
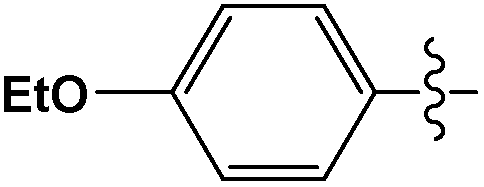
A mechanistic study of cross-coupling reactions catalyzed by palladium nanoparticles supported on polyaniline nanofibers†
William M.
Lemke
,
Richard B.
Kaner
and
Paula L.
Diaconescu
*
Department of Chemistry & Biochemistry, University of California, Los Angeles, California 90095, USA. E-mail: pld@chem.ucla.edu
First published on 7th November 2014
Abstract
Palladium nanoparticles supported by polyaniline nanofibers were used as pre-catalysts for C–C cross-coupling reactions of aryl chlorides in water. Using regular techniques, such as scanning electron microscopy (SEM), transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS) in conjunction with reactivity studies, we determined that a homogeneous mechanism is operating. The appearance of palladium nanoparticles and polyaniline nanofibers changes after each reaction accompanied by changes in the palladium oxidation state. Our methods could not rule out the contribution of a heterogeneous mechanism or indicate its extent.
Introduction
Palladium pre-catalysts for Suzuki,1,2 Heck,3–6 and Sonogashira7,8 C–C cross-coupling reactions have evolved immensely since initial reports in 1971.9–12 Early catalysts were homogeneous13,14 and contained ligands such as phosphines,15,16 pincer,17,18 or N-heterocyclic carbenes.19,20 A shift toward palladium nanoparticles has been apparent in the past decade because they offer the benefits of heterogeneous catalysis.21–24 Although their role in catalysis is not well understood, metal nanoparticles provide advantages specific to both heterogeneous and homogeneous catalysts and because of that are sometimes called “semi-heterogeneous” catalysts.21 These advantages relate to the fact that small clusters of metals in low oxidation states are stabilized in solution, and include recoverability and reusability as well as high activity and selectivity.When immobilized on a support, palladium nanoparticles can be recovered and recycled like heterogeneous catalysts, but also provide selectivity and low loadings like homogeneous catalysts.21 The support acts as a stabilizing agent to prevent aggregation of the nanoparticles and, usually, has a high surface area and porosity to allow maximum coverage with nanoparticles; graphene derivatives,25,26 carbon spheres,27 and silica28,29 have been intensely studied in this respect.
Recently, polymers have been used as supports.21,22,30,31 In general, a reducing agent is required for palladium nanoparticle formation.21,22 Polyaniline (PANI),32–35 however, provides both stabilization and reducing properties. PANI has been used for various applications due to its electrical conductivity.35–46 PANI has three main oxidation states, which enables it to readily undergo redox reactions (Fig. 1). Doping the half oxidized emeraldine base form of PANI with a strong acid leads to the electrically conducting emeraldine salt form.
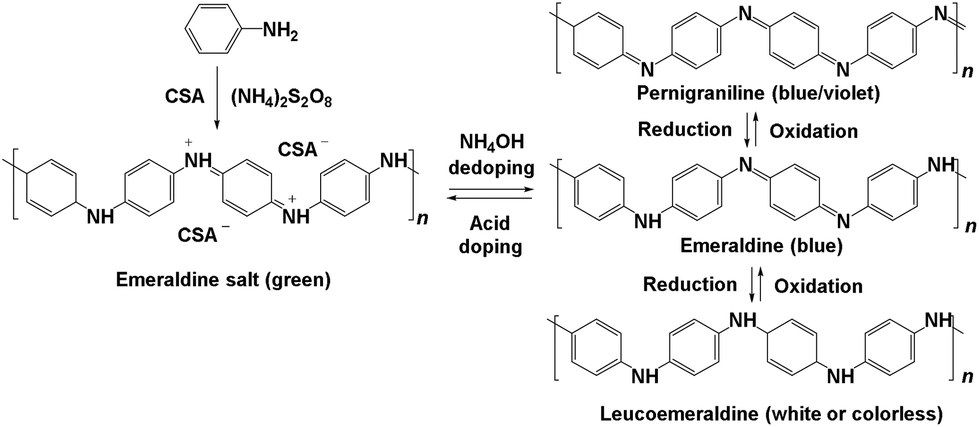 | ||
| Fig. 1 The three main redox states of dedoped polyaniline along with the acid doped salt form. | ||
We47 and others48,49 previously reported that palladium nanoparticles supported on PANI nanofibers work well for Suzuki coupling in water. In our case, the palladium nanoparticles showed a bimodal distribution with particles of 2 and 50 nm in diameter being present. The catalyst showed excellent yields in Suzuki coupling reactions using the less expensive and less reactive aryl chloride substrates, rather than aryl bromides and iodides. These reactions were carried out in water, which is the least expensive solvent as well as the most environmentally friendly, and the entire system was recyclable. The simplicity of our system is what makes it attractive; there is no extra step to create the nanoparticles before mixing with the support. Palladium nanoparticles on polyaniline nanofibers give good conversions at about half the time of other systems.25,27 Our system is also advantageous because it does not need a phase transfer agent like tetrabutylammonium bromide to help with the hydrophobicity of aryl chlorides.48,49 At the time of our initial report, we hypothesized that a dissolution and redeposition mechanism was operating.
Over the past decade, there have been contradicting claims on the true type of catalysis that takes place with palladium nanoparticles.50,51 The most accepted interpretation for the Heck reaction, which has been better studied than the Suzuki reaction, is that C–C coupling is achieved by soluble palladium species through a mechanism of dissolution/redeposition.17,50,52–59 Various proposals exist with respect to the details of the Suzuki reaction: (1) oxidative addition occurs with palladium ions dissolved in solution, catalysis takes place in solution, driving the dissolution equilibrium towards the generation of molecular clusters,60 and, once the aryl halide is consumed, palladium is redeposited either on the surface of the support or on the metallic clusters that are still present;60,61 (2) oxidative addition takes place on the surface of colloidal palladium nanoparticles formed in solution; and (3) oxidative addition takes place on the surface of palladium nanoparticles present on the support. Formation of Pd(II) species induces dissolution, the next steps occur in solution, and once the starting material is consumed, the palladium(0) formed gets redeposited.62 The difference between the three proposals is related to the first step of the reaction, also considered the rate-determining step for both homogeneous and heterogeneous catalysts in Suzuki coupling.17,50,52 For the more reactive aryl halides (i.e. iodides), reports favor, the first explanation;63 in such cases, various ligands and additives increase the lifetime of palladium(0) in solution. This mechanism is similar to that for the Heck reaction, for which a homogeneous pathway is accepted for all types of aryl halides.52 The biggest difference between the Heck and Suzuki reactions is that the former requires the presence of additional empty coordination sites, such that the olefinic substrate can bind to palladium, and, therefore, the catalytically active palladium species is in solution, where ligand coordination and dissociation can occur readily.52 In order to explain the activity towards aryl bromides and chlorides of certain catalysts in the Suzuki reaction, heterogeneous pathways (mechanisms 2 and 3) are proposed to contribute increasingly (the less reactive the aryl halide the higher the contribution of the heterogeneous mechanism) to the overall reaction. It is important to note that an operando X-ray absorption spectroscopy study in combination with other techniques of a Suzuki coupling of 4-iodoanisole catalyzed by polyvinylpyrrolidone-stabilized palladium nanoparticles at 60 °C found that the mechanism of this reaction is heterogeneous (note the relatively low temperature of the reaction compared to usual reaction temperatures of ca. 100 °C).64,65 The difference in behavior between various systems arises because different ligands/supports could favor the oxidative addition step by electron donation, similar to situations of homogeneous catalysis, where electron-donating ligands are used to increase the rate of oxidative additions.66 Distinguishing between these three possibilities is difficult and no simple test has yet been proposed. Using a multitude of experiments allows more subtle interpretations, which usually explain the mechanism as a combination of homogeneous and heterogeneous pathways,67 but it is rarely possible to differentiate between mechanisms 2 and 3.68,69
Herein, we report our mechanistic studies on the cross-coupling reaction of aryl chlorides catalyzed by palladium nanoparticles supported on polyaniline nanofibers. Using regular techniques, such as scanning electron microscopy (SEM), transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS) in conjunction with reactivity studies, we could determine that a homogeneous mechanism is operating. However, our methods could not rule out the contribution of a heterogeneous mechanism or indicate its extent.
Results and discussion
Synthesis and characterization of pre-catalysts
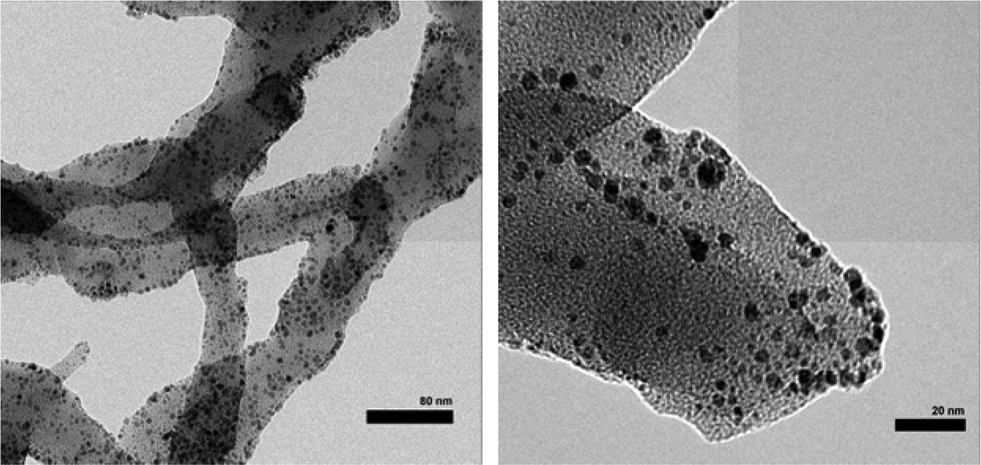
Chemical oxidative polymerization by the rapid-mixed pathway32,33 was used to synthesize PANI nanofibers. The nanofibers undergo a colour change from green to violet when dedoped with base (ammonium hydroxide). The PANI nanofibers as-synthesized had a diameter of 50 nm as shown by transmission electron microscopy (TEM, Fig. 2). The diameter of the nanofibers can be controlled by the acid dopant during polymerization, in this case, camphorsulfonic acid (CSA). A water dispersion of PANI was mixed with a palladium(II) nitrate solution to form the pre-catalyst. Purification of the pre-catalyst was carried out by dialysis. | ||
| Fig. 2 TEM of palladium nanoparticles supported on PANI nanofibers. | ||
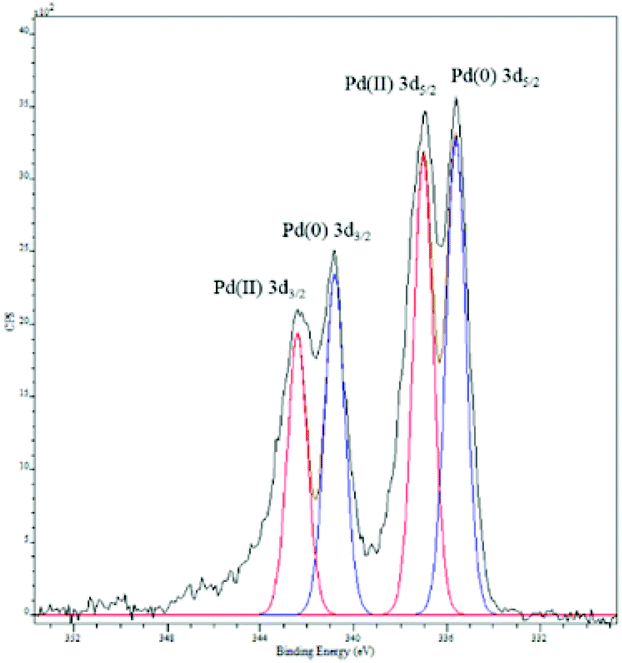
Palladium deposition on the fibers was imaged using scanning electron microscopy (SEM) and quantified by inductive coupled plasma optical emission spectrometry (ICP-OES) and energy dispersive spectroscopy (EDS, Fig. S1†). The solution had a palladium concentration of 6 mM. Palladium nanoparticles deposited on polyaniline nanofibers had an average diameter of 2 nm (Fig. 2), with a unimodal distribution, unlike what was observed with the system previously reported by us.47 The deposition likely occurs by a redox mechanism with the emeraldine form of PANI that leaves palladium in the metallic state. However, X-ray photoelectron spectroscopy (XPS) showed that palladium is in the +2 oxidation state with Pd 3d5/2 at 337 eV and Pd 3d3/2 at 342 eV (Fig. 3).70 It is possible that a redox reaction occurs to form Pd(0) nanoparticles and since the pernigraniline form of PANI is unstable at room temperature, it is reduced over time causing the palladium nanoparticles to be oxidized. To determine if palladium aggregation occurs over time, the composite was used over several months. TEM showed that nanoparticles remained the same size (Fig. S3†) and the activity was constant.
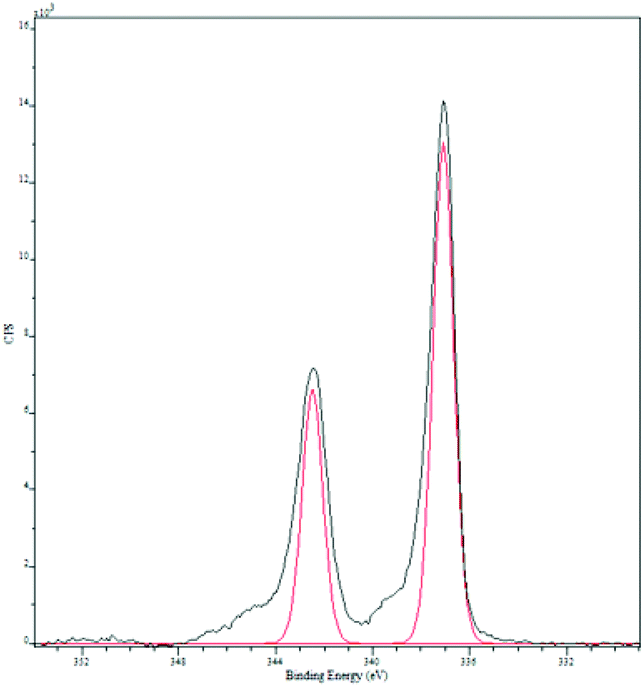 | ||
| Fig. 3 X-ray photoelectron spectroscopy of palladium nanoparticles supported on PANI nanofibers. The red trace is the standard for Pd(II).70 | ||
C–C bond formation reactions
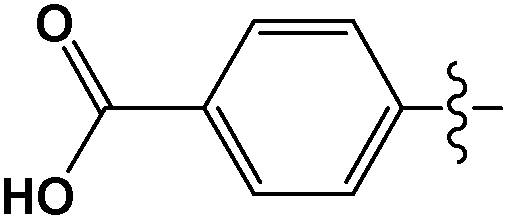
Cross-coupling reactions with aryl chlorides in air (a test reaction under N2 gave similar results) resulted in high conversion values (Table 1). Most reactions were completed within 4–6 h at 100 °C. Aryl chlorides with electron withdrawing groups (Table 1, entries 1–3) provided the highest conversions. 4-Chloroanisole and other reactions with electron donating groups (Table 1, entries 4–5) also provided significant conversion values. The products were isolated by a standard workup procedure. The isolated yields were much lower than what would be expected based on conversion values (Table 1). It is possible that some of the product is trapped by the polyaniline support; various workup conditions did not have a large influence on the isolated yield and no by-products were identified with the exception of 4-chlorobenzaldehyde (26% of the homo-coupled product as identified by 1H NMR spectroscopy). The trapping of the product might be responsible for the decrease in activity observed in subsequent runs (see below). It is noteworthy that the results obtained with the present palladium/PANI nanocomposites differ somewhat from those reported by us previously.47 As mentioned above, the identity of the palladium nanoparticles is not the same: we consistently obtain a unimodal distribution in the present systems although we observed two types of palladium nanoparticles previously. After numerous synthetic attempts, we concluded that the batch of palladium nitrate originally used must have contained some impurity that determined the bimodal distribution of the resulting nanoparticles. All other batches of palladium nitrate, from the same and other vendors, give a unimodal distribution of the resulting nanoparticles.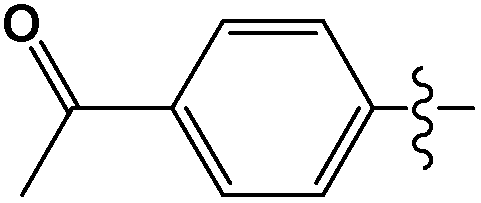


Mechanistic studies
The substrate 4-chloroacetophenone was used to test the recyclability of the catalyst. The nanocomposite was used up to four times; a significant decrease in yield was observed after the second run (Table 2). Leaching of palladium in solution increased slightly after each reaction, but was low compared to the total palladium content (Table 2). It is important to note that although it has been discussed that supporting palladium in the +2 oxidation state renders the system more prone to leaching,71–73 this is not the case in our studies. A redox interplay between PANI and palladium might be responsible for these observations similar to how redox-active ligands cooperate with organometallic complexes.74,75 On the other hand, the role of PANI as a good ligand (basic/nucleophilic properties) for palladium(II), without invoking its redox activity, cannot be discarded.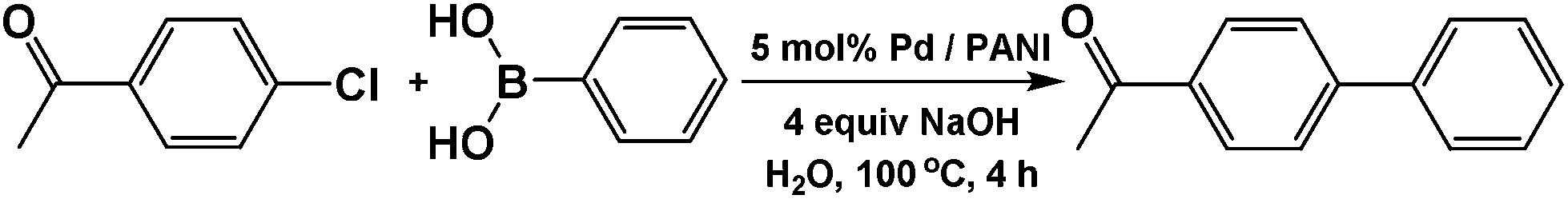
Palladium nanoparticles showed an increase in size to 5–10 nm after the fourth reaction (Fig. 4). Results from XPS studies show that the composite starts with Pd(II) and after 30 min is still Pd(II) (Fig. S7†), meaning that within the first 30 min of the reaction, palladium gets reduced and is reoxidized by the aryl halide substrate. However, after the first reaction was completed, the composite showed palladium in both +2 and 0 oxidation states (Fig. S8†).70 This result was similar to that obtained after the fourth run (Fig. 5). These data indicate that once palladium(0) forms on the surface of polyaniline nanofibers, it cannot be brought into solution by reactants. This interpretation indicates that leaching of palladium(II) in solution has to occur before its reduction, therefore, favouring mechanism 2 discussed in the introduction.
 | ||
| Fig. 4 TEM of the palladium/PANI pre-catalyst after four consecutive runs. | ||
 | ||
| Fig. 5 X-ray photoelectron spectroscopy of palladium nanoparticles supported on PANI nanofibers after four consecutive runs. The red trace is the standard for Pd(II) and the blue trace is the standard for Pd(0).70 | ||
A filtration and three-phase test60,76 were used to determine if the catalyst truly operates in a heterogeneous manner. Separation of the catalyst from the reaction mixture was achieved by filtering the solution through a 2 cm pad of Celite on a medium frit while still hot. After 30 min into the reaction, the solution was filtered and used with new reactants. No product formation was observed after running this new reaction for the usual four hour time. This suggests that palladium is acting heterogeneously. However, experiments reported by Lipshutz77 showed that it is possible that the metal deposits back on the support in the short time period required to filter the reaction. Indeed, TEM showed that some of the fibers filtered through Celite, therefore, a three-phase test was then applied.
A solid-bound reactant was formed by combining 4-bromobenzoyl bromide and aminopropyl bound on silica.78 A three-phase test would show whether palladium acts as a heterogeneous or homogenous catalyst. If the reaction with the solid-bound reactant is inactive, then oxidative addition occurs on the surface of palladium nanoparticles deposited on PANI nanofibers, consistent with a heterogeneous mechanism. If reactivity is detected in the presence of functionalized silica, then palladium leaches into solution and then undergoes oxidative addition. As a control, no reaction was observed when the reaction was performed in the presence of the solid-supported substrate without the Pd/PANI catalyst. In the presence of Pd/PANI, 100% conversion of the solid-bound reactant was determined by 1H NMR spectroscopy. This test shows that the present system is homogeneous during catalysis and that palladium leaches. Once palladium leaches into solution, it is reduced to the active form that likely performs catalysis through the accepted Suzuki mechanism (Fig. 6). This proposal is consistent with the results from XPS studies that leaching of palladium(II) in solution has to occur before its reduction.
 | ||
| Fig. 6 Proposed mechanism of the Suzuki reaction of aryl chlorides catalyzed by palladium nanoparticles supported on PANI nanofibers. | ||
Conclusions
Palladium-PANI composites represent unique catalysts for C–C cross-coupling reactions since PANI nanofibers can provide multiple functions such as support, ligand, and redox agent. Using regular techniques, such as scanning electron microscopy (SEM), transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS) in conjunction with reactivity studies, we could only determine with certainty that a homogeneous mechanism is operating. The main information was obtained by imaging the catalyst system at various stages during the reaction and recycling attempts. Although there was a small amount of palladium lost through leaching, deactivation was observed and was attributed to Ostwald ripening. In addition, the appearance of palladium nanoparticles and polyaniline nanofibers changes after each reaction accompanied by changes in the palladium oxidation state. Our methods could not rule out the contribution of a heterogeneous mechanism or indicate its extent. XPS studies of aliquots taken from recycling experiments show that the composite starts with Pd(II) and after 30 min is still Pd(II), meaning that within the first 30 min of the reaction, palladium gets reduced and is reoxidized by the aryl halide substrate. However, after the first reaction was completed, the composite showed palladium in both +2 and 0 oxidation states, a situation that persists even after the fourth run. These data indicated that once palladium(0) forms on the surface of polyaniline nanofibers, it could not be brought into solution by reactants. Considering all the data discussed herein, our mechanistic proposal is most consistent with mechanism 2 discussed in the introduction, i.e. leaching of palladium(II) in solution has to occur before its reduction.Experimental section
General considerations
All experiments were performed under air unless otherwise specified. Palladium(II) nitrate was purchased from Alfa Aesar and kept in a glove box. Functionalized silica was purchased from Fisher and used as received. Aniline was purchased from Sigma Aldrich and distilled before use. Camphor sulfonic acid (CSA) was purchased from Sigma Aldrich and used as received. Ammonium peroxydisulfate was purchased from Alfa Aesar and used as received. The functionalized silica substrate used for the three-phase test was prepared following a reported procedure.781H NMR spectra were recorded on a Bruker300 in CDCl3 as the solvent. The Pd/PANI solution was diluted 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and then drop-cast onto a holy carbon grid for TEM analysis (T12 Quick CryoEM and CryoET (FEI) located at the Electron Imaging Center for NanoMachines (EICN) in the California NanoSystems Institute (CNSI)). Samples were digested with HNO3 and then diluted for ICP-OES (TJA Radial IRIS 1000 ICP-OES). Samples for XPS (Kratos XPS Axis Ultra DLD located at Materials Lab at The Molecular Instrumentation Center (MIC)) were concentrated solutions that were drop-cast onto silica wafers.
:1 and then drop-cast onto a holy carbon grid for TEM analysis (T12 Quick CryoEM and CryoET (FEI) located at the Electron Imaging Center for NanoMachines (EICN) in the California NanoSystems Institute (CNSI)). Samples were digested with HNO3 and then diluted for ICP-OES (TJA Radial IRIS 1000 ICP-OES). Samples for XPS (Kratos XPS Axis Ultra DLD located at Materials Lab at The Molecular Instrumentation Center (MIC)) were concentrated solutions that were drop-cast onto silica wafers.
Polyaniline nanofibers
Preparation of PANI nanofibers was carried out using the rapid-mixing method.32,33 A 4:1 molar ratio of aniline to ammonium peroxydisulfate was used (2.981 g of distilled aniline diluted to 100 mL with a 1 M camphor sulfonic acid, CSA). 1.827 g of ammonium persulfate was dissolved in 100 mL of 1 M CSA. After mixing, 200 mL of PANI solution was obtained. The two solutions were combined and mixed for 30 s. The mixture was left on a bench top for 24 h for the polymerization to take place. The fibers were washed and dedoped by centrifugation with H2O, 2 × 0.1 M ammonium hydroxide, and 2 × H2O. The fibers were then diluted to a concentration of 2 g L−1.
Palladium-PANI nanofibers composite
A 10 mM solution of palladium(II) nitrate was mixed in a 1:4 molar ratio with the PANI solution. The solution was shaken for 2 min and left to sit for 24 h. The solution was purified by dialysis for 2 days.
Several different methods were employed in order to synthesize more active palladium nanoparticles than those described above. Hydrochloric acid was used to dope polyaniline instead of camphorsulfonic acid to make small nanofibers. The system gave a similar conversion to the CSA/polyaniline system described above. A longer incubation time of palladium and polyaniline also gave comparable results. Other methods, such as stirring palladium with polyaniline or adding palladium before the polymerization of aniline led to nanocomposites with lower conversions.
General reaction
4-Chloro substituted reactant (1 equiv.), 0.141 g phenylboronic acid (1.5 equiv.), and 0.104 g sodium hydroxide (4 equiv.) were mixed in a round bottom flask with 20 mL of water. The flask was heated to 100 °C, then 6 mL of Pd/PANI was added (5 mol%). The reaction proceeded for 4–6 h and then was cooled to room temperature. The mixture was acidified for the carboxylic reactant by adding HCl (20% v/v). The product was extracted with 3 × 10 mL of ethyl acetate and washed with 3 × 7 mL of water, then it was dried with MgSO4 and the solvent was removed using a rotovap. The obtained product was a whitish-brown solid. Conversion was measured by comparing peaks of reactants versus product in 1H NMR spectra. Yield was determined by weighing out dry product.Recycling procedure
After extraction, the water layers were combined. They were heated in a round-bottom flask at 50 °C to boil off volatiles. Once cooled down, the mixture was centrifuged for 30 min at 4500 rpm. The solution was then decanted by pipette and washed with water. It was centrifuged for another 30 min and decanted again to 10 mL. It was then added to the next reaction the following day.Three-phase test: formation of bromide functionalized silica78
1.150 g (5.25 mmol) of 4-bromobenzoyl chloride was dissolved in 10 mL of dry THF. The solution was then added to a flask with 1 g of silica functionalized with an aminopropyl group. 0.4 mL (5 mmol) of pyridine was then added to the flask. The solution was stirred for 12 h under nitrogen. The silica was then purified by centrifugation with 3 × HCl, 2 × K2CO3, 2 × water, and 2 × ethanol. It was filtered and left to dry in air overnight. The functionalized silica was analyzed by elemental analysis and it was found that 1.1 mmol of substrate per g of silica was appended.
Three-phase test: Suzuki coupling reaction
0.250 g bromide functionalized silica, 0.099 g phenylboronic acid (1.5 equiv.), and 0.298 g potassium carbonate (4 equiv.) were mixed in a round-bottom flask with 10 mL of water. To this mixture, 4.2 mL Pd/PANI was added (5 mol%) and the flask was inserted into an oil bath at 100 °C. The reaction proceeded for 6 h and was cooled to room temperature. Once cooled, the mixture was filtered through an empty medium frit. A Soxhlet extraction with ethanol of the solid was performed. After that, the solid was hydrolyzed with 2 M KOH at 100 °C for 3 days. The resulting mixture was filtered and the filtrate was acidified with HCl (20% v/v). The solution was then extracted with ethyl acetate (3 × 10 mL), the filtrate dried with MgSO4, and the volatiles were removed with a rotovap. A whitish-brown solid remained. The product was analyzed by 1H NMR spectroscopy and conversion shows that only product remains.Acknowledgements
This research was supported by the National Science Foundation (CAREER Grant 0847735 to PLD and CHE-1048804 for NMR spectroscopy). We thank the California NanoSystems Institute (CNSI) for the use of a T12 TEM instrument, Jessica Wang for assistance in TEM imaging, and Brian McVerry for assistance with polyaniline nanofiber synthesis.Notes and references
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. Suzuki, Chem. Commun., 2005, 4759–4763 RSC.
- R. F. Heck, in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon, New York, 1991, vol. 4 Search PubMed.
- R. F. Heck, Org. React., 1982, 27, 345–390 CAS.
- T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581–581 CrossRef CAS.
- R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- K. Sonogashira, in Metal-Catalyzed Cross-Coupling Reactions, eds. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998, p. ch. 4 Search PubMed.
- K. Sonogashira, in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon, New York, 1991, vol. 3, p. chapter 2.4 Search PubMed.
- M. Tamura and J. K. Kochi, J. Am. Chem. Soc., 1971, 93, 1487–1489 CrossRef CAS.
- K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374–4376 CrossRef CAS.
- R. J. P. Corriu and J. P. Masse, J. Chem. Soc., Chem. Commun., 1972, 144a–144a, 10.1039/C3972000144A.
- M. Yamamura, I. Moritani and S.-I. Murahashi, J. Organomet. Chem., 1975, 91, C39–C42 CrossRef CAS.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- S. Klaus, H. Neumann, A. Zapf, D. Strübing, S. Hübner, J. Almena, T. Riermeier, P. Groß, M. Sarich, W.-R. Krahnert, K. Rossen and M. Beller, Angew. Chem., Int. Ed., 2006, 45, 154–158 CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, J. Organomet. Chem., 2004, 689, 4055–4082 CrossRef CAS PubMed.
- E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CrossRef CAS PubMed.
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed.
- V. P. W. Böhm, C. W. K. Gstöttmayr, T. Weskamp and W. A. Herrmann, J. Organomet. Chem., 2000, 595, 186–190 CrossRef.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- A. Ohtaka, Chem. Rec., 2013, 13, 274–285 CrossRef CAS PubMed.
- C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494–503 CrossRef CAS PubMed.
- D. Astruc, Inorg. Chem., 2007, 46, 1884–1894 CrossRef CAS PubMed.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mülhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS PubMed.
- A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, J. Catal., 2011, 279, 1–11 CrossRef CAS PubMed.
- C. B. Putta and S. Ghosh, Adv. Synth. Catal., 2011, 353, 1889–1896 CrossRef CAS.
- R. B. Bedford, U. G. Singh, R. I. Walton, R. T. Williams and S. A. Davis, Chem. Mater., 2005, 17, 701–707 CrossRef CAS.
- K.-i. Shimizu, S. Koizumi, T. Hatamachi, H. Yoshida, S. Komai, T. Kodama and Y. Kitayama, J. Catal., 2004, 228, 141–151 CrossRef CAS PubMed.
- S. Navalón, M. Álvaro and H. García, ChemCatChem, 2013, 5, 3460–3480 CrossRef.
- K. Bester, A. Bukowska and W. Bukowski, Appl. Catal., A, 2012, 443–444, 181–190 CrossRef CAS PubMed.
- J. Huang and R. B. Kaner, J. Am. Chem. Soc., 2004, 126, 851–855 CrossRef CAS PubMed.
- J. Huang and R. B. Kaner, Chem. Commun., 2006, 367–376 RSC.
- D. Li and R. B. Kaner, Chem. Commun., 2005, 3286–3288 RSC.
- D. Li, J. Huang and R. B. Kaner, Acc. Chem. Res., 2008, 42, 135–145 CrossRef PubMed.
- A. Z. Sadek, W. Wlodarski, K. Shin, R. B. Kaner and K. Kalantar-zadeh, Nanotechnology, 2006, 17, 4488–4492 CrossRef CAS.
- S. Virji, R. B. Kaner and B. H. Weiller, J. Phys. Chem. B, 2006, 110, 22266–22270 CrossRef CAS PubMed.
- S. Virji, J. D. Fowler, C. O. Baker, J. Huang, R. B. Kaner and B. H. Weiller, Small, 2005, 1, 624–627 CrossRef CAS PubMed.
- S. Virji, R. B. Kaner and B. H. Weiller, Chem. Mater., 2005, 17, 1256–1260 CrossRef CAS.
- J. Huang, S. Virji, B. H. Weiller and R. B. Kaner, Chem. – Eur. J., 2004, 10, 1314–1319 CrossRef CAS PubMed.
- S. Virji, J. Huang, R. B. Kaner and B. H. Weiller, Nano Lett., 2004, 4, 491–496 CrossRef CAS.
- J. Huang, S. Virji, B. H. Weiller and R. B. Kaner, J. Am. Chem. Soc., 2003, 125, 314–315 CrossRef CAS PubMed.
- B. T. McVerry, J. A. T. Temple, X. Huang, K. L. Marsh, E. M. V. Hoek and R. B. Kaner, Chem. Mater., 2013, 25, 3597–3602 CrossRef CAS.
- Y. Liao, X.-G. Li, E. M. V. Hoek and R. B. Kaner, J. Mater. Chem., 2013, 1, 15390–15396 RSC.
- Y. Liao, C. Zhang, X. Wang, X.-G. Li, S. J. Ippolito, K. Kalantar-zadeh and R. B. Kaner, J. Phys. Chem. C, 2011, 115, 16187–16192 CAS.
- E. Spain, R. Kojima, R. B. Kaner, G. G. Wallace, J. O'Grady, K. Lacey, T. Barry, T. E. Keyes and R. J. Forster, Biosens. Bioelectron., 2011, 26, 2613–2618 CrossRef CAS PubMed.
- B. J. Gallon, R. W. Kojima, R. B. Kaner and P. L. Diaconescu, Angew. Chem., Int. Ed., 2007, 46, 7251–7254 CrossRef CAS PubMed.
- B. M. Choudary, M. Roy, S. Roy, M. L. Kantam, B. Sreedhar and K. V. Kumar, Adv. Synth. Catal., 2006, 348, 1734–1742 CrossRef CAS.
- M. L. Kantam, M. Roy, S. Roy, B. Sreedhar, S. S. Madhavendra, B. M. Choudary and R. L. De, Tetrahedron, 2007, 63, 8002–8009 CrossRef CAS PubMed.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- A. F. Schmidt and A. A. Kurokhtina, Kinet. Catal., 2012, 53, 714–730 CrossRef CAS.
- F. Alonso, I. P. Beletskaya and M. Yus, Tetrahedron, 2005, 61, 11771–11835 CrossRef CAS PubMed.
- J. G. De Vries, Dalton Trans., 2006, 421–429 RSC.
- M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 1559–1563 RSC.
- C. C. Cassol, A. P. Umpierre, G. Machado, S. I. Wolke and J. Dupont, J. Am. Chem. Soc., 2005, 127, 3298–3299 CrossRef CAS PubMed.
- M. B. Thathagar, J. E. t. Elshof and G. Rothenberg, Angew. Chem., Int. Ed., 2006, 45, 2886–2890 CrossRef CAS PubMed.
- M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165–168 CrossRef CAS.
- A. Biffis, M. Zecca and M. Basato, J. Mol. Catal. A: Chem., 2001, 173, 249–274 CrossRef CAS.
- A. Biffis, M. Zecca and M. Basato, Eur. J. Inorg. Chem., 2001, 2001, 1131–1133 CrossRef.
- I. W. Davies, L. Matty, D. L. Hughes and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 10139–10140 CrossRef CAS.
- F. Zhao, M. Shirai, Y. Ikushima and M. Arai, J. Mol. Catal. A: Chem., 2002, 180, 211–219 CrossRef CAS.
- M. B. Thathagar, P. J. Kooyman, R. Boerleider, E. Jansen, C. J. Elsevier and G. Rothenberg, Adv. Synth. Catal., 2005, 347, 1965–1968 CrossRef CAS.
- A. F. Schmidt, A. A. Kurokhtina and E. V. Larina, Kinet. Catal., 2012, 53, 84–90 CrossRef CAS.
- A. F. Lee, P. J. Ellis, I. J. S. Fairlamb and K. Wilson, Dalton Trans., 2010, 39, 10473–10482 RSC.
- P. J. Ellis, I. J. S. Fairlamb, S. F. J. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820–1824 CrossRef CAS PubMed.
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, Inc., Hoboken, New Jersey, 2005 Search PubMed.
- A. A. Kurokhtina, E. V. Larina, A. F. Schmidt, A. Malaika, B. Krzyżyńska, P. Rechnia and M. Kozłowski, J. Mol. Catal. A: Chem., 2013, 379, 327–332 CrossRef CAS PubMed.
- R. Narayanan, C. Tabor and M. El-Sayed, Top. Catal., 2008, 48, 60–74 CrossRef CAS.
- M. Trilla, R. Pleixats, M. W. C. Man, C. Bied and J. J. E. Moreau, Adv. Synth. Catal., 2008, 350, 577–590 CrossRef CAS.
- J. Wang, K. G. Neoh and E. T. Kang, J. Colloid Interface Sci., 2001, 239, 78–86 CrossRef CAS PubMed.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed.
- M. Dams, L. Drijkoningen, D. De Vos and P. Jacobs, Chem. Commun., 2002, 1062–1063, 10.1039/B201180H.
- K. Okumura, K. Nota, K. Yoshida and M. Niwa, J. Catal., 2005, 231, 245–253 CrossRef CAS PubMed.
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794–795 CrossRef CAS PubMed.
- J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 2012, 363–375 CrossRef CAS.
- J. Rebek, Tetrahedron, 1979, 35, 723–731 CrossRef CAS.
- S. Tasler and B. H. Lipshutz, J. Org. Chem., 2003, 68, 1190–1199 CrossRef CAS PubMed.
- C. Baleizão, A. Corma, H. García and A. Leyva, J. Org. Chem., 2003, 69, 439–446 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: EDS, SEM, TEM, and XPS images and NMR spectra. See DOI: 10.1039/c4qi00130c |
| This journal is © the Partner Organisations 2015 |