
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlow synthesis of ethyl isocyanoacetate enabling the telescoped synthesis of 1,2,4-triazoles and pyrrolo-[1,2-c]pyrimidines†
Marcus
Baumann
a,
Antonio M.
Rodriguez Garcia
ab and
Ian R.
Baxendale
*a
aDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: i.r.baxendale@durham.ac.uk
bUniversidad de Castilla-La Mancha, Departamento de Química Orgánica, Facultad de Ciencias y Tecnologías Químicas, Avd. Camilo José Cela, 10, 13071 Ciudad Real, Spain
First published on 26th February 2015
Abstract
The efficient flow synthesis of important heterocyclic building blocks based on the 1,2,4-triazole and pyrrolo[1,2-c]pyrimidine scaffold has been achieved. Crucially, a telescoped continuous flow process was developed based on the reaction of N-formylglycine with triphosgene to deliver a stream of ethyl isocyanoacetate in situ, which subsequently yielded the desired heterocyclic entities in a telescoped reaction. Additionally, the functionalisation of the pyrrolo[1,2-c]pyrimidine core via subsequent SEAr reactions was studied revealing insight into a ‘halogen dance’ phenomenon associated with these medicinally relevant architectures.
Introduction
The efficient preparation of novel heterocyclic building blocks functionalised with suitable derivatisation sites is of paramount importance to the successful discovery of new bioactive molecules.1 Certainly the most effective pharmaceutical and agrochemical strategy for the generation of propriety leads is through screening of diverse core structures, which upon strategic functionalisation engenders desirable physiological properties and yields focused libraries of advanced drug-like structures. Consequently, the conformational presentation displayed by the core template impacts strongly on the available binding and contact points that can be interacted upon within the biological environment. It is therefore not surprising that most of today's top-selling drugs contain at least one heterocyclic core often acting as its pharmacophore.2 In order to efficiently access new chemical scaffolds medicinal chemists have been increasingly exploiting enabling technologies to allow access to novel chemical space.3 Amongst these technologies flow chemistry approaches have gained widespread acceptance as a powerful means to overcome limitations regarding scale-up, reproducibility, safety and accessible chemical space with numerous publications detailing the results of such studies.4One particular area where flow chemistry has led to significantly improved safety profiles compared to traditional batch synthesis is the in situ generation and subsequent use of highly reactive, yet hazardous species. Examples include, but are not limited to, the generation and use of ozone,5 azides,6 diazonium species,7 or elemental fluorine8 as well as cryogenic9 or high temperature10 reactions.
In this paper we wish to disclose our latest results towards the flow synthesis of valuable heterocyclic architectures such as 1,2,4-triazoles and pyrrolo[1,2-c]pyrimidines based on the in situ generation of ethyl isocyanoacetate through the dehydration of N-formylglycine using reactive triphosgene (Scheme 1).
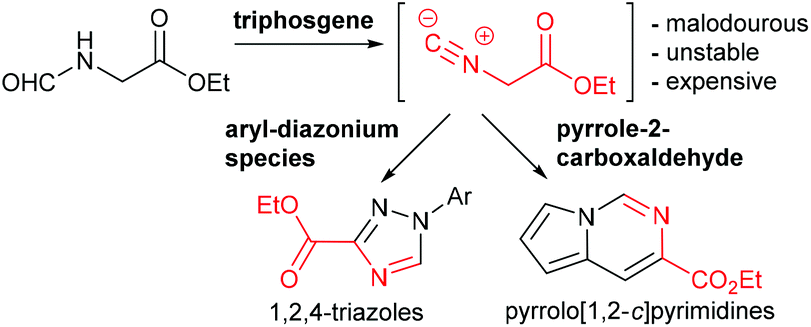 | ||
| Scheme 1 Multi-step flow approach towards valuable heterocyclic scaffolds. | ||
Results and discussion
Synthesis of ethyl isocyanoacetate in flow
Ethyl isocyanoacetate (3) is a commercially available high boiling liquid characterised by its pungent odour as well as its sensitivity towards light and moisture. Despite this it is frequently used as a key building block in multicomponent reactions and heterocycle synthesis.11 Because of these features we decided to develop a convenient in situ flow synthesis of ethyl isocyanoacetate starting from readily available N-formylglycine (1).12 As the dehydrating agent we choose triphosgene (2), a commercially available crystalline solid.13 This material is not only easier to handle than many other dehydrating agents (e.g. COCl2, POCl3, SOCl2etc.), but also comprises three equivalents of active dehydrating agent per molecule. Furthermore, only HCl and CO2 are generated as by-products through its use. In an initial study we used a commercially available R2+ Vapourtec flow reactor which was configured to mix two reagent streams via a T-piece: stream A contained N-formylglycine (DCM, 0.25 M, 1.0 equiv.), DIPEA (2 equiv.) and DMAP (0.3 equiv.) and stream B contained triphosgene (DCM, 0.0875 M, 0.35 equiv.). Using 1H-NMR analysis of aliquots of the product stream it was quickly established that the desired ethyl isocyanoacetate forms almost quantitatively at room temperature within a 20 min residence time (Scheme 2).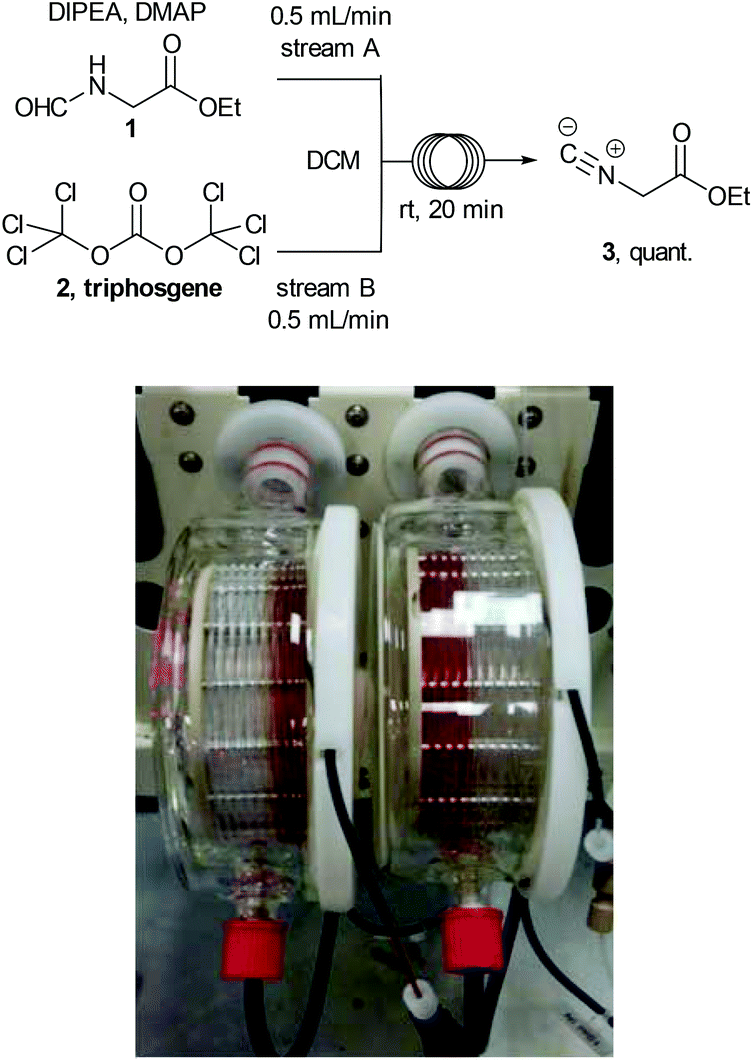 | ||
| Scheme 2 Flow synthesis of ethyl isocyanoacetate (Reactor flow pathway left to right). | ||
Synthesis of 1,2,4-triazoles in flow
Having developed an efficient protocol for the synthesis of ethyl isocyanoacetate in flow, we next turned our attention to its reaction with various aryl diazonium species, also prepared in situ, to furnish a convergent synthesis of 1,2,4-triazoles.14To facilitate the reaction, four stock solutions (solutions 1–4) were prepared, along with a small collection of aniline starting materials (solution 5). To perform the multi-step flow sequence an integrated flow reactor configured as shown in Scheme 3 was constructed. This comprised a Uniqsis FlowSyn with an ALF delivery and control system, a Vapourtec R2+/R4 unit, two auxiliary Knauer K100 HPLC pumps and two Polar bear plus flow reactors (see ESI† for additional details). Using this setup a continuous flow stream of intermediate 3 was prepared as described (see above) and was united with a solution containing the corresponding diazonium coupling partner 6 as a convergent assembly process.
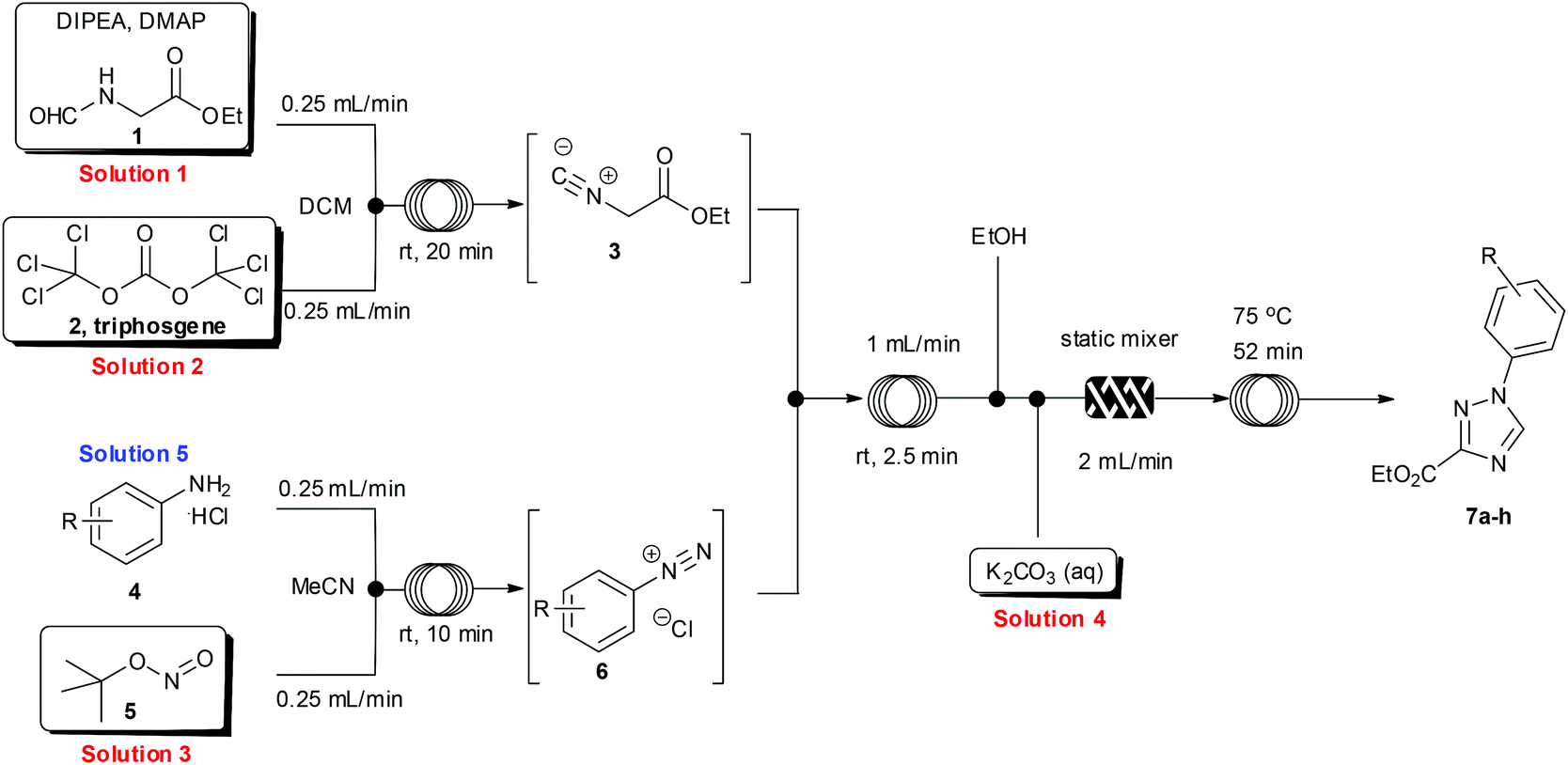 | ||
| Scheme 3 Multi-step flow approach towards 1,2,4-triazoles. | ||
The diazonium component 6 was readily prepared from the precursor aniline or its mono-hydrochloride salts (in MeCN) by treatment with tert-butyl nitrite at ambient temperature.15 For the subsequent cycloaddition reaction the two flow streams containing 3 and 6 were combined and then in succession diluted with a stream of pure ethanol and treated with an aqueous feed of potassium carbonate (5 equiv.). A static mixer was placed in-line to thoroughly blend the flow stream before it passed into a heated reaction coil maintained at 75 °C. The reactor output was collected and worked up by evaporation of the solvent followed by neutralisation with dilute hydrochloric acid and partitioning into ethyl acetate. The products were isolated in high yield and purity after solvent evaporation and trituration of the crude product with a mixture of diethyl ether–ethanol 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Pleasingly the above flow protocol proved general and was applied to the synthesis of a small selection of 1,2,4-triazole products (7a–h) (Fig. 1).
:1. Pleasingly the above flow protocol proved general and was applied to the synthesis of a small selection of 1,2,4-triazole products (7a–h) (Fig. 1).
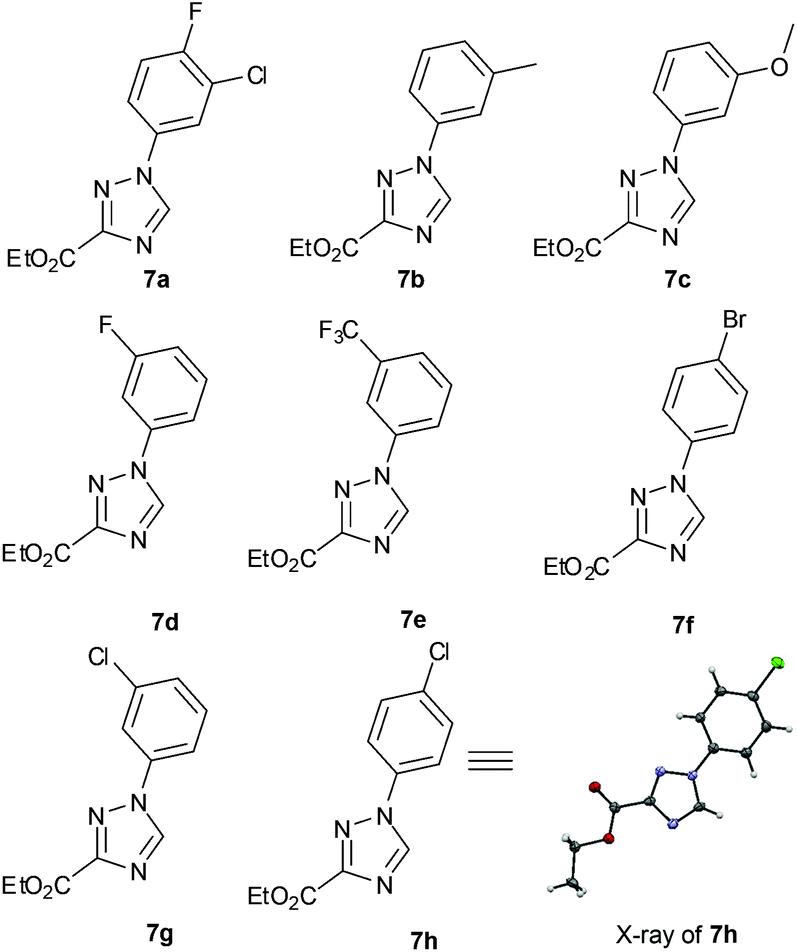 | ||
| Fig. 1 Representative [1,2,4]-triazoles prepared in flow. | ||
Derivatisation of 1,2,4-triazoles
In order to briefly evaluate the feasibility of these [1,2,4]-triazoles (7a–h) to serve as building blocks towards future research efforts we decided to specifically study the functionalisation of the 5-position of the heterocyclic core. To this end regioselective bromination was affected by treating 7h with a mixture of NaH and NBS in dry THF16 (Scheme 4). Pleasingly, after 10 h reaction time, complete consumption of the starting material and clean formation of the desired product 8 was observed. After purification 8 was subjected to a small number of Suzuki cross coupling reactions in order to synthesise 9a–e.17 Although this reaction required prolonged reaction times at elevated temperatures (8–10 h at 120 °C) the desired Suzuki cross coupling products were formed chemoselectively and isolated in reasonable yield allowing further efforts to be directed towards the diversification of these scaffolds.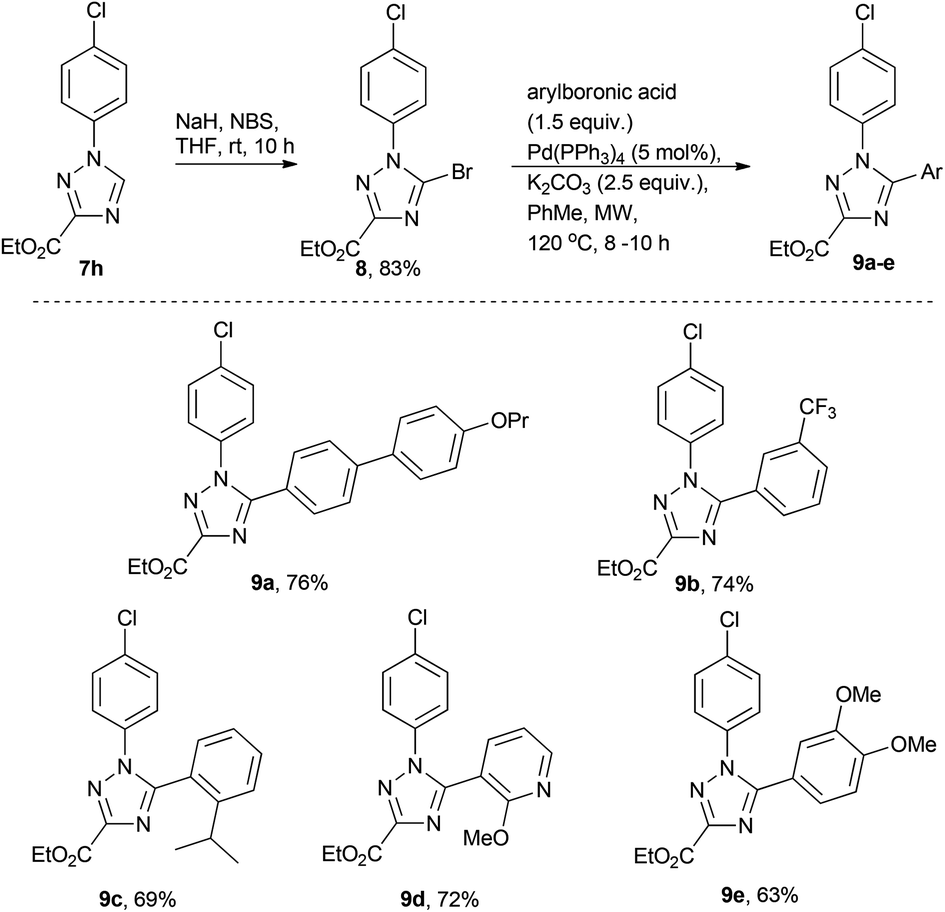 | ||
| Scheme 4 Functionalization study of triazole 7h. | ||
Synthesis of pyrrolo[1,2-c]pyrimidines in flow
After accomplishing a successful multi-step flow synthesis of various 1,2,4-triazoles we decided to diversify the protocol towards the synthesis of another underexploited scaffold – the pyrrolo[1,2-c]pyrimidine. This structure has shown value for example as the core structure of various nicotinic acetylcholine receptor agonists developed to treat CNS disorders18 and can be prepared by reacting ethyl isocyanoacetate with readily available pyrrole-2-carboxaldehyde.19 After a short optimisation study we found that treatment of a flow stream of ethyl isocyanoacetate with a secondary stream containing pyrrole-2-carboxaldehyde (10, 0.25 M, 1.0 equiv.) and piperidine (6 equiv.) in DCM afforded the desired pyrrolo[1,2-c]pyrimidine structure 11 after passing through a flow coil maintained at slightly elevated temperature (85 °C, 26 min residence time, Scheme 5; Fig. 2).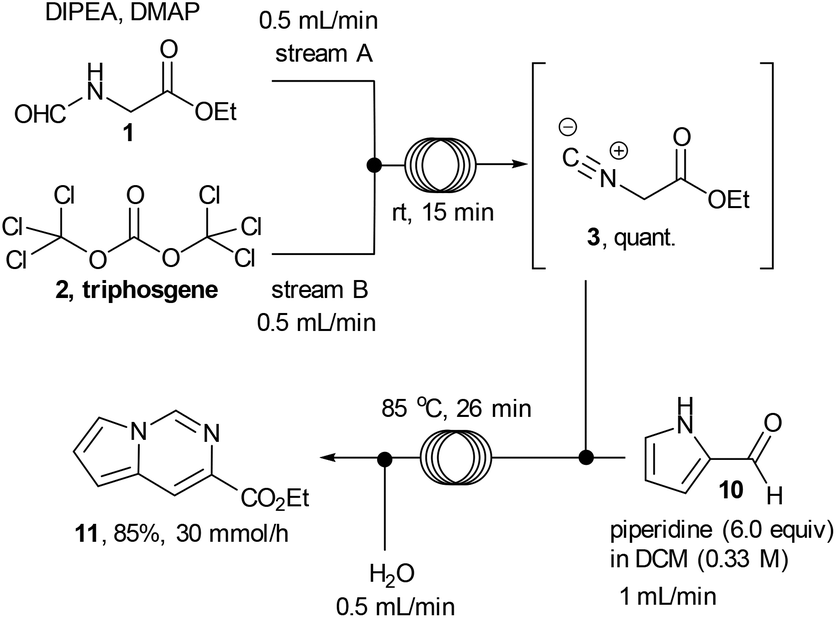 | ||
| Scheme 5 Multi-step flow approach towards pyrrolo[1,2-c]pyrimidine. | ||
 | ||
| Fig. 2 Flow reactor used for the synthesis of pyrrolo[1,2-c]pyrimidine. | ||
In order to avoid any issues of precipitate formation within the flow system (i.e. piperidine-HCl salts) a working concentration of 0.25 M was targeted resulting in a theoretical throughput of 5.71 g h−1. The desired pyrrolo[1,2-c]pyrimidine product could be isolated in 85% yield and high purity (>95% by 1H-NMR) as a beige solid after extraction and evaporation of the volatiles. This crude material was of sufficient quality to be used in further reactions however an analytical sample was further purified by column chromatography to furnish a yellow solid (78%). The reactor could be run under semi-continuous operation delivering ∼30 g over the course of a standard working day (8 h) including start-up/shut down and cleaning of the system.
Having established convenient access to multigram quantities of this pyrrolo[1,2-c]pyrimidine core (11) we decided to evaluate its functionalisation via electrophilic aromatic substitution reactions. Specifically we intended to establish whether this scaffold could be decorated in a regioselective manner allowing access to versatile architectures for future synthesis programmes. We therefore performed a number of Density Functional Theory (DFT) calculations to compare the intermediates originating from addition to the most likely C7 and C5 positions of the pyrrole subunit (Scheme 6, Table 1). The geometries of the species were optimised with the 6-311++G(2df,2pd) basis set (for iodine the cc-pVTZ-PP basis set20 with the Stuttgarts pseudopotential21 was used) employing Truhlar functionals, M06-2X,22 which have been shown to successfully describe the thermochemistry, kinetics, and noncovalent interactions of such systems. The computational calculations were performed using Gaussian 09 program suite;23 frequency analysis was also performed to confirm the nature of the stationary points and to obtain zero-point energies (ZPEs). Solvent effects on molecular geometries and energies were estimated by means of Polarization Continuum Models (PCM)24 with DCM as the solvent.
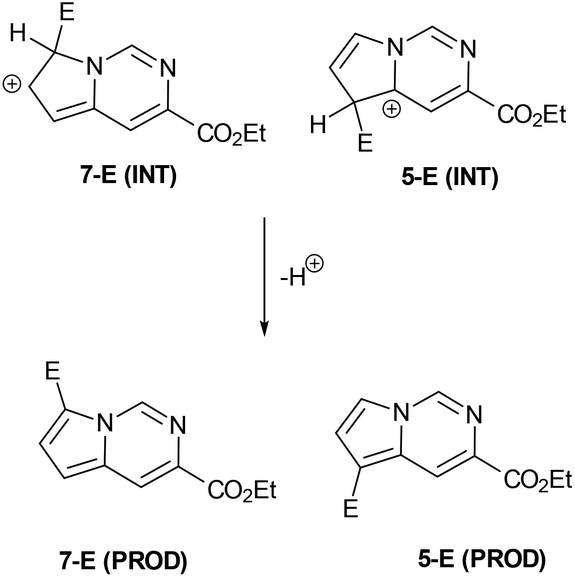 | ||
| Scheme 6 General schematic of the DFT calculations performed on the electrophilic attack on the pyrrolopyrimidine moiety. | ||
The data correlated in Table 1, implies a distinct kinetic preference for reaction at the C7 over C5 position due to enhanced delocalisation into the adjacent ring, however the most stable products are those with substitution at C5. Further DFT studies, also confirmed the nucleophilic character of the specified C5 and C7 positions of the pyrrolo[1,2-c]pyrimidine (Fig. 3). We were therefore confident that a regioselective functionalisation of the pyrrolopyrimidine could be achieved.
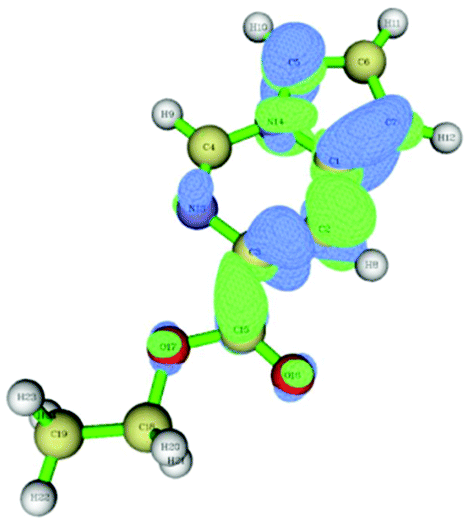 | ||
| Fig. 3 Dual descriptor Fukui function of pyrrolo[1,2-c]pyrimidine (11). (green electrophilic regions; blue nucleophilic regions). | ||
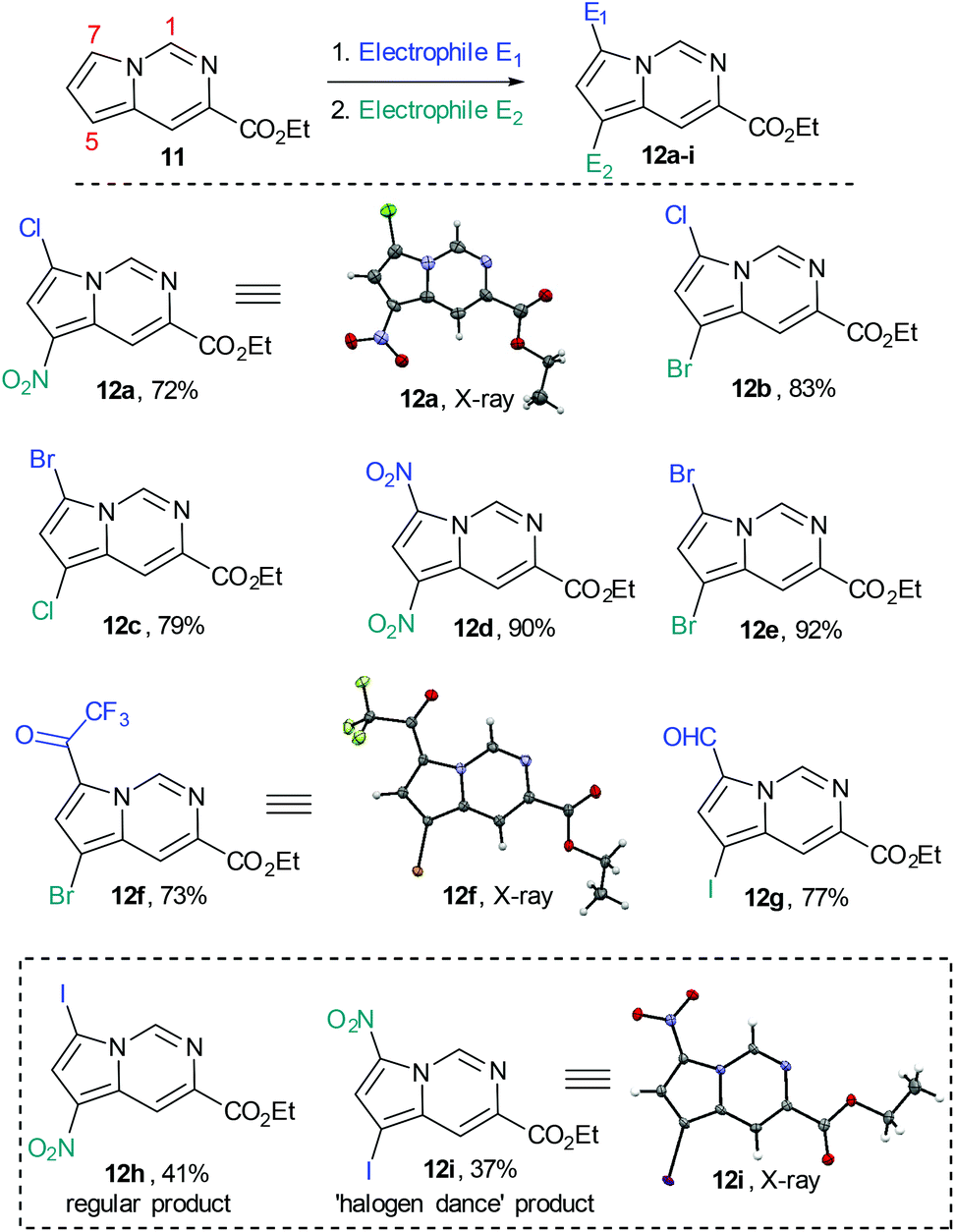
We subsequently prepared a number of pyrrolo[1,2-c]pyrimidine derivatives (12a–i) confirming that the most reactive site was indeed the C7 position of the pyrrole moiety followed by the C5 position. This analysis was largely enabled by single crystal X-ray diffraction experiments as 1H- and 13C-NMR experiments were not always conclusive (Scheme 7).
 | ||
| Scheme 7 Functionalization of the pyrrolo[1,2-c]pyrimidine core by SEAr reactions (overall isolated yields for 2 steps). | ||
Whilst this strategy allowed for the efficient and regioselective monofunctionalisation at C7 via halogenation (Cl, Br. I), formylation and acetylation reactions, it was noted that the subsequent introduction of a second electrophilic substituent did not always furnish the predicted product.
The nitration or bromination of the chlorinated pyrrolo[1,2-c]pyrimidine intermediate did yield the expected reaction products 12a and 12b. However, preparation of the dihalo-structure 12c was only possible when halogenating (use of NBS or NCS) under neutral conditions. Conducting the chlorination step in the presence of HCl or for prolonged reaction times (i.e. >10 h) resulted in the formation of a mixture of 12b and 12c.
Additionally, when attempting nitration of substrate 8 it was noted that a mixture of two mono-nitrated products as well as the di-nitrated scaffold 12d formed very rapidly. Furthermore, attempted nitration of the 7-mono-iodinated pyrrolo[1,2-c]pyrimidine core yielded a mixture of nitrated products (12h, 12i) that after longer reaction time (12 h) would also generate small quantities (∼15%) of the previously prepared di-nitrated product 12d. As the identity of 12i had been confirmed by single crystal X-ray diffraction experiments, we suggest the occurrence of a ‘halogen dance’ phenomenon in cases where brominated or iodinated monofunctionalisation products are subjected to a second SEAr reaction under strongly acidic conditions. We thus propose that under acidic conditions the second electrophile is again first introduced at C7 generating a tetrahedral intermediate 13b (Scheme 8). This structure can then undergo a [1,3]-shift in which the most weakly bound halogen migrates to the 5-position of the pyrrolo[1,2-c]pyrimidine scaffold (i.e.13c) thus resembling other ‘halogen dance’ phenomena reported in the literature.25 Overall, the strategic use of this rearrangement could lead to otherwise difficult to access structures and thus opens avenues towards specifically functionalised pyrrolo[1,2-c]pyrimidine scaffolds. Further research is now required to fully evaluate and develop these initial findings.
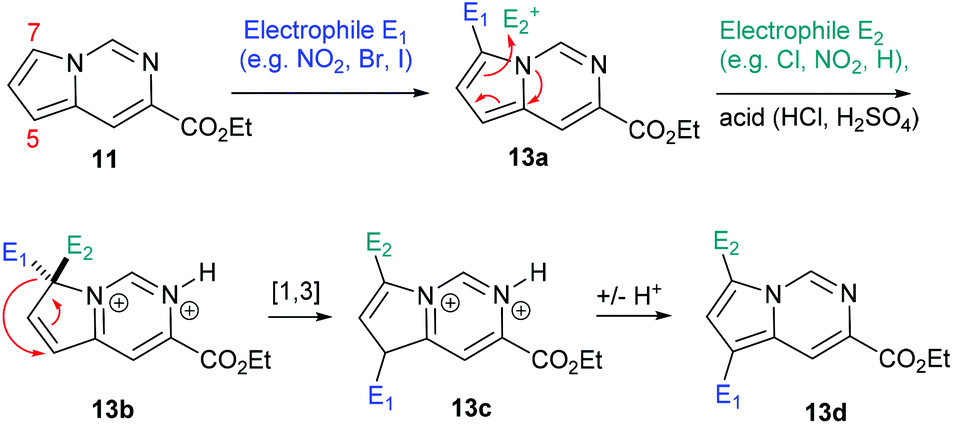 | ||
| Scheme 8 Proposed mechanism for ‘halogen dance’ under acidic conditions. | ||
Conclusions
In conclusion, we have successfully developed a valuable flow protocol for the synthesis of ethyl isocyanoacetate via dehydration of N-formylglycine using triphosgene. The versatility of generating this isocyanide building block in situ was further exploited by multi-step sequences towards various 1,2,4-triazoles as well as the pyrrolo[1,2-c]pyrimidine scaffold, whose further functionalisation by means of SEAr reactions has been demonstrated. These studies have also allowed insights in the occurrence of halogen dance phenomena during the elaboration of the pyrrolo[1,2-c]pyrimidine scaffold. We believe that this study demonstrates the benefits of modern flow techniques in the generation and use of hazardous, yet versatile intermediates towards medicinally relevant structures.Experimental section
General experimental section
1H-NMR spectra were recorded on either Bruker Avance-400, Varian VNMRS-600 or Varian VNMRS-700 instruments and are reported relative to residual solvent: CHCl3 (δ 7.26 ppm) or DMSO (δ 2.50 ppm). 13C-NMR spectra were recorded on the same instruments and are reported relative to CHCl3 (δ 77.16 ppm) or DMSO (δ 39.52 ppm). Data for 1H-NMR are reported as follows: chemical shift (δ/ppm) (integration, multiplicity, coupling constant (Hz)). Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br. s = broad singlet, app = apparent. Data for 13C-NMR are reported in terms of chemical shift (δ/ppm) and multiplicity (C, CH, CH2 or CH3). Data for 19F-NMR were recorded on the above instruments at a frequency of 376 MHz using CFCl3 as external standard. DEPT-135, COSY, HSQC, HMBC and NOESY experiments were used in the structural assignment. IR spectra were obtained by use of a Perkin Elmer RX1 spectrometer (neat, ATR sampling) with the intensities of the characteristic signals being reported as weak (w, <20% of tallest signal), medium (m, 21–70% of tallest signal) or strong (s, >71% of tallest signal). Low and high resolution mass spectrometry was performed using the indicated techniques on either Waters LCT Premier XE or Waters TQD instruments equipped with Acquity UPLC and a lock-mass electrospray ion source. For accurate mass measurements the deviation from the calculated formula is reported in ppm. Melting points were recorded on an Optimelt automated melting point system with a heating rate of 1 °C min−1 and are uncorrected.Single crystal X-ray data were collected at 120.0 K on a Bruker SMART 6000 (sealed tube, graphite monochromator) (compounds 7h and SI12f′) and Bruker D8 Venture (Photon 100 CMOS detector, IμS microsource, focusing mirrors) (compounds 12a, 12f and 12i) diffractometers (λMoKα, λ = 0.71073 Å) equipped with Cryostream (Oxford Cryosystems) open flow nitrogen cryostates. The structures were solved by direct methods and refined by full-matrix least squares on F2 for all data using SHELX26 and OLEX227 software. All non-hydrogen atoms were refined with anisotropic displacement parameters, the H-atoms in the structures 12a, 12f and 12i were placed in calculated positions and refined in “riding” mode. The H atoms in the other structures were found in the difference Fourier maps and refined isotropically. Crystallographic data and parameters of the refinement are given in Table 1 (ESI†). Crystallographic data for the structures have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC-1039437–1039441.
Acknowledgements
We gratefully acknowledge financial support through the Royal Society (M.B. and I.R.B.) and MEC & MINECO for a FPU fellowship (A.M.R.G.). Furthermore we are very grateful to Dr D. S. Yufid and Dr A. S. Batsanov (Department of Chemistry, Durham University) for solving several X-ray crystal structures.Notes and references
- (a) J. J. Li, Heterocyclic Chemistry in Drug Discovery, Wiley, 2013. ISBN: 978-1-118-14890-7 Search PubMed; (b) R. B. Silverman, The Organic Chemistry of Drug Design and Drug Action, Elsevier Academic Press, Burlington, MA, 2004 Search PubMed; (c) H. Wild, D. Heimbach and C. Huwe, Angew. Chem., Int. Ed., 2011, 50, 7452 CrossRef CAS PubMed.
- (a) M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, Beilstein J. Org. Chem., 2011, 7, 442 CrossRef CAS PubMed; (b) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265 CrossRef PubMed; (c) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- (a) L. Malet-Sanz and F. Susanne, J. Med. Chem., 2012, 55, 4062 CrossRef CAS PubMed; (b) M. Baumann, I. R. Baxendale and S. V. Ley, Mol. Diversity, 2011, 15, 613 CrossRef CAS PubMed; (c) R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502 CrossRef CAS PubMed; (d) C. Wiles and P. Watts, Green Chem., 2012, 14, 38 RSC; (e) V. Hessel, B. Cortese and M. H. J. M. de Croon, Chem. Eng. Sci., 2011, 66, 1426 CrossRef CAS PubMed; (f) J. Yoshida, H. Kim and A. Nagaki, ChemSusChem, 2010, 4, 331 CrossRef PubMed; (g) M. D. Hopkin, I. R. Baxendale and S. V. Ley, Org. Biomol. Chem., 2013, 11, 1822 RSC.
- (a) I. R. Baxendale, L. Brocken and C. J. Mallia, Green Process Synth., 2013, 2, 211 CAS; (b) I. R. Baxendale, J. Chem. Technol. Biotechnol., 2013, 88, 4 CrossRef, 519; (c) J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17 CrossRef CAS; (d) J. C. Pastre, D. L. Browne, M. O'Brien and S. V. Ley, Org. Process Res. Dev., 2013, 17, 1183 CrossRef CAS; (e) M. A. Kabeshov, B. Musio, P. R. D. Murray, D. L. Browne and S. V. Ley, Org. Lett., 2014, 16, 4618 CrossRef CAS PubMed; (f) J. Wegner, S. Ceylan and A. Kirschning, Org. Biomol. Chem., 2012, 10, 7721 RSC; (g) P. R. D. Murray, D. L. Browne, J. C. Pastre, C. Butters, D. Guthrie and S. V. Ley, Org. Process Res. Dev., 2013, 17, 1192 CrossRef CAS.
- (a) M. O'Brien, I. R. Baxendale and S. V. Ley, Org. Lett., 2010, 12, 1596 CrossRef PubMed; (b) C. Battilochio, I. R. Baxendale, M. Biava, M. O. Kitching and S. V. Ley, Org. Process Res. Dev., 2012, 16, 798 CrossRef; (c) M. Irfan, T. N. Glasnov and C. O. Kappe, Org. Lett., 2011, 13, 984 CrossRef CAS PubMed.
- (a) C. J. Smith, C. D. Smith, N. Nikbin, S. V. Ley and I. R. Baxendale, Org. Biomol. Chem., 2011, 9, 1927 RSC; (b) H. R. Sahoo, J. G. Kralj and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 5704 CrossRef CAS PubMed; (c) L. Kupracz, J. Hartwig, J. Wegner, S. Ceylan and A. Kirschning, Beilstein J. Org. Chem., 2011, 7, 1441 CrossRef CAS PubMed; (d) M. Baumann, I. R. Baxendale, S. V. Ley, N. Nikbin, C. D. Smith and J. P. Tierney, Org. Biomol. Chem., 2008, 6, 1577 RSC; (e) C. J. Smith, N. Nikbin, S. V. Ley, H. Lange and I. R. Baxendale, Org. Biomol. Chem., 2011, 9, 1938 RSC; (f) P. B. Palde and T. F. Jamison, Angew. Chem., Int. Ed., 2011, 50, 3525 CrossRef CAS PubMed; (g) I. R. Baxendale, S. V. Ley, A. C. Mansfield and C. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 4017 CrossRef CAS PubMed; (h) F. R. Bou-Hamdan, F. Lévesque, A. G. O'Brien and P. H. Seeberger, Beilstein J. Org. Chem., 2011, 7, 1124 CrossRef CAS PubMed; (i) C. D. Smith, I. R. Baxendale, S. Lanners, J. J. Hayward, S. C. Smith and S. V. Ley, Org. Biomol. Chem., 2007, 5, 1559 RSC; (j) C. J. Smith, N. Nikbin, C. D. Smith, S. V. Ley and I. R. Baxendale, Org. Biomol. Chem., 2011, 9, 1927 RSC; (k) M. M. E. Delville, P. J. Nieuwland, P. Janssen, K. Koch, J. C. M. van Hest and F. P. J. T. Rutjes, Chem. Eng. J., 2011, 167, 556 CrossRef CAS PubMed.
- (a) D. X. Hu, M. O'Brien and S. V. Ley, Org. Lett., 2012, 14, 4246 CrossRef CAS PubMed; (b) L. Malet-Sanz, J. Madrzak, S. V. Ley and I. R. Baxendale, Org. Biomol. Chem., 2010, 8, 3312 RSC.
- (a) C. McPake, C. B. Murray and G. Sandford, Tetrahedron Lett., 2009, 50, 1674 CrossRef CAS PubMed; (b) C. McPake and G. Sandford, Org. Process Res. Dev., 2012, 16, 844 CrossRef CAS; (c) J. R. Breen, G. Sandford, D. S. Yufit, J. A. K. Howard, J. Fray and B. Patel, Beilstein J. Org. Chem., 2011, 7, 1048 CrossRef CAS PubMed.
- (a) D. L. Browne, M. Baumann, B. H. Harji, I. R. Baxendale and S. V. Ley, Org. Lett., 2011, 13, 3312 CrossRef CAS PubMed; (b) J. A. Newby, D. W. Blaylock, P. M. Witt, J. C. Pastre, M. K. Zacharova, S. V. Ley and D. L. Browne, Org. Process Res. Dev., 2014, 18, 1211 CrossRef CAS; (c) A. Nagaki, A. Kenmoku, Y. Moriwaki, A. Hayashi and J. Yoshida, Angew. Chem., Int. Ed., 2010, 49, 7543 CrossRef CAS PubMed; (d) A. Nagaki, E. Takizawa and J. Yoshida, Chem. – Eur. J., 2010, 16, 14149 CrossRef CAS PubMed.
- (a) J. Hartwig, S. Ceylan, L. Kupracz, L. Coutable and A. Kirschning, Angew. Chem., Int. Ed., 2013, 52, 9813 CrossRef CAS PubMed; (b) J. Hartwig, L. Kupracz and A. Kirschning, Chem. Lett., 2012, 41, 562 CrossRef; (c) B. Reichart and C. O. Kappe, Tetrahedron Lett., 2012, 53, 952 CrossRef CAS PubMed; (d) J. Yoshida, Flash Chemistry: Fast Organic Synthesis in Microsystems, John Wiley & Sons, Chichester, UK, 2008 Search PubMed.
- (a) K. Matsumoto and M. Suzuki, Ethyl Isocyanoacetate; Encyclopedia of Reagents for Organic Synthesis, 2001 Search PubMed; (b) M. Suginome and Y. Ito, Sci. Synth., 2004, 19, 445 Search PubMed; (c) S. Kotha and S. Halder, Synlett, 2010, 337 CrossRef CAS PubMed; (d) M. Baumann, I. R. Baxendale, S. V. Ley, C. D. Smith and G. K. Tranmer, Org. Lett., 2006, 8, 5231 CrossRef CAS PubMed.
- N-formylglycine is commercially available (CAS 3154-51-6).
- S. Fuse, N. Tanabe and T. Takahashi, Chem. Commun., 2011, 47, 12661 RSC.
- K. Matsumoto, M. Suzuki, M. Tomie, N. Yoneda and M. Miyoshi, Synthesis, 1975, 609 CrossRef CAS.
- D. L. Browne, I. R. Baxendale and S. V. Ley, Tetrahedron, 2011, 67, 10296–10303 CrossRef CAS PubMed.
- M. Zibinsky and V. V. Fokin, Org. Lett., 2011, 13, 4870 CrossRef CAS PubMed.
- R. Romagnoli, P. G. Baraldi, O. Cruz-Lopez, C. Lopez Cara, M. D. Carrion, A. Brancale, E. Hamel, L. Chen, R. Bortolozzi, G. Basso and G. Viola, J. Med. Chem., 2010, 53, 4248 CrossRef CAS PubMed.
- For instance WO 2003070732; WO 2004039366; WO 2004064836; WO 2004052461.
- (a) J. M. Minguez, J. J. Vaquero, J. L. García-Navio and J. Alvarez-Builla, Tetrahedron Lett., 1996, 37, 4263 CrossRef CAS; (b) J. M. Minguez, J. J. Vaquero, J. Alvarez and O. Catano, J. Org. Chem., 1999, 64, 7788 CrossRef CAS.
- K. A. Peterson, B. C. Shepler, D. Figgen and H. Stoll, J. Phys. Chem. A, 2006, 110, 13877 CrossRef CAS PubMed.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113 CrossRef CAS PubMed.
- (a) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed; (b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision D.01), Gaussian Inc., Wallingford CT, 2009, 2009 Search PubMed.
- (a) S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef; (b) J. Tomasi, R. Cammi, B. Mennucci, C. Cappelli and S. Corni, Phys. Chem. Chem. Phys., 2002, 4, 5697 RSC; (c) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- (a) M. Vicinius Nora de Souza, Curr. Org. Chem., 2007, 11, 637 CrossRef; (b) M. Schnürch, M. Spina, A. Farooq Khan, M. D. Mihovilovic and P. Stanetty, Chem. Soc. Rev., 2007, 36, 1046 RSC; (c) M. Schnürch, Recent Progress on the Halogen Dance Reaction on Heterocycles, in Halogenated Heterocycles, Topics in Heterocyclic Chemistry, Springer, 2012, vol. 27, pp. 185–218 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1039437, 1039438, 1039439, 1039440 and 1039441. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob00245a |
| This journal is © The Royal Society of Chemistry 2015 |