2H), 7.54 (br s, 2H), 4.83 (s, 4H), 3.47 (m, 4H), 1.51 (m, 4H), 1.30 (m, 8H), 1.23 (m, 2H); 13C NMR (75 MHz, DMSO-d6, ppm) δ: 183, 147.4, 146.3, 143.9, 120.6, 113.4, 43.6, 41.2, 28.2 28.0, 25.8; MS (ESI+): m/z 536 [M + Na]+; Tm: 271–272 °C (decomposition upon melting); FT-IR (cm−1): 3231, 3062, 2925, 2852, 1551.
2,3,4,7,9,20,22,25,26,27,32-Undecaazatetracyclo[26.3.1.12,5.124,27]tetratriaconta-1(32),3,5(34),24(33),25,28,30-heptaene-8,21-dithione (6e)
Synthesis according to general procedure 1: 0.100 g 5 (0.28 mmol), 0.048 g 1,10-diaminodecane (0.28 mmol). 0.027 g (18% yield) of receptor 6e was obtained.1H NMR (300 MHz, DMSO-d6, ppm) δ: 8.99 (s, 2H), 8.40 (t, 1H, J = 8 Hz), 8.22 (d, 2H, J = 8 Hz), 7.81 (br s, 2H), 7.53 (br s, 2H), 4.82 (s, 4H), 3.39 (m, 4H), 1.48 (m, 4H), 1.27 (m, 12H); 13C NMR (75 MHz, DMSO-d6, ppm) δ: 183, 147.4, 146.1, 143.9, 143.9, 120.6, 113.3, 43.6, 41.2, 28.2 28.0, 25.8; MS (ESI+): m/z 550 [M + Na]+; Tm: 260–262 °C (decomposition upon melting); FT-IR (cm−1): 3288, 2923, 2851,1559.
2,3,4,7,9,22,24,27,28,29,34-Undecaazatetracyclo[28.3.1.12,5.126,29]hexatriaconta-1(34),3,5(36),26(35),27,30,32-heptaene-8,23-dithione (6f)
Synthesis according to general procedure 1: 0.100 g 5 (0.28 mmol), 0.056 g 1,12-diaminododecane (0.28 mmol). 0.084 g (54% yield) of receptor 6f was obtained.1H NMR (300 MHz, DMSO-d6, ppm) δ: 9.01 (s, 2H), 8.41 (t, 1H, J = 8 Hz), 8.24 (d, 2H, J = 8 Hz), 7.84 (br s, 2H), 7.53 (br s, 2H), 4.82 (s, 4H), 3.45 (m, 4H), 1.48 (m, 4H), 1.23 (m, 16H); 13C NMR (75 MHz, DMSO-d6, ppm) δ: 183, 147.4, 146.0, 143.9, 143.9, 120.7, 113.2, 43.6, 41.2, 28.3 28.1, 25.9; MS (ESI+): m/z 578 [M + Na]+; Tm: 268–270 °C (decomposition upon melting); FT-IR (cm−1): 3288, 3076, 2923, 2850,1535.
Acknowledgements
The authors thank the European Cooperation in Science and Technology (COST) action CM1005 “Supramolecular chemistry in water” for funding. PAG thanks the EPSRC for funding (EP/J009687/1) and for access to the crystallographic facilities at the University of Southampton and thanks the Royal Society and the Wolfson Foundation for a Royal Society Wolfson Research Merit Award. This work was also financially supported by the KU Leuven, the Ministerie voor Wetenschapsbeleid and the FWO-Vlaanderen. We thank Diamond Light Source for access to beamline I19 (proposal number: 8521) that contributed to the results presented here.Notes and references
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC.
- G. W. Gokel and N. Barkey, New J. Chem., 2009, 33, 947–963 RSC.
- J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC.
- C. J. E. Haynes and P. A. Gale, Chem. Commun., 2011, 47, 8203–8209 RSC.
- F. M. Ashcroft, Ion channels and disease, Academic Press, San Diego, 2000 CrossRef CAS PubMed; S. K. Ko, S. K. Kim, A. Share, V. M. Lynch, J. Park, W. Namkung, W. Van Rossom, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Nat. Chem., 2014, 6, 885–892 CrossRef CAS PubMed.
- S. M. Rowe, S. Miller and E. J. Sorscher, N. Engl. J. Med., 2005, 352, 1992–2001 CrossRef CAS PubMed.
- D. C. Gadsby, P. Vergani and L. Csanady, Nature, 2006, 440, 477–483 CrossRef CAS PubMed.
- M. H. Akabas, J. Biol. Chem., 2000, 275, 3729–3722 CrossRef CAS PubMed.
- B. P. Hay and V. S. Bryantsev, Chem. Commun., 2008, 2417–2428 RSC.
- C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520–563 RSC.
- K. P. McDonald, Y. Hua and A. H. Flood, Top Heterocycl. Chem., 2010, 24, 341–366 Search PubMed; J. P. Byrne, J. A. Kitchen and T. Gunnlaugsson, Chem. Soc. Rev., 2014, 43, 5302–5325 RSC; M. G. Fisher, P. A. Gale, J. R. Hiscock, M. B. Hursthouse, M. E. Light, F. P. Schmidtchen and C. C. Tong, Chem. Commun., 2009, 3017–3019 RSC; M. Yano, C. C. Tong, M. E. Light, F. P. Schmidtchen and P. A. Gale, Org. Biomol. Chem., 2010, 8, 4356–4363 Search PubMed . See also: S. J. Moore, M. G. Fisher, M. Yano, C. C. Tong and P. A. Gale, Chem. Commun., 2011, 47, 689–691 RSC.
- M. H. Palmer, R. H. Findlay and A. J. Gaskell, J. Chem. Soc., Perkin Trans. 2, 1974, 420–428 RSC.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- T. Merckx, P. Verwilst and W. Dehaen, Tetrahedron Lett., 2013, 54, 4237–4240 CrossRef CAS PubMed.
- G. Han, M. Tamaki and V. Hruby, J. Pept. Res., 2001, 58, 338–341 CrossRef CAS.
- C. Frassineti, S. Ghelli, P. Gans, A. Sabatini, M. S. Moruzzi and A. Vacca, Anal. Biochem., 1995, 231, 374–382 CrossRef CAS PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- (a) B. D. Smith and T. N. Lambert, Chem. Commun., 2003, 2261–2268 RSC; (b) A. V. Koulov, T. N. Lambert, R. Shukla, M. Jain, J. M. Boon, B. D. Smith, H. Y. Li, D. N. Sheppard, J. B. Joos, J. P. Clare and A. P. Davis, Angew. Chem., Int. Ed., 2003, 42, 4931–4933 CrossRef CAS PubMed.
- A. V. Hill, Biochem. J., 1913, 7, 471–480 CAS.
- T. P. W. McMullen, R. N. A. H. Lewis and R. N. McElhaney, Curr. Opin. Colloid Interface Sci., 2004, 8, 459–468 CrossRef CAS PubMed; J. C. M. Holthuis, G. van Meer and K. Huitema, Mol. Membr. Biol., 2003, 20, 231–241 CrossRef; W. F. D. Bennett, J. L. MacCallum and D. P. Tieleman, J. Am. Chem. Soc., 2009, 131, 1972–1978 CrossRef PubMed.
- (a) V. Saggiomo, S. Otto, I. Marques, V. Felix, T. Torroba and R. Quesada, Chem. Commun., 2012, 48, 5274–5276 RSC; (b) C. J. E. Haynes, S. J. Moore, J. R. Hiscock, I. Marques, P. J. Costa, V. Felix and P. A. Gale, Chem. Sci., 2012, 3, 1436–1444 RSC.
- N. Busschaert, S. J. Bradberry, M. Wenzel, C. J. E. Haynes, J. R. Hiscock, I. L. Kirby, L. E. Karagiannidis, S. J. Moore, N. J. Wells, J. Herniman, G. J. Langley, P. N. Horton, M. E. Light, I. Marques, P. J. Costa, V. Félix, J. G. Frey and P. A. Gale, Chem. Sci., 2013, 4, 3036–3045 RSC.
- C. C. Tong, R. Quesada, J. L. Sessler and P. A. Gale, Chem. Commun., 2008, 6321–6323 RSC.
- N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernandez, R. Perez-Tomas and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed; N. Busschaert, L. E. Karagiannidis, M. Wenzel, C. J. E. Haynes, N. J. Wells, P. G. Young, D. Makuc, J. Plavec, K. A. Jolliffe and P. A. Gale, Chem. Sci., 2014, 5, 1118–1127 RSC.
- N. Busschaert, P. A. Gale, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis and W. A. Harrell, Chem. Commun., 2010, 46, 6252–6254 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1029618–1029620. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob02236j |
| ‡ Data were collected either on a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum rotating anode generator with HF Varimax optics (100 μm focus) (tetraethylammonium chloride and 3-ethyl-1-methylimidazolium chloride complexes of compound 6a) or collected at Station I19 of the Diamond Light Source synchrotron on a Crystal Logics kappa-geometry goniometer equipped with a Rigaku Saturn 724+ CCD detector (1-ethylpyridinium chloride complex of compound 6a).18 Standard procedures were followed although specific refinement issues can be found in the _olex2_refinement_description of the corresponding CIF. Crystal data for the tetraethylammonium chloride complex of compound 6a. CCDC 1029618: M = 637.32, Monoclinic, a = 17.3691(11), b = 11.8081(8), c = 17.9776(13) Å, α = 90.00°, β = 118.4010(10)°, γ = 90.00°, U = 3243.4(4) Å3, T = 100(2) K, space group P21/n, Z = 4, 28788 reflections measured, 7417 unique reflections (Rint = 0.0226). The final R1 values were 0.0314 (I > 2σ(I)). The final wR(F2) values were 0.0816 (I > 2σ(I)). The final R1 values were 0.0378 (all data). The final wR(F2) values were 0.0854 (all data). The goodness of fit on F2 was 1.047. Crystal data for the 1-ethylpyridinium chloride complex of compound 6a. CCDC 1029619: M = 624.24, Triclinic, a = 10.391(2), b = 16.944(4), c = 18.900(4) Å, α = 107.255(2)°, β = 90.906(2)°, γ = 96.740(2)°, U = 3151.4(12) Å3, T = 100(2) K, space group P 3-Ethyl-1-methylimidazolium chloride complex of compound 6a. CCDC 1029620: M = 618.24, Triclinic, a = 10.1456(7), b = 15.6738(11), c = 19.0361(13) Å, α = 91.480(5)°, β = 97.780(5)°, γ = 94.128(5)°, U = 2989.5(4) Å3, T = 100(2) K, space group P21/n, Z = 4, 38182 reflections measured, 13640 unique reflections (Rint = 0.0420). The final R1 values were 0.0797 (I > 2σ(I)). The final wR(F2) values were 0.2159 (I > 2σ(I)). The final R1 values were 0.1201 (all data). The final wR(F2) values were 0.2484(all data). The goodness of fit on F2 was 1.025. |
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ,
, | This journal is © The Royal Society of Chemistry 2015 |