
A structural and biological study on the new 3,5-diacetyl-1,2,4-triazol bis(p-chlorophenylthiosemicarbazone) ligand and its bimetallic complexes†
A. I.
Matesanz
*a,
P.
Albacete
a,
J.
Perles
b and
P.
Souza
a
aDpto. de Química Inorgánica (Módulo 07), Facultad de Ciencias, Universidad Autónoma de Madrid, c/ Francisco Tomás y Valiente n° 7, 28049-Madrid, Spain. E-mail: ana.matesanz@uam.es
bServicio Interdepartamental de Investigación (Módulo 13), Facultad de Ciencias, Universidad Autónoma de Madrid, c/ Francisco Tomás y Valiente n° 7, 28049-Madrid, Spain
First published on 19th November 2014
Abstract
Preparation and characterization of the new ligand 3,5-diacetyl-1,2,4-triazol bis(4N-p-chlorophenylthiosemicarbazone), H5L1, and its bimetallic complexes [Pd(μ-H3L1)]2 and [Pt(μ-H3L1)]2, are described. The molecular structure of the complexes, determined by single crystal X-ray crystallography, reveals that each ligand coordinates, in an asymmetric dideprotonated form, to the metal ions in a square planar geometry. The new compounds synthesized have been evaluated for antiproliferative activity in vitro against NCI-H460, T-47D, A2780 and A2780cisR human cancer cell lines. The cytotoxicity data suggest that these compounds may be endowed with important antitumor properties, especially H5L1, since they exhibit excellent antiproliferative activity surpassing the activity of cisplatin against three of the four tumor cell lines studied. The DNA binding ability of H5L1 and [Pt(μ-H3L1)]2 with calf thymus DNA in Tris-HCl buffer solution (pH = 7.2) was explored by UV-Vis absorption spectroscopy and viscosity measurements. These data indicated that both compounds bind to DNA by a groove binding mode.
Introduction
Cisplatin is one of the most active chemotherapeutic agents available for the treatment of a number of solid tumors. The main cellular target of the drug is assumed to be DNA, specifically the formation of 1,2-intrastrand cross-link adducts that force the DNA to kink toward the major groove. These DNA distortions can effectively block the cell division and finally trigger cell death.1–3Unfortunately the clinical utility of cisplatin is restricted due to the frequent development of drug resistance, the limited spectrum of tumors against which this drug is active and also the severe normal tissue toxicity. These disadvantages have driven the development of improved platinum-based anticancer drugs whose structure and mode of action differ from that of cisplatin, especially those that interact with specific molecular targets as for example are the processes associated with DNA: transcription, replication and repair.4–8
In this regard, of particular interest are compounds targeting ribonucleotide reductase (RR), an enzyme that catalyzes the reduction of ribonucleotides to deoxyribonucleotides, which are the building blocks for the de novo DNA synthesis in all living cells. Cancer cells require increased RR activity to meet the demand for deoxyribonucleotides that are needed to support their rapid proliferation. Thus inhibition of RR activity leads to inhibition of DNA synthesis and repair, and also induces cell cycle arrest and apoptosis.9–11
The α-(N)-heterocyclic thiosemicarbazones have been reported to be among the most effective RR inhibitors identified yet. They constitute an important and versatile class of chelate ligands with highly interesting biological properties due to the important number of free ligands and derived metal complexes that have shown antimicrobial, antiviral or antitumor activity.12–16 For thiosemicarbazone metal complexes multiple antitumor mechanisms of action have been proposed which in addition to the inhibition of RR include (a) covalent binding to the nitrogen bases of DNA hindering or blocking base replication, (b) creation of lesions in DNA strands by oxidative rupture and (c) binding with DNA through non-covalent interactions such as H-bonding, intercalation, electrostatic interactions or groove binding.17–21
Investigations in our laboratory have led to the development of a series of 3,5-diacetyl-3,5-triazol bis(4N-monosubstituted thiosemicarbazones) and in vitro antiproliferative experiments have shown that some of them, particularly those bearing cyclohexyl, phenyl and tolyl groups, exhibit important cytotoxic activity in various human cell lines derived from different types of solid tumors.22–25 These results encouraged us to further investigate their antitumor properties as well as those of novel derivatives.
Thus, this work is aimed to describe the synthesis and chemical characterization of the new 3,5-diacetyl-1,2,4-triazol bis(4N-p-chlorophenylthiosemicarbazone) ligand, H5L1, and its bimetallic palladium(II) and platinum(II) complexes (Scheme 1).
 | ||
| Scheme 1 Structure of the 3,5-diacetyl-1,2,4-triazol bis(4N-p-chlorophenylthiosemicarbazone) ligand and its metal complexes synthesized. | ||
The cytotoxic activity of the new compounds synthesized, the related 3,5-diacetyl-1,2,4-triazol bis(4N-p-tolylthiosemicarbazone) ligand, H5L3, its platinum(II) derivative, di{3,5-diacetyl-1,2,4-triazol bis(4N-p-tolylthiosemicarbazonato) platinum(II)}, and cisplatin (assumed as the reference antitumor drug) has been studied against four human cancer cell lines: NCI-H460 (non-small cell lung cancer), T-47D (ductal breast epithelial cancer), A2780 and A2780cisR (epithelial ovarian cancer). DNA interaction abilities of H5L1 and [Pt(μ-H3L1)]2 have been investigated by absorption spectroscopy and viscosity measurements and also their binding constants (Kb) have been determined.
Results and discussion
Synthesis and spectroscopic characterization
A new multidentate ligand, H5L1, has been synthesized with high purity and acceptable yield. This air and moisture stable yellow powder was obtained as a monohydrate and was characterized by elemental analysis (C, H, N, S), mass spectrometry and IR and 1H NMR spectroscopy.The reaction of the flexible binucleating 3,5-diacetyl-1,2,4-triazol bis(4N-p-chlorophenylthiosemicarbazone) ligand with [MCl4]2− ions (M = Pd or Pt) led to the isolation of the neutral dimer complexes [Pd(μ-H3L1)]2 and [Pt(μ-H3L1)]2 in which each triazol-bis(thiosemicarbazone) behaves as a dianionic ligand with deprotonation of one hydrazine (2NH) and the triazole ring protons.
The air and moisture stable solids obtained were characterized by analytical and spectroscopic studies (selected IR and UV–vis bands are listed in the Experimental section). The positive ionization mass spectra, ESI(+)-MS, in acetonitrile show a small peak at m/z = 1250.88 for [Pd(μ-H3L1)]2 and at m/z = 1428 for [Pt(μ-H3L1)]2 which confirm the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ligand to metal stoichiometry. As shown in Fig. 1, the experimental isotopic patterns of this ion fit perfectly with the theoretical mass distribution for the proposed [Pd(μ-H3L1)]2 species.
:2 ligand to metal stoichiometry. As shown in Fig. 1, the experimental isotopic patterns of this ion fit perfectly with the theoretical mass distribution for the proposed [Pd(μ-H3L1)]2 species.
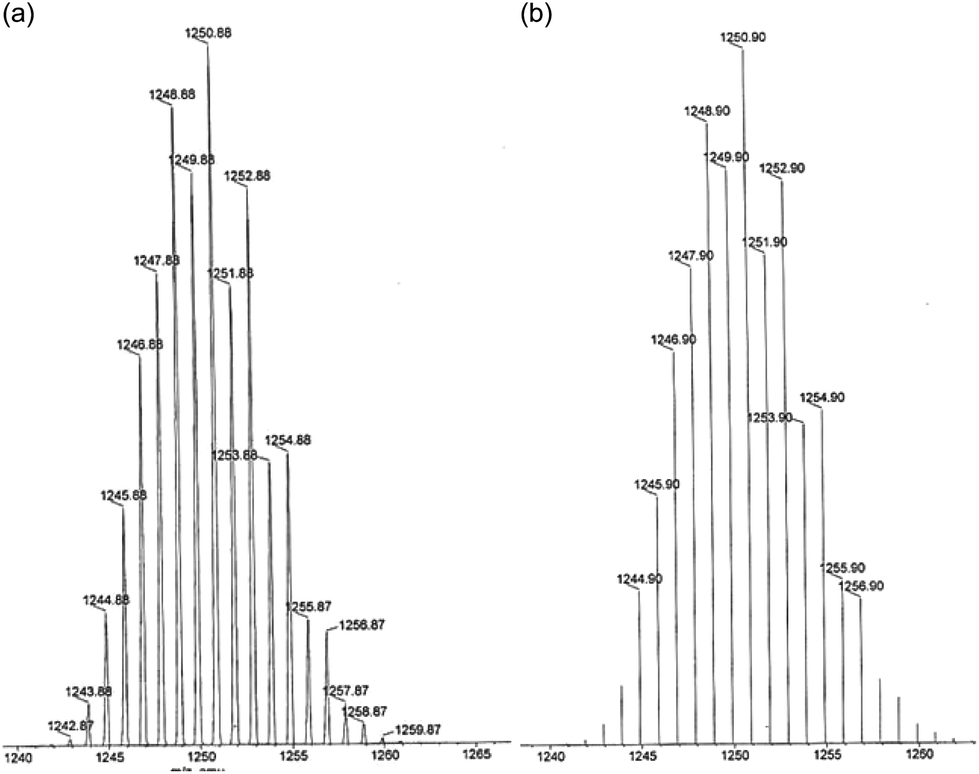 | ||
| Fig. 1 ESI(+)-MS spectra in acetonitrile solution of the fragment [Pd(μ-H3L1)]2+ (a) experimental and (b) theoretical isotopic distribution. | ||
The infrared spectral bands most useful for determining the mode of coordination of this ligand are the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) iminic and ν(CS) thioamide IV vibrations. However, the high delocalization and the asymmetric coordination only induce minor changes in these bands hindering the IR analysis. Specifically, in the spectra of metal complexes the ν(CN) band appears slightly shifted to higher wavenumbers and the ν(CS) thioamide IV band has decreased in intensity.
N) iminic and ν(CS) thioamide IV vibrations. However, the high delocalization and the asymmetric coordination only induce minor changes in these bands hindering the IR analysis. Specifically, in the spectra of metal complexes the ν(CN) band appears slightly shifted to higher wavenumbers and the ν(CS) thioamide IV band has decreased in intensity.
The 1H NMR spectrum of the free ligand shows various peaks at low fields: δ = 14.75 ppm assigned to the triazolic proton, δ ∼ 11.3 ppm assigned to the >CN–2NH– protons and δ ∼ 10 ppm assigned to the –C(S)–4NH– protons. In the 1H NMR spectra of the complexes the absence of any signals above 13 ppm together with the presence of only one signal assigned to >CN–2NH– protons is consistent with the asymmetric dideprotonation of the ligands.
The electronic absorption spectra of both the H5L1 ligand and [Pd(μ-H3L1)]2 and [Pt(μ-H3L1)]2 complexes exhibit two intense bands in the region 250–400 nm, which can be ascribed to ligand-centered n→π* and π→π* transitions. In addition the spectra of metal complexes exhibit other less energetic bands assigned to a ligand to metal (LMCT) and a metal to ligand charge transfer (MLCT) transition.26
Description of the crystal structures
Good quality crystals, suitable for single crystal X-ray diffraction analysis, were obtained for [Pd(μ-H3L1)]2 and [Pt(μ-H3L1)]2 compounds by recrystallization in dimethylsulfoxide (DMSO). The molecular structure of the [Pd(μ-H3L1)]2 complex (Fig. 2) consists of discrete [Pd(μ-H3L1)]2·2.5DMSO·H2O molecules (the H atoms of the H2O molecule were not located in the Fourier difference map).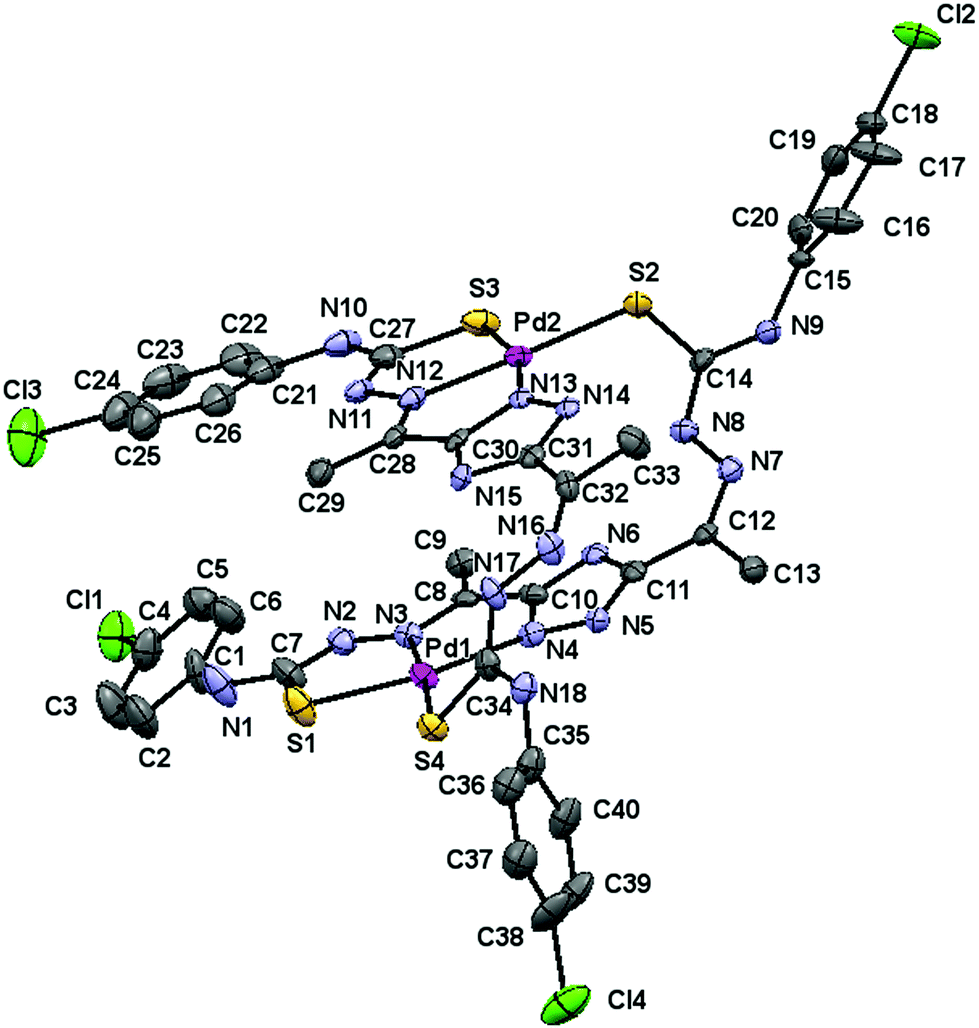 | ||
| Fig. 2 Molecular structure of the [Pd(μ-H3L1)]2·2.5DMSO·H2O complex, hydrogen atoms and solvent molecules are omitted for clarity. The displacement ellipsoids are drawn at the 50% probability. | ||
This neutral Pd(II) complex crystallizes in the monoclinic P21/n space group with Z = 4 and its crystallographic analysis reveals unambiguously a dimeric structure, which results from the pairing of two metal centers through two thiosemicarbazone bridging moieties.
Each Pd(II) center is four-coordinated with a [N2S2] donor environment, via one triazole nitrogen atom and the azomethine nitrogen, one sulfur atom belonging to the deprotonated arm of a ligand, and a sulfur atom from the neutral arm of the other ligand. Thus, the deprotonated thiosemicarbazone arm behaves as bidentate and the neutral one behaves as monodentate, so that the ligand acts as a bridge.
The bond angle data (Table 1) indicate that the stereochemistry around each palladium(II) ion is almost planar. The angles deviate slightly from that expected for a regular square planar geometry, although this distortion may be attributed to the restricted bite angle of the tridentate moieties. Coordination results in the formation of two five-membered (PdSCNN and PdNCCN) chelate rings for each palladium(II) ion, which are coplanar with the deprotonated triazole ring.
| Bond distances (Å) | |||
|---|---|---|---|
| Pd1–S1 | 2.261(3) | Pd2–S2 | 2.316(3) |
| Pd1–S4 | 2.309(3) | Pd2–S3 | 2.268(3) |
| Pd1–N3 | 2.015(9) | Pd2–N12 | 2.012(9) |
| Pd1–N4 | 2.064(9) | Pd2–N13 | 2.080(9) |
| C7–S1 | 1.80(1) | C21–N10 | 1.441(17) |
| C14–S2 | 1.74(1) | C27–N10 | 1.345(15) |
| C27–S3 | 1.77(1) | C28–N12 | 1.295(14) |
| C34–S4 | 1.72(1) | C32–N16 | 1.289(15) |
| C7–N1 | 1.339(16) | C34–N18 | 1.353(14) |
| C7–N2 | 1.304(15) | N2–N3 | 1.359(13) |
| C8–N3 | 1.322(14) | N7–N8 | 1.403(13) |
| C12–N7 | 1.294(14) | N11–N12 | 1.386(12) |
| C14–N8 | 1.319(14) | N16–N17 | 1.402(13) |
| C14–N9 | 1.332(14) | ||
| Bond angles (°) | |||
|---|---|---|---|
| N3–Pd1–N4 | 80.6(3) | N12–Pd2–N13 | 79.7(3) |
| N3–Pd1–S1 | 83.8(3) | N12–Pd2–S3 | 84.0(3) |
| S1–Pd1–S4 | 91.9(1) | S3–Pd2–S2 | 92.2(1) |
| N4–Pd1–S4 | 103.7(3) | N13–Pd2–S2 | 104.0(2) |
| N4–Pd1–S1 | 164.4(3) | N13–Pd2–S3 | 163.7(3) |
| N3–Pd1–S4 | 175.6(3) | N12–Pd2–S2 | 176.2(3) |
The Pd–N [2.012(9)–2.080(9) Å] and Pd–S [2.261(3)–2.316(3) Å] bond distances are slightly shorter than the sum of covalent radii of Pd and N or Pd and S (2.10 and 2.44 Å respectively) and are comparable to those reported for other Pd(II) thiosemicarbazone complexes.24 It is important to note that upon coordination, the deprotonated arms undergo certain evolution from the thione to the thiol form [C7–S1 = 1.80(1) Å and C27–S3 = 1.77(1) Å], while the neutral thiosemicarbazone arms present shorter C–S bond lengths [C14–S2 = 1.74(1) Å and C34–S4 = 1.72(1) Å]. The C–N and N–N bond distances are intermediate between formal single and double bonds, pointing to extensive delocalization over the entire 3,5-diacetyl-1,2,4-triazole bis(thiosemicarbazone) skeleton.
Interestingly, the flexibility of the ligand originating from the free rotation of the two thiosemicarbazone arms allows each ligand the coordination to two metal ions in a twisted conformation generating two parallel coordination planes. The interplanar separation between the two triazole moieties is 3.302 Å, which is considered optimal for π–π interactions (intramolecular stacking).27 This arrangement is reinforced by double intramolecular hydrogen bonds between the 2NH of the bridging thiosemicarbazone moieties and the uncoordinated triazole nitrogen atoms.
The supramolecular association involves intermolecular hydrogen bonds between the 4NH and the oxygen atoms from DMSO and water solvent molecules and the arrangement in the crystal can be described as a packing of columns of [Pd(μ-H3L1)]2 molecules parallel to the [100] direction.
The molecular structure of the complex [Pt(μ-H3L1)]2·3DMSO is shown in Fig. 3 and selected bond lengths and angles are given in Table 2.
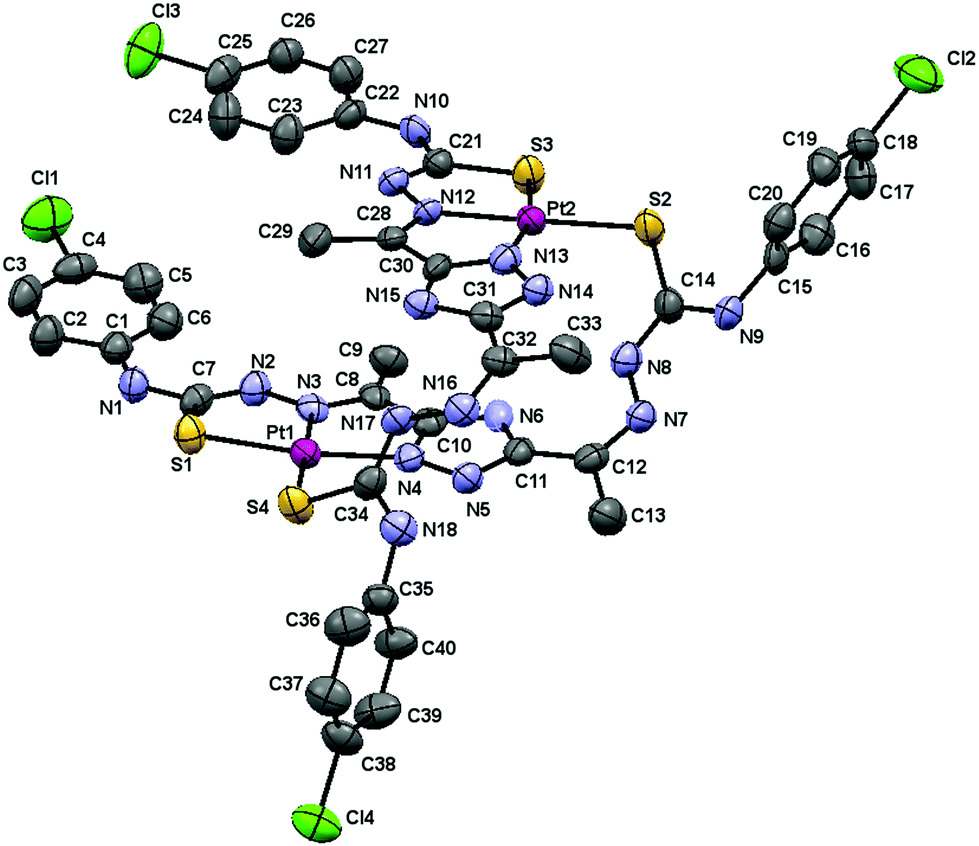 | ||
| Fig. 3 Molecular structure of the [Pt(μ-H3L1)]2·3DMSO complex, hydrogen atoms and solvent molecules are omitted for clarity. The displacement ellipsoids are drawn at the 50% probability. | ||
| Bond distances (Å) | |||
|---|---|---|---|
| Pt1–S1 | 2.260(3) | Pt2–S2 | 2.281(3) |
| Pt1–S4 | 2.292(2) | Pt2–S3 | 2.250(2) |
| Pt1–N3 | 1.994(7) | Pt2–N12 | 2.000(8) |
| Pt1–N4 | 2.026(8) | Pt2–N13 | 2.017(7) |
| C7–S1 | 1.773(9) | C21–S3 | 1.75(1) |
| C14–S2 | 1.716(7) | C34–S4 | 1.73(1) |
| C7–N1 | 1.356(11) | C30–N15 | 1.343(11) |
| C7–N2 | 1.320(11) | C32–N16 | 1.272(10) |
| C8–N3 | 1.312(11) | C34–N17 | 1.321(10) |
| C12–N7 | 1.270(11) | C34–N18 | 1.338(11) |
| C14–N8 | 1.326(11) | N2–N3 | 1.383(10) |
| C14–N9 | 1.314(11) | N7–N8 | 1.391(10) |
| C21–N11 | 1.309(11) | N11–N12 | 1.393(9) |
| C28–N12 | 1.304(11) | N16–N17 | 1.385(10) |
| C30–N13 | 1.330(11) | ||
| Bond angles (°) | |||
|---|---|---|---|
| N3–Pt1–N4 | 79.5(3) | N12–Pt2–N13 | 79.8(3) |
| N3–Pt1–S1 | 84.6(2) | N12–Pt2–S3 | 84.7(2) |
| S1–Pt1–S4 | 94.7(1) | S3–Pt2–S2 | 95.08(9) |
| N4–Pt1–S4 | 101.3(2) | N13–Pt2–S2 | 100.5(2) |
| N4–Pt1–S1 | 164.0(2) | N13–Pt2–S3 | 164.4(2) |
| N3–Pt1–S4 | 179.2(2) | N12–Pt2–S2 | 178.9(2) |
This dimeric compound crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with Z = 2. In a similar manner to the above described palladium dimer, the two 3,5-diacetyl-1,2,4-triazol bis(4N-p-chloro phenylthiosemicarbazone) ligands coordinate in a dideprotonated form to the Pt(II) ions with one arm in a tridentate fashion (SNN) and with the other in a monodentate S-bridging mode.
space group with Z = 2. In a similar manner to the above described palladium dimer, the two 3,5-diacetyl-1,2,4-triazol bis(4N-p-chloro phenylthiosemicarbazone) ligands coordinate in a dideprotonated form to the Pt(II) ions with one arm in a tridentate fashion (SNN) and with the other in a monodentate S-bridging mode.
The Pt–N and Pt–S bond distances are very similar to those found in [Pd(μ-H3L1)]2 as would be expected based on the covalent radii similarity of palladium and platinum (1.39 and 1.36 Å respectively).
There is also a π–π interaction between the two triazole rings (centroid distance of 3.394 Å), as well as intramolecular hydrogen bonds involving the 2NH of thiosemicarbazone moieties and the uncoordinated triazole nitrogen atoms. However, in this case, the two planes extend parallel up to the terminal phenyl rings so that a π–π stacking interaction between phenyl rings is also observed.
The supramolecular association is achieved through hydrogen interactions between 4NH and oxygen atoms from the DMSO molecules and the arrangement in the solid state can be described as a packing of columns of [Pt(μ-H3L1)]2 molecules parallel to the [010] direction.
These two structures with tridentate and monodentate bonding modes in the two arms, rather than bis-bidentate in both, result from the preferential binding of sulfur over nitrogen to palladium(II) and platinum(II) metal ions as well as the high stability of the tricyclic ring system of the tridentate moieties.
In vitro antiproliferative activity
To assess the antitumor potential of the synthesized compounds, its antiproliferative activity (in powder solid form) was tested in vitro against a panel of human cancer cell lines containing lung (NCI-H460), breast (T-47D) and ovarian (A2780 and A2780cisR) cancer cells. For comparison purposes, the cytotoxicity of cisplatin was always evaluated under the same experimental conditions.Out of an initial screening of the three compounds synthesized, both the free ligand and the platinum(II) derivative showed relevant antiproliferative activity with IC50 values falling in the low-micromolar range.
The remaining palladium(II) complex showed, at the maximum concentration tested, 100 μM, very low cellular growth inhibition (<50%) and therefore its cytotoxicity could not be evaluated. IC50 values obtained for these two compounds together with those for the related ligand 3,5-diacetyl-1,2,4-triazol bis(4N-p-tolylthiosemicarbazone), H5L3, and its platinum(II) complex [Pt(μ-H3L3)]2 are shown in Table 3. Specifically, the new ligand synthesized, H5L1, exhibits excellent antiproliferative activity surpassing the activity of cisplatin against three of the four tumor cell lines studied.
| IC50 ± SD (μM) | ||||
|---|---|---|---|---|
| A2780 | A2780cisR | NCI-H460 | T-47D | |
| a The IC50 values are averages of two independent measurements. b Values taken from ref. 23. | ||||
| H5L1 | 3.10 ± 0.02 | 3.51 ± 0.03 | 7.07 ± 0.07 | 3.37 ± 0.1 |
| [Pt(μ-H3L1)]2 | 35 ± 1 | >100 | >100 | 49 ± 1 |
| H5L3 | 3.7 ± 0.02b | 3.8 ± 0.4b | 13 ± 2b | 5.33 ± 0.05 |
| [Pt(μ-H3L3)]2 | 2.6 ± 0.04b | 4.9 ± 0.06b | 6.3 ± 0.26b | 7.08 ± 0.08 |
| Cisplatin | 0.88 ± 0.01 | 7.77 ± 0.10 | 7.25 ± 0.56 | 12 ± 1 |
Of particular relevance is the value of the resistance factor RF (defined as IC50 in A2780cisR/IC50 in A2780) of 1.1 versus 8.8 for cisplatin. An RF value of <2 is considered to denote non-cross-resistance and therefore this compound is able to circumvent cisplatin resistance.28,29 This ability to overcome cisplatin resistance in A2780cisR cells is in agreement with our previous studies on H5L3 suggesting that the mechanisms of resistance to cisplatin – most likely reduced drug transport, enhanced DNA repair and elevated GSH levels – are scarcely effective toward these 3,5-diacetyl-1,2,4-triazol bis(4N-substituted-thiosemicarbazones). Upon complexation, the [Pt(μ-H3L1)]2 complex demonstrated a moderate inhibitory effect against A2780 and T-47D cells while the related [Pt(μ-H3L3)]2 retained the activity in the four cell lines assayed. Interestingly, H5L1, H5L3 and [Pt(μ-H3L3)]2 compounds were more cytotoxic than cisplatin against T-47D cells.
DNA interaction studies
In order to initially address whether any direct interaction with DNA is part of the mechanism of action of the compounds, UV-visible absorption spectra in the absence and presence of calf thymus DNA (CT-DNA) were obtained for H5L1 and [Pt(μ-H3L1)]2 compounds.The UV-Vis absorption spectrum of the typical B-form DNA exhibits a characteristic π→π* band at 260 nm as a consequence of the chromophoric groups in purine and pyrimidine moieties. Compounds binding with DNA through intercalation are consistent with hypochromism (decrease in DNA band absorption), resulting from a stacking interaction between the aromatic ligand chromophore and the base pair of DNA. In the case of compounds binding with DNA through external contact (including groove binding and electrostatic attraction) usually hyperchromism (increase in DNA band absorption) is observed which is attributed to contraction and overall damage of the secondary structure of DNA.30
Thus, the absorption spectra of CT-DNA in the presence of H5L1 and [Pt(μ-H3L1)]2 were recorded, by keeping constant the CT-DNA concentration (5.45 × 10−5 M) in diverse [CT-DNA]/[compound] mixing ratios and monitoring the change in the absorption intensity of the typical CT-DNA spectral band at 260 nm. As shown in Fig. 4 and 5, when the concentration of H5L1 or [Pt(μ-H3L1)]2 is gradually increased a significant increase in absorption of the DNA band occurs with the percentage of hyperchromism observed [% hyperchromism = (ADNA bound − ADNA free)/ADNA bound] about 22% for H5L1 and 37% for [Pt(μ-H3L1)]2.
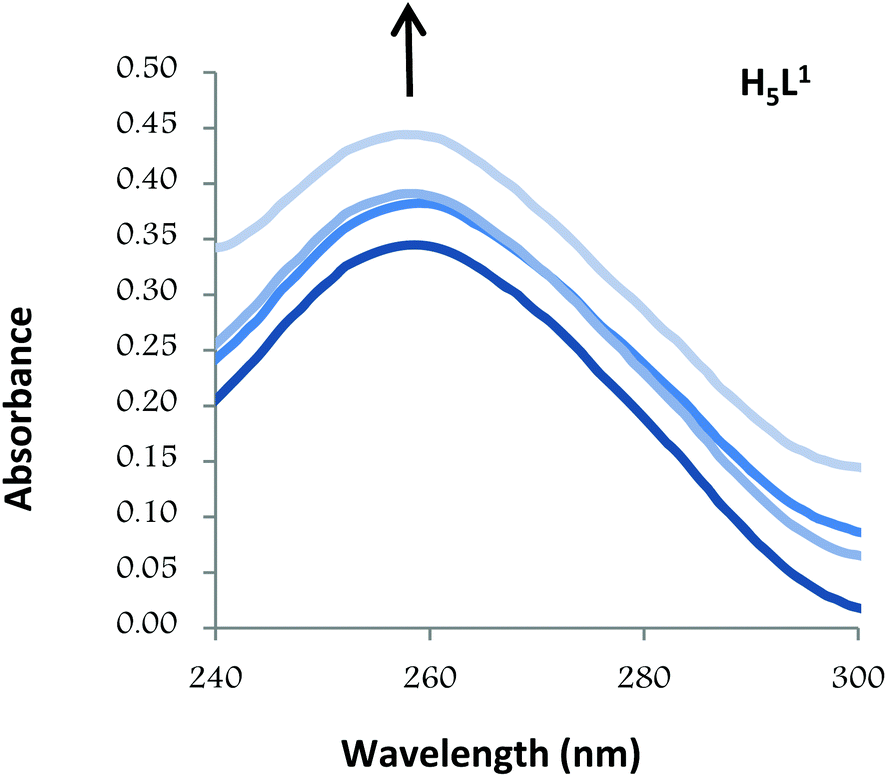 | ||
| Fig. 4 UV absorption spectra of CT-DNA in the absence and presence of increasing amounts of the compound H5L1 at diverse R values (R = [DNA]/[compound]): 0.0, 1.5, 2.0, 2.5. | ||
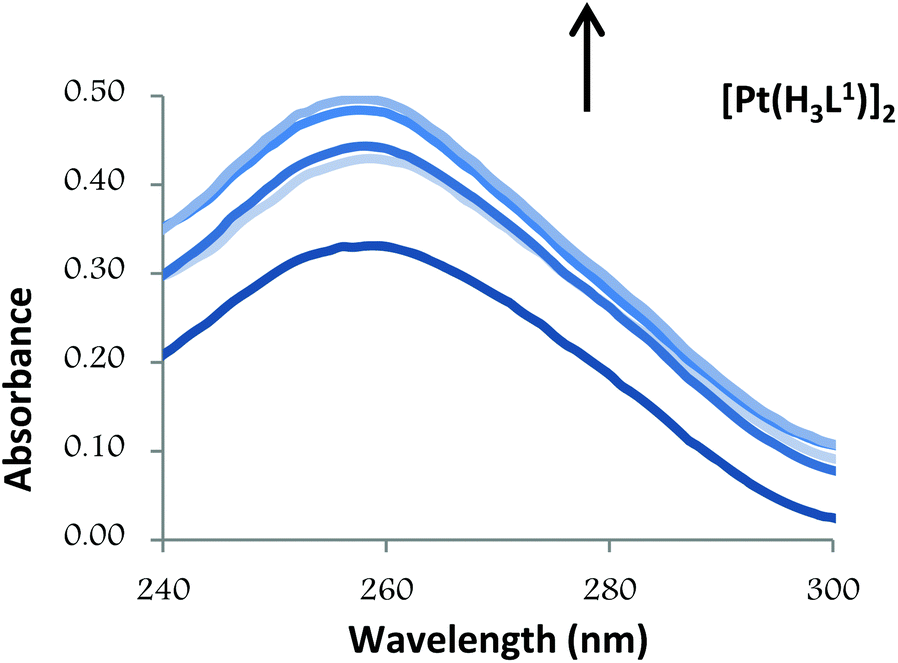 | ||
| Fig. 5 UV absorption spectra of CT-DNA in the absence and presence of increasing amounts of the compound [Pt(H3L1)]2 at diverse R values (R = [DNA]/[compound]): 0.0, 3.0, 3.5, 4.0, 5.0. | ||
The observed hyperchromic effect suggests that both compounds bind to CT-DNA by external contact.31 These characteristics are comparable to those previously reported for various neutral bis(thiosemicarbazone)palladium and platinum complexes and for which a non-covalent binding along outside of the DNA helix surface (major or minor groove) was proposed.24,32
In the UV region, H5L1 and [Pt(μ-H3L1)]2 exhibit, in a DMSO–Tris buffer (2.5:100) mixture, intense absorption bands attributable to intraligand transitions which could be perturbed in the case of an interaction with CT DNA. Thus, in order to determine the intrinsic binding constant (Kb), absorption titration experiments were performed by maintaining a constant concentration of the desired compound while gradually increasing the concentration of CT DNA and monitoring the change in the absorption intensity of one of the intraligand charge transfer bands (for the H5L1 ligand the band at λmax 320 nm was used whereas for [Pt(μ-H3L1)]2 with less resolved bands the band at λmax 255 nm was chosen). While measuring the absorption spectra, an equal amount of CT DNA was added to both the test solution and the reference solution to eliminate the absorbance of CT DNA itself. The data were then fitted to the following equation33:
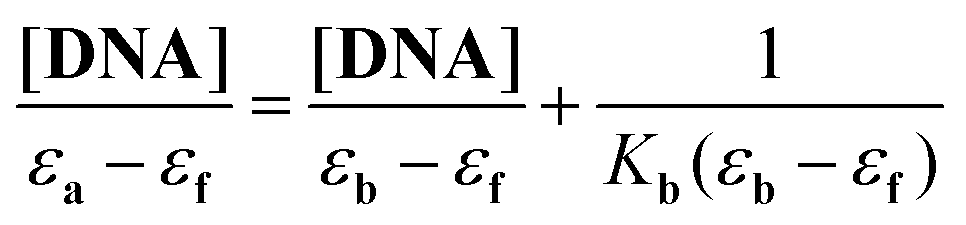
A plot of [DNA]/(εa − εf) versus [DNA] gives a slope of 1/(εb − εf) and a Y-intercept equal to 1/{Kb(εb − εf)}. The intrinsic binding constant Kb is calculated as the ratio of the slope to the Y-intercept.
The UV spectra of H5L1 and [Pt(μ-H3L1)]2, Fig. 6 and 7 respectively, display an increase in the absorptivity upon addition of increasing amounts of CT DNA (0–50 μM), which is indicative of interactions between the electronic states of the ligand chromophores with that of DNA bases.
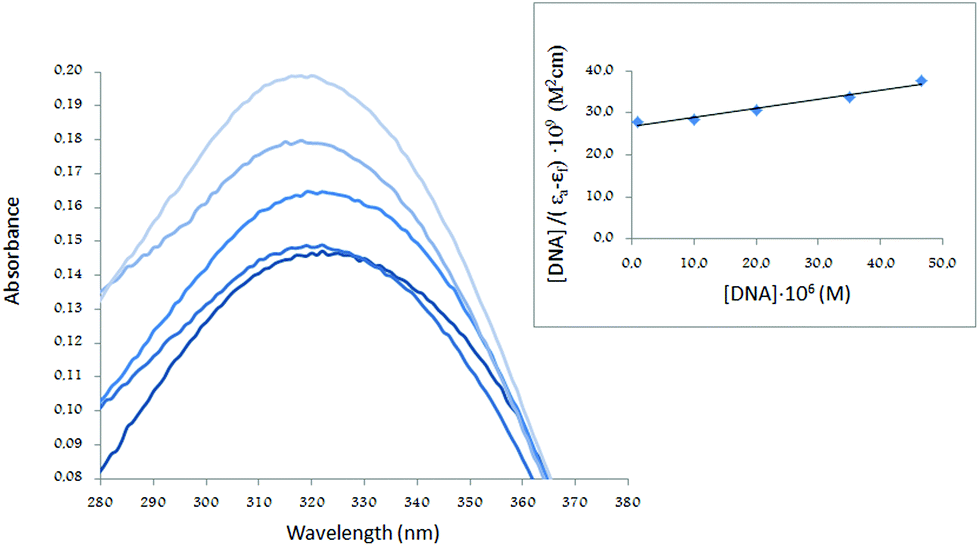 | ||
| Fig. 6 UV absorption spectra of H5L1 in the presence of increasing amounts of CT-DNA. Data were collected for [H5L1] = 5 × 10−6 M and [CT-DNA] = 1.25 × 10−6, 5 × 10−6, 20 × 10−6, 25 × 10−6, 50 × 10−6 M. The inset shows a fitting of the absorbance data used to obtain the binding constants. | ||
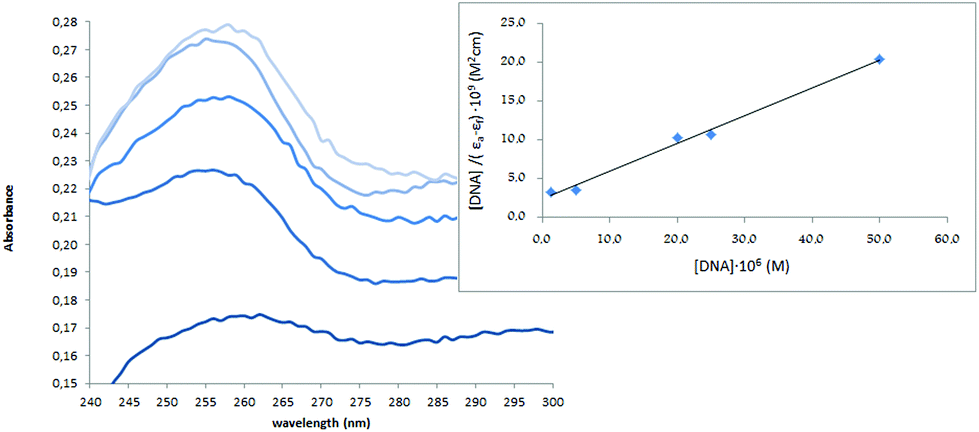 | ||
| Fig. 7 UV absorption spectra of [Pt(μ-H3L1)]2 in the presence of increasing amounts of CT-DNA. Data were collected for [Pt(μ-H3L1)]2 = 5 × 10−6 M and [CT-DNA] = 1.25 × 10−6, 5 × 10−6, 20 × 10−6, 25 × 10−6, 50 × 10−6 M. The inset shows a fitting of the absorbance data used to obtain the binding constants. | ||
This change in absorbance values was used to calculate the magnitude of the intrinsic binding constant, Kb, for H5L1 and [Pt(μ-H3L1)]2 compounds, the values are 8.06 × 103 M−1 (correlation coefficient R2 = 0.97) and 1.51 × 105 M−1 (correlation coefficient R2 = 0.99) respectively.
The higher value of Kb obtained for [Pt(μ-H3L1)]2 in comparison with that calculated for the free ligand, H5L1, suggests that the coordination to Pt(II) ions enhances significantly the ability to bind to CT DNA. On the other hand, no significant shift is observed in the UV absorption band of the [Pt(μ-H3L1)]2 complex indicating that there is no change of the coordination environment of the metal ion and this consequently discards covalent binding.
The Kb values obtained here are lower than that reported for double-stranded DNA intercalators while they are comparable to other classical groove binders.34,35 Therefore, these findings support the interaction of both compounds with DNA via groove binding.
Taking into account that both the free ligand and the platinum complex have demonstrated similar cytotoxic activity, it seems that DNA interactions are not uniquely responsible for inhibition of cell proliferation. Further studies and more practical experiments are required to elucidate the biochemical mechanisms involved in their biological activity.
A classical intercalator molecule like ethidium bromide causes a significant increase in the viscosity of the DNA solution due to an increase in the separation of base pairs at the interaction site and an increase in the overall double helix length, whereas a groove binder agent like Hoechst 33258 does not appreciably lengthen the DNA helix and therefore causes less pronounced changes (positive or negative) or no changes in the viscosity of DNA solutions. On the other hand, cisplatin, which is known to fold DNA through covalent binding, causes a decrease in the relative viscosity of the DNA solution.36
The values of cube root of the relative specific viscosity, (η/η0)1/3, versus the ratio of the complex concentration to DNA, 1/R (where R = [CT-DNA]/[compound]), in the absence and in the presence of compounds H5L1 and [Pt(μ-H3L1)]2 are plotted in Fig. 8. Both compounds exhibit similar patterns: initially, at a low concentration of the compound, a clear diminution of the viscosity is observed suggesting a bending of the DNA chain probably due to strong hydrogen bonding interactions; however, when the concentration of the compound increases, a slight increase of the viscosity is observed pointing to groove binding interactions as the dominant DNA interactions.37,38 Thus, these results provide further support for the groove binding mode between these compounds and DNA.
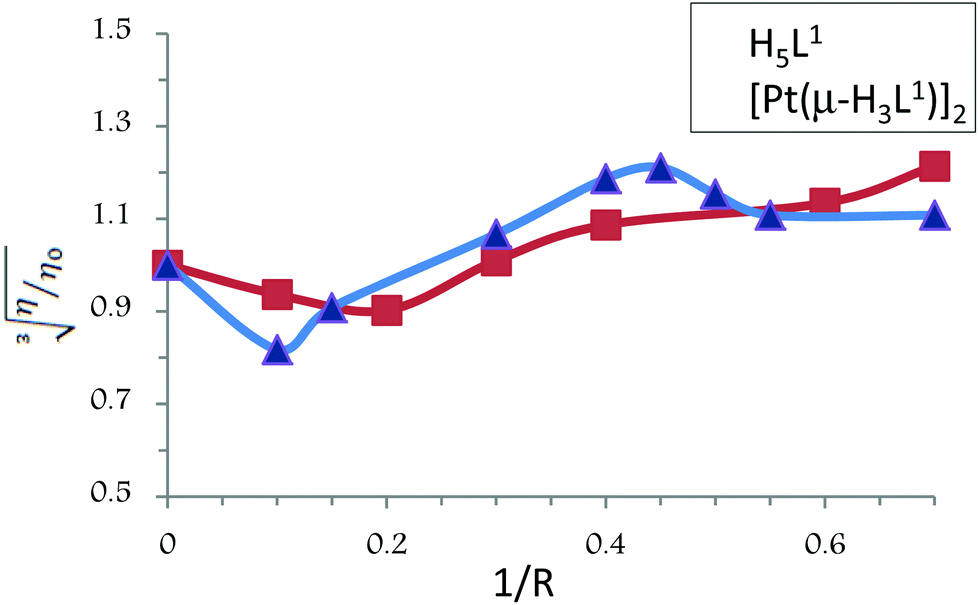 | ||
| Fig. 8 Effect of increasing amounts of compounds H5L1 and [Pt(μ-H3L1)]2 on the viscosity of CT-DNA. The data were collected for [DNA] = 5.45 × 10−5 M at diverse 1/R ratios (R = [DNA]/[compound]). | ||
Experimental section
Measurements
Elemental analyses were performed on a LECO CHNS-932 microanalyzer. Fast atom bombardment (FAB) mass spectroscopy (MS) was performed on a VG AutoSpec spectrometer and Electrospray Ionization (ESI) mass spectroscopy was carried out on a QSTAR mass spectrometer. Nuclear Magnetic Resonance (NMR) spectra were recorded on a BRUKER AMX-300 spectrometer. All cited physical measurements were conducted by the Servicio Interdepartamental de Investigación (SIdI) of the Universidad Autónoma de Madrid.Melting points were determined with Stuart Scientific SMP3 apparatus. The pH measurements were carried out with a Crison BASIC 20+ pH-meter equipped with a combined Crison glass electrode. Infrared spectra were recorded on a Jasco FT/IR-410 spectrophotometer. Electronic spectra were recorded on a Thermo Scientific Evolution 260 Bio UV-visible (UV-VIS) spectrophotometer. Viscosity experiments were conducted on an automated AND viscometer model SV-1A.
Materials
Solvents were purified and dried according to standard procedures. Ultrapure MIlli Q water was used for all biological experiments. Hydrazine hydrate, L-lactic acid, p-chlorophenyl isothiocyanate, palladium(II) chloride, lithium chloride and potassium tetrachloridoplatinate(II) were commercially available.Synthesis of compounds
Yield (75%), mp 188 °C. Elemental analysis found, C, 44.50; H, 4.15; N, 24.50; S 11.85; C20H19N9S2Cl2·H2O requires C, 44.61; H, 4.07; N, 24.22; S 12.32%. MS (FAB+ with mNBA: nitrobenzyl alcohol matrix) m/z 520 (100%) for [C20H19N9S2Cl2 + H]+. IR (KBr pellet): ν/cm−1 3263 (s, NH-triazol), 3128 and 3089 (s, 2NH and 4NH), 1584 (m, CN), 756 (w, CS-thioamide IV band). 1H NMR (300.14 MHz, DMSO-d6): δ (ppm) 14.75 (s, NH-triazol, 1H), 11.30 (s, 2NH, 1H), 11.29 (s, 2NH, 1H), 10.23 (s, 4NH, 1H), 10.00 (s, 4NH, 1H), 7.68–7.28 (m, aromatic-thiosemicarbazide, 8H), 2.45 and 2.44 (s, CH3-triazol, 3H).
:1 molar ratio. The reaction mixture was stirred for 5 h at room temperature and the resulting orange solid obtained was filtered, washed with methanol and diethyl ether, and dried in vacuo.
Single crystal X-ray diffraction
Data were collected on a Bruker Kappa Apex II diffractometer. Crystallographic data are listed in Table 4.| [Pd(μ-H3L1)]2·2.5DMSO·H2O | [Pt(μ-H3L1)]2·3DMSO | |
|---|---|---|
| Chemical formula | C45H49Cl4N18O3.50Pd2S6.50 | C46H52Cl4N18O3Pt2S7 |
| Formula weight | 1461.01 | 1661.46 |
| Temperature | 100(2) K | 296(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å |
| Crystal size | 0.02 × 0.02 × 0.21 mm | 0.02 × 0.04 × 0.14 mm |
| Crystal habit | Clear orange needles | Clear orange prismatic |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21/n |
P |
| Unit cell dimensions | a = 8.123(3) Å | a = 14.6278(2) Å |
| b = 29.002(9) Å | b = 15.6060(2) Å | |
| c = 28.87(1) Å | c = 15.6896(3) Å | |
| α = 90° | α = 66.436(1)° | |
| β = 93.92(2)° | β = 81.756(1)° | |
| γ = 90° | γ = 68.305(1)° | |
| Volume | 6786.(5) Å3 | 3050.39(8) Å3 |
| Z | 4 | 2 |
| Density (calculated) | 1.430 Mg cm−3 | 1.809 Mg cm−3 |
| Absorption coefficient | 0.937 mm−1 | 5.052 mm−1 |
| Theta range for data collection | 1.41–25.33° | 1.42–25.35° |
| Index ranges | −9 ≤ h ≤ 9 | −17 ≤ h ≤ 17 |
| −34 ≤ k ≤ 31 | −18 ≤ k ≤ 18 | |
| −34 ≤ l ≤ 26 | −18 ≤ l ≤ 18 | |
| Reflections collected | 38026 |
79411 |
| Independent reflections | 12166 [R(int) = 0.0376] |
11149 [R(int) = 0.0728] |
| Coverage of independent reflections | 98.1% | 99.7% |
| Data/restraints/parameters | 12166/0/740 |
11149/0/731 |
| Goodness-of-fit on F2 | 1.048 | 1.032 |
| Final R indices | 9274 data; I > 2σ(I) | 7715 data; I > 2σ(I) |
| R 1 = 0.1086, wR2 = 0.2948 | R 1 = 0.0429, wR2 = 0.1108 | |
| all data | all data | |
| R 1 = 0.1367, wR2 = 0.3212 | R 1 = 0.0807, wR2 = 0.1439 | |
| Largest diff. peak and holes | 6.233 and −2.253 e Å−3 | 2.893 and −2.688 e Å−3 |
The software package SHELXTL was used for space group determination, structure solution, and refinement.40 The structures were solved by direct methods, completed with difference Fourier syntheses, and refined with anisotropic displacement parameters. DMSO solvent molecules present a high degree of disorder, especially in the palladium derivative. The collection of diffraction data at 100 K did not improve this positional disorder.
In vitro antiproliferative activity
The human cancer cells, A2780 and A2780cisR (epithelial ovarian cancer), T-47D (breast cancer) and NCI-H460 (non-small cell lung cancer), were grown in RPMI-1640 medium supplemented with 10% foetal bovine serum (FBS) and 2 mM L-glutamine under an atmosphere of 5% CO2 at 37 °C.Cell proliferation was evaluated by the sulforhodamine B assay. Cells were plated in 96-well sterile plates at a density of 1.5 × 104 (for NCI-H460), 4 × 103 (for A2780 and A2780cisR) or 0.5 × 103 (for T-47D) cells per well with 100 μL of medium and were then incubated for 24 h (A2780, A2780cisR and NCI-H460) or 48 h (T-47D). After attachment to the culture surface the cells were incubated with various concentrations of the tested compounds freshly dissolved in DMSO (1 mg mL−1) and diluted in the culture medium (DMSO final concentration 1%) for 48 h (for NCI-H460) or 96 h (for A2780, A2780cisR and T-47D). The cells were fixed by adding 50 μL of 30% trichloroacetic acid (TCA) per well.
The plates were incubated at 4 °C for 1 h and then washed five times with distilled water. The cellular material fixed with TCA was stained with 0.4% sulforhodamine B dissolved in 1% acetic acid for 10 min. The unbound dye was removed by rinsing with 0.1% acetic acid. The protein-bound dye was extracted with 10 mM unbuffered Tris base for determination of the optical density (at 515 nm) using a Tecan Ultra Evolution spectrophotometer.
The effects of compounds were expressed as corrected percentage inhibition values according to the following equation:
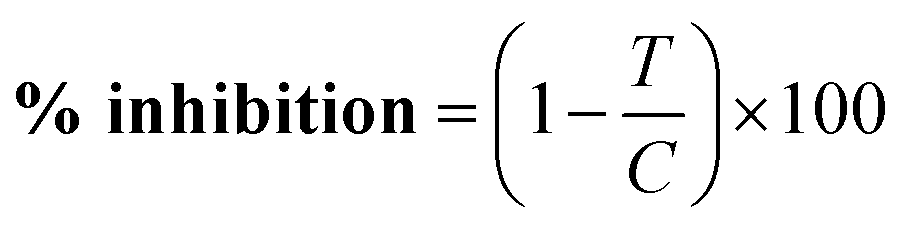
The inhibitory potential of compounds was measured by calculating concentration–percentage inhibition curves, these curves were fitted to the following equation:

These experiments were carried out at the Unidad de Evaluación de Actividades Farmacológicas de Compuestos Químicos (USEF), Universidad de Santiago de Compostela.
DNA-binding experiments
CT-DNA stock solution was prepared by dissolving the lyophilized sodium salt in Tris-buffer (NaCl 50 mM, Tris-HCl 5 mM, pH was adjusted to 7.2 with NaOH 0.5 M) by stirring for 5 hours. The CT-DNA solution was standardized spectrophotometrically42 by using its known molar absorption coefficient at 260 nm (6600 M−1 cm−1). The ratio of the UV absorbance at 260 and 280 nm, A260/A280, was ca. 1.9, indicating that the DNA was sufficiently free of proteins. The stock solution was kept frozen until the day of the experiment.Concentrated stock solutions (5 × 10−3 M and 5 × 10−6 M) of H5L1 and [Pt(μ-H3L1)]2 compounds were prepared by dissolving the compound in DMSO. From these stock solutions, for all experiments the desired concentration of the compound was achieved by dilution with Tris-buffer (NaCl 50 mM, Tris-HCl 5 mM, pH was adjusted to 7.2 with NaOH 0.5 M) to give homogeneous solutions with the DMSO content of less than 2.5%.
To investigate the binding mode, spectrophotometric titrations were performed, at a fixed DNA concentration equal to 5.45 × 10−5 M, with increasing concentrations of compounds (R = [CT-DNA]/[compound]) and the absorbance change was monitored at the wavelength maximum of 260 nm after incubation (10 min at 37 °C).
To calculate the binding parameters, spectrophotometric titrations were performed with increasing concentrations of DNA (0–50 μM) at a fixed compound concentration equal to 5 μM and the absorbance change was monitored in one characteristic charge transfer band of the compound after incubation (10 min at 37 °C).
The viscosity experiments were carried out, at a constant temperature of 25.0 ± 0.1 °C in a thermostated water bath using a water jacket accessory, at a fixed DNA concentration equal to 5.45 × 10−5 M with increasing concentrations of compounds to ratios (1/R = [compound]/[CT-DNA]) in the range 0–0.7. For all of the experiments, the desired concentration of the compound was achieved by dilution with Tris-buffer and DMSO to give homogeneous solutions containing a final concentration of 2.0% of DMSO.
Conclusions
A new family of Pt(II) and Pd(II) bis(thiosemicarbazone) compounds of the 3,5-diacetyl-1,2,4-triazol series containing an aryl ring with an electron withdrawing substituent (p-chlorophenyl group) has been successfully prepared and characterized.According to the molecular structure, determined by single crystal X-ray diffraction, the two complexes display a dimeric structure as a result of the pairing of two metal centers through two thiosemicarbazone bridging moieties.
Investigation of potential biological properties (cytotoxicity and DNA binding studies) of the newly synthesized compounds indicated that H5L1 showed the strongest antiproliferative activity surpassing the activity of the conventional standard cisplatin against three of the four tumor cell lines assayed. However the [Pt(μ-H3L1)]2 complex exhibited better binding with CT-DNA which was inferred from the greater magnitude of the binding, in the range of classical groove binders. Thus our results suggest that non-covalent interactions with DNA are not the unique molecular target for these compounds.
Acknowledgements
We are grateful to Ministerio de Economía y Competitividad, Instituto de Salud Carlos III (PI1100659) of Spain for financial support.Notes and references
- F. Arnesano, M. Losacco and G. Natile, Eur. J. Inorg. Chem., 2013, 2701–2711 CrossRef CAS.
- J. D. Hoeschele, Dalton Trans., 2009, 10648–10650 CAS.
- J. Reedijk, Eur. J. Inorg. Chem., 2009, 1303–1312 CrossRef CAS.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- Z. Guo and P. J. Sadler, Angew. Chem., Int. Ed., 1999, 38, 1512–1531 CrossRef CAS.
- M. S. Razzaque, Nephrol., Dial., Transplant., 2007, 1–5 CAS.
- M. Okuda, K. Masaki, S. Fukatsu, Y. Hashimoto and K. Inui, Biochem. Pharmacol., 2000, 59, 195–201 CrossRef CAS.
- J. Shao, X. Liu, L. Zhu and Y. Yen, Expert Opin. Ther. Targets, 2013, 17, 1423–1437 CrossRef CAS PubMed.
- D. S. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2005, 57, 547–583 CrossRef CAS PubMed.
- C. Kunos, T. Radivoyevitch, F. W. Abdul-Karim, J. Fanning, O. Abulafia, A. J. Bonebrake and L. Usha, J. Transl. Med., 2012, 10, 79 CrossRef CAS PubMed.
- J. G. da Silva, L. S. Azzolini, S. M. S. V. Wardell, J. L. Wardell and H. Beraldo, Polyhedron, 2009, 28, 2301–2305 CrossRef CAS PubMed.
- G. L. Parrilha, J. G. da Silva, L. F. Gouveia, A. K. Gasparoto, R. P. Dias, W. R. Rocha, D. A. Santos, N. L. Speziali and H. Beraldo, Eur. J. Med. Chem., 2011, 46, 1473–1482 CrossRef CAS PubMed.
- P. Genova, T. Varadinova, A. I. Matesanz, D. Marinova and P. Souza, Toxicol. Appl. Pharmacol., 2004, 197, 107–112 CrossRef CAS PubMed.
- P. J. Jansson, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2010, 53, 5759–5769 CrossRef CAS PubMed.
- C. R. Kowol, R. Trondl, V. B. Arion, M. A. Jakupec, I. Lichtscheidl and B. K. Keppler, Dalton Trans., 2010, 39, 704–706 RSC.
- A. I. Matesanz and P. Souza, Mini-Rev. Med. Chem., 2009, 9, 1389–1396 CrossRef CAS.
- S. Chandra, S. Parmar and Y. Kumar, Bioinorg. Chem. Appl., 2009, 851316, DOI:10.1155/2009/851316.
- B. García, J. Garcia-Tojal, R. Ruíz, R. Gil-García, S. Ibeas, B. Donnadieu and J. M. Leal, J. Inorg. Biochem., 2008, 102, 1892–1900 CrossRef PubMed.
- F. Bisceglie, S. Pinelli, R. Alinovi, P. Tarasconi, A. Buschini, F. Mussi, A. Mutti and G. Pelosi, J. Inorg. Biochem., 2012, 116, 195–203 CrossRef CAS PubMed.
- K. Abdi, H. Hadadzadeh, M. Salimi, J. Simpson and A. D. Khalaji, Polyhedron, 2012, 44, 101–112 CrossRef CAS PubMed.
- A. I. Matesanz and P. Souza, J. Inorg. Biochem., 2007, 101, 245–253 CrossRef CAS PubMed.
- A. I. Matesanz, C. Hernández, A. Rodríguez and P. Souza, Dalton Trans., 2011, 40, 5738–5745 RSC.
- A. I. Matesanz, C. Hernández, A. Rodríguez and P. Souza, J. Inorg. Biochem., 2011, 105, 1613–1622 CrossRef CAS PubMed.
- A. I. Matesanz, J. Perles and P. Souza, Dalton Trans., 2012, 41, 12538–12547 RSC.
- A. A. Ali, H. Nimir, C. Aktas, V. Huch, U. Rauch, K. Schäfer and M. Veith, Organometallics, 2012, 31, 2256–2262 CrossRef CAS.
- J. W. Steed and J. L. Atwood, in Supramolecular Chemistry, John Wiley & Sons, Chichester, 2nd edn, 2009, pp. 28–35 Search PubMed.
- L. R. Kelland, C. F. J. Barnard, K. J. Mellish, M. Jones, P. M. Goddard, M. Valenti, A. Bryant, B. A. Murrer and K. R. Harrap, Cancer Res., 1994, 54, 5618–5622 CAS.
- J. Ruiz, C. Vicente, C. Haro and D. Bautista, Inorg. Chem., 2013, 52, 974–982 CrossRef CAS PubMed.
- F. Arjmand and M. Aziz, Eur. J. Med. Chem., 2009, 44, 834–844 CrossRef CAS PubMed.
- P. Kalaivani, R. Prabhakaran, E. Vaishnavi, T. Rueffer, H. Lang, P. Poornima, R. Renganathan, V. Vijaya Padma and K. Natarajan, Inorg. Chem. Front., 2014, 1, 311–324 RSC.
- A. I. Matesanz, C. Hernández and P. Souza, J. Inorg. Biochem., 2014, 138, 16–23 CrossRef CAS PubMed.
- B. Pedras, R. M. F. Batista, L. Tormo, S. P. G. Costa, M. M. M. Raposo, G. Orellana, J. L. Capelo and C. Lodeiro, Inorg. Chim. Acta, 2012, 381, 95–103 CrossRef CAS PubMed.
- H. K. H. Fong and B. R. Copp, Mar. Drugs, 2013, 11, 274–299, DOI:10.3390/md11020274.
- N. Shahabadi, S. Hadidi and A. Taherpour, Appl. Biochem. Biotechnol., 2014, 172, 2436–2454 CrossRef CAS PubMed.
- D. Suh and J. B. Chaires, Bioorg. Med. Chem., 1995, 3, 723–728 CrossRef CAS.
- E. Escribano, M. Font-Bardia, T. Calvet, J. Lorenzo, P. Gámez and V. Moreno, Inorg. Chim. Acta, 2013, 394, 65–76 CrossRef CAS PubMed.
- J. Hernández-Gil, S. Ferrer, N. Cabedo, M. P. López-Gresa, A. Castiñeiras and F. Lloret, J. Inorg. Biochem., 2013, 125, 50–63 CrossRef PubMed.
- C. D. Brandt, J. A. Kitchen, U. Beckmann, N. G. White, G. B. Jameson and S. Brooker, Supramol. Chem., 2007, 19, 17–27 CrossRef CAS.
- SHELXTL-NT version 6.12, Structure Determination Package, Bruker-Nonius XS, Madison, Wisconsin, USA, 2001 Search PubMed.
- GraphPad Prism, version 2.01, GraphPad Software, Inc., San Diego, CA, 1996 Search PubMed.
- M. A. Ali, A. H. Mirza, A. L. Tan, L. K. Wei and P. V. Bernhardt, Polyhedron, 2004, 23, 2037–2043 CrossRef CAS PubMed.
Footnote |
| † CCDC 997270 and 997271 contain the supplementary crystallographic data for [Pd(μ-H3L1)]2·2.5DMSO·H2O and [Pt(μ-H3L1)]2·3DMSO respectively. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00128a |
| This journal is © the Partner Organisations 2015 |