
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of bottlebrush polymers via transfer-to and grafting-through approaches using a RAFT chain transfer agent with a ROMP-active Z-group†
Scott C.
Radzinski‡
,
Jeffrey C.
Foster‡
and
John B.
Matson
 *
*
Department of Chemistry and Macromolecules and Interfaces Institute, Virginia Tech, Blacksburg, Virginia 24061, USA. E-mail: jbmatson@vt.edu
First published on 14th January 2015
Abstract
A novel dithiocarbamate chain transfer agent (CTA1) with a directly polymerizable Z-group was synthesized for use in reversible addition–fragmentation chain transfer polymerization (RAFT). This CTA effectively mediated RAFT polymerization of styrenic and acrylic monomers with dispersities (Đ) < 1.08. Utilizing the polymerizable Z-group on the ω-chain end that is inherited from the RAFT process, bottlebrush polymers were synthesized via ring-opening metathesis polymerization (ROMP) in a grafting-through process. The effect of a number of parameters on the grafting process was studied, and optimized conditions yielded bottlebrush polymers of controllable molecular weights, narrow molecular weight distributions, and high conversions (>90%). Bottlebrush polymers made by a transfer-to strategy were also synthesized from CTA1. In this case, ROMP was first carried out to produce poly(CTA1) (PCTA1), then RAFT was performed from the PCTA1 backbone. This technique allows for the preparation of high molecular weight bottlebrush polymers without radical coupling between bottlebrush polymers. Lastly, regardless of the synthetic method, all bottlebrush polymers produced using CTA1 are composed of polymeric side chains that are attached to the bottlebrush backbone through a labile dithiocarbamate linkage that can be cleaved in the presence of nucleophiles such as amines. The unique combination of these capabilities allows for the study of bottlebrush polymer formation by both transfer-to and grafting-through strategies using a single agent.
Introduction
Over the past two decades, bottlebrush polymers have become an increasingly relevant polymer topology as a result of their well-defined structures, shape persistence, nanoscopic dimensions, and unique mechanical and rheological properties.1 Bottlebrush polymers are comprised of polymeric side-chains grafted to a polymer backbone, and given sufficient grafting density, steric repulsion between polymeric neighbors causes the backbone to adopt a chain-extended conformation.2,3 Consequently, cylindrical,4,5 spherical,6,7 or worm-like,8 morphologies can be realized in single macromolecules by tuning polymer composition, grafting density, or side-chain molecular weight. Additionally, important polymeric properties, such as amphiphilicity or stimuli-responsiveness, can be extrapolated to bottlebrush systems by tuning the chemical composition of the polymeric side-chains.9–11 In terms of mechanical properties, bottlebrush polymers differ from linear polymers primarily due to their inability to interact via chain entanglements—a phenomenon from which many of the physical properties of linear polymers arise. Therefore, bottlebrush polymers have garnered interest for use in applications such as rheology modifiers and super-soft elastomers.12 Lastly, their size and shape-persistence makes bottlebrush polymers well suited for the in vivo delivery of therapeutic agents.13Bottlebrush polymers can be prepared via one of four approaches: (1) the grafting-from strategy, whereby polymer side chains are grown from a polymeric backbone decorated with initiating functionalities; (2) the grafting-to methodology involving the attachment of pre-formed polymers to reactive sites on a polymer backbone; (3) the grafting-through or macromonomer (MM) approach, in which polymers fitted with a polymerizable moiety are utilized as MMs in an subsequent polymerization; and, (4) the transfer-to strategy (sometimes called the RAFT Z-group approach). Transfer-to is a unique hybrid of the grafting-from and grafting-to strategies in which polymeric radicals detach from the bottlebrush backbone, propagate freely in solution, and return to the backbone through a chain-transfer reaction with a pendant CTA.14 While the grafting-from, grafting-to, and transfer-to strategies yield macromolecules with a bottlebrush topology, the grafting process is hindered by steric interactions between adjacent polymer chains, resulting in low grafting densities.15 However, despite this shortcoming, grafting-from (and to a greater extent transfer-to) can be employed to synthesize bottlebrush polymers with relatively higher molecular weights (on the order of ≥106 Da) than are possible with grafting-through. In contrast, grafting-through results in “perfectly grafted” (i.e. the highest possible grafting density) bottlebrush polymers, as each repeat unit bears a polymeric side chain. In view of the high grafting density and synthetic versatility of the grafting-through technique, recent efforts have focused on its application.11,16–31
To prepare well-defined bottlebrush polymers via the grafting-through strategy, reversible-deactivation radical polymerization techniques such as atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain transfer polymerization (RAFT) are often employed.1 Generally, semi-telechelic MMs of predefined MW and low dispersity (Đ) are synthesized via one of these techniques. In a second step, the resulting MMs are functionalized with a polymerizable moiety in a post polymerization reaction, and the MMs are subsequently polymerized using an additional polymerization method in the third and final step.20,23 Alternatively, in a two-step method, MM synthesis can be conducted in the presence of an initiator or chain-transfer agent (CTA) containing an orthogonal functionality.17,21,22 For example, norbornene and derivatives thereof have been coupled to dithioester and trithiocarbonate CTAs.19 In this case, RAFT was utilized to prepare the MM grafts, and ring-opening metathesis polymerization (ROMP) was employed in a subsequent grafting-through step. ROMP is particularly well suited for this purpose because of the high functional group tolerance and rapid propagation rate of several ruthenium-based olefin metathesis catalysts (Fig. 1).16 Bottlebrush polymers have been prepared via a combination of these two polymerization techniques in separate steps as described above, or more simply via a one-pot strategy wherein a ROMP catalyst is added to a terminated RAFT reaction mixture.18
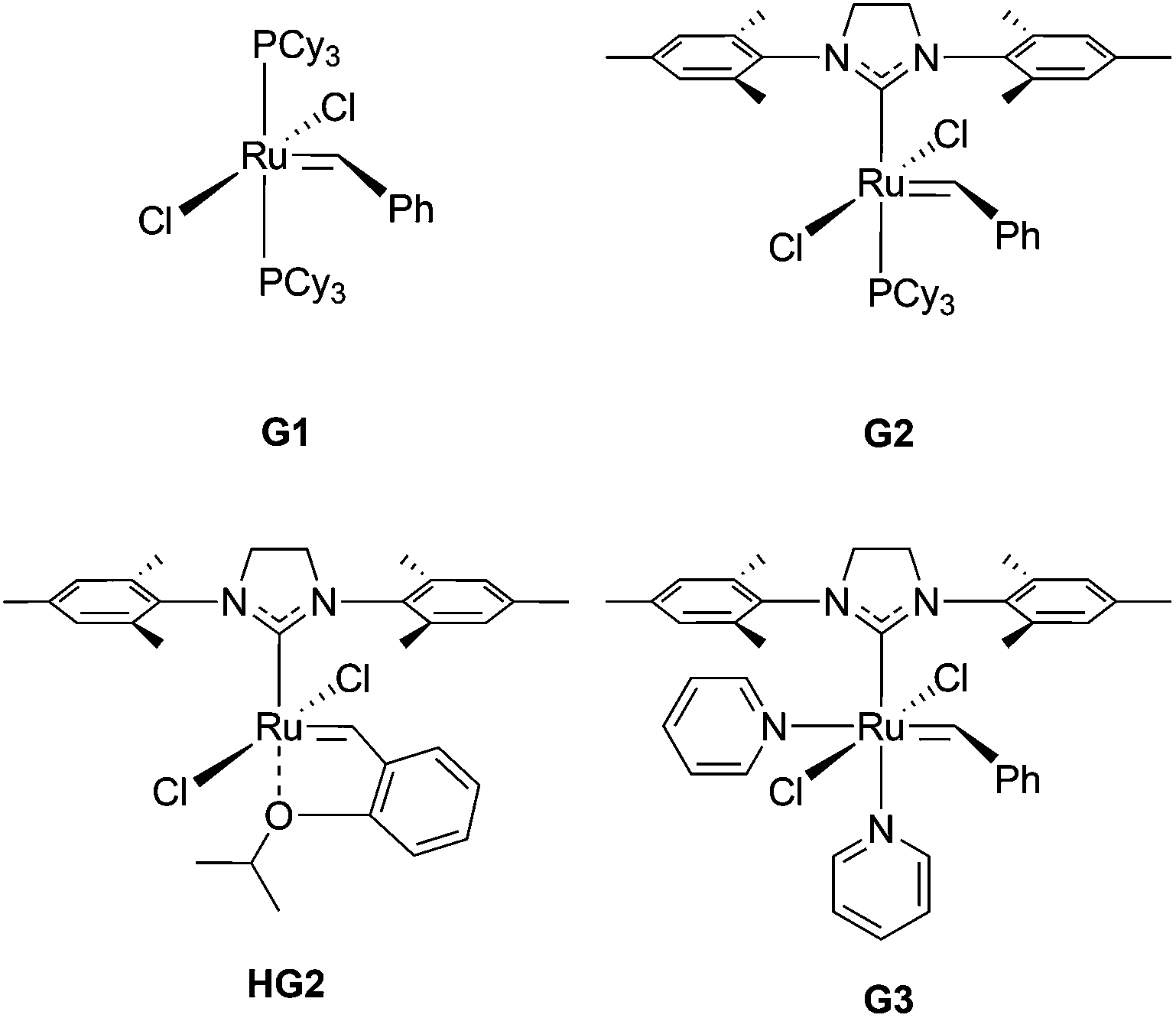 | ||
| Fig. 1 ROMP catalysts used in this work. | ||
RAFT polymerization is mediated by a thiocarbonylthio-containing agent with an activating Z-group and a leaving group (R-group) that is comparable in radical stability to that of the monomer-derived radical. A number of R- and Z-groups have been utilized to gain control over the RAFT polymerization of a wide range of vinyl monomers.32 However, to our knowledge, there exists no report in the literature on the incorporation of a directly polymerizable Z-group such as exo-norbornene imide into a RAFT agent (Scheme 1). We envisioned that such a CTA (CTA1) could be employed in both RAFT transfer-to and ROMP grafting-through methodologies.
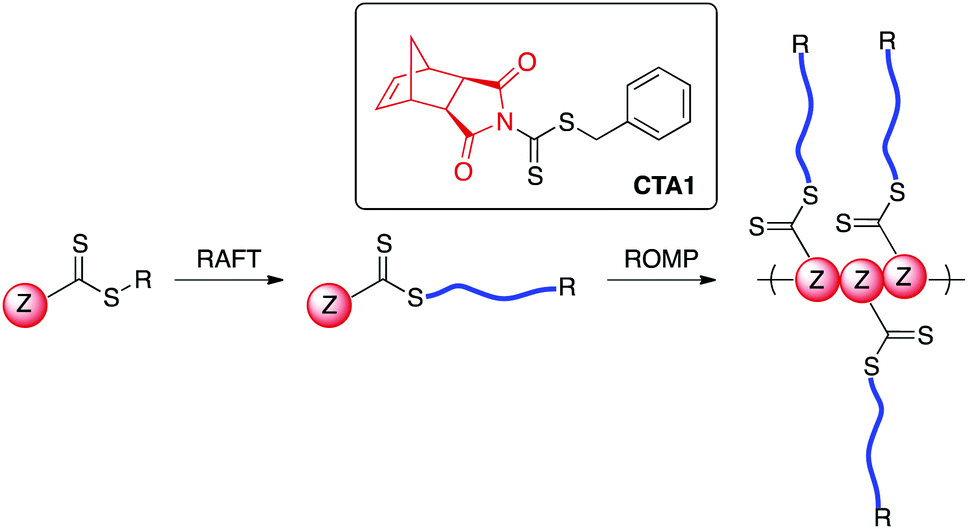 | ||
| Scheme 1 Proposed CTA structure with directly polymerizable Z-group. | ||
While directly polymerizable R-groups have been incorporated into RAFT CTAs,18,19 a directly polymerizable Z-group would experience the benefits inherent in the Z-group, or transfer-to, approach.33–36 Such benefits are a result of the RAFT mechanism when applied in a graft polymerization. During a RAFT transfer-to polymerization, growing polymer chains detach from the bottlebrush polymer backbone during propagation and then add back to the backbone through reaction with a pendant thiocarbonylthio group. Because the growing “arms” are free in solution, coupling between growing adjacent “arms” attached to the bottlebrush backbone does not occur. In addition, radical coupling between growing bottlebrush polymers cannot occur, as the propagating radical resides on the detached polymeric side chains. As a result, the transfer-to approach affords bottlebrush polymers with lower dispersities and higher possible conversions relative to conventional RAFT grafting-from using the R group approach.36,37 An additional advantage of the incorporation of a directly polymerizable Z-group is the location of the thiocarbonylthio group in the bottlebrush polymer. In the case of bottlebrush polymers prepared from CTA1, the thiocarbonylthio group would link the polymeric arms to the backbone polymer, in contrast to systems using directly polymerizable R-groups, which leave the thiocarbonylthio group on the bottlebrush surface. Given the wealth of literature concerning the post-polymerization removal of RAFT end groups,38 we envisioned the possibility of thiocarbonylthio degradation-driven side chain dissociation. Herein, we investigate the efficacy of CTA1 as a mediator of RAFT polymerization and the availability of the norbornene functionality for ROMP. Additionally, we evaluate the preparation of bottlebrush polymers via both transfer-to and grafting-through strategies. Finally, we explore side-chain cleavage by aminolysis to investigate side chain molecular weights and molecular weight distributions.
Materials and methods
Materials
All reagents were obtained from commercial vendors and used as received unless otherwise stated. Styrene and n-butyl acrylate were passed through small columns of basic alumina prior to use. ROMP catalysts (PCy3)2(Cl)2Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh (G1), (H2IMes)(Cl)2(PCy3)RuCHPh (G2) and (H2IMes)(Cl)2(PCy3)RuCH(2-OiPrPh) (HG2) were obtained as a generous gift from Materia. ROMP catalyst (H2IMes)(pyr)2(Cl)2RuCHPh (G3) was prepared from G2 according to literature procedures.39,40
CHPh (G1), (H2IMes)(Cl)2(PCy3)RuCHPh (G2) and (H2IMes)(Cl)2(PCy3)RuCH(2-OiPrPh) (HG2) were obtained as a generous gift from Materia. ROMP catalyst (H2IMes)(pyr)2(Cl)2RuCHPh (G3) was prepared from G2 according to literature procedures.39,40
Methods
NMR spectra were measured on Agilent 400 MHz or Bruker 500 MHz spectrometers. 1H and 13C NMR chemical shifts are reported in ppm relative to internal solvent resonances. Yields refer to chromatographically and spectroscopically pure compounds unless otherwise stated. Size exclusion chromatography (SEC) was carried out in THF at 1 mL min−1 at 30 °C on two Agilent PLgel 10 μm MIXED-B columns connected in series with a Wyatt Dawn Helios 2 light scattering detector and a Wyatt Optilab Rex refractive index detector. No calibration standards were used, and dn/dc values were obtained by assuming 100% mass elution from the columns. Atomic force microscopy (AFM) was conducted using a Veeco BioScope II AFM in tapping mode in air at room temperature using Nano World Pointprobe-silicon SPM Sensor tips (spring constant = 7.4 N m−1, resonance frequency = 160 kHz). Dynamic light scattering (DLS) was conducted using a Malvern Zetasizer Nano operating at 25 °C. Polymer solutions were prepared at 1 mg mL−1 and were filtered with a 0.25 μm filter prior to scanning. The calculations of the particle size distributions and distribution averages were conducted using CONTIN particle size distribution analysis routines. All measurements were made in triplicate and errors reflect standard deviations.Synthesis of CTA1
KOH (0.60 g, 10.7 mmol) was ground to a fine powder with a mortar and pestle and placed in a 100 mL round bottom flask. To the flask was added exo-norbornene imide (1.33 g, 8.15 mmol) followed by 30 mL of DMF. This mixture was stirred for 5 min, followed by dropwise addition of CS2 (2.46 mL, 40.8 mmol). The solution developed a deep red color. After an additional 3 h of stirring, benzyl bromide (4.84 mL, 40.8 mmol) was added dropwise, and the reaction mixture was stirred at rt for 12 h. The following day, the reaction mixture was diluted with diethyl ether (∼50 mL) and washed with H2O (3 × 150 mL) and brine. The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified on a silica gel column, eluting with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2–hexanes, to give 1.30 g of CTA1 as a yellow solid (48% yield). 1H NMR (CDCl3): δ 1.57 (m, 2H), 2.82 (s, 2H), 3.39 (s, 2H), 4.50 (s, 2H), 6.33 (s, 2H), 7.35 (m, 5H). 13C NMR (CDCl3): δ 199.31, 173.75, 138.29, 132.93, 129.34, 128.95, 128.26, 48.27, 46.47, 43.62, 43.22. HRMS (m/z): calculated 330.0617, found 330.0627.
:1 CH2Cl2–hexanes, to give 1.30 g of CTA1 as a yellow solid (48% yield). 1H NMR (CDCl3): δ 1.57 (m, 2H), 2.82 (s, 2H), 3.39 (s, 2H), 4.50 (s, 2H), 6.33 (s, 2H), 7.35 (m, 5H). 13C NMR (CDCl3): δ 199.31, 173.75, 138.29, 132.93, 129.34, 128.95, 128.26, 48.27, 46.47, 43.62, 43.22. HRMS (m/z): calculated 330.0617, found 330.0627.
Synthesis of polystyrene MMs
A typical polymerization procedure of styrene is as follows: to an oven-dried Schlenk tube equipped with a magnetic stir bar was added CTA1 (29.0 mg, 87.0 μmol), 2,2′-azobis(2-methylpropionitrile) (AIBN) (1.43 mg, 8.73 μmol), styrene (1 mL, 8.7 mmol), and 1 mL of THF. The reaction mixture was deoxygenated by three freeze–pump–thaw cycles. The Schlenk tube was then backfilled with N2 and submerged in an oil bath maintained at 75 °C. Samples were removed periodically by N2-purged syringe to monitor molecular weight evolution by SEC and conversion by 1H NMR spectroscopy. The polymerization was quenched by submerging the tube into liquid N2 and exposing the reaction mixture to air. The resulting polystyrene was purified via precipitation from MeOH (3×).Synthesis of poly(n-butyl acrylate) (nBA) MMs
A typical polymerization procedure of nBA is as follows: to an oven-dried Schlenk tube equipped with a magnetic stir bar was added CTA1 (46.0 mg, 140 μmol), AIBN (2.29 mg, 14.0 μmol), nBA (2 mL, 14 mmol), and 2 mL of THF. The reaction mixture was deoxygenated by three freeze–pump–thaw cycles. The Schlenk tube was then backfilled with N2 and submerged in an oil bath maintained at 60 °C. Samples were removed periodically by N2-purged syringe to monitor molecular weight evolution by SEC and conversion by 1H NMR spectroscopy. The polymerization was quenched by submerging the tube into liquid N2 and exposing the reaction solution to air. The resulting poly(nBA) was purified via precipitation from hexanes (3×).Synthesis of poly(CTA1) (PCTA1)
A typical polymerization procedure of CTA1 is as follows: CTA1 (109 mg, 329 μmol) was dissolved in 1.5 mL of anhydrous CH2Cl2 in a 1-dram vial. A solution of G3 in anhydrous CH2Cl2 was prepared at 9.6 mg mL−1 in a second vial. 0.5 mL of this G3 soln was added rapidly to the vial containing CTA1. The polymerization was quenched after 20 min by adding 1–3 drops of ethyl vinyl ether. The polymer was isolated via precipitation from hexanes and dried under vacuum to yield 93 mg of pure polymer as an off-white powder (85% yield).Synthesis of poly(CTA1-g-styrene) by RAFT transfer-to
To an oven-dried Schlenk tube equipped with a magnetic stir bar was added poly(CTA1) (22.0 mg, 873 μmol, Mn = 25100 g mol−1), AIBN (0.014 mg, 0.087 μmol), styrene (5 mL, 43 mmol), and 5 mL of THF. The reaction mixture was deoxygenated by three freeze–pump–thaw cycles. The Schlenk tube was then backfilled with N2 and submerged in an oil bath maintained at 75 °C. Samples were removed periodically by N2-purged syringe to monitor molecular weight evolution by SEC and conversion by 1H NMR spectroscopy. The polymerization was quenched by submerging the tube into liquid N2 and exposing the reaction solution to air. The resulting poly(CTA1-g-styrene) was purified via precipitation from MeOH.
Synthesis of poly(CTA1-g-styrene) by RAFT grafting-through
A typical grafting-through polymerization procedure is as follows: to a vial containing MM in anhydrous CH2Cl2 was added rapidly a soln of G3 in anhydrous CH2Cl2 to make a final polymer concentration of 100 mg mL−1. The polymerization was stirred at rt. After 1 h, the polymerization was quenched by adding 1–3 drops of ethyl vinyl ether. The resulting bottlebrush polymer was isolated via precipitation from a MeOH–H2O mixture and dried under vacuum.Aminolysis of bottlebrush polymers
Bottlebrush polymer was dissolved in 0.3 mL of THF in a 1-dram vial equipped with a stir bar. To the vial was added 0.3 mL of a 40 w/v% soln of methylamine in H2O. The reaction mixture was stirred at rt in air for 72 h to ensure complete thiol oxidation. A few drops of THF were added to dissolve the precipitated solids, and the resulting aminolyzed side chains were isolated via precipitation from MeOH and were dried under vacuum overnight.Results and discussion
CTA1 synthesis
CTA1 was synthesized in one-pot starting from exo-norbornene imide (see ESI† for further details) as shown in Scheme 2. The reaction proceeds via the potassium hydroxide-assisted attack of the imide nitrogen on carbon disulfide, forming a dithiocarbamate salt. In the second step, the dithiocarbamate intermediate reacts with benzyl bromide in a substitution reaction to yield the desired product.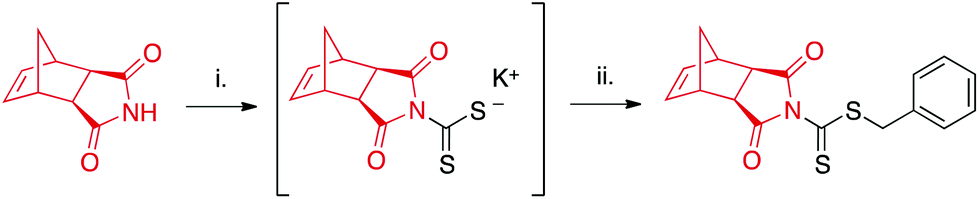 | ||
| Scheme 2 One-pot synthesis of CTA1. aExperimental conditions: (i) CS2, KOH, DMF; (ii) benzyl bromide. | ||
Column purification was required to remove unreacted starting materials, and isolated yields ranged from 35–48%. While this range is lower than reported values for the synthesis of other dithiocarbamates,41,42 many such CTAs are derived from electron-rich secondary amines in contrast to the relatively electron-deficient imide employed in our study. This electron-deficiency results in the limited nucleophilicity of the imide nitrogen,43 hindering its addition to electrophiles such as CS2. Longer reaction times or increased temperatures were not found to increase yields.
RAFT polymerization
The success of a RAFT polymerization is tied to many factors, chief among which is the matching of monomer reactivity to that of the CTA.44–48 More activated monomers (MAMs) (i.e., methacrylates, acrylates, styrenes) require electron deficient CS bonds, such as those present in dithioester and trithiocarbonate RAFT agents, while less activated monomers (LAMs) (i.e., vinyl esters and amides) can only be polymerized in the presence of more electron rich thiocarbonyl-containing compounds such as xanthates. In fact, reaction of LAMs with dithiobenzoate or certain trithiocarbonate CTAs can result in complete inhibition of polymerization.49 The use of N-pyrrolyl and N-phthalimidyl moieties as Z-groups in dithiocarbamate RAFT CTAs was first reported by Cheifari and coworkers.50 These CTAs are suitable for controlling the polymerization of many MAMs.47,51–53 Therefore, we envisioned that CTA1 could be employed to mediate the polymerization of this class of vinyl monomers.
To evaluate our hypothesis, RAFT polymerizations of styrene and n-butyl acrylate (nBA) were carried out in the presence of CTA1 (Scheme 3). The polymerizations were conducted in THF (1:1 v/v% THF–monomer) at 75 °C for styrene or 60 °C for nBA in the presence of 2,2′-azobis(isobutyronitrile) (AIBN). To maintain a high level of chain end fidelity, a [CTA]/[AIBN] ratio of 10:1 was chosen.54 Kinetic analysis was performed by removing aliquots of the polymerization solution at various time points via N2-purged syringe. The polymerizations were quenched by exposing the reaction mixture to air and submerging the reaction vessel into liquid N2. Molecular weight (MW) and dispersity (Đ) were determined by size-exclusion chromatography (SEC), and conversions were measured by 1H NMR spectroscopy. CTA1-mediated RAFT polymerization of styrene and nBA yielded polymers of controllable molecular weights with narrow molecular weight distributions (Đ < 1.08). As previously reported for similar dithiocarbamate CTAs,55 polymerization of methyl methacrylate was uncontrolled. On the opposite end of the monomer reactivity spectrum, polymerization of vinyl acetate was completely inhibited in the presence of CTA1.
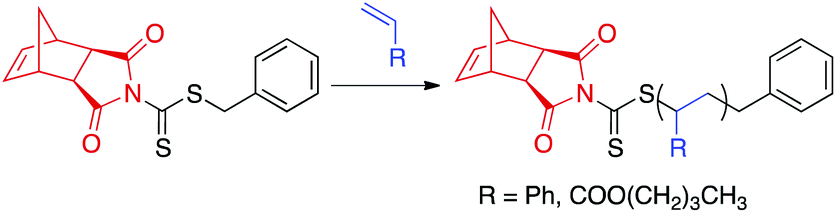 | ||
| Scheme 3 CTA1-mediated RAFT polymerization of styrene or nBA. aExperimental conditions: AIBN, THF, 75 °C for styrene or 60 °C for nBA. | ||
Kinetic analysis of CTA1-mediated polymerization of styrene and nBA is shown in Fig. 2. Molecular weight distributions determined by SEC were monomodal with low Đ, indicative of a well-controlled polymerization and a high chain transfer efficiency of CTA1 under the conditions investigated. Conversion increased linearly with time for nBA, and Đ decreased over the course of the reaction for both nBA and styrene. In the case of styrene, a non-linear relationship was observed in the semi-logarithmic plot (Fig. 2C) and a maximum conversion of only 48% was obtained after 24 h, indicative of the occurrence of termination reactions during the polymerization. The polymerization was repeated under more dilute conditions (2:1 v/v% THF–styrene), resulting in a reduction of termination reactions and a higher terminal conversion (81% after 24 h). For both monomers, the linear relationship between MW and conversion corroborated the controlled nature of the polymerization (Fig. 2D).
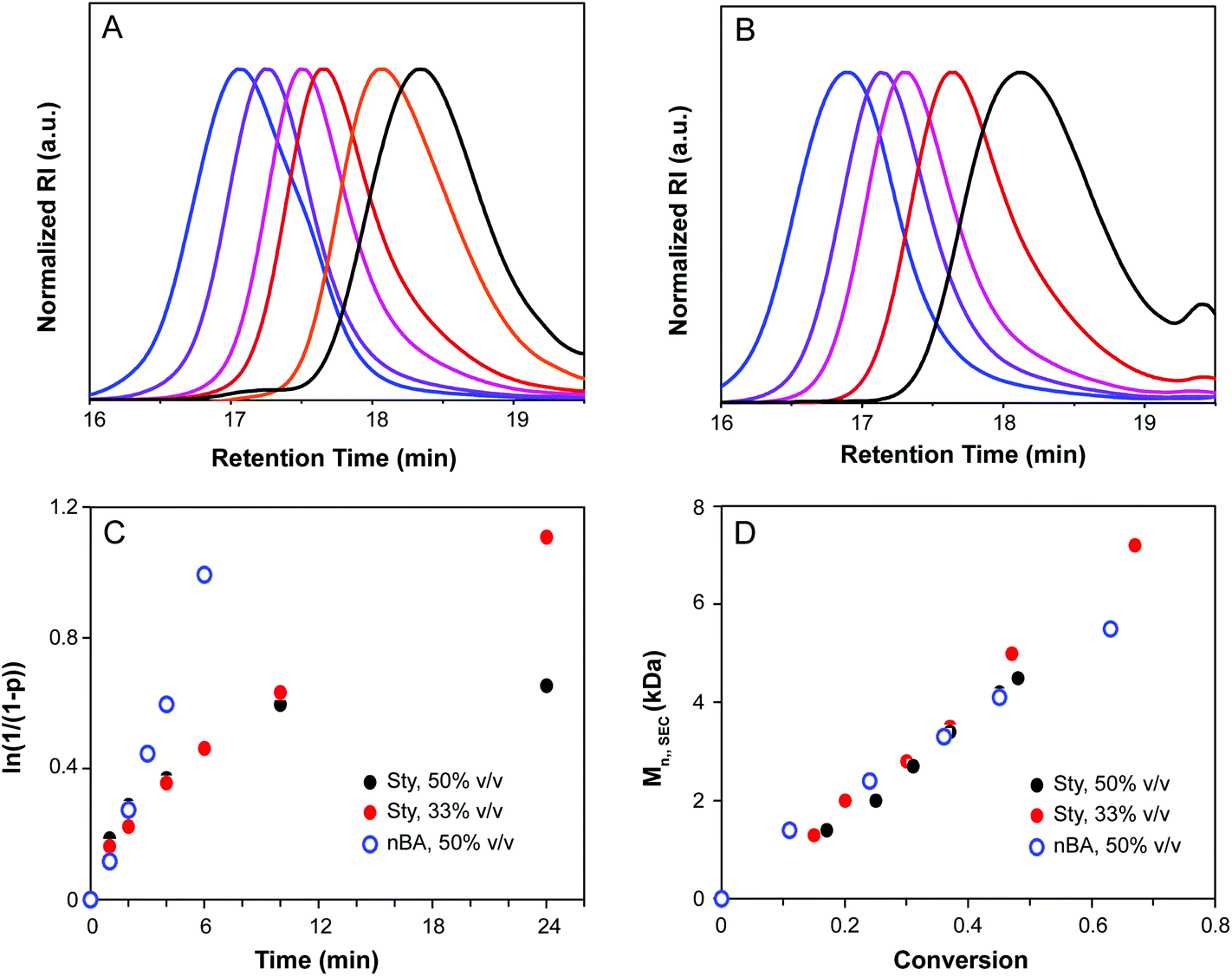 | ||
| Fig. 2 Kinetic analysis of CTA1-mediated RAFT polymerization. (A and B) SEC traces show evolution of MW during the polymerizations of styrene (A) and nBA (B). (C) ln(1/(1 − p)) vs. time for the polymerization of styrene ([styrene]/[CTA]/[I] = 100:1:0.1, 50% v/v in THF, 75 °C, black circles; [styrene]/[CTA]/[I] = 100:1:0.1, 33% v/v in THF, 75 °C, red circles) and nBA ([nBA]/[CTA]/[I] = 100:1:0.1, 50% v/v in THF, 60 °C, blue open circles). (D) Mn as a function of conversion for the polymerization of styrene and nBA. | ||
Chain extension of a polymer prepared using CTA1 was attempted to further validate its capability to mediate controlled RAFT polymerization. For the first block, poly(nBA) of MW = 6900 Da (Đ = 1.06) was synthesized using CTA1. This polymerization yielded a macroCTA with a dithiocarbamate at the ω-chain end. The poly(nBA) macroCTA was utilized in a second step to control the polymerization of styrene ([M]/[CTA]/[AIBN] = 1000:1:0.1), ultimately resulting in the formation of a block copolymer. After 4 h, the polymer had grown to 27.0 kDa as measured by SEC (Đ = 1.05) (Fig. S9†). This measured MW agreed with the expected value of 26.6 kDa based on conversion as measured by 1H NMR spectroscopy.
To prepare macromonomers for ROMP grafting-through, a series of polymers of differential MW were synthesized, and the isolated polymers were characterized by SEC and 1H NMR spectroscopy (Fig. S8 and S10†). A summary of our analysis is provided in Table 1. The subscripts in the polymer name assignments refer to the degree of polymerization of the polystyrene component of the macromonomers. Polymers with relatively narrow molecular weight distributions were obtained. In addition, MWs measured by SEC were in good agreement with those calculated from conversion using 1H NMR spectroscopy.
| Polymera | M n (SEC) (Da)b |
M
n (theo)c (Da) |
Đ |
|---|---|---|---|
| a Average degree of polymerization shown as a subscript calculated from SEC data using the formula DP = (Mn − MWCTA)/MWstyrene. b Measured by SEC using absolute MW determined by light scattering. c Determined by 1H NMR spectroscopy using the formula Mn (theo) = MWstyrene × ([styrene]/[CTA]) × % conv. | |||
| MM29 | 3300 | 3100 | 1.05 |
| MM52 | 5700 | 6000 | 1.02 |
| MM113 | 12100 |
13300 |
1.04 |
An important outcome of CTA1-mediated RAFT polymerization is the preservation of the reactive norbornene olefin moiety on the ω-end of the resulting polymer chain, allowing for subsequent bottlebrush formation via ROMP grafting-through. The orthogonality of the norbornene olefin with reversible-deactivation radical polymerization has been previously shown.56 The poor reactivity of the internal norbornene olefin in RAFT relative to the terminal, vinyl groups of acrylic monomers is attributed to their differences in electronics. 1H NMR spectroscopic analysis of the pure MMs confirmed the preservation of the norbornene olefin, evident as a singlet at 6.2 ppm (Fig. S8†).
RAFT transfer-to
There exist four methodologies for preparing bottlebrush polymers, and careful consideration is warranted to select the best strategy for a given system. For example, adoption of a grafting-through approach is appropriate when high grafting density is needed. However, bottlebrush formation via grafting-through is hindered by high MW macromonomers, limiting the use of this strategy to applications in which bottlebrushes with relatively short side chains are acceptable.57 In contrast, the transfer-to technique allows for the synthesis of bottlebrush polymers with high MW side chains, but can potentially suffer from broad side chain dispersities and lower than “perfect” grafting densities due to termination reactions during the polymerization.58
CTA1 can be utilized for both transfer-to and grafting-through strategies, creating a unique opportunity to evaluate the differences between these two techniques while using identical chemistry. To evaluate the ability of CTA1 to control the growth of high MW side chains from a polymeric backbone by RAFT transfer-to polymerization, CTA1 was first polymerized by ROMP (Scheme 4 left) using Grubbs’ 3rd generation catalyst ((H2IMes)(Cl)2(pyr)2RuCHPh) (G3) (50:1 [CTA1]/[G3]) to yield a poly(CTA1) (PCTA1) consisting of a poly(norbornene) backbone with a dithiocarbamate group on each repeat unit (Fig. S7†). Following our characterization of the resulting polymeric CTA, PCTA1 was then subjected to RAFT polymerization conditions ([styrene]/[macroCTA]/[AIBN] = 50000:1:0.1, THF, 75 °C), and the progress of the reaction was monitored by SEC (Fig. 3). Molecular weight distributions of the growing bottlebrush polymer were monomodal, and the corresponding polymers exhibited successively shorter retention times, indicative of increasing MW. After 91 h, the polymer had grown from 25.1 kDa (for PCTA1) to 1250 kDa, with Đ remaining low (<1.02) throughout the course of the polymerization. A second peak in the SEC traces at ∼15 min corresponds to dead polymer arising from termination reactions between detached polymeric radicals. This phenomenon is discussed in further detail below.
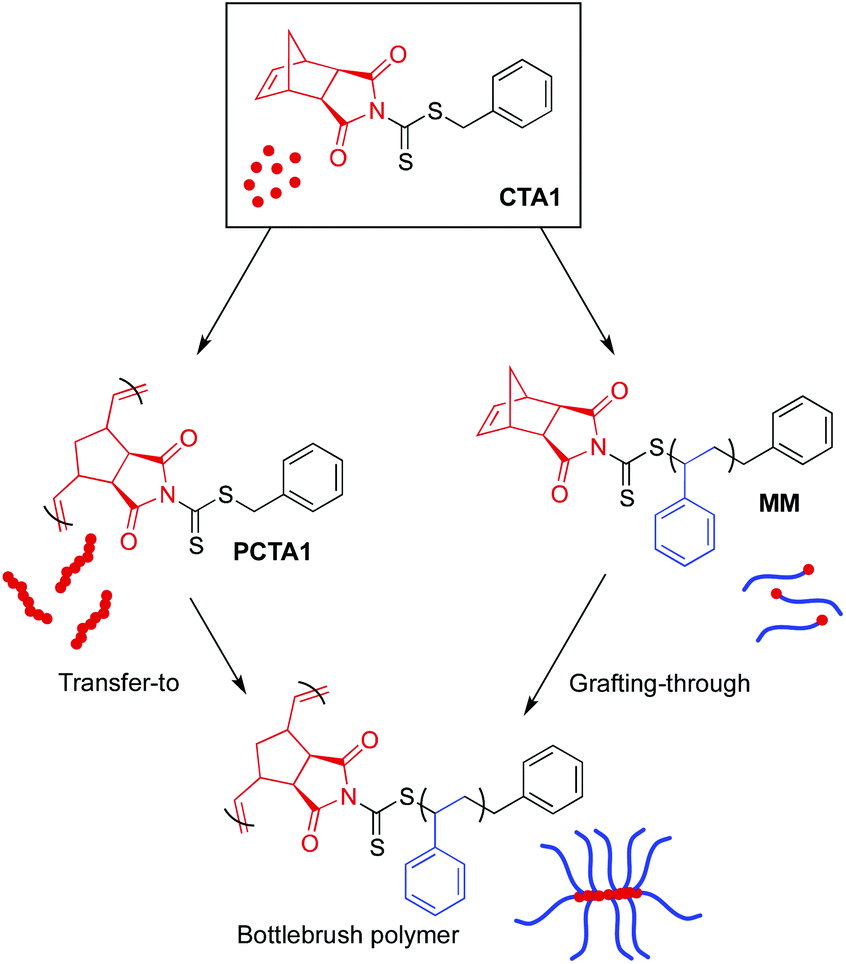 | ||
| Scheme 4 Preparation of bottlebrush polymers from CTA1 by transfer-to or grafting-through. | ||
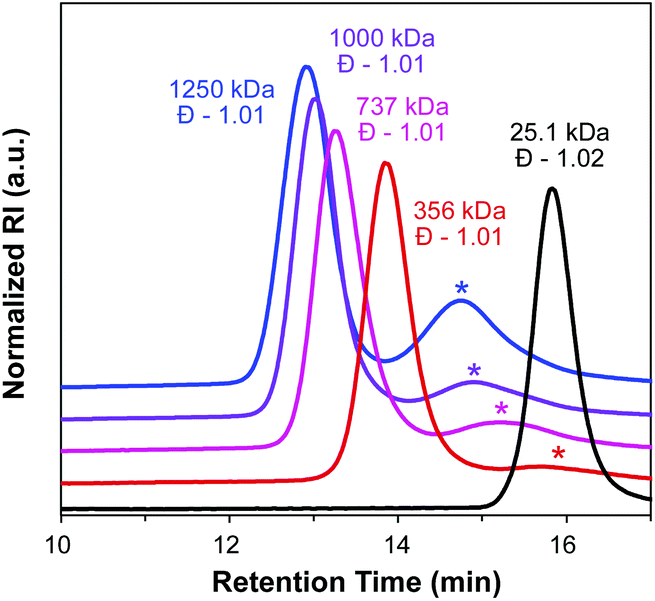 | ||
| Fig. 3 RAFT transfer-to mediated by PCTA1 (black trace), sampled at 19 h (red trace), 42 h (magenta trace), 67 h (purple trace), and 91 h (blue trace). Starred peaks correspond to “dead” polymer. Baselines are shifted for clarity. | ||
ROMP grafting-through
The presence of a strained, cyclic olefin at the ω-chain end of polymers prepared via RAFT using CTA1 allows for the direct synthesis of bottlebrush polymers by ROMP. In order to prepare bottlebrush polymers by ROMP, polystyrene MMs were precipitated three times from THF into methanol to ensure the complete removal of styrene, which can participate in olefin metathesis in the presence of Ru catalysts.59 The existence of residual styrene during ROMP has been shown to result in chain transfer and a broadening of the molecular weight distribution.60 Therefore, MM purity was confirmed by monitoring the disappearance of styrene olefin peaks at 5.5 and 5.0 ppm by 1H NMR spectroscopy between precipitations.A series of Ru-based metathesis catalysts were evaluated for their efficacy towards the grafting-through polymerization of CTA1-derived polystyrene (MM29) (Scheme 4 right). Toward this end, ROMP of MM29 was conducted in the presence of various ROMP catalysts at a [MM]/[catalyst] ratio of 50:1 (Fig. S11†). Grafting-through polymerizations initiated by Grubbs’ 1st ((PCy3)2(Cl)2RuCHPh) (G1) and 2nd generation ((H2IMes)(Cl)2(PCy3)RuCHPh) (G2) catalysts showed poor conversion to the corresponding bottlebrush polymer after 1 h in CH2Cl2 (Table 2, entries 1 and 2, respectively). The Hoveyda-Grubbs’ 2nd generation catalyst ((H2IMes)(Cl)2(PCy3)RuCH(2-OiPrPh)) (HG2) effected higher conversion to bottlebrush polymer (78%); however, the resulting polymer had a broad molecular weight distribution (Đ = 1.50) (Table 2, entry 3). Grafting-through by the modified Grubbs’ 2nd generation catalyst (G3) showed 91% conversion to bottlebrush polymer with Đ = 1.03 and a monomodal molecular weight distribution (Table 2, entry 5). Based on this analysis, G3 was utilized for further experiments.
| Entry | MM | MM Mn,SECa (kDa) |
Cat. | [MM]/[I] | [MM] (mg mL−1) | % Conv. to BBb | BB Mn,theoc (kDa) |
BB Mn,SECa (kDa) |
BB Đa |
|---|---|---|---|---|---|---|---|---|---|
| a Measured by SEC using absolute MW determination by light scattering. b Determined from SEC by comparing the integrations of BB and MM peaks. c Calculated using the formula % conv. × ([M]/[I]) × Mn,MM. All polymerizations were conducted for 1 h in CH2Cl2. | |||||||||
| 1 | MM29 | 3.3 | G1 | 50:1 |
100 | 8 | 13.2 | 21.1 | 1.11 |
| 2 | MM29 | 3.3 | G2 | 50:1 |
100 | 16 | 26.4 | 353 | 1.48 |
| 3 | MM29 | 3.3 | HG2 | 50:1 |
100 | 78 | 129 | 334 | 1.50 |
| 4 | MM29 | 3.3 | G3 | 25:1 |
100 | 91 | 75.0 | 85.6 | 1.04 |
| 5 | MM29 | 3.3 | G3 | 50:1 |
100 | 91 | 150 | 152 | 1.03 |
| 6 | MM29 | 3.3 | G3 | 75:1 |
100 | 89 | 220 | 240 | 1.14 |
| 7 | MM29 | 3.3 | G3 | 100:1 |
100 | 87 | 330 | 340 | 1.25 |
| 8 | MM29 | 3.3 | G3 | 50:1 |
25 | 70 | 116 | 100 | 1.10 |
| 9 | MM29 | 3.3 | G3 | 50:1 |
50 | 80 | 132 | 125 | 1.09 |
| 10 | MM52 | 5.6 | G3 | 50:1 |
100 | 65 | 182 | 190 | 1.09 |
| 11 | MM113 | 12.1 | G3 | 50:1 |
100 | 40 | 242 | 180 | 1.16 |
Macromonomer MW was found to influence ROMP grafting-through polymerization (Table 2, entries 5, 10–11, Fig. S12†). Comparing the grafting-through polymerization of three MMs of differing MW (3.3 KDa, 5.6 KDa, and 12 KDa) revealed an inverse correlation between MW and conversion. This observed dependence of conversion on MW is attributed to steric factors.57 The MW of the isolated bottlebrush polymer made from MM29 was 152 kDa (Table 2, entry 5), which is in good agreement with the expected molecular weight of 150 kDa. Despite lower conversions, polymerizations of MM52 and MM113 showed reasonable agreement between theoretical and observed Mn values and maintained fairly low dispersities.
To further confirm the livingness of grafting-through polymerization of CTA1-derived MMs, the [MM29]/[G3] ratio was varied from 25:1 to 100:1 (Table 2, entries 4–7). The resulting bottlebrush polymers were monomodal and their SEC traces exhibited the expected inverse relationship between MW and retention time (Fig. 4A). Mn values determined by SEC are in good agreement with theoretical MWs. A plot of Mnvs. [M]/[I] showed the anticipated linear trend (Fig. 4B).61 Additionally, dispersity values were low, ranging from 1.04–1.25 throughout the series.
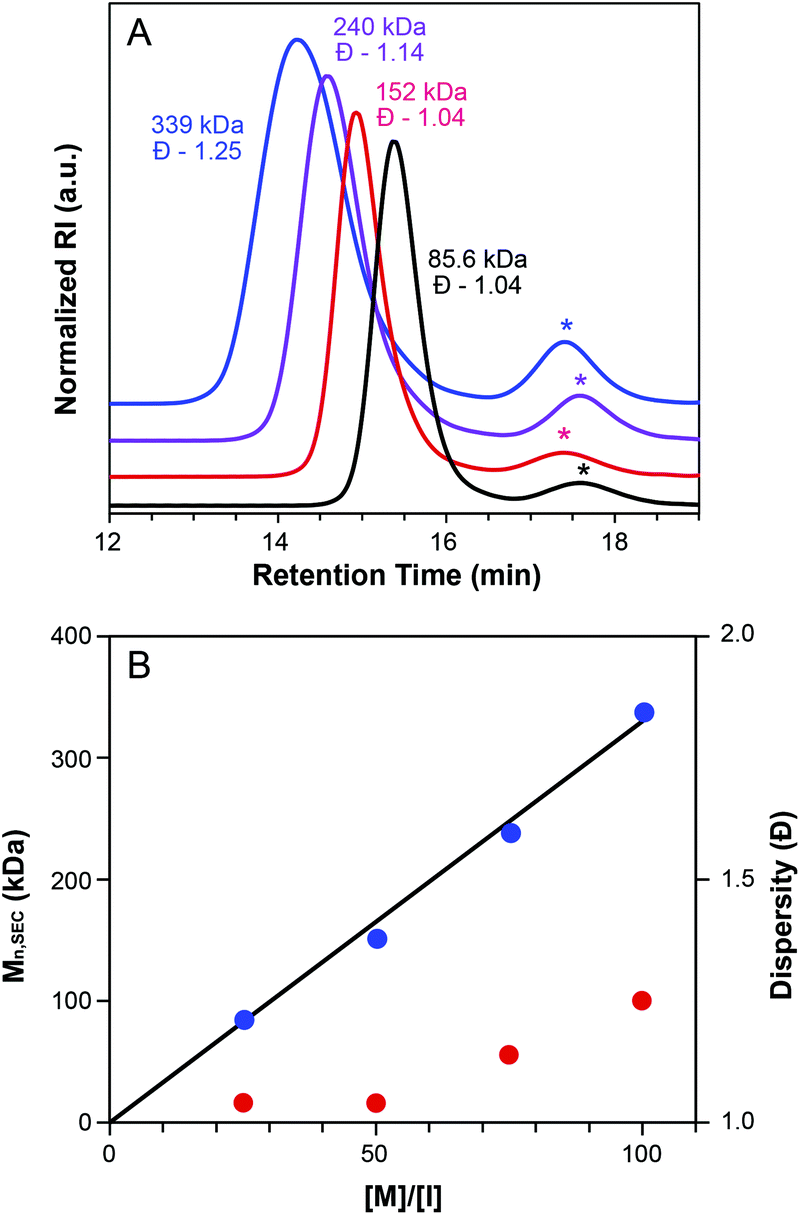 | ||
| Fig. 4 Assessment of the livingness of ROMP grafting-through of MM29. (A) SEC traces illustrating the increase in MW with increasing [MM]/[G3] ratio (black = [25]:[1], red = [50]:[1], purple = [75]:[1], blue = [100]:[1]). Starred peaks are assigned to residual MMs. Baselines are shifted for clarity. (B) Plot of MW vs. [M]/[I] ratio. The black line represents the theoretical MW for each [MM]/[G3]. | ||
Macromonomer concentration had a small but significant influence on the conversion to bottlebrush polymer (Table 2, entries 5, 8–9, Fig. S13†). Measured conversions varied from 70% to 80% to 91% for the evaluated concentration range (25 mg mL−1 to 100 mg mL−1), with increasing concentration resulting in higher conversion to BB.
It is important to note that MM conversion could not be increased beyond 91%, even under optimized conditions. Given the wealth of literature regarding the dependence of RAFT chain-end fidelity on a number of factors including [M]/[I] and [CTA]/[I] ratios,49 it can be hypothesized that the observed conversion limit of 91% originates from the existence of “non-living” chains, or those that do not possesses a thiocarbonylthio group on the ω-chain end, in the MM samples. It has been shown that ca. 8% of polymer chains prepared by RAFT polymerization under similar conditions are of this “non-living” type.54,62 Polymers of this “non-living” variety do not possess a norbornene on the chain end and thus will not polymerize during ROMP grafting-through.
Aminolysis of bottlebrush polymers
The dithiocarbamate linkage is susceptible to reaction with nucleophiles such as amines.38 Reaction of this functional group proceeds via an addition-elimination mechanism, resulting in detachment of polymeric side chains and their replacement by the nucleophile. The displaced side chains bear thiol groups at their ω-chain ends, which oxidize in air to form disulfide linkages between polymer chains.38,63 Therefore, nucleophilic displacement initially yields a mixture of free side chains, side chain dimers, and the poly(norbornene) bottlebrush backbone. Over time, the remaining thiol-terminated polymers become quantitatively oxidized. Disulfide reduction is possible, but this process generally requires elevated temperatures and long reaction times.64The presence of the dithiocarbamate moiety adjacent to the bottlebrush backbone allowed for the cleavage of the polystyrene side chains (Scheme 5). A series of bottlebrush polymers prepared by RAFT transfer-to were dissolved in THF and exposed to a 40% w/v solution of methylamine in H2O for 72 h. The resulting mixture of backbone and dimerized side chain polymers was separated from residual reactants via precipitation. SEC analysis of the aminolyzed bottlebrush polymers showed a clean shift of the molecular weight distribution to a longer retention time, indicating that the side chains had been quantitatively cleaved (Fig. S14†). Interestingly, the measured MWs of the dissociated side chains were higher than the expected values calculated by dividing the experimental BB MW by the DP of PCTA1 (Table 3). We attribute this deviation to three independent phenomena. First, oxidation of ω-chain end thiols liberated during aminolysis resulted in disulfide bond formation, as has been previously reported.63 This reaction doubles the observed molecular weight of the side chains. Second, increased steric crowding of the dithiocarbamate CTAs near the bottlebrush backbone by the attached polystyrene sidechains likely led to radical termination reactions between detached polymeric radicals during the transfer-to process, as has also been observed.65,66 This side reaction not only yielded a significant amount of dead polymer, evident as a low MW peak in the SEC trace (Fig. 3), but also likely resulted in lower than perfect grafting density and a higher than expected average side chain MW. Lastly, “dead” polymer arising during the RAFT polymerization is also incorporated into this sample. Although the concentration of “dead” polymer is surely eclipsed by the more abundant aminolyzed sidechains, these “dead” polymer chains could explain the observed broadening of the molecular weight distributions of the aminolyzed polymers relative to those of the bottlebrush polymers prior to aminolysis. While ultrahigh MW polymers can be obtained via RAFT transfer-to, the limitations of this methodology (i.e., limited control over grafting density) are made clear by this aminolysis experiment.
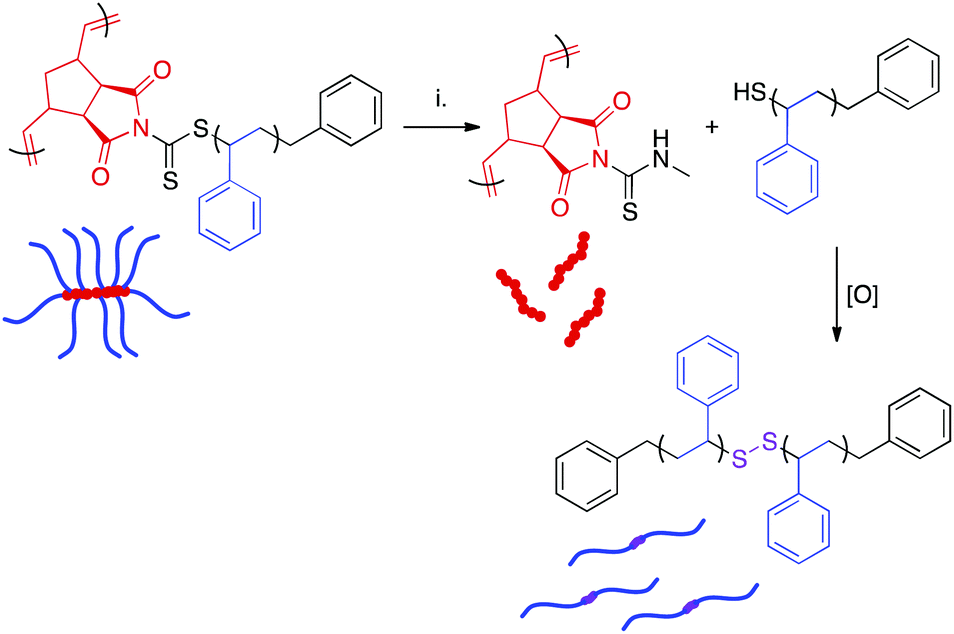 | ||
| Scheme 5 Displacement of polymeric side chains by methylamine. Experimental conditions: (i) CH3NH2, H2O–THF. | ||
| Polymerization time (h) |
M
n,SECa (kDa) |
Đ |
M
n,sidechains theob (kDa) |
M
n,sidechains SECa (kDa) |
Đ
aminolysisa |
|---|---|---|---|---|---|
| a Measured by SEC using absolute MW by light scattering. b Determined using the formula: Mn,sidechains = Mn,BB/DPMCTA1. | |||||
| 0 | 25.1 | 1.02 | — | — | — |
| 19 | 356 | 1.01 | 4.7 | 19.5 | 1.15 |
| 42 | 737 | 1.01 | 9.9 | 35.5 | 1.21 |
| 67 | 1000 | 1.01 | 13.2 | 47.4 | 1.23 |
| 91 | 1250 | 1.01 | 16.5 | 56.9 | 1.28 |
To further evaluate bottlebrush polymers prepared by the grafting-through approach, we subjected these polymers to aminolysis as well. ROMP of MM29 at a [MM]/[I] ratio of 50:1 was carried out to give a BB with degradable side chain linkages. A MW of 164 kDa was determined for the BB, with Đ = 1.07. Aminolysis of the BB proceeded rapidly and quantitatively at rt in the presence of an excess of methylamine in THF–H2O. SEC analysis of the precipitated reaction mixture revealed a narrow molecular weight distribution and a MW approximately double that of the starting MM (5400 Da, Đ = 1.01), as expected for polystyrene side chain disulfide dimers (Fig. 5). Quantitative aminolysis was confirmed by the complete disappearance of the bottlebrush peak in the SEC trace. Additionally, DLS analysis of the cleaved bottlebrushes exhibited a shift in the size of the macromolecules from 11.8 ± 3.1 nm for the BB to 2.6 ± 0.4 nm for the dissociated side chain dimers (original MM29 = 2.0 ± 0.5 nm) (Fig. S15†).
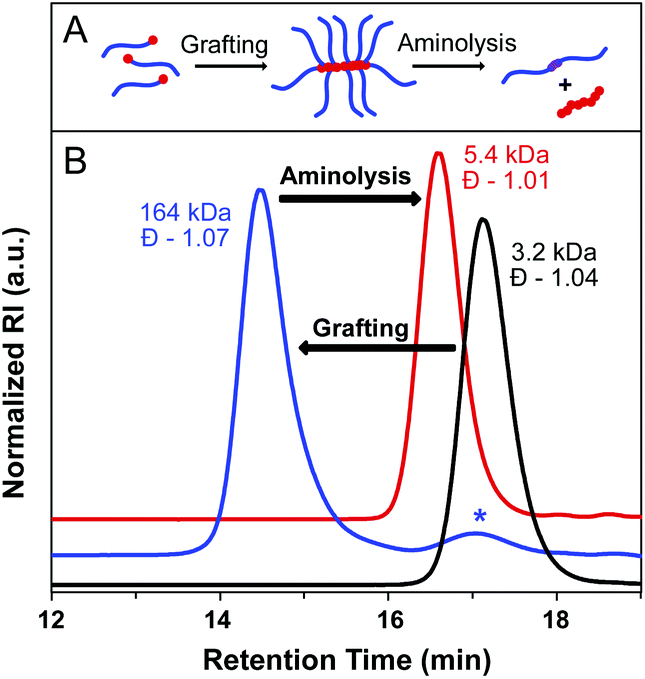 | ||
| Fig. 5 (A) Graphical representation of grafting-through and aminolysis. (B) BB synthesis by ROMP grafting-through (blue trace) from MM29 (black trace) and subsequent aminolysis with methylamine (red trace). The starred peak corresponds to residual MM. Baselines are shifted for clarity. | ||
Conclusions
A novel RAFT CTA with a directly polymerizable Z-group was prepared in a one-pot synthesis from an exo-norbornene imide. CTA1-mediated RAFT polymerizations of styrene and nBA were conducted successfully, yielding polymers of controllable MW and low dispersity bearing a polymerizable norbornene moiety on the ω-chain end. We demonstrated that CTA1 could be utilized effectively for transfer-to and grafting-through methodologies, with the former resulting in high MW bottlebrush polymers (1250 kDa). Polystyrene MMs prepared using CTA1 were polymerized by ROMP via a grafting-through strategy. In general, ROMP polymerization of CTA1-derived MMs proceeded efficiently when catalyzed by Grubbs’ 3rd generation catalyst, with conversions on the order of 70–90%. Macromonomer MW, concentration, and the [MM]/[catalyst] ratio of the polymerization were found to influence the conversion to BB. In general, an inverse relationship between MW of the MM and the conversion to BB was observed, with the smallest MM (3300 Da) resulting in the highest conversion. Increasing the MM concentration from 25 to 100 mg mL−1 also enhanced conversion, while increasing the [MM]/[G3] ratio from 25:1 to 100:1 resulted in decreased conversion and a broadening of the molecular weight distribution. Side chain scission via aminolysis was quantitative and revealed significant differences between the bottlebrush polymers prepared by the two different approaches. CTA1 is unique in that it allows for the preparation of bottlebrush polymers utilizing functionality built into the chain-transfer agent. We expect CTA1 will prove to be useful for the facile preparation of bottlebrush polymers possessing inherent side chain lability via the transfer-to or grafting-through approach.
Acknowledgements
This work was supported by the Virginia Tech Department of Chemistry. We also thank Oak-Ridge Associated Universities for partial support of this work (Powe Junior Faculty Enhancement Award).References
- S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759 CrossRef CAS PubMed.
- C. Grigoriadis, A. Nese, K. Matyjaszewski, T. Pakula, H.-J. Butt and G. Floudas, Macromol. Chem. Phys., 2012, 213, 1311 CrossRef CAS.
- J. Pietrasik, B. S. Sumerlin, H.-i. Lee, R. R. Gil and K. Matyjaszewski, Polymer, 2007, 48, 496 CrossRef CAS PubMed.
- J. Yuan, Y. Lu, F. Schacher, T. Lunkenbein, S. Weiss, H. Schmalz and A. H. E. Muller, Chem. Mater., 2009, 21, 4146 CrossRef CAS.
- J. Bolton and J. Rzayev, ACS Macro Lett., 2012, 1, 15 CrossRef CAS.
- S. L. Pesek, X. Li, B. Hammouda, K. Hong and R. Verduzco, Macromolecules, 2013, 46, 6998 CrossRef CAS.
- S. J. Dalsin, M. A. Hillmyer and F. S. Bates, ACS Macro Lett., 2014, 3, 423 CrossRef CAS.
- M. Wintermantel, M. Gerle, K. Fischer, M. Schmidt, I. Wataoka, H. Urakawa, K. Kajiwara and Y. Tsukahara, Macromolecules, 1996, 29, 978 CrossRef CAS.
- H.-i. Lee, J. Pietrasik, S. S. Sheiko and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 24 CrossRef CAS PubMed.
- A. Nese, Y. Li, S. Averick, Y. Kwak, D. Konkolewicz, S. S. Sheiko and K. Matyjaszewski, ACS Macro Lett., 2011, 1, 227 CrossRef.
- Y. Li, E. Themistou, J. Zou, B. P. Das, M. Tsianou and C. Cheng, ACS Macro Lett., 2011, 1, 52 CrossRef.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276 CrossRef CAS PubMed.
- J. A. Johnson, Y. Y. Lu, A. O. Burts, Y.-H. Lim, M. G. Finn, J. T. Koberstein, N. J. Turro, D. A. Tirrell and R. H. Grubbs, J. Am. Chem. Soc., 2010, 133, 559 CrossRef PubMed.
- B. S. Sumerlin, ACS Macro Lett., 2011, 1, 141 CrossRef.
- S. Minko, in Polymer Surfaces and Interfaces, ed. M. Stamm, Springer, Berlin, Heidelberg, 2008, p. 215 Search PubMed.
- C. W. Bielawski and R. H. Grubbs, Angew. Chem., Int. Ed., 2000, 112, 3025 CrossRef.
- C. J. Hawker, D. Mecerreyes, E. Elce, J. Dao, J. L. Hedrick, I. Barakat, P. Dubois, R. Jérôme and W. Volksen, Macromol. Chem. Phys., 1997, 198, 155 CrossRef CAS.
- A. Li, J. Ma, G. Sun, Z. Li, S. Cho, C. Clark and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1681 CrossRef CAS.
- Z. Li, J. Ma, C. Cheng, K. Zhang and K. L. Wooley, Macromolecules, 2010, 43, 1182 CrossRef CAS.
- E. Masuda, S. Kishiro, T. Kitayama and K. Hatada, Polym. J., 1991, 23, 847 CrossRef CAS.
- A. O. Moughton, T. Sagawa, W. M. Gramlich, M. Seo, T. P. Lodge and M. A. Hillmyer, Polym. Chem., 2013, 4, 166 RSC.
- D. Neugebauer, Y. Zhang, T. Pakula, S. S. Sheiko and K. Matyjaszewski, Macromolecules, 2003, 36, 6746 CrossRef CAS.
- Y. Xia, J. A. Kornfield and R. H. Grubbs, Macromolecules, 2009, 42, 3761 CrossRef CAS.
- K. O. Kim and T.-L. Choi, Macromolecules, 2013, 46, 5905 CrossRef CAS.
- K. O. Kim, S. Shin, J. Kim and T.-L. Choi, Macromolecules, 2014, 47, 1351 CrossRef CAS.
- J. Zou, G. Jafr, E. Themistou, Y. Yap, Z. A. P. Wintrob, P. Alexandridis, A. C. Ceacareanu and C. Cheng, Chem. Commun., 2011, 47, 4493 RSC.
- S. Jha, S. Dutta and N. B. Bowden, Macromolecules, 2004, 37, 4365 CrossRef CAS.
- A. C. Engler, J. M. W. Chan, K. Fukushima, D. J. Coady, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2013, 2, 332 CrossRef CAS.
- C. Li, N. Gunari, K. Fischer, A. Janshoff and M. Schmidt, Angew. Chem., Int. Ed., 2004, 43, 1101 CrossRef CAS PubMed.
- A. Nese, N. V. Lebedeva, G. Sherwood, S. Averick, Y. Li, H. Gao, L. Peteanu, S. S. Sheiko and K. Matyjaszewski, Macromolecules, 2011, 44, 5905 CrossRef CAS.
- M. H. Stenzel, L. Zhang and W. T. S. Huck, Macromol. Rapid Commun., 2006, 27, 1121 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079 CrossRef CAS PubMed.
- Y. Tsujii, M. Ejaz, K. Sato, A. Goto and T. Fukuda, Macromolecules, 2001, 34, 8872 CrossRef CAS.
- M. H. Stenzel, T. P. Davis and A. G. Fane, J. Mater. Chem., 2003, 13, 2090 RSC.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539 CrossRef CAS.
- M. Hernández-Guerrero, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Eur. Polym. J., 2005, 41, 2264 CrossRef PubMed.
- C. Barner-Kowollik, T. P. Davis, J. P. A. Heuts, M. H. Stenzel, P. Vana and M. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 365 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149 RSC.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035 CrossRef CAS.
- J. Liu, A. X. Gao and J. A. Johnson, J. Visualized Exp., 2013, e50874 Search PubMed.
- J. Skey and R. K. O'Reilly, Chem. Commun., 2008, 4183 RSC.
- N. Azizi, F. Aryanasab and M. R. Saidi, Org. Lett., 2006, 8, 5275 CrossRef CAS PubMed.
- M. Breugst, T. Tokuyasu and H. Mayr, J. Org. Chem., 2010, 75, 5250 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985 CrossRef CAS.
- G. Moad, T. A. Mayadunne Roshan, E. Rizzardo, M. Skidmore and H. Thang San, in Advances in Controlled/Living Radical Polymerization, American Chemical Society, 2003, vol. 854, p. 520 Search PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669 CrossRef CAS.
- M. S. Donovan, A. B. Lowe, B. S. Sumerlin and C. L. McCormick, Macromolecules, 2002, 35, 4123 CrossRef CAS.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496 RSC.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, Y. K. Chong, G. Moad and S. H. Thang, Macromolecules, 1999, 32, 6977 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458 CrossRef CAS PubMed.
- D. Zhou, X. Zhu, J. Zhu and H. Yin, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4849 CrossRef CAS.
- J. Vandenbergh and T. Junkers, Macromolecules, 2014, 47, 5578 CrossRef.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321 CrossRef CAS.
- Z.-m. Dong, X.-h. Liu, X.-l. Tang and Y.-s. Li, Macromolecules, 2009, 42, 4596 CrossRef CAS.
- S. Hilf and A. F. M. Kilbinger, Macromol. Rapid Commun., 2007, 28, 1225 CrossRef CAS.
- K. L. Beers, S. G. Gaynor, K. Matyjaszewski, S. S. Sheiko and M. Möller, Macromolecules, 1998, 31, 9413 CrossRef CAS.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360 CrossRef CAS PubMed.
- W. E. Crowe, J. P. Mitchell, V. C. Gibson and R. R. Schrock, Macromolecules, 1990, 23, 3534 CrossRef CAS.
- M. W. Wagaman and R. H. Grubbs, Macromolecules, 1997, 30, 3978 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 Search PubMed.
- Z. Wang, J. He, Y. Tao, L. Yang, H. Jiang and Y. Yang, Macromolecules, 2003, 36, 7446 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 3087 CrossRef CAS.
- Y. Zhao and S. Perrier, Macromolecules, 2006, 39, 8603 CrossRef CAS.
- M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4498 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py01567c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |