
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing the anti-inflammatory activity of chalcones by tuning the Michael acceptor site†
Hannelore
Rücker
a,
Nafisah
Al-Rifai
a,
Anne
Rascle
b,
Eva
Gottfried
c,
Lidia
Brodziak-Jarosz
de,
Clarissa
Gerhäuser
e,
Tobias P.
Dick
d and
Sabine
Amslinger
*a
aInstitute of Organic Chemistry, University of Regensburg, Universitätsstraße 31, 93053 Regensburg, Germany. E-mail: sabine.amslinger@chemie.uni-regensburg.de
bInstitute of Immunology, University of Regensburg, Franz-Josef-Strauss-Allee 11, 93053 Regensburg, Germany
cDepartment of Internal Medicine III, University Hospital of Regensburg, Franz-Josef-Strauß Allee 11, 93053 Regensburg, Germany
dDivision of Redox Regulation, German Cancer Research Center (DKFZ), DKFZ-ZMBH Alliance, Im Neuenheimer Feld 280, 69120 Heidelberg, Germany
eDivision of Epigenomics and Cancer Risk Factors, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 280, 69120 Heidelberg, Germany
First published on 14th January 2015
Abstract
Inflammatory signaling pathways orchestrate the cellular response to infection and injury. These pathways are known to be modulated by compounds that alkylate cysteinyl thiols. One class of phytochemicals with strong thiol alkylating activity is the chalcones. In this study we tested fourteen chalcone derivatives, α-X-substituted 2′,3,4,4′-tetramethoxychalcones (α-X-TMCs, X = H, F, Cl, Br, I, CN, Me, p-NO2-C6H4, Ph, p-OMe-C6H4, NO2, CF3, COOEt, COOH), for their ability to modulate inflammatory responses, as monitored by their influence on heme oxygenase-1 (HO-1) activity, inducible nitric oxide synthase (iNOS) activity, and cytokine expression levels. We confirmed that the transcriptional activity of Nrf2 was activated by α-X-TMCs while for NF-κB it was inhibited. For most α-X-TMCs, anti-inflammatory activity was positively correlated with thiol alkylating activity, i.e. stronger electrophiles (X = CF3, Br and Cl) being more potent. Notably, this correlation did not hold true for the strongest electrophiles (X = CN and NO2) which were found to be ineffective as anti-inflammatory compounds. These results emphasize the idea that chemical fine-tuning of electrophilicity is needed to achieve and optimize desired therapeutic effects.
Introduction
The expression of anti-inflammatory proteins, such as heme oxygenase-1 (HO-1), and proinflammatory proteins, such as inducible NO synthase (iNOS), can be modulated by protein thiol modifications. In particular, the oxidation of protein thiols or their alkylation by electrophiles, especially α,β-unsaturated carbonyl compounds (often referred to as Michael acceptors since they are able to undergo 1,4-additions with thiols, see Fig. 2A)1 leads to either activation or inhibition of individual transcription factors. Alkylation of Keap12 releases the transcription factor Nrf2 and thus induces HO-1 expression (Fig. 2B). The inactivation of NF-κB3 by Michael acceptors4 inhibits expression of proinflammatory proteins (e.g. iNOS, TNF, IL-6, IL-8) and appears to be mediated by either inhibition of IκBα kinase (IKK) (Fig. 2C, Inhibition I)5 or by alkylation of NF-κB cysteinyl residues within the DNA-binding domain6 (Fig. 2C, Inhibition II). For example, the natural chalcone xanthohumol was found to covalently modify cysteinyl residues of both NF-κB p65 and IKK.5 However, the activation of Nrf2 and inactivation of NF-κB by protein thiol modifications may also be caused more indirectly through glutathione (GSH) depletion or oxidation.7The anti-inflammatory activity of chalcones is of significant biomedical interest. Precise tuning of chalcone thiol reactivity may lead to derivatives with improved therapeutic potential. To modulate the chemical reactivity in thia-Michael additions a substituent can be introduced in the α-position of the α,β-unsaturated carbonyl system (Fig. 2A). Compounds with different substituents in the α-position can then be screened for desired biological activities.8 We previously established a panel of fourteen α-X-substituted 2′,3,4,4′-tetramethoxychalcones (α-X-TMCs, Fig. 1) exhibiting a wide range of chemical reactivity. We observed that increased chalcone thiol reactivity correlated with stronger induction of HO-1 and stronger repression of iNOS.8b Using a selection of cell-based assays, we now further demonstrate that α-X-TMCs differentially affect the Nrf2 and NF-κB inflammatory response pathways within living cells.
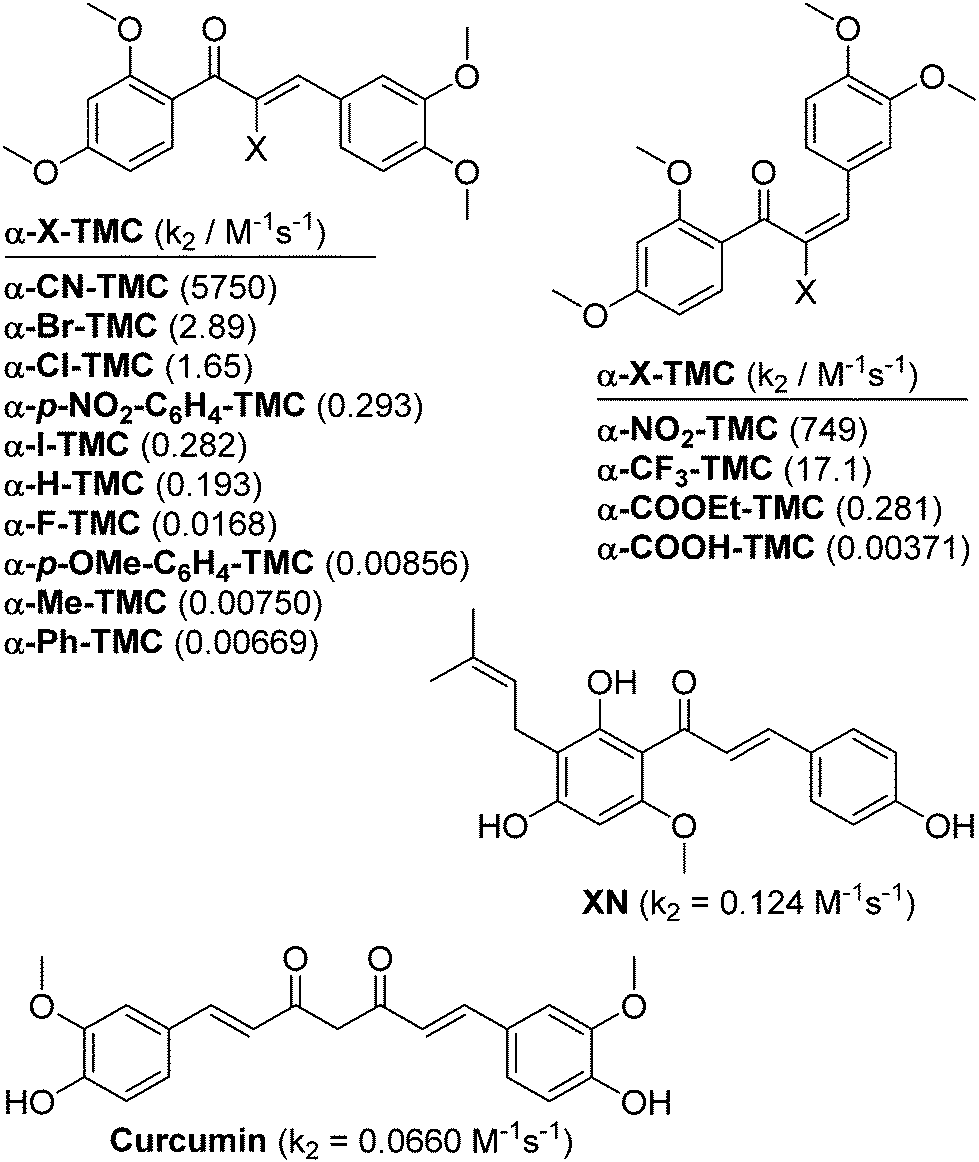 | ||
| Fig. 1 Structures of α-X-2′,3,4,4′-tetramethoxychalcones (α-X-TMCs) tested in this study together with the natural products xanthohumol (XN) and curcumin which served as positive controls. Second-order rate constants (k2) for the reaction with cysteamine are given in parentheses.8 | ||
 | ||
| Fig. 2 Influence of α,β-unsaturated carbonyl compounds, acting as Michael acceptors, on transcription factors involved in inflammation. (A) Michael addition leading to protein-electrophile adducts; possible influence of Michael acceptors (α,β) on the Keap1-Nrf2 pathway (B) and on the NF-κB pathway (C). α,β, α,β-unsaturated carbonyl compound; Keap1, Kelch-like ECH-associated protein 1; Nrf2, nuclear factor-erythroid 2-related factor 2; ARE/EpRE, antioxidant response element/electrophile response element; HO-1, heme oxygenase-1; IκBα, inhibitory factor κBα; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; IKK, inhibitor of nuclear factor κ-B kinase; IL-6, IL-8, interleukin-6, interleukin-8; iNOS, inducible NO synthase; TNF, tumor necrosis factor. | ||
Results and discussion
α-X-TMCs induce HO-1 activity in RAW264.7 macrophages
Using our recently developed ELISA-based HO-1 assay,9 α-X-TMCs were tested for their ability to induce HO-1 activity in RAW264.7 macrophages (Fig. 3). All α-X-TMCs were used at a concentration of 1 μM, except for α-CF3-TMC, which is cytotoxic at this concentration8b and therefore was used at 0.5 μM. HO-1 activity was determined after 3, 6 and 24 h of treatment with α-X-TMCs. Most of the α-X-TMCs stimulated HO-1 activity weakly, with a maximal increase of 2.7 fold after 6 h of treatment in the case of α-CF3-TMC. The derivatives α-CF3-TMC, α-Br-TMC, α-H-TMC and α-Cl-TMC exhibited the most significant effect (2.1–2.7 fold of control). Induced HO-1 activity peaked at 6 h, with the exception of α-I-TMC which induced even higher activity at 24 h (1.9 fold induction). Overall, these findings agree well with our previous study, which showed that α-X-TMCs with intermediate to high electrophilicity (i.e. α-CF3-TMC, α-Br-TMC, α-Cl-TMC and α-H-TMC) efficiently induce HO-1 protein expression at the same concentrations (0.5, 1 μM).8b Likewise, as seen previously,8b the strongest electrophiles, α-CN-TMC and α-NO2-TMC did not cause significant induction of HO-1 activity at 1 μM within the same time frame. This may be due to a direct reaction with GSH,10 potentially competing with the modification of protein thiols.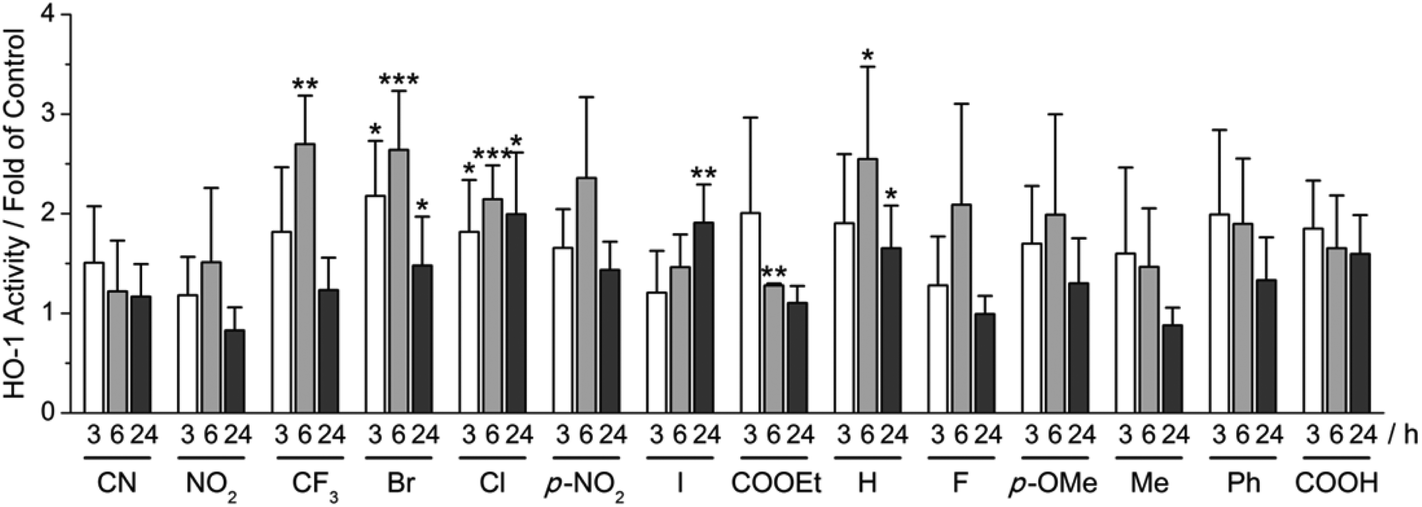 | ||
| Fig. 3 Influence of α-X-TMCs on HO-1 activity. RAW264.7 cells were treated with different α-X-TMCs at 1 μM (0.5 μM for α-CF3-TMC) for the indicated times (3, 6 and 24 h). Labels indicate the substituent X in the α-position of the α,β-unsaturated carbonyl moiety of the chalcone (p-NO2 is short for p-NO2-C4H6 and p-OMe for p-OMe-C4H6). HO-1 activity was determined by an ELISA-based bilirubin quantification assay. At least three independent measurements were performed. Levels of significance: ***, p < 0.001; **, p < 0.01; *, p < 0.05. | ||
α-X-TMCs induce Nrf2 activity in AREc32 cells
Having observed induction of HO-1 activity by α-X-TMCs (Fig. 3), we then asked whether this effect was caused by the activation of Nrf2, a key transcription factor for HO-1 expression. Nrf2 activation was quantified in human breast cancer MCF7 cells expressing a luciferase reporter construct under the control of the human ARE (antioxidant response element) core sequence (AREc32 cells).11 The α-X-TMCs were applied for 24 h at concentrations ranging from 1.56 to 100 μM. In parallel, cytotoxicity was assessed by sulforhodamine B (SRB) staining.12 Only compound concentrations causing less than 50% cytotoxicity were included in the data analysis. Values were normalized to the established Nrf2-inducer xanthohumol (XN) which in our assay activated Nrf2 maximally at 25 μM with 42 ± 11 fold induction (taken for normalization as 100%) relative to the solvent control (Fig. 4). The best Nrf2 activators were α-CF3-TMC (219% of XN at 3.13 μM), α-Br-TMC (317% of XN at 6.25 μM) and α-Cl-TMC (180% of XN at 6.25 μM), which mirrors their effectiveness in inducing HO-1 expression and activity. Similar values of maximal Nrf2 induction showed α-H-TMC (147% of XN) and α-I-TMC (206% of XN), however at a higher concentration (25 μM). Weak electrophiles (α-COOH-TMC, α-p-OMe-C6H4-TMC, α-F-TMC, and α-Ph-TMC) were less effective than XN (16–44%), even at higher concentrations (25, 50 or 100 μM). Overall, and similar to HO-1 activity, k2 values (Fig. 1)8 correlated with the ability of α-X-TMCs to induce Nrf2 activity (Fig. 4). In agreement with our previous results on HO-1 expression,8b the α-X-aromatic compound α-p-NO2-C6H4-TMC activated Nrf2 only at very high concentrations (161% of XN at 100 μM). While α-NO2-TMC was slightly active (31% of XN at 50 μM), α-CN-TMC was completely inactive, again suggesting that the two strongest electrophiles in our panel are ineffective in inducing Nrf2 dependent gene expression. | ||
| Fig. 4 Maximum Nrf2 induction of α-X-TMCs relative to the maximum Nrf2 induction of the positive control xanthohumol (XN*) (set to 100%). Activities were tested by incubating α-X-TMCs at different concentrations (1.56–100 μM) for 24 h with AREc32 cells. α-CN-TMC showed no effect and thus was not included. Data points represent the mean of 2–3 independent measurements. | ||
Some of the compounds enhanced Nrf2 activity by 10 fold at a very low concentration, as quantified by their C10 values (Table S1†). α-CF3-TMC (C10 = 0.495 μM) was most potent, followed by α-Br-TMC (1.39 μM), α-Cl-TMC (2.46 μM), α-I-TMC (5.94 μM), α-H-TMC (7.23 μM) and XN (10.4 μM). C10 values strongly correlated with maximal Nrf2 activation (Fig. 4) and with thiol reactivity k2 values (Fig. 1).
The unexpectedly high C10 values of α-p-NO2-C6H4-TMC (14.8 μM) and α-COOEt-TMC (18.3 μM) may indicate additional intracellular interactions caused by the substituent X, potentially interfering with the electrophilic reactions that lead to Nrf2 activation.
α-X-TMCs inhibit iNOS-dependent NO production in LPS-stimulated RAW264.7 macrophages
Next we asked to what extent the different α-X-TMCs inhibit NF-κB-dependent gene expression. To this end we quantified iNOS-dependent NO production by LPS-stimulated RAW264.7 macrophages using the Griess assay13 (Table 1). In our previous study we already observed a significant correlation between k2 values and the ability of α-X-TMCs to inhibit iNOS activity.8b This correlation was established for the series X = CF3, Br, Cl, I, H, and F, which are the α-X-TMCs with intermediate to strong, but not very strong electrophilicity, and for which X is not expected to engage in extra H bonding or π-interactions.| α-X-TMC | Toxicity limita [μM] | Maximum inhibition of NO production [%] (conc [μM])b | IC50 (NO production) [μM] |
|---|---|---|---|
| a Toxicity of chalcones was measured with the MTT assay in the absence of LPS (no differences were found in the presence of LPS). Concentrations causing cell viabilities > 80% after 24 h incubation were assigned as non-toxic. b Stimulation with α-X-TMCs for 24 h in the presence of 10 ng mL−1 LPS. ns, non-significant (no significant NO inhibition observed); nd, not determined. | |||
| CN | 25 | 16.4 ± 8.7 (1) | nd |
| NO2 | 10 | 26.9 ± 7.9 (10) | nd |
| CF3 | 0.5 | 84.6 ± 8.9 (0.5) | 0.120 ± 0.062 |
| Br | 1 | 67.4 ± 11.7 (1) | 0.640 ± 0.179 |
| Cl | 1 | 47.2 ± 14.6 (1) | 0.992 ± 0.482 |
| p-NO2-C6H4 | 10 | 35.8 ± 2.2 (10) | nd |
| I | 5 | 72.2 ± 9.7 (5) | 3.15 ± 0.70 |
| COOEt | 10 | 66.8 ± 8.4 (10) | 6.65 ± 0.53 |
| H | 10 | 96.9 ± 11.9 (10) | 4.44 ± 1.29 |
| F | 25 | 86.6 ± 18.9 (25) | 12.9 ± 5.6 |
| p-OMe-C6H4 | 10 | ns | nd |
| Me | 10 | −34.7 ± 15.2 (5) | nd |
| Ph | 25 | 27.9 ± 13.4 (25) | nd |
| COOH | 25 | 20.8 ± 3.7 (25) | nd |
Using different non-toxic concentrations we determined the IC50 values for the inhibition of NO production (Table 1). With the exception of α-Me-TMC and α-p-OMe-C6H4-TMC, all α-X-TMCs inhibited NO generation. α-p-OMe-C6H4-TMC was completely inactive, while α-Me-TMC even enhanced NO production by 35%. Most compounds revealed a concentration-dependent inhibitory effect on NO production, with the exception of α-CN-TMC (see ESI Fig. S1†). While the strongest inhibition of NO release was achieved by treatment with 10 μM α-H-TMC (97% inhibition, Table 1), the most potent chalcone was α-CF3-TMC causing 85% inhibition at a concentration of 0.5 μM.
For seven α-X-TMCs it was possible to calculate IC50 values for the inhibition of NO production. These correlated very well with the corresponding k2 values, in the following order: α-CF3-TMC (IC50 = 120 nM) < α-Br-TMC (640 nM) < α-Cl-TMC (992 nM) < α-I-TMC (3.15 μM) < α-H-TMC (4.44 μM) < α-F-TMC (12.9 μM). The IC50 value for α-COOEt-TMC (6.65 μM) did not match the expected correlation. In addition, α-p-NO2-C6H4-TMC was considerably less active than one would have predicted from its k2 value. However, these observations are in agreement with our previous study and may be explained by electrophilicity-independent intracellular interactions or reactions. The relatively weak inhibition of NO production by α-CN-TMC and α-NO2-TMC (Table 1) could result from their very strong electrophilicity, possibly causing their inactivation.
α-X-TMCs inhibit TNF secretion by LPS-stimulated primary human macrophages
Having observed consistent effects in different assays, we selected four α-X-TMCs with anti-inflammatory activity (X = CF3, Br, Cl, and H) and two inactive α-X-TMCs (X = CN, NO2) for further study. We wanted to ascertain that anti-inflammatory effects can also be observed with primary human cells, not just with cell lines. Thus we tested the influence of the compounds on TNF secretion by LPS-stimulated primary human macrophages (Fig. 5A).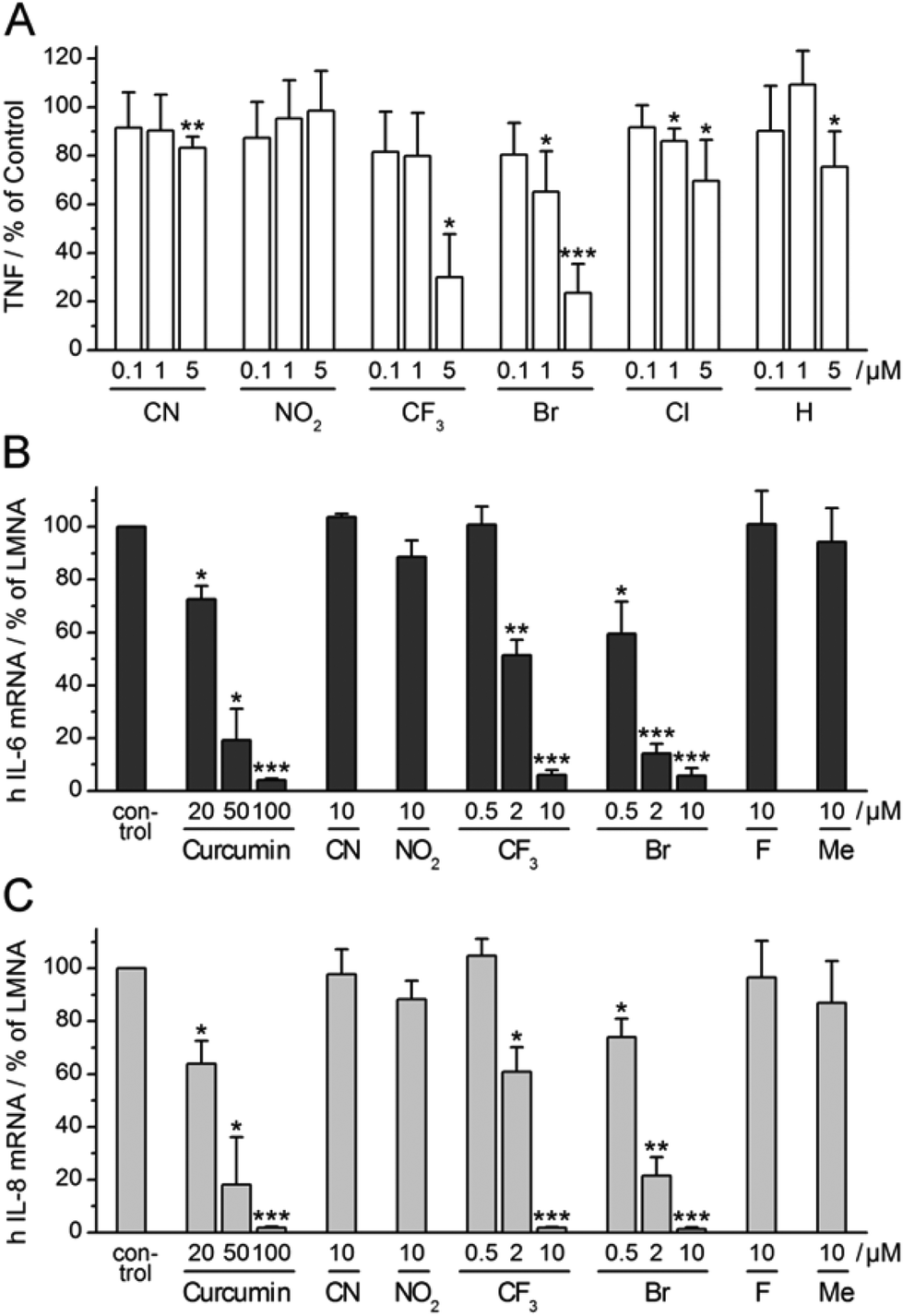 | ||
| Fig. 5 Influence of selected α-X-TMCs on LPS-induced cytokine expression. (A) TNF production of LPS-stimulated (100 ng mL−1) human primary macrophages after 24 h incubation without (control) or with α-X-TMCs; independent experiments with cells from four individual donors were performed. TNF was determined in cellular supernatants by (B) ELISA. (C) RT-PCR analysis of IL-6 and IL-8 mRNA levels in human HeLa cells relative to LMNA (human Lamin A/C). HeLa cells were pre-incubated with α-X-TMCs or DMSO (control) for 45–60 min prior to a 60 min stimulation with 30 ng mL−1 recombinant human TNF: (B) IL-6, (C) IL-8. Data represent four (A) or three (B), (C) independent experiments. Levels of significance: ***, p < 0.001; **, p < 0.01; *, p < 0.05. | ||
TNF levels in the cellular supernatant were measured by ELISA after 24 h. Using α-X-TMCs at a concentration of 5 μM, the following percentages of inhibition were found: α-Br-TMC 76%, α-CF3-TMC 70%, α-Cl-TMC 30%, α-H-TMC 25%, α-CN-TMC 17%, and α-NO2-TMC 0%. At a concentration of 1 μM only α-Br-TMC (35%) and α-Cl-TMC (14%) inhibited TNF release. The α-CF3-TMC response was highly variable amongst the four donors tested and therefore appeared less pronounced. We confirmed that decreases in TNF production were not caused by cell death (data not shown). This result demonstrated once again that α-Br-TMC, α-CF3-TMC and, to lesser extent, α-Cl-TMC interfere with proinflammatory pathways – in this case inhibiting TNF release.
α-X-TMCs inhibit expression of IL-6 and IL-8 in TNF-stimulated HeLa cells
Expression of the cytokines interleukin-6 (IL-6) and interleukin-8 (IL-8) is stimulated by NF-κB transcriptional activity and therefore predicted to be influenced by treatment with α-X-TMCs. We tested the two strongest (α-CN-TMC, α-NO2-TMC), two moderately strong (α-CF3-TMC and α-Br-TMC) and two poor to very poor electrophiles (α-F-TMC and α-Me-TMC). As expected, using TNF-stimulated HeLa cells, we found a strong reduction of IL-6 and IL-8 mRNA levels upon treatment with either α-CF3-TMC or α-Br-TMC (Fig. 5B and C). At 10 μM both compounds almost completely abolished the induction of IL-6 and IL-8 (94–99% inhibition). Whilst α-Br-TMC still showed a pronounced effect at 0.5 μM, α-CF3-TMC gave similar inhibition ratios at 2 μM, but not below. Both compounds were about 10 fold more active than the established NF-κB inhibitor curcumin14 which served as a positive control and reference (Fig. 5B and C).α-Br-TMC inhibits TNF-induced nuclear translocation of NF-κB p65 in HeLa cells
Having observed that NF-κB-dependent transcription of IL-6 and IL-8 was inhibited by α-CF3-TMC and α-Br-TMC, we aimed to confirm that this effect was due to the inhibition of NF-κB activation. We therefore monitored nuclear translocation of NF-κB in TNF stimulated HeLa cells in the presence or absence of 10 μM α-Br-TMC.While cytosolic levels of NF-κB p65 were largely unaffected by α-Br-TMC or TNF, TNF-induced nuclear translocation of NF-κB p65 was significantly reduced after pre-treatment with α-Br-TMC (Fig. 6). This result demonstrated that α-Br-TMC inhibits the NF-κB activation pathway upstream of nuclear translocation.
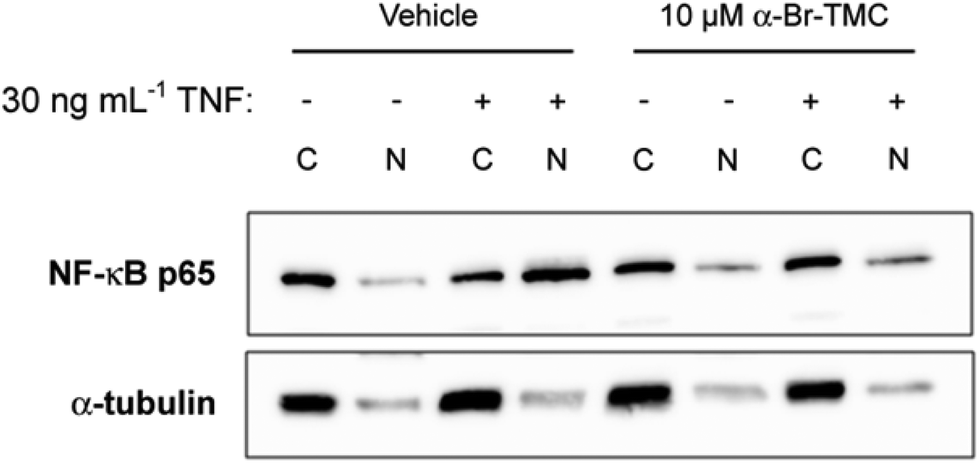 | ||
| Fig. 6 Inhibition of TNF-induced nuclear translocation of NF-κB p65 by α-Br-TMC. Western blot analysis of cytosolic (C) and nuclear (N) extracts stained with an anti-NF-κB p65 antibody. HeLa cells were pre-incubated with α-Br-TMC or DMSO (vehicle) for 45 min prior to a 15 min stimulation with 30 ng mL−1 recombinant human TNF. One representative blot out of three independent experiments is shown. Immunostaining of α-tubulin served as a quality control for cellular fractionation. | ||
Experimental
Chemicals and reagents
α-X-TMCs were prepared as described previously.8 Compounds were purchased from the following commercial sources and used without further purification: curcumin (B6938), hemin and gelatin (from cold water fish skin) from Sigma (Germany), NADPH from AppliChem (Germany), OPD (ortho-phenylenediamine dihydrochloride) from Acros Organics (Belgium), bilirubin from Frontier Scientific (UK), Triton X-100 from Merck (Germany), xanthohumol (XN) from Carl Roth (Germany) or, alternatively, XN was obtained as hop-derived ethanolic extract with 80% purity (Hallertauer Hopfenverwertungsgesellschaft, Mainburg, Germany) and purified by silica gel column chromatography with n-hexane–ethyl acetate as previously described (purity (HPLC) > 99%).15Cell viability test (MTT assay) on RAW264.7 cells
Murine macrophage RAW264.7 cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum (FCS) and 2 mM glutamine (Biochrom, Germany) at 37 °C in humidified air containing 5% CO2. In order to exclude cytotoxicity of the compounds a MTT assay was performed and the effect of the compounds on cell viability compared to control cells was determined. The results were published in a previous study of the α-X-chalcones.8bELISA-based HO-1 activity assay on RAW264.7 cells
The assay was performed as previously reported via in situ formation of bilirubin by HO-1/BVR activity and quantification of bilirubin by ELISA.9Briefly, RAW264.7 macrophages (8 × 104 cells per well) were placed in 96-well plates for 24 h and then treated with compounds for 3, 6 and 24 h. Controls received no compounds. After cell lysis (40 mM TRIS-HCl, pH 7.4, 250 mM sucrose, 137 mM NaCl, 10% (v/v) glycerin, 2 mM EDTA, 0.1% (v/v) Triton X-100, complete protease inhibitor cocktail, Roche Diagnostics, Germany) the HO-1 reaction mixture (40 mM TRIS-HCl, pH 7.4, 250 mM sucrose, 0.3 mM NADPH, 1.0 ng BVR (biliverdin reductase, Stressgen, Assay Designs, USA) and 2.5 μM hemin) was applied for 1 h. Bilirubin standards (0.50–2500 × 10−9 M bilirubin) were prepared in 40 mM TRIS-HCl, pH 7.4, 250 mM sucrose and combined with supernatant of whole cell lysates from control cells. Bilirubin was quantified by using an excess of the anti-bilirubin antibody 24G7 from mouse (Shino-Test, Japan, 0.57 μg μL−1 in 1% G-PBS with 0.5 mM sodium salicylate) and subsequently analyzing the unbound 24G7 by ELISA. To trap 24G7 an immunoplate (Nunc, Denmark) coated with a bilirubin-BSA conjugate (0.35 μg protein per well) was utilized. Detection was done using a HRP-conjugated anti-mouse antibody from goat (Rockland, USA; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10000) with a freshly prepared substrate solution (0.40 g mL−1 OPD and 0.40 μL mL−1 30% H2O2 in citrate buffer, pH 5.0). After quenching with aqueous 3 M H2SO4 the absorbance was measured at 492 nm (Multiskan Spectrum, Thermo, Finland). The sigmoidal calibration curve was fit to a four parameter logistic equation to determine unknown bilirubin concentrations. HO activity was calculated as pmol bilirubin formed per hour and per milligram of protein (pmol BR h−1 mg−1) and expressed as fold HO-1 activity compared to control cells.
:10000) with a freshly prepared substrate solution (0.40 g mL−1 OPD and 0.40 μL mL−1 30% H2O2 in citrate buffer, pH 5.0). After quenching with aqueous 3 M H2SO4 the absorbance was measured at 492 nm (Multiskan Spectrum, Thermo, Finland). The sigmoidal calibration curve was fit to a four parameter logistic equation to determine unknown bilirubin concentrations. HO activity was calculated as pmol bilirubin formed per hour and per milligram of protein (pmol BR h−1 mg−1) and expressed as fold HO-1 activity compared to control cells.
AREc32 (human mammary epithelial adenocarcinoma MCF7) cells
AREc32 is a reporter cell line derived from the human mammary epithelial adenocarcinoma MCF7 cells. It contains a luciferase reporter gene construct under the control of eight copies of the Nrf2 binding site ARE.11 AREc32 cells were cultured at 37 °C and 5% CO2 in a humidified atmosphere in complete DMEM medium supplemented with 10% heat-inactivated FCS, 2 mM glutamine, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (medium and all supplements from Gibco, USA), and maintained under antibiotic selection (0.8 mg mL−1 G418 (Sigma, Germany)).Cytotoxicity assay on α-X-TMC-stimulated AREc32 cells
Biomass content was assessed by sulforhodamine B (SRB, Sigma, Germany) staining.12 Briefly, AREc32 cells were seeded and treated as for the luciferase reporter assay. Detached cells in the supernatant were discarded and attached cells were fixed with 10% trichloroacetic acid for 30 min at 4 °C. Then, the plate was rinsed with distilled water, dried and stained for 15 min with a 0.4% solution of SRB in 1% acetic acid, washed several times with 1% acetic acid and dried again. The dye was eluted with 10 mM TRIS base followed by the measurement of OD at 490 nm and 515 nm (SpectraMax M5e Microplate reader, Molecular Devices, USA).Luciferase reporter assay on α-X-TMC-stimulated AREc32 cells
AREc32 cells were seeded at a density of 9 × 103 per well in 50 μL growth medium in 384-well plates. After 22 h compounds were added to obtain final concentrations in the range between 1.56–100 μM. Each concentration was tested in quadruplicate. An equal volume of vehicle was added to the control wells (0.016–1% DMSO). The reporter assay was performed as described previously.11 For normalization all values were divided by vehicle control values, yielding the fold change relative to the control. Further, the luciferase activity was normalized to biomass content.iNOS inhibition test (Griess assay) on RAW264.7 cells
The produced NO which is accumulated as nitrite in the culture medium was quantified using the Griess reaction. RAW264.7 macrophages (8 × 104 cells per well) were plated in 96-well plates and allowed to attach for 24 h. Stock solutions of compounds were prepared in DMSO (100 mM) and stored at −20 °C. Test concentrations were freshly prepared by diluting the stock solution in culture medium in presence and absence of 10 ng mL−1 LPS and then incubated with the cells for 24 h. The final concentration of DMSO was ≤0.025% and the total assay volume was 100 μL. 50 μL of culture medium was mixed with 50 μL of Griess reagent (0.1% NED (N-1-naphtylethylenediamine dihydrochloride), 1% sulfanilamine, 0.35% phosphoric acid in water) and incubated for 15 min at room temperature. The absorbance was measured at 560 nm (Multiskan Spectrum, Thermo, Finland) at 25 °C and nitrite concentrations were calculated from a standard curve established with serial dilutions of NaNO2 (0/5/10/25/50 μM) in culture medium. Nitrite production was expressed as percent compared to control cells incubated with 10 ng mL−1 of LPS.Quantification of TNF in LPS-stimulated human macrophages treated with α-X-TMCs
Macrophages were differentiated from human monocytes, obtained by leukapheresis of healthy donors, followed by density gradient centrifugation over Ficoll/Hypaque and separation by countercurrent centrifugation (J6M-E centrifuge, Beckmann, Germany) as described previously.16 The purity (> 85%) of the monocytes was determined by morphology and the expression of CD14 antigen. For the differentiation of macrophages, monocytes were cultured on teflon foils for 7 days at a concentration of 1 × 106 cells mL−1 in RPMI 1640, 10% human AB-serum (both from PAN Biotech, Germany), 2 mM glutamine and 50 U mL−1 penicillin–50 mg mL−1 streptomycin (all from Gibco/Life Technologies, Germany) at 5% CO2 and 37 °C. Thereafter, macrophages were harvested, counted and 0.2 × 106 cells in 1 mL incubated overnight in 24-well plates with 100 ng mL−1 LPS (Enzo Life Sciences, Germany) for maturation with or without the addition of α-X-TMCs with X = CN, NO2, CF3, Br, Cl, H (0.5, 1, 5 μM). After 24 h, supernatants were harvested, frozen and later analyzed for cytokine production according to the manufacturer's instructions (DuoSet ELISA, R&D Systems, Germany).Inhibition of NF-κB activation assay in human HeLa cervical cancer cells
HeLa cells (a kind gift from Daniela Männel, Institute of Immunology, University of Regensburg) were grown in RPMI 1640 supplemented with 10% heat-inactivated FCS and 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (all PAN-Biotech, Germany). The potential inhibitory effect of synthetic chalcones and curcumin on NF-κB activation was measured by incubating HeLa cells for 45–60 min with the indicated concentration of compounds or DMSO as vehicle, prior to stimulation with recombinant human TNF (30 ng mL−1; gift from Daniela Männel) for 15 min (Western blot analysis) or 60 min (gene expression analysis). DMSO was adjusted to 0.01% (Western blot analysis) or 0.1% (gene expression analysis) in all conditions. Following TNF stimulation, cells were washed in cold PBS and harvested for either gene expression or Western blot analysis. For gene expression analysis, cells were lysed in the iScript RT-qPCR sample preparation reagent (Bio-Rad Laboratories, Germany) and subjected to cDNA synthesis using the iScript cDNA Synthesis kit (Bio-Rad Laboratories, Germany), following the manufacturer's instructions. Quantitative PCR was performed on a RotorGene Q (Qiagen, Germany) with SYBR Green I and HotStarTaq (Qiagen, Germany). Data were normalized to human Lamin A/C (LMNA) mRNA as previously described,17 and expressed as relative mRNA levels (% of vehicle or control). Forward and reverse real-time PCR primers specific for human LMNA, IL-6 and IL-8 have been described.18 Data shown represent the mean of three independent experiments. For Western blot analysis, cells were scrapped off mechanically; nuclear and cytosolic protein fractionation and NF-κB p65 immunoblotting were performed as previously described.18 Quality of the cytosolic and nuclear extraction was verified by immunoblotting with an antibody directed against the cytosolic marker α-tubulin (Santa-Cruz Biotechnology, Germany; 1:200). Chemoluminescence detection was performed using Amersham ECL Prime (GE Healthcare Life Sciences, Germany) and images were captured on an ImageQuant LAS 4000 mini imaging system (GE Healthcare Life Sciences, Germany). The immunoblot shown is representative of three independent experiments.
Data evaluation/statistical analysis
The data is presented as the mean ± SD of several independent experiments (as indicated in figure captions) carried out in duplicates, triplicates or quadruplicates depending on the assay. A sigmoidal logistic function was used to fit dose–response curves and determine IC50 values using a Microsoft Excel calculation sheet (Ed50plus, v1.0, April 2000, Mario H. Vargas, Instituto Nacional de Enfermedades, Mexico). Comparison between groups was made using a two-sided paired Student's t test. A p value < 0.05 was considered statistically significant. Levels of significance: ***, p < 0.001; **, p < 0.01; *, p < 0.05.Conclusion
The direct covalent modification of protein thiols by alkylating agents can activate Nrf2 and inhibit NF-κB and hence lead to the dampening of inflammatory responses.4 To exploit this phenomenon for therapeutic interventions, fine tuning of electrophilicity is desirable. If the electrophilicity is too low, the compound is unlikely to be effective at concentrations that are pharmacologically accessible. If the electrophilicity is too high, the compound may react non-specifically with a broad range of protein thiols or may be neutralized by glutathione conjugation. In contrast, compounds with intermediate electrophilicity may avoid rapid clearance and target a relatively narrow range of proteins, namely those with exposed and/or activated thiolate residues poised to be involved in protein redox regulation, like those in Keap1 or NF-κB.Thus, it appears critical to hit the right thiol reactivity window.19 We pursued the strategy to first classify electrophiles by in vitro kinetics (second order rate constant determination for the reaction with cysteamine), followed by probing the same electrophiles for their cellular effects. A similar approach used theoretical chemistry calculations to predict the electrophilicity of aryl nitriles, successfully explaining structure activity relationships for the inhibition of cysteine proteases.20
Out of fourteen α-X-TMCs, three compounds stood out as potent anti-inflammatory agents, namely α-CF3-TMC, α-Br-TMC and α-Cl-TMC. These were highly efficient in activating Nrf2 and inhibiting NF-κB, with corresponding effects on their respective transcriptional gene products. Notably, suppression of iNOS activity was observed with IC50 values in the nanomolar range. Although alternative electrophilicity-independent modes of action cannot be strictly ruled out at this point, the inactivity of the most electrophilic compounds α-CN-TMC and α-NO2-TMC can be rationalized by their predicted rapid reaction with intracellular GSH, which is present at millimolar concentrations.21
As a scaffold the chalcones facilitate straight forward access to libraries of fine-tuned electrophiles, allowing detailed studies of the relationship between electrophilicity and biological activity. In future work such electrophiles may be linked to molecules containing non-covalent recognition motifs, thus directing the electrophilic warhead22 to particular protein targets.23
Acknowledgements
We thank Dita Fritsch, Susanne Brueggemann and Monika Wehrstein for technical assistance and the Fonds der Chemischen Industrie (Liebig scholarship to S.A.) and the DAAD (Ph. D. scholarship to N.A.) for financial support. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement n° 215009 (L.B.-J.), the Deutsche Krebshilfe (Grant n° 109750 to A.R.) and the Deutsche Forschungsgemeinschaft (Grant n° RA 2010/2-1 to A.R.). The AREc32 cell line used in this study was provided by Roland Wolf and Cancer Research UK.Notes and references
- (a) R. Foresti, M. Hoque, D. Monti, C. J. Green and R. Motterlini, J. Pharmacol. Exp. Ther., 2005, 312, 686–693 CrossRef CAS PubMed; (b) H. Abuarqoub, R. Foresti, C. J. Green and R. Motterlini, Am. J. Physiol. Cell Physiol., 2006, 290, C1092–C1099 CrossRef CAS PubMed; (c) S. Lee, J. Kim, G. Seo, Y. C. Kim and D. Sohn, Inflammation Res., 2009, 58, 257–262 CrossRef CAS PubMed; (d) Y. Nakamura, C. Yoshida, A. Murakami, H. Ohigashi, T. Osawa and K. Uchida, FEBS Lett., 2004, 572, 245–250 CrossRef CAS PubMed.
- (a) A. T. Dinkova-Kostova, M. A. Massiah, R. E. Bozak, R. J. Hicks and P. Talalay, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3404–3409 CrossRef CAS PubMed; (b) A. T. Dinkova-Kostova, W. D. Holtzclaw, R. N. Cole, K. Itoh, N. Wakabayashi, Y. Katoh, M. Yamamoto and P. Talalay, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11908–11913 CrossRef CAS PubMed.
- A. Rahman and F. Fazal, Proc. Am. Thor. Soc., 2011, 8, 497–503 CrossRef CAS PubMed.
- V. Pande, S. F. Sousa and M. J. Ramos, Curr. Med. Chem., 2009, 16, 4261–4273 CrossRef CAS.
- K. B. Harikumar, A. B. Kunnumakkara, K. S. Ahn, P. Anand, S. Krishnan, S. Guha and B. B. Aggarwal, Blood, 2009, 113, 2003–2013 CrossRef CAS PubMed.
- M. B. Toledano, D. Ghosh, F. Trinh and W. J. Leonard, Mol. Cell. Biol., 1993, 13, 852–860 CAS.
- A. J. L. Chia, C. E. Goldring, N. R. Kitteringham, S. Q. Wong, P. Morgan and B. K. Park, Biochem. Pharmacol., 2010, 80, 410–421 CrossRef CAS PubMed.
- (a) S. Amslinger, N. Al-Rifai, K. Winter, K. Wörmann, R. Scholz, P. Baumeister and M. Wild, Org. Biomol. Chem., 2013, 11, 549–554 RSC; (b) N. Al-Rifai, H. Rücker and S. Amslinger, Chem. – Eur. J., 2013, 19, 15384–15395 CrossRef CAS PubMed.
- H. Rücker and S. Amslinger, Free Radicals Biol. Med., 2015, 78, 135–146 CrossRef PubMed.
- T. J. Schmidt, Bioorg. Med. Chem., 1997, 5, 645–653 CrossRef CAS.
- X. J. Wang, J. D. Hayes and C. R. Wolf, Cancer Res., 2006, 66, 10983–10994 CrossRef CAS PubMed.
- V. Vichai and K. Kirtikara, Nat. Protocols, 2006, 1, 1112–1116 CAS.
- (a) E. Park, M. R. Quinn, C. E. Wright and G. Schullerlevis, J. Leukocyte Biol., 1993, 54, 119–124 CAS; (b) B. Kraus, H. Wolff, E. F. Elstner and J. Heilmann, Naunyn–Schmiedeberg's Arch. Pharmacol., 2010, 381, 541–553 CrossRef CAS PubMed.
- T. Esatbeyoglu, P. Huebbe, I. M. A. Ernst, D. Chin, A. E. Wagner and G. Rimbach, Angew. Chem., Int. Ed., 2012, 51, 5308–5332 CrossRef CAS PubMed.
- C. Gerhäuser, A. Alt, E. Heiss, A. Gamal-Eldeen, K. Klimo, J. Knauft, I. Neumann, H.-R. Scherf, N. Frank, H. Bartsch and H. Becker, Mol. Cancer Ther., 2002, 1, 959–969 Search PubMed.
- R. Andreesen, C. Scheibenbogen, W. Brugger, S. Krause, H.-G. Meerpohl, H.-G. Leser, H. Engler and G. W. Löhr, Cancer Res., 1990, 50, 7450–7456 CAS.
- (a) A. Rascle, J. A. Johnston and B. Amati, Mol. Cell. Biol., 2003, 23, 4162–4173 CrossRef CAS; (b) A. Rascle and E. Lees, Nucleic Acids Res., 2003, 31, 6882–6890 CrossRef CAS PubMed; (c) B. Basham, M. Sathe, J. Grein, T. McClanahan, A. D'Andrea, E. Lees and A. Rascle, Nucleic Acids Res., 2008, 36, 3802–3818 CrossRef CAS PubMed.
- A. Rascle, T. Neumann, A.-S. Raschta, A. Neumann, E. Heining, J. Kastner and R. Witzgall, Exp. Cell Res., 2009, 315, 76–96 CrossRef CAS PubMed.
- D. S. Johnson, E. Weerapana and B. F. Cravatt, Future Med. Chem., 2010, 2, 949–964 CrossRef CAS.
- V. Ehmke, J. E. Q. Quinsaat, P. Rivera-Fuentes, C. Heindl, C. Freymond, M. Rottmann, R. Brun, T. Schirmeister and F. Diederich, Org. Biomol. Chem., 2012, 10, 5764–5768 CAS.
- B. Ketterer, B. Coles and D. J. Meyer, Environ. Health Perspect., 1983, 49, 59–69 CrossRef CAS.
- S. Serim, U. Haedke and S. H. L. Verhelst, ChemMedChem, 2012, 7, 1146–1159 CrossRef CAS PubMed.
- J. Jacobs, V. Grum-Tokars, Y. Zhou, M. Turlington, S. A. Saldanha, P. Chase, A. Eggler, E. S. Dawson, Y. M. Baez-Santos, S. Tomar, A. M. Mielech, S. C. Baker, C. W. Lindsley, P. Hodder, A. Mesecar and S. R. Stauffer, J. Med. Chem., 2012, 56, 534–546 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: inhibition of NO production and Table S1: C10 values of Nrf2 induction. See DOI: 10.1039/c4ob02301c |
| This journal is © The Royal Society of Chemistry 2015 |