
Diketopyrrolopyrrole-based copolymers bearing highly π-extended donating units and their thin-film transistors and photovoltaic cells†
Min Ju
Cho‡
a,
Jicheol
Shin‡
a,
Tae Ryang
Hong
a,
Hyun Ah
Um
a,
Tae Wan
Lee
a,
Gyeong Woo
Kim
b,
Jang Hyuk
Kwon
b and
Dong Hoon
Choi
*a
aDepartment of Chemistry, Research Institute for Natural Sciences, Korea University, Sungbuk-gu, Seoul 136-701, South Korea. E-mail: dhchoi8803@korea.ac.kr; Fax: +82-2-925-4284; Tel: +82-2-3290-3140
bDepartment of Information Display, Kyung Hee University, Dongdaemoon-gu, Seoul, 130-701, Korea
First published on 9th September 2014
Abstract
New diketopyrrolopyrrole (DPP)-based π-extended conjugated polymers containing relatively long conjugated donor monomers were successfully synthesized in order to investigate their physical properties and device performance in thin-film transistors and photovoltaic cells. The solubility of the polymers was improved via side-chain engineering, and relatively high molecular weights were achieved by modifying the synthetic procedures. The highest occupied molecular orbital and the lowest unoccupied molecular orbital and optical bandgaps of the polymers were easily tuned by incorporating different donor monomers with different donating abilities. All polymers were highly crystalline with a predominant edge-on orientation on the substrate. The polymers displayed different hole mobilities in TFTs depending on the donor monomer. The highest mobility was 4.17 cm2 V−1 s−1, which was shown by the TFT fabricated with a terselenophene-containing DPP polymer after thermal annealing. Furthermore, bulk heterojunction organic photovoltaic cells made from a blend film of the polymers and (6,6)-phenyl C71-butyric acid methyl ester demonstrated promising device performance with power conversion efficiencies in the range of 3.48–5.05%.
Introduction
Because donor (D)–acceptor (A) type conjugated polymers exhibit preferred crystalline morphology and highly efficient light-harvesting properties with strong absorption in the visible to near-infrared wavelength region, numerous new structured polymers have been designed and applied to organic thin film transistors (OTFTs)1,2 and organic photovoltaics (OPVs).3,4 Moreover, D–A copolymers with good solution processability and film-forming properties have been suggested as the best organic semiconductors for future large-scale electronic and optoelectronic device fabrication. Recently, among the many D–A type conjugated copolymers, diketopyrrolopyrrole (DPP)-based copolymers have been identified as highly promising materials for future flexible OTFTs5,6 and OPVs7 because they usually have significantly high carrier mobilities in TFTs and high power conversion efficiencies (PCEs) in OPVs.The DPP unit in the repeating group of those conjugated copolymers is used as a strong, fused-ring acceptor to impose coplanarity and enhance intermolecular interactions. The solubility of these copolymers with a highly rigid backbone can easily be controlled by side-chain engineering, i.e., introducing various branched alkyl groups into the two N-positions of the DPP unit.8 In addition, incorporating a relatively long π-conjugated donating monomer between DPP accepting units can extend the conjugation length of the copolymer chain and reduce the steric hindrance of the bulky alkyl side chains.9
Various thiophene-based donating units, such as thienothiophene (TT),10 bithiophene (BT),11 and dithienothiophene (DTT),12 have been employed in DPP-based copolymers reported in the literature. These copolymers exhibited good performance in OTFTs and OPVs via high carrier mobility and power conversion efficiency, respectively.
Besides DPP-based copolymers bearing thiophene-based donor monomers, the copolymers containing stronger electron-donating selenophene moieties, such as selenophene,13 biselenophene,14 and (E)-1,2-di(selenophen-2-yl)ethene,15 were also reported to show significantly higher carrier mobilities in TFTs. The high performance in TFTs made of selenophene-containing copolymers is known to be attributed to the favorable quinoidal character and well-ordered arrangement of polymer chains on the substrates.13–15
Very recently, a few groups have reported that DPP-based copolymers that contain longer π-extended β-unsubstituted oligothiophene and oligoselenophene units display good performance in TFTs with markedly high carrier mobilities. For example, Liu's group reported that a DPP-based copolymer containing β-unsubstituted terthiophene as a donor unit and 2-octyldodecyl side-chain groups exhibited a hole mobility of 3.46 cm2 V−1 s−1.16
In our previous work, we demonstrated that a DPP-based copolymer containing a β-unsubstituted terselenophene unit showed good solubility in chloroform after introduction of a longer branched 2-decyltetradecyl group in place of the frequently used 2-octyldodecyl group at the two N-positions of the DPP unit. As a result, the copolymer exhibits high crystallinity in the film state and an enhanced maximum hole mobility of 5.0 cm2 V−1 s−1.17 TFT devices based on these polymers displayed a much higher hole mobility than devices containing DPP-based copolymers with relatively short mono- or bithiophene/biselenophene units. However, the number of papers published to date on DPP-based conjugated copolymers containing various longer β-unsubstituted thiophene/selenophene and benzene-based donating monomers is insignificant compared to the volume of comparable work on DPP-based polymers with relatively short donor monomers.11,13,14,18 Therefore, the effect of the variation of the longer donating groups on physical properties and device performance through the incorporation of three-component aromatic donating units without alkyl chains in a DPP-based copolymer backbone is worth studying. Moreover, investigating the general and photophysical properties of these copolymers, such as solubility, molecular weight, crystallinity, and absorption behavior, is valuable for determining the correlation between donor structures and corresponding TFT and PV device performances.
In this work, we designed and synthesized a new series of DPP-based copolymers containing π-extended donating groups, including 2,5-di(thiophen-2-yl)selenophene (TST), terthiophene (TTT), and 1,4-di(thiophen-2-yl)benzene (TPhT) besides β-unsubstituted terselenophene (SSS).17 The target copolymers enable us to study the correlation between the four different donor structures and various physical properties. Moreover, the device performances of OTFTs and OPVs made of these copolymers were also investigated. The solubility of these copolymers was ensured by introducing 2-decyltetradecyl groups into the two N-positions of the DPP unit, and their moderately high molecular weight was achieved by controlling the reaction time and the type of Pd polymerization catalyst. All copolymers showed p-type semiconducting behavior; TFTs containing the copolymers exhibited high carrier mobilities of 1.28–4.17 cm2 V−1 s−1. Bulk heterojunction (BHJ) photovoltaic cells containing these copolymers and (6,6)-phenyl C71-butyric acid methyl ester (PC71BM) have promising power conversion efficiencies. In particular, a OPV device containing P(DPP-TPhT) and PC71BM showed the highest power conversion efficiency of 5.05%.
Experimental
Materials
All reagents were purchased from Acros, TCI, and Sigma-Aldrich Co. and used without further purification unless stated otherwise. The reagent-grade solvents used in this study were freshly dried using standard distillation methods. Compound 1, 2,5-bis(trimethylstannyl)selenophene, and P(DPP-SSS) were prepared using literature methods.17Synthesis of 3,6-di(2,2′-bithiophen-5-yl)-2,5-bis(2-decyltetradecyl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione (2)
Tetrakis(triphenylphosphine)palladium(0) (70 mg, 0.1 mmol) was added to a solution of compound 1 (2.26 g, 2.00 mmol) and 2-(tributylstannyl)thiophene (1.86 g, 5.00 mmol) in toluene (20 mL) at room temperature under an argon atmosphere. The mixture was heated at 90 °C for 24 h and cooled to room temperature. The reaction mixture was poured into water and extracted with methylene chloride. The organic phase was dried over MgSO4. The compound was purified using silica-gel chromatography (eluent: dichloroethane–hexane = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v) to yield 1.94 g (85%) of the desired product as a dark-purple powder. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.91 (d, J = 3.9 Hz, 2H), 7.3–7.33 (m, 6H), 7.07 (d, J = 3.9 Hz, 2H), 4.04 (d, 4H), 1.96 (m, 2H), 1.15–1.43 (m, 80H), 0.83–0.88 (m, 12H). MALDI-TOF MS exact mass calcd for C70H108N2O2S4 [M] 1136.729, found 1136.304.
:3 v/v) to yield 1.94 g (85%) of the desired product as a dark-purple powder. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.91 (d, J = 3.9 Hz, 2H), 7.3–7.33 (m, 6H), 7.07 (d, J = 3.9 Hz, 2H), 4.04 (d, 4H), 1.96 (m, 2H), 1.15–1.43 (m, 80H), 0.83–0.88 (m, 12H). MALDI-TOF MS exact mass calcd for C70H108N2O2S4 [M] 1136.729, found 1136.304.
Synthesis of 3,6-bis(5′-bromo-2,2′-bithiophen-5-yl)-2,5-bis(2-decyltetradecyl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione (3)
N-Bromosuccinimide (NBS) (0.53 g, 2.98 mmol) was added to a solution of compound 2 (1.7 g, 1.49 mmol) in chloroform (20 mL) under an argon atmosphere; the resulting solution was protected from light and stirred at room temperature for 12 h. The mixture was poured into 200 mL of methanol and then filtered. The precipitate was purified using silica-gel chromatography (eluent: dichloroethane–hexane = 1:3 v/v) to yield 1.56 g (81%) of the desired product as a dark-purple powder. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.87 (d, J = 3.9 Hz, 2H), 7.22 (d, J = 3.9 Hz, 2H), 7.04 (d, J = 3.9 Hz, 2H), 7.02 (d, J = 3.9 Hz, 2H), 4.01 (d, 4H), 1.94 (m, 2H), 1.17–1.45 (m, 80H), 0.83–0.89 (m, 12H). MALDI-TOF MS exact mass calcd for C70H106Br2N2O2S4 [M] 1292.550, found 1292.091.
Synthesis of P(DPP-TST)
Tris(dibenzylideneacetone)dipalladium(0) (9.2 mg, 10.0 μmol) and tri(o-tolyl)phosphine (6.1 mg, 20.0 μmol) were added to a solution of compound 3 (0.259 g, 0.2 mmol) and 2,5-bis(trimethylstannyl)selenophene (0.117 g, 0.2 mmol) in toluene (20 mL) at room temperature under an argon atmosphere. The mixture was heated at 100 °C and held until some precipitate appeared (∼5 h). After cooling it to room temperature, the mixture was added to vigorously stirred methanol (300 mL), filtered, and washed with methanol. The crude copolymer was purified by Soxhlet extraction with methanol, acetone, THF, and chloroform, successively. The chloroform fraction was evaporated and then the copolymer was precipitated in methanol, filtered, and dried to give the P(DPP-TST) copolymer (0.18 g, 71%) (Mn = 197 kDa, Mw = 561 kDa, PDI = 2.85). Anal. Calcd for (C74H108N2O2S4Se)n: C, 70.27; H, 8.61; N, 2.21; S, 10.14. Found: C, 69.87; H, 8.52; N, 2.29; S, 10.73.Synthesis of P(DPP-TTT)
Tetrakis(triphenylphosphine)palladium(0) (11.4 mg, 10.0 μmol) was added to a solution of compound 3 (0.259 g, 0.2 mmol) and 2,5-bis(trimethylstannyl)thiophene (0.098 g, 0.2 mmol) in toluene (20 mL) at room temperature under an argon atmosphere. The mixture was heated at 100 °C for 48 h. After cooling the mixture to room temperature, it was added to vigorously stirred methanol (300 mL), filtered, and washed with methanol. The crude polymer was purified by Soxhlet extraction with acetone, THF, and chloroform successively. The chloroform fraction was evaporated and then the copolymer was precipitated in methanol, filtered, and dried to give the P(DPP-TTT) copolymer (0.15 g, 62%) (Mn = 68 kDa, Mw = 258 kDa, PDI = 3.79). Anal. Calcd for (C74H108N2O2S5)n: C, 72.97; H, 8.94; N, 2.30; S, 13.10. Found: C, 71.59; H, 8.45; N, 2.42; S, 13.31.Synthesis of P(DPP-TPhT)
Tris(dibenzylideneacetone) dipalladium(0) (9.2 mg, 10.0 μmol), tri(o-tolyl)phosphine (6.1 mg, 20.0 μmol), and Aliquat 336 were added to a solution of compound 3 (0.259 g, 0.2 mmol) and 1,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene (0.66 g, 0.2 mmol) in toluene (15 mL) and 2 M K2CO3 (5 mL) at room temperature under an argon atmosphere. The mixture was heated at 100 °C until some precipitate appeared (∼7 h). After cooling the mixture to room temperature, it was added to vigorously stirred methanol (300 mL), filtered, and washed with methanol. The crude polymer was purified by Soxhlet extraction with acetone, THF, and chloroform successively. The chloroform fraction was evaporated and then the copolymer was precipitated in methanol, filtered, and dried to give the P(DPP-TPhT) copolymer (0.13 g, 54%) (Mn = 254 kDa, Mw = 1094 kDa, PDI = 4.31). Anal. Calcd for (C76H110N2O2S4)n: C, 75.32; H, 9.15; N, 2.31; S, 10.58. Found: C, 74.87; H, 8.91; N, 2.14; S, 11.02.Instruments
1H NMR spectra were recorded on a Varian Mercury NMR 300 MHz spectrometer using deuterated chloroform purchased from Cambridge Isotope Laboratories, Inc. Elemental analyses were performed using an EA1112 (Thermo Electron Corp.) elemental analyzer. The molecular weights of the polymers were measured by gel permeation chromatography (Waters GPC, Waters 515 pump, Waters 410 RI, 2× PLgel Mixed-B) at 35 °C using polystyrene as the standard and chloroform as an eluent. Thermal properties were investigated on a Mettler differential scanning calorimetry (DSC) 821e instrument under a nitrogen atmosphere. Absorption spectra of samples in solution and film state were obtained using a UV-Vis absorption spectrometer (HP 8453, PDA type, λ = 190–1100 nm). The redox potentials of the DPP-copolymers were measured by cyclic voltammetry (model: EA161 eDAQ). The electrolyte solution employed was 0.10 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) in freshly dried acetonitrile. Ag/AgCl and Pt wire (0.5 mm in diameter) electrodes were utilized as the reference and counter electrodes, respectively. The scan rate was 50 mV s−1. Grazing incidence X-ray diffraction (GI-XRD) measurements were performed at the 3C (SAXS I) beam lines (energy = 10.0 keV, pixel size = 79.6 μm, wavelength = 1.17 Å) at the Pohang Accelerator Laboratory (PAL). The measurements were obtained in a 2θ scanning interval between 0° and 25°; the components of the scattering vector parallel (qxy) and perpendicular (qz) to the substrate were determined using the following equation: q = (4π/λ)sinθ, where θ is the half scattering angle and λ is the wavelength of the incident radiation. The film samples were fabricated by spin-casting onto OTS-SiO2/Si followed by drying at 50 °C under vacuum (solvent: chloroform; concentration: 3 mg mL−1). Atomic force microscopy (AFM, Advanced Scanning Probe Microscope, XE-100, PSIA) in tapping mode with a silicon cantilever was used to characterize the surface morphologies of the polymer films.
TFT device fabrication
To characterize the TFT performance, bottom-contact/bottom-gate (BCBG) device geometries were employed. Surface modification was carried out with OTS to generate a hydrophobic insulator surface. In the case of the BCBG device, source and drain gold electrodes were thermally evaporated (100 nm) onto an OTS-treated heavily n-doped Si/SiO2 substrate through a shadow mask with a channel width and length of 1500 and 100 μm, respectively. Polymer thin films were spin-coated using a chloroform solution (0.3 wt%) that was stirred for three days at room temperature. After depositing the polymer film on the OTS-SiO2/Si substrate, the samples were annealed on a hot plate at suitable temperatures for 10 min (annealing temperatures: 180 °C for P(DPP-SSS), 160 °C for P(DPP-TTT), 160 °C for P(DPP-TST) and 180 °C for P(DPP-TPhT)). The field effect mobilities were extracted in the saturation regime using the following relationship: μsat = (2IDSL)/(WC(VG − VT)2), where IDS is the saturation drain current, C is the capacitance (∼11.5 nF cm−2) of the SiO2 dielectric, VG is the gate bias, and VT is the threshold voltage. The device performance was measured in air using a 4200-SCS parameter analyzer system (Keithley Instruments Inc.). Mobility data were collected from more than 10 different devices and averaged.Polymer solar cell fabrication
Polymer solar cells were fabricated on commercially available patterned indium-tin oxide (ITO) glass substrates. A 30 nm layer of PEDOT:PSS was spin-coated on top of the ITO and baked at 150 °C for 10 min. Successively, the photoactive layer of polymer:PC71BM (1:2 wt%) was spin-coated at 1200 rpm on top of the PEDOT:PSS layer. Finally, the devices were completed with the deposition of ∼100 nm LiF/Al by using a thermal evaporator (vacuum at 10−6–10−7 Torr) to produce a 0.04 cm2 active area using a mask. The J–V characteristics were measured using a computerized Keithley 2400 source meter in the dark and under AM 1.5 G illumination at 100 mW cm−2 supplied by a solar simulator (Oriel, 1000 W). The spectral response was measured using a tungsten–halogen light source combined with a monochromator (Spectra Pro 2300, Acton Research). The monochromatic light was filtered by a monochromator to obtain UV, visible, and IR wavelength ranges to remove harmonics and was modulated using an optical chopper at 350 Hz. The resulting photocurrent on the photovoltaic cell was measured using a lock-in amplifier (SR-830, Stanford Research). The power of the monochromatic light was measured using a calibrated optical power meter and a photodetector (Coherent Fieldmax II with UV to IR sensor heads and a Thorlabs PM100). All equipment and data acquisition were controlled using LabView. PCE data were collected from more than 10 different devices and averaged.
Results and discussion
Synthesis and characterization
The general procedures for the synthesis of the DPP-based monomer, 3, and copolymers are outlined in Scheme 1. Monomer 3, which comprises extended thiophene units, was readily synthesized according to our previously reported procedure.17 P(DPP-TTT) and P(DPP-TST) copolymers were synthesized via Stille coupling of monomer 3 with 2,5-bis(trimethylstannyl)thiophene and 2,5-bis(trimethyl stannyl)selenophene in the presence of Pd(PPh3)4 and Pd2(dba)3, respectively. Similarly, P(DPP-TPhT) was prepared via Suzuki coupling polymerization of monomer 3 with 1,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene. The polymerization reaction was quenched when insoluble precipitates were evident in the reaction mixture; this strategy enables an increased reaction yield with fewer insoluble high molecular weight fragments and ensures the solubility required for thin film fabrication using a solution-based method. | ||
| Scheme 1 Synthetic scheme for DPP-based copolymers. (i) Pd(PPh3)4, 2-(tributylstannyl)thiophene, toluene, 90 °C, (ii) NBS, chloroform, (iii) Pd2(dba)3, P(o-tolyl)3, 2,5-bis(trimethylstannyl)selenophene, toluene, 100 °C, 5 h, (iv) Pd(PPh3)4, 2,5-bis(trimethylstannyl)thiophene, toluene, 100 °C, 48 h, (v) Pd2(dba)3, P(o-tolyl)3, 2,5-1,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene, 2 M K2CO3, toluene, 100 °C, 7 h. The synthetic procedure for P(DPP-SSS) was described in ref. 17. | ||
For example, the polymerization yields of P(DPP-SSS) and P(DPP-TTT) containing homogeneous donating units were very low when the Pd2(dba)3 polymerization catalyst was used because this catalyst results in higher molecular weight polymers. The resulting two polymers were not as soluble after polymerization as those produced using Pd(PPh3)4. However, soluble P(DPP-TST) and P(DPP-TPhT) polymers, which contain heterogeneous donating units, were generated using the Pd2(dba)3 catalyst. The synthesized crude copolymers were purified via Soxhlet extraction using methanol, acetone, tetrahydrofuran (THF), and chloroform in succession. All the copolymers showed unique solvent selectivity: good solubility in chloroform, but poor solubility in toluene, monochlorobenzene, and o-dichlorobenzene at room temperature. In particular, the solubilities in chloroform were greater than 10 mg mL−1 for P(DPP-TTT) and P(DPP-SSS) and around 5 mg mL−1 for P(DPP-TST) and P(DPP-TPhT). The molecular weights and polydispersity indices (PDIs) of the polymers were measured using gel permeation chromatography (GPC) with chloroform as the eluent and polystyrene as the standard (Table 1). The resulting molecular weights (Mn) are 47, 197, 68, and 254 kDa for P(DPP-SSS), P(DPP-TST), P(DPP-TTT), and P(DPP-TPhT), respectively. In brief, we successfully fabricated copolymers with suitable solubilities and molecular weights for the generation of possibly high-performance TFT and PV devices through solution processing.
| Polymer | M n (kDa) | PDI | λ max (nm) | λ cut-off (nm) |
E
optg
(eV) |
E ox (V) | HOMOd (eV) | LUMOe (eV) | |
|---|---|---|---|---|---|---|---|---|---|
| Solutionb | Film | ||||||||
| a Data of P(DPP-SSS) are cited from ref. 17. b Data were measured in chloroform solution. c The optical bandgap energies were calculated from the absorption edges (λcut-off), Eoptg = 1240/λcut-off. d First oxidation potentials and HOMO energies were measured by CV. e LUMO energy was calculated from the difference between the HOMO and optical bandgap energies. | |||||||||
| P(DPP-SSS)a | 47 | 2.97 | 823 | 827 | 910 | 1.36 | 0.88 | −5.15 | −3.79 |
| P(DPP-TST) | 197 | 2.85 | 773 | 789 | 860 | 1.44 | 0.90 | −5.17 | −3.73 |
| P(DPP-TTT) | 68 | 3.79 | 762 | 785 | 868 | 1.43 | 0.92 | −5.19 | −3.76 |
| P(DPP-TPhT) | 254 | 4.31 | 746 | 746 | 812 | 1.53 | 0.94 | −5.21 | −3.68 |
The DSC thermograms of synthesized copolymers are illustrated in Fig. S5.† The transition temperatures of P(DPP-SSS), P(DPP-TST), P(DPP-TTT), and P(DPP-SSS) as an endothermic peak were around 306, 275, 303, and 327 °C, respectively, which is presumably due to the melting behavior of the π-conjugated polymer backbone. However, the glass transition temperatures of the copolymers were not observed in the measuring range.
Optical and electrochemical properties
UV-Vis absorption spectra of the synthesized DPP-based copolymers in chloroform solutions and as thin films are presented in Fig. 1. The measured parameters in the spectra and the corresponding electrochemical properties are summarized in Table 1. In the solution states, all polymers showed strong absorption in far-red and near-infrared ranges, which correspond to absorption maximum wavelengths in the range of 746 to 823 nm. In the film state, the absorption spectra shifted bathochromically due to significant intermolecular interactions and specific aggregation between polymer chains. Replacing the terthiophene in P(DPP-TTT) with terselenophene to form P(DPP-SSS) results in slightly broader and more red-shifted absorption spectra both in the solution and film states; this is likely due to the fact that terselenophene, which is a more electron-rich group, induces stronger intramolecular charge transfer and well-ordered polymer chain arrangement in the solid state.15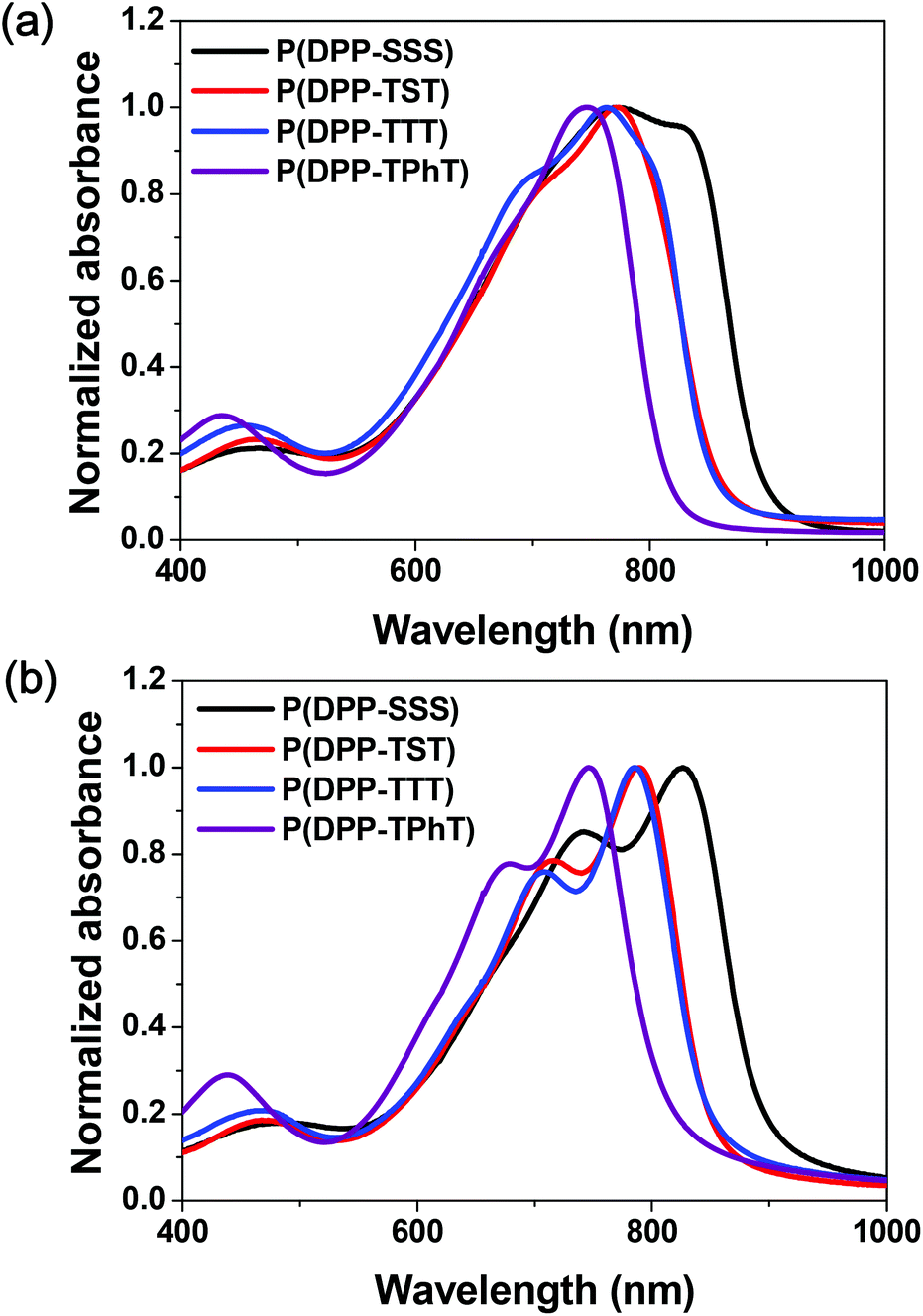 | ||
| Fig. 1 UV-Vis absorption spectra of the prepared DPP-based copolymers in (a) chloroform solution and (b) thin films. | ||
However, relative to that shown by P(DPP-TTT), the P(DPP-TST) copolymer which contains one selenophene moiety between the thiophene fragments in the donor monomer, did not feature a significant change in the spectral shift and shape in the solution and film states; this indicates that, unlike the TTT unit in P(DPP-TTT), the single selenophene in the TST unit does not sufficiently affect the conjugative effect of P(DPP-TST). Among these four copolymers, P(DPP-TPhT) has the largest optical bandgap (Eopt.g = 1.53 eV) and P(DPP-SSS) has the smallest (1.36 eV) because of the effects of the terselenophene moiety.
Cyclic voltammetry (CV) was performed to electrochemically evaluate the molecular energy levels of the copolymers. Fig. 2 shows cyclic voltammograms of the four DPP-based polymer films on a platinum flat electrode. The onset points of the oxidative waves were determined to be 0.88, 0.90, 0.92, and 0.94 V for P(DPP-SSS), P(DPP-TST), P(DPP-TTT), and P(DPP-TPhT), respectively; these results correspond to the highest occupied molecular orbital (HOMO) levels of −5.15, −5.17, −5.19, and −5.21 eV, respectively. To obtain lower energy HOMOs in DPP-based copolymers containing three-component donating units, it is more amenable to combine thiophene and phenyl groups as a donor monomer unit.
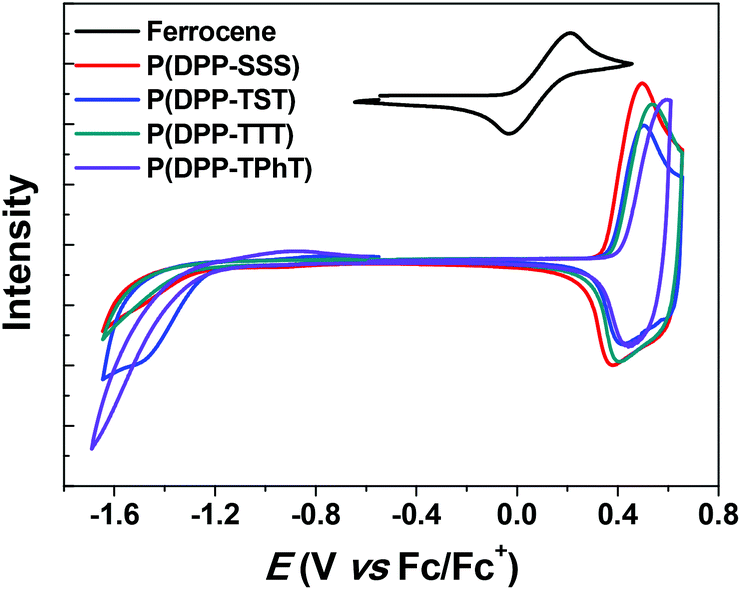 | ||
| Fig. 2 Cyclic voltammograms of the DPP-based copolymers; samples were thin films on a Pt electrode. | ||
To determine the lowest unoccupied molecular orbital (LUMO) levels, we combined the HOMO values with the Eopt.g derived from the absorption edges in their absorption spectra. Using these results, the HOMO and LUMO levels of the copolymers were easily tuned by varying the donor structure.
To investigate the geometry and molecular energy levels, theoretical calculations with density functional theory (DFT) using the B3LYP functional were applied to simplified molecular structures derived from the polymer backbone. The 6-31G(d) basis set was used to calculate the molecular orbital energy levels (Fig. S6†). All calculations were carried out using the Spartan program ('10). From the calculated results, the SSS, TST, and TTT units feature highly planar structures, whereas the TPhT unit has a dihedral angle of 22.3° between the phenyl and thienyl groups. Therefore, the molecular plane of the repeating group in P(DPP-TPhT) could be slightly twisted, while the others would be planar. With respect to the bandgap energies, all the simplified molecules showed very similar values to those obtained experimentally for copolymers via absorption spectral analysis. Additionally, the properties of P(DPP-TST) are estimated to be quite similar to those of P(DPP-TTT) from the absorption spectroscopy, CV, and theoretical calculation results.
Grazing incidence X-ray diffraction (GI-XRD) analysis
The microstructures and orientations of the copolymer chains in thin films were investigated via GI-XRD (Fig. 3). Thin films were prepared via spin-coating onto an n-octyltrichlorosilane (OTS)-treated SiO2/Si substrate. The GI-XRD analysis data for the copolymers are summarized in Table S1.† Similar to the reported results for the P(DPP-SSS) copolymer, annealed thin films of P(DPP-TST), P(DPP-TTT), and P(DPP-TPhT) also showed highly prominent peaks up to the fourth order in the out-of-plane profiles; this indicates that the polymer chains are regularly arranged with highly ordered lamellar planes perpendicular to the substrate. It should be noted that annealed films of the four copolymers exhibited a predominant edge-on orientation according to the high intensity (010) diffraction pattern and the disappearance of the (100) diffraction peak in the in-plane profiles. The π–π stacking distances (d(010)) of the four copolymers are in the range of 3.62–3.75 Å (Fig. 3e). The GI-XRD results clearly reveal that the four copolymers that bear relatively long conjugated donor moieties predominantly show high crystallinity and an edge-on orientation on the OTS-SiO2/Si substrate.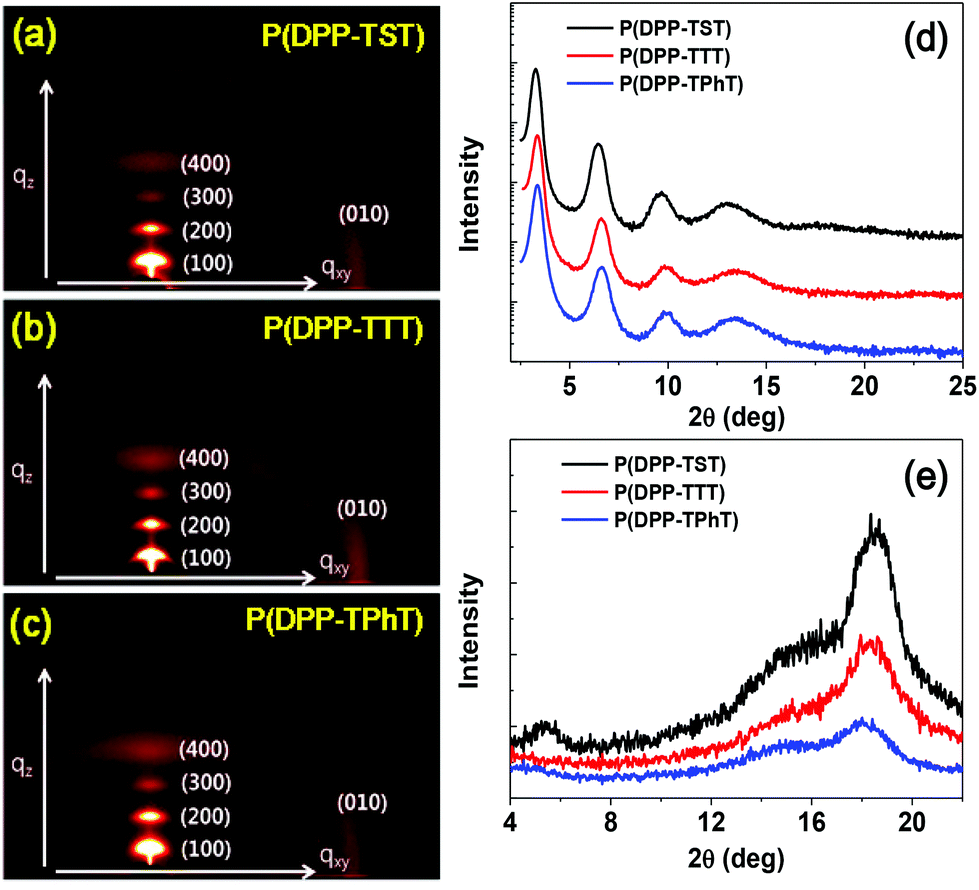 | ||
| Fig. 3 (a, b, c) 2D XRD patterns and 1D (d) out-of-plane and (e) in-plane modes of XRD patterns of P(DPP-TST), P(DPP-TTT), and P(DPP-TPhT). The diffraction patterns of P(DPP-SSS) are available in ref. 17. | ||
Performance of TFT devices
Despite the incorporation of unsubstituted long conjugated donor units into the repeating group of the copolymer, the good solubility of the copolymers in chloroform led us to fabricate the TFT devices via the spin-coating method. To investigate the charge transport properties of the new DPP-based copolymers, bottom-contact/bottom-gate (BCBG) TFT devices were successfully fabricated with the polymer films via the spin-coating method. The mobilities were obtained from the source–drain current–gate voltage curves (IDSversus VG) in well-resolved saturation regions (Fig. 4). The corresponding TFT performances of the copolymers are summarized in Table 2. Maximum hole mobilities were obtained from the thermally annealed films and are 4.17, 2.38, 3.98, and 1.28 cm2 V−1 s−1 for P(DPP-SSS), P(DPP-TST), P(DPP-TTT), and P(DPP-TPhT), respectively, at different annealing temperatures. The mobilities are 2–3 times higher than those of devices using as-spun films (Fig. S7 and S8†).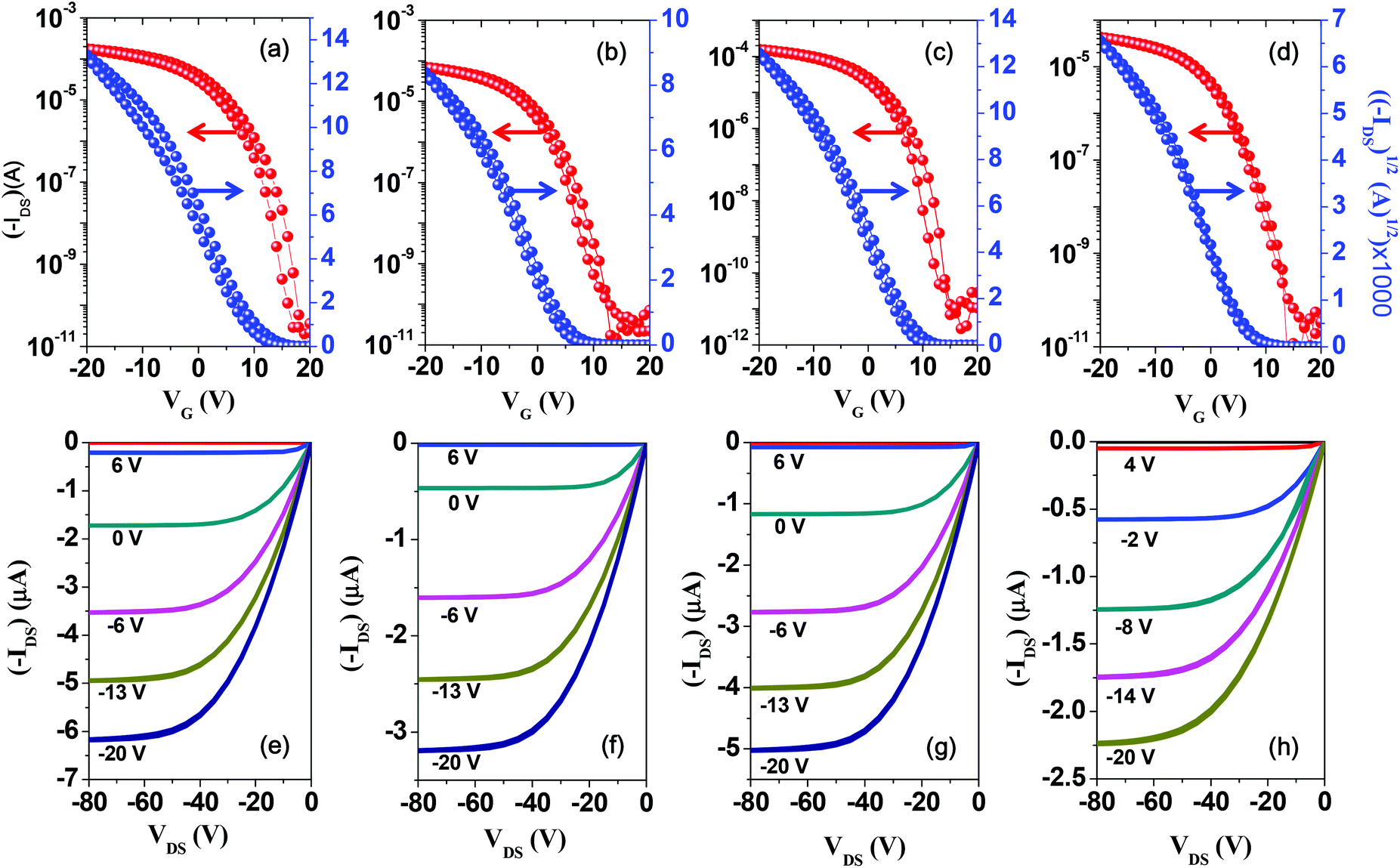 | ||
| Fig. 4 Transfer (top) and output curves (bottom) of TFT devices containing the DPP-based copolymer films after thermal annealing. (a, e): P(DPP-SSS), (b, f): P(DPP-TST): (c, g): P(DPP-TTT), (d, h): P(DPP-TPhT). | ||
| Polymer | T annealing (°C) | μ (cm2 V−1 s−1)a | I on/Ioff | V T (V) | Polymer:PC71BM (wt%) |
J sc (mA cm−2) | V oc (V) | FF | PCE (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Gate voltage ranges for determining the saturated mobilities: (i) −5 V to 5 V for P(DPP-SSS) film before annealing; −3 V to 7 V for annealed P(DPP-SSS) film. (ii) −10 V to 0 V for P(DPP-TST) film before annealing; −4 V to 2 V for annealed P(DPP-TST) film. (iii) −10 V to 5 V for P(DPP-TTT) film before annealing; −5 V to 5 V for annealed P(DPP-TTT) film. (i) −30 V to 20 V for P(DPP-TPhT) film before annealing; −5 V to 5 V for annealed P(DPP-TPhT) film. b Average value. | |||||||||
| P(DPP-SSS) | As-cast | 2.28 (2.15)b | >106 | 10 | 1:2 |
11.81 | 0.59 | 0.50 | 3.48 (3.36)b |
| 180 | 4.17 (4.07)b | 12 | |||||||
| P(DPP-TST) | As-cast | 0.62 (0.56)b | >106 | 6 | 1:2 |
13.14 | 0.60 | 0.53 | 4.18 (4.02)b |
| 160 | 2.38 (2.31)b | 5 | |||||||
| P(DPP-TTT) | As-cast | 1.66 (1.59)b | >106 | 8 | 1:2 |
15.63 | 0.62 | 0.51 | 4.94 (4.82)b |
| 160 | 3.98 (3.85)b | 10 | |||||||
| P(DPP-TPhT) | As-cast | 0.54 (0.51)b | >106 | 7 | 1:2 |
14.72 | 0.66 | 0.52 | 5.05 (5.01)b |
| 180 | 1.28 (1.22)b | −3 | |||||||
All the DPP-based copolymers containing highly π-extended donating groups show higher hole mobility than reported DPP-based copolymers bearing shorter conjugated donating units, such as mono/bithiophene, mono/biselenophene, and benzene.11,13,14,18 In particular, P(DPP-SSS) and P(DPP-TTT), which contain homogeneous donating units, exhibit much higher hole mobility than both P(DPP-TST) and P(DPP-TPhT), which contain heterogeneous donating units; this is attributed to more facile packing of the polymer chains. However, the difference in the TFT performances is difficult to explain via the molecular energy levels or GI-XRD results because of their similar crystalline behavior.
To better understand the origin of the TFT performance, atomic force microscopy (AFM) images of the polymer film surfaces were recorded. As shown in Fig. 5, the height images of the thermally annealed P(DPP-SSS) and P(DPP-TTT) copolymer films show a highly fine fibrous morphology, which can support enhanced charge transport in the active channel of TFT devices relative to that of P(DPP-TST) and P(DPP-TPhT) copolymer films. In addition, compared to P(DPP-TTT), P(DPP-SSS) which has terselenophene donors was reported to enhance hole mobility through stronger intermolecular interactions and a more favorable quinoidal nature.15
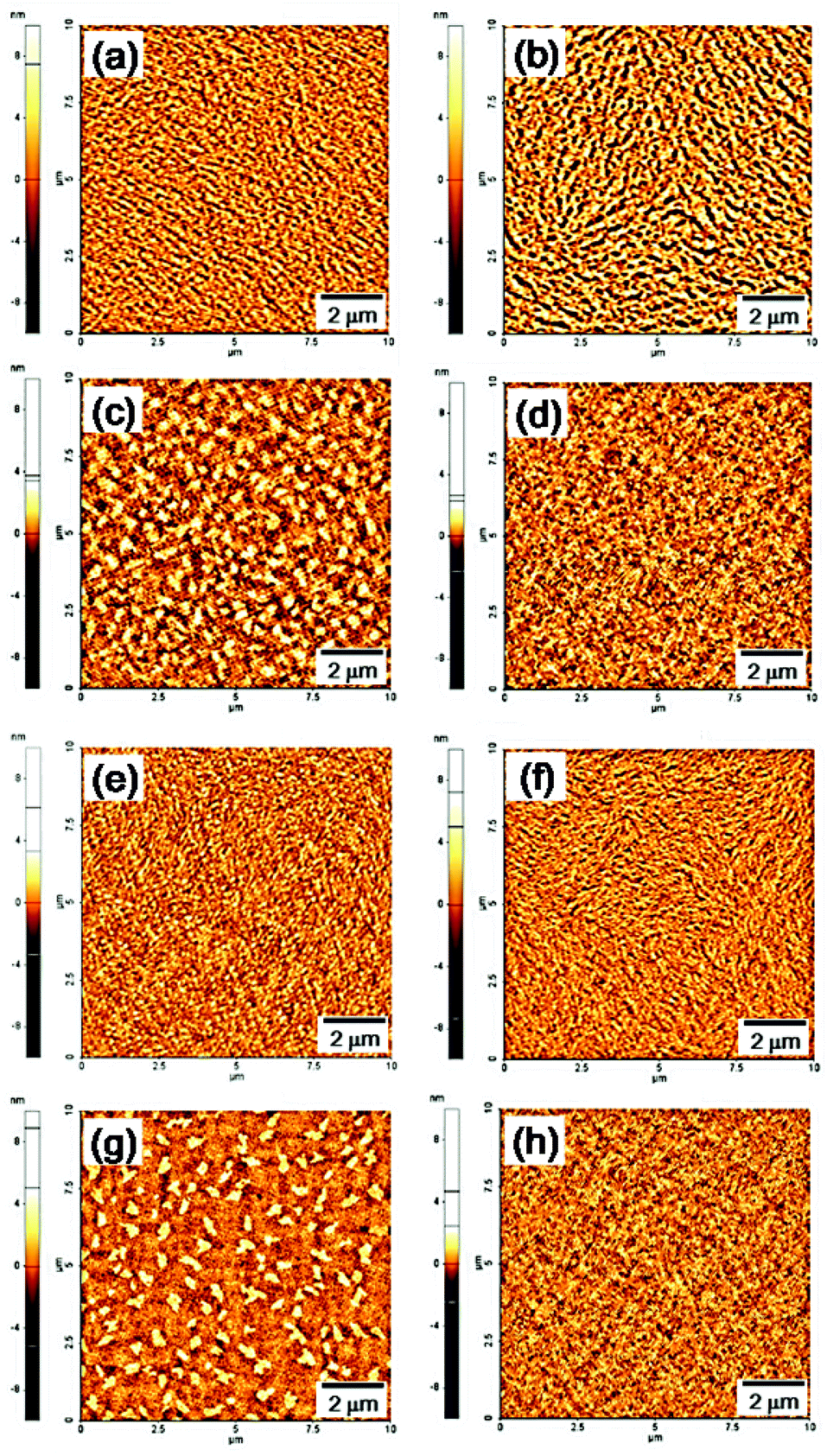 | ||
| Fig. 5 AFM height images (10 × 10 μm) of the polymer films before and after thermal annealing: (a, b) P(DPP-SSS);17 (c, d) P(DPP-TST); (e, f) P(DPP-TTT); (g, h) P(DPP-TPhT); (a, c, e, g) as-spun films; (b, d, f, h) thermally annealed films. | ||
Photovoltaic properties
Bulk heterojunction PV cells were fabricated using synthesized DPP-based copolymers as the photoactive layers by spin-coating a blend solution of the polymer and PC71BM (1:2 wt%) in an o-DCB–chloroform (1:4 v/v) co-solvent. Optimal performance was obtained with the following device configuration: ITO/PEDOT:PSS (30 nm)/polymer:PC71BM/LiF (1 nm)/Al (100 nm) (Fig. 6a). The device performance data are summarized in Table 2.
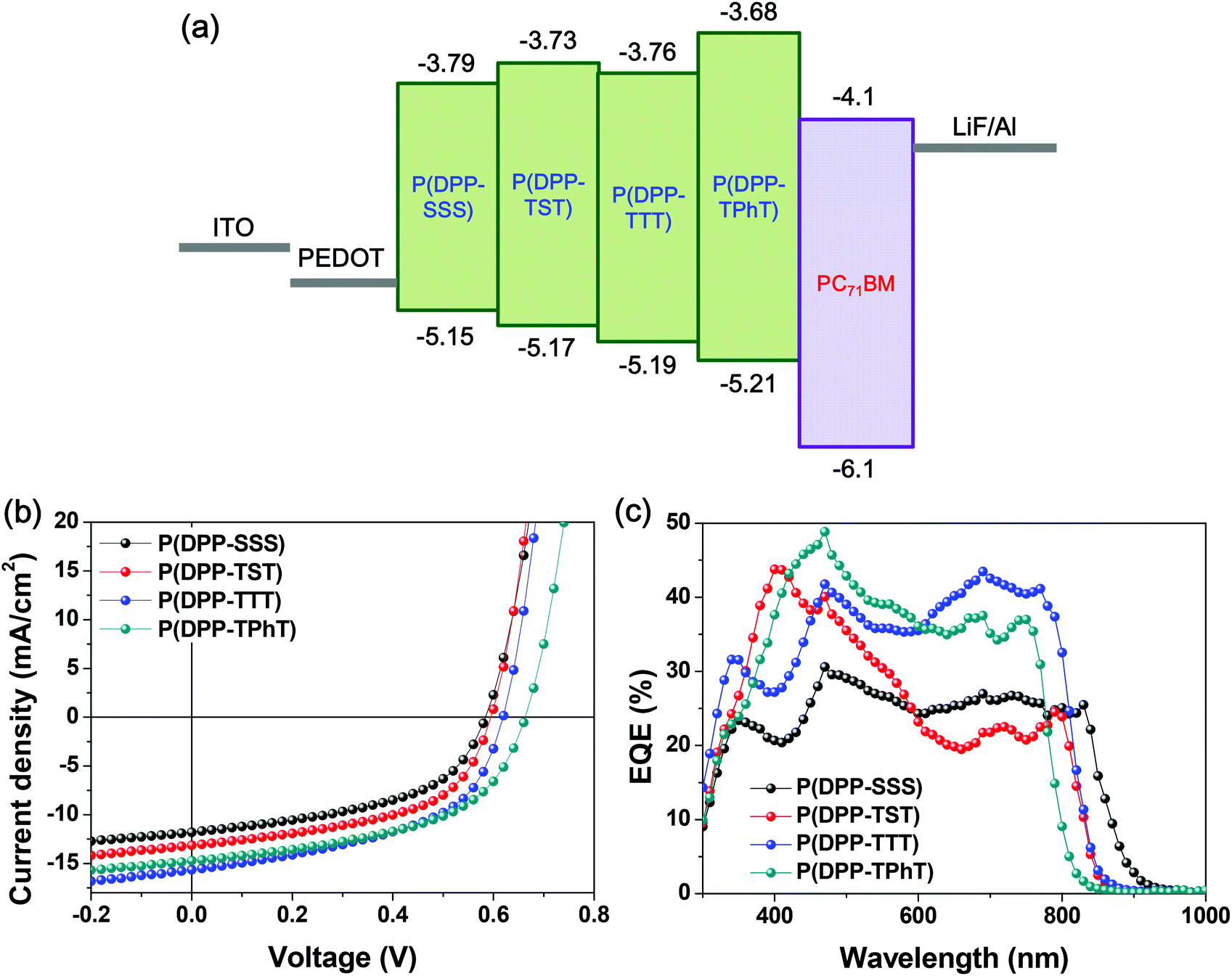 | ||
| Fig. 6 (a) Energy level alignment and OPV device structure based on the polymer:PC71BM blend. (b) J–V characteristics of BHJ OPVs fabricated from four copolymers under illumination with simulated AM 1.5 G solar irradiation at 100 mW cm−2. (c) EQE spectra of the OPVs. | ||
Fig. 6b and c show the current density–voltage (J–V) and external quantum efficiency (EQE) characteristics obtained under simulated AM 1.5 G solar irradiation (100 mW cm−2). Conventional devices fabricated with P(DPP-SSS), P(DPP-TST), P(DPP-TTT), and P(DPP-TPhT) demonstrate power conversion efficiencies (PCEs) of 3.48, 4.18, 4.94, and 5.05%, respectively, and open-circuit voltages (Vocs) of 0.59, 0.60, 0.62, and 0.66 V, respectively. The Voc values of the four copolymers are slightly different, which is well consistent with the differences between the HOMO levels of the copolymers and LUMO level of PC71BM.
The EQE spectra of the devices fabricated with the four polymers show a significant contribution to photocurrent generation in the wavelength range of 400–950 nm (Fig. 6b).
Of the AFM images of the PC71BM blend films with copolymers, that of the P(DPP-SSS) copolymer shows a very coarse domain with a root mean square (RMS) of 2.14 nm that might hamper effective exciton diffusion and migration of separated charges in the blend film, thus decreasing the Jsc of the P(DPP-SSS):PC71BM device (Fig. 7). The other three copolymer films with PC71BM feature much finer domains without the addition of additives such as 1,8-diiodooctane and 1-chloronaphthalene, which results in higher Jsc values (13.14–15.63 mA cm−2).
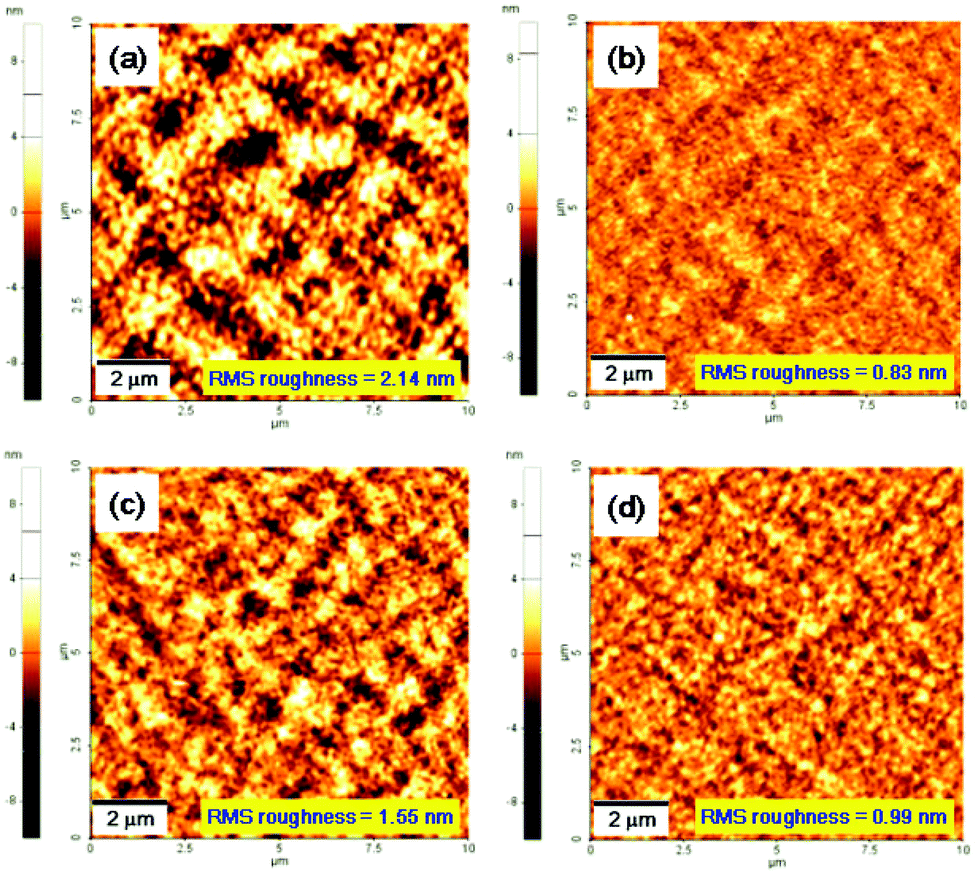 | ||
| Fig. 7 AFM height images (10 × 10 μm) of the blend film. (a) P(DPP-SSS) with PC71BM, (b) P(DPP-TST) with PC71BM, (c) P(DPP-TTT) with PC71BM, and (d) P(DPP-TPhT) with PC71BM. Polymer:PC71BM = 1:2 weight ratio. | ||
In particular, the PV device fabricated with PC71BM and P(DPP-TPhT) shows the highest power conversion efficiency (5.05%), a high Jsc of 14.72 mA cm−2, and the highest Voc of 0.66 V owing to its fine film morphology having nanophase separation and lower-lying HOMO value.
Conclusions
We designed and synthesized four DPP-based polymers with π-extended donating units of different lengths, including three-component homogeneous aromatic rings (SSS and TTT) and heterogeneous aromatic rings (TST and TPhT). The absorption ranges and energy levels of the polymers were easily tuned through the introduction of these donating units. Thermally annealed thin films of the polymers exhibited high crystallinity and edge-on orientation on OTS-treated SiO2/Si substrates. TFT devices containing the thermally annealed P(DPP-SSS) and P(DPP-TTT) films exhibited high hole mobilities of 4.17 and 3.98 cm2 V−1 s−1, respectively, because of their crystalline fibrous surface morphology. PV devices fabricated with P(DPP-TTT) and P(DPP-TPhT) in combination with PC71BM displayed larger Voc and Jsc values and higher PCEs. Using P(DPP-TPhT), which was identified as the optimal donor material, we obtained the highest PCE value of 5.05% without solvent additives, thermal treatment, and favorable interlayers. Interestingly, the four polymers showed selective solubility in organic solvents, which can lead us to fabricate multi-layered devices through solution processing. These results unambiguously demonstrate the effect of various π-extending donating units on the physical, electronic, and optoelectronic properties of DPP-based copolymers.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF2012R1A2A1A01008797) and by Priority Research Centers Program through the NRF funded by the Ministry of Education, Science and Technology (NRF201200020209). MJ Cho particularly acknowledges the support from a Korea University grant (2013–2014).Notes and references
-
(a) H. N. Tsao, D. Cho, J. Andreasen, W. A. Rouhanipour, D. W. Breiby, W. Pisula and K. Müllen, Adv. Mater., 2009, 21, 209 CrossRef CAS
; (b) H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. Shakya Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. J. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2011, 133, 3272 CrossRef CAS PubMed
-
(a) H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605 CrossRef CAS PubMed
-
(a) Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792 CrossRef CAS PubMed
-
(a) M.-S. Su, C.-Y. Kuo, M.-C. Yuan, U. S. Jeng, C.-J. Su and K.-H. Wei, Adv. Mater., 2011, 23, 3315 CrossRef CAS PubMed
-
(a) Z. Chen, M. J. Lee, R. S. Ashraf, Y. Gu, S. Albert-Seifried, M. M. Nielsen, B. Schroeder, T. D. Anthopoulos, M. Heeney, I. McCulloch and H. Sirringhaus, Adv. Mater., 2012, 24, 647 CrossRef CAS PubMed
-
(a) D. S. Yang, K. H. Kim, M. J. Cho, J.-I. Jin and D. H. Choi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1457 CrossRef CAS
-
(a) L. Dou, J. Gao, E. Richard, J. B. You, C. C. Chen, K. C. Cha, Y. J. He, G. Li and Y. Yang, J. Am. Chem. Soc., 2012, 134, 10071 CrossRef CAS PubMed
-
(a) T. W. Lee, D. H. Lee, J. Shin, M. J. Cho and D. H. Choi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5280 CrossRef CAS
-
(a) X. Zhang, L. J. Richter, D. M. DeLongchamp, R. J. Kline, M. R. Hammond, I. McCulloch, M. Heeney, R. S. Ashraf, J. N. Smith, T. D. Anthopoulos, B. Schroeder, Y. H. Geerts, D. A. Fischer and M. F. Toney, J. Am. Chem. Soc., 2011, 133, 15073 CrossRef CAS PubMed
-
(a) Y. Li, S. P. Singh and P. Sonar, Adv. Mater., 2010, 22, 4862 CrossRef CAS PubMed
-
(a) Y. Li, P. Sonar, S. P. Singh, M. S. Soh, M. van Meurs and J. Tan, J. Am. Chem. Soc., 2011, 133, 2198 CrossRef CAS PubMed
-
(a) J. W. Jung, F. Liu, T. P. Russell and W. H. Jo, Energy Environ. Sci., 2012, 5, 6857 RSC
- H.-W. Lin, W.-Y. Lee and W.-C. Chen, J. Mater. Chem., 2012, 22, 2120 RSC
- J. S. Ha, K. H. Kim and D. H. Choi, J. Am. Chem. Soc., 2011, 133, 10364 CrossRef CAS PubMed
- I. Kang, T. K. An, J.-A. Hong, H.-J. Yun, R. Kim, D. S. Chung, C. E. Park, Y.-H. Kim and S.-K. Kwon, Adv. Mater., 2013, 25, 524 CrossRef CAS PubMed
- Z. Yi, X. Sun, Y. Zhao, Y. Guo, X. Chen, J. Qin, G. Yu and Y. Liu, Chem. Mater., 2012, 24, 4350 CrossRef CAS
- M. J. Cho, J. Shin, S. H. Yoon, T. W. Lee, M. Kaur and D. H. Choi, Chem. Commun., 2013, 49, 7132 RSC
-
(a) J. C. Bijleveld, A. P. Zoombelt, S. G. J. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. J. Janssen, J. Am. Chem. Soc., 2009, 131, 16616 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR, GPC, computation, and OTFT results of the DPP-based copolymers. See DOI: 10.1039/c4py01112k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |