
Role of 4,4′-bipyridine versus longer spacers 4,4′-azobipyridine, 1,2-bis(4-pyridyl)ethylene, and 1,2-bis(pyridin-3-ylmethylene)hydrazine in the formation of thermally labile metallophosphate coordination polymers†
Palanisamy
Rajakannu‡
a,
Rana
Howlader‡
a,
Alok Ch.
Kalita
 a,
Raymond J.
Butcher
b and
Ramaswamy
Murugavel
*a
a,
Raymond J.
Butcher
b and
Ramaswamy
Murugavel
*a
aDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400 076, India. E-mail: rmv@chem.iitb.ac.in; Fax: +91 22 2576 7152; Tel: +91 22 2576 7163
bDepartment of Chemistry, Howard University, Washington, DC 20059, USA
First published on 21st November 2014
Abstract
One dimensional metallophosphate coordination polymers {[M(dtbp)2(azopy)(H2O)2]·(azopy)}x (M = Mn (1); Co (2); Cu (3); Cd (4)) have been synthesized from the reaction of a suitable metal precursor with di-tert-butylphosphate (dtbp-H) in the presence of ditopic linker 4,4′-azobipyridine (azopy) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2 stoichiometric ratio. Isostructural compounds 1–4 have been characterized by analytical and spectroscopic methods and single crystal X-ray diffraction studies. Single crystal X-ray diffraction measurements further reveal that compounds 1–3 (all C2/c) and 4 (P
:2:2 stoichiometric ratio. Isostructural compounds 1–4 have been characterized by analytical and spectroscopic methods and single crystal X-ray diffraction studies. Single crystal X-ray diffraction measurements further reveal that compounds 1–3 (all C2/c) and 4 (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) are linear 1D coordination polymers. The uncoordinated 4,4′-azobipyridine in the lattice is responsible for the conversion of these 1D coordination polymers into 3D supramolecular assemblies through O–H⋯N hydrogen bonding interactions between coordinated water and N-centers of an azopy ligand. Similar reactions carried out using 1,2-bis(4-pyridyl)ethylene (bpe) as the linker yielded compounds having the formula {[M(dtbp)2(bpe)(H2O)2]·(bpe)}x (M = Mn (5); Co (6); Cu (7); Ni (8)). Compounds 5–8 have been characterized by analytical and spectroscopic methods. Preliminary single crystal X-ray diffraction studies carried out on poorly diffracting crystals of 5 and 6 establish their isostructural nature to 1–4, also displaying a similar supramolecular aggregation behaviour. A longer ditopic N,N′-donor ligand, 1,2-bis(pyridin-3-ylmethylene)hydrazine (bph), has been used in place of bpe/azoby to synthesize 1-dimensional coordination polymers [{M(bph)(H2O)4}{(dtbp)2}] (M = Ni (9) and M = Co (10)) and [Cd(bph)3(dtbp)2]n (11) which have completely different structural motifs when compared to 1–4.
) are linear 1D coordination polymers. The uncoordinated 4,4′-azobipyridine in the lattice is responsible for the conversion of these 1D coordination polymers into 3D supramolecular assemblies through O–H⋯N hydrogen bonding interactions between coordinated water and N-centers of an azopy ligand. Similar reactions carried out using 1,2-bis(4-pyridyl)ethylene (bpe) as the linker yielded compounds having the formula {[M(dtbp)2(bpe)(H2O)2]·(bpe)}x (M = Mn (5); Co (6); Cu (7); Ni (8)). Compounds 5–8 have been characterized by analytical and spectroscopic methods. Preliminary single crystal X-ray diffraction studies carried out on poorly diffracting crystals of 5 and 6 establish their isostructural nature to 1–4, also displaying a similar supramolecular aggregation behaviour. A longer ditopic N,N′-donor ligand, 1,2-bis(pyridin-3-ylmethylene)hydrazine (bph), has been used in place of bpe/azoby to synthesize 1-dimensional coordination polymers [{M(bph)(H2O)4}{(dtbp)2}] (M = Ni (9) and M = Co (10)) and [Cd(bph)3(dtbp)2]n (11) which have completely different structural motifs when compared to 1–4.
Introduction
The growing interest in supramolecular chemistry of transition-metal complexes and coordination polymers (CPs) exhibiting different types of networks or cage topologies can be traced to potential applications of these materials arising out of their solvent-inclusion, gas-adsorption, electronic, or nonlinear optical properties.1,2 While the majority of CPs and metal–organic frameworks (MOFs) are based on the coordination of multitopic carboxylate ligands with transition metal ions,3 recent research has established the importance of both phosphonic acids4,5 and phosphate esters4 as building blocks for rationally designed CPs and MOFs. Metal phosphates have in particular been studied in much detail as they appear in nature as minerals or as constituents of living animals and play an important role in energy conversion cycles, apart from being similar to silicates from a structural point of view.6 The other reasons which have made metallophosphate chemistry very relevant in recent times are their usefulness as models for secondary building units (SBUs) of zeolites7 and their ability to function as single-source precursors for the preparation of ceramic materials,8 apart from the facile structural transformations metal phosphates undergo under mild conditions.9 Our earlier studies, which were initially directed towards use of di-tert-butylphosphate derived one-dimensional coordination polymers [M(dtbp)2]n as ceramic precursors for M(PO3)2,8 have been later extended to studies on structural transformations effecting facile dimensionality changes at room temperature. For example, [M(dtbp)2]n (M = Mn, Co, Cu, Cd) can be readily transformed to two-dimensional grid structures by the addition of 4,4-bipyridine at room temperature as schematically depicted in Scheme 1.9 Subsequently, a variety of supra-molecular structures and coordination polymers exhibiting different types of network topologies has been prepared in our laboratory to understand the formation of these superstructures and the dimensionality changes.10,11 In order to probe whether other types of CPs can be formed by the dtbp ligand in the presence of other ditopic spacers, we have employed in the present study not only longer but also functional ditopic N-donor spacers 4,4′-azobipyridine (azopy), 1,2-bis(4-pyridyl)ethylene (bpe) and 1,2-bis(pyridin-3-ylmethylene)hydrazine (bph) to prepare newer metallophosphate coordination polymers exhibiting completely different supramolecular aggregation compared to those observed for 4,4′-bipyridine.9 | ||
| Scheme 1 One-pot (path A) and stepwise (path B) syntheses of two-dimensional 4,4′-bipyridine derived metallophosphate polymers [M(dtbp)2(bpy)2]x exhibiting a grid architecture. | ||
Results and discussion
Synthesis and characterization of coordination polymers 1–4 using 4,4′-azobipyridine
Metallophosphates of the formula {[M(dtbp)2(azopy)(H2O)2]·(azopy)}x (M = Mn (1); Co (2); Cu (3); Cd (4)) have been synthesized by the reaction of metal acetates or perchlorates with dtbp-H and azopy in methanol and DMF (Scheme 2). Alternatively the compounds can also be synthesized from the dtbp polymers [M(dtbp)2]n by mixing azopy at room temperature. The products, which are air and moisture stable and soluble in polar organic solvents, have been characterized by elemental analysis, FT-IR, UV-Vis, NMR and EPR spectroscopy and TG-DTA analysis. | ||
| Scheme 2 Synthesis of metallophosphate polymers 1–8. | ||
Elemental analysis values obtained for 1–4 are consistent with the formula {[M(dtbp)2(azopy)(H2O)2](azopy)}, which has later been verified by single crystal X-ray diffraction studies (vide infra). The FT-IR spectrum of 1 shows characteristic absorptions at 2977, 1184 and 1069 cm−1 corresponding to the νasy(C–H), ν(P–O–O) and νM–O–P vibrations. The presence of a broad νO–H absorption at 3388 cm−1 is indicative of the presence of water either in the coordination sphere of the metal or in the lattice. The FT-IR spectra of 2–4 are similar to that of 1 (Fig. S9–S12†). While compounds 1–3 are paramagnetic, the diamagnetic 4 yields a single resonance in the 31P NMR spectrum at −5.87 ppm indicating the presence of only one type of phosphorus in the complex (Fig. 1). The 1H NMR spectrum of 4 shows well separated peaks for the protons of azopy and dtbp ligands (Fig. 2). Two doublets appearing at 7.91 and 8.85 ppm correspond to the C–H protons of the azopy ligand. The tert-butyl protons of dtbp appear as a singlet at 1.38 ppm.
 | ||
| Fig. 1 31P NMR spectrum of 4 in CD3OD. | ||
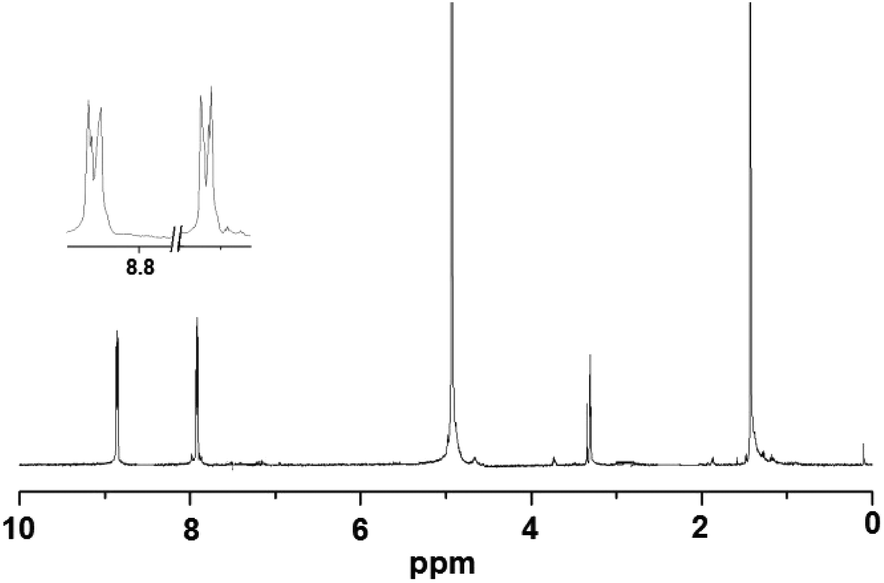 | ||
| Fig. 2 1H NMR spectrum of 4 in CD3OD. | ||
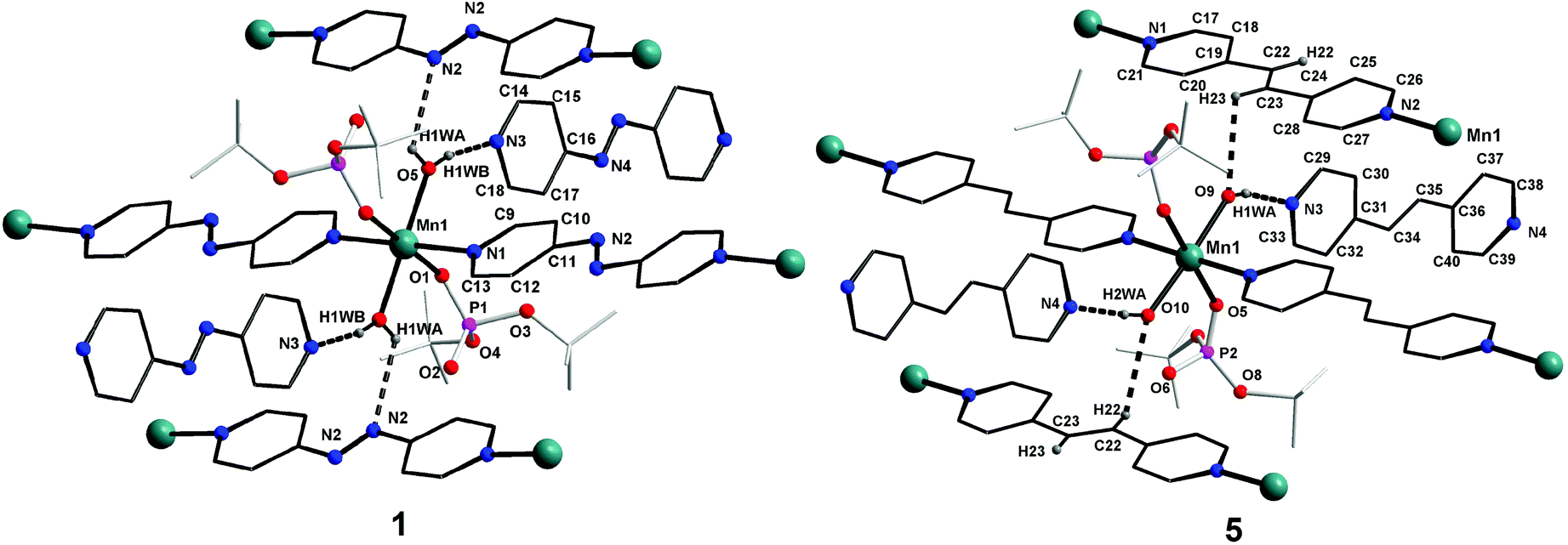
space group, is also isostructural to compounds 1–3. The coordination environment around the central metal ion for compounds 1–4 is shown in Fig. 3. Selected structural parameters for compounds 1–4 are listed in Table 1. The hydrogen bond parameters for 1–4 are given in Table S1.† Due to the close resemblance of structures of 1–4, a detailed description of the structure of compound 1 alone is presented here. The ESI† contains other details. The asymmetric part of the unit cell in 1 contains one manganese ion, two metal bound terminal unidentate dtbp ligands, two coordinated water molecules and two azopy ligands (of which only one is coordinated to the metal and the other remains uncoordinated). Both the azopy ligands are found in trans configuration in all the four crystal structures.
 | ||
| Fig. 3 Repeating units in the molecular structure of compounds 1–4. | ||
| Parameters | 1 (M = Mn) | 2 (M = Co) | 3 (M = Cu) | 4 (M = Cd) | 5 (M = Mn) |
|---|---|---|---|---|---|
| M–N/Å | 2.313(2) | 2.078(4) | 2.090(2) | 2.3617(18) | 2.239(12), 2.297(11) |
| M–O (P)/Å | 2.1136(18) | 1.938(3) | 1.931(2) | 2.2504(15) | 2.136(8), 2.143(6) |
| M–OH2/Å | 2.1882(19) | 2.406(4) | 2.406(2) | 2.2931(15) | 2.167(6), 2.186(7) |
| H2O–M–OH2/° | 180.0 | 180.0 | 180.0 | 180.0 | 177.0(3) |
| PO–M–OP/° | 180.0 | 180.0 | 180.0 | 180.0 | 178.6(4) |
| N–M–N/° | 180.0 | 180.0 | 180.0 | 180.0 | 179.2(4) |
| O–M–N/° | 86.95(7) | 92.21(14) | 87.70(9) | 85.43(6) | 91.0(4), 87.7(3) |
| 93.05(7) | 87.79(14) | 92.30(9) | 94.57(6) | 90.3(4), 91.9(4) | |
| 90.29(7) | 90.78(13) | 90.81(9) | 91.33(6) | 88.8(4), 92.5(3) | |
| 89.71(7) | 89.22(13) | 89.19(9) | 88.67(6) | 90.5(3), 87.3(4) | |
| H2O–M–OP/° | 90.29(7) | 88.15(14) | 91.86(9) | 91.69(6) | 89.2(3), 90.5(2) |
| 89.71(7) | 91.85(14) | 88.14(9) | 88.31(6) | 92.8(3), 87.6(3) |
The central manganese adopts a nearly ideal octahedral geometry as shown in Fig. 3 as can been seen from the bond angles listed in Table 1. The M–N, M–OH2, and M–OP distances found for compounds 1–4 are very similar to those observed for other metal phosphate complexes that have a similar set of ligands, especially those with the grid framework structures formed by the dtbp ligand.7 It is of interest to examine in detail the formation of the final framework in the structure of 1. For a better understanding of the framework formation, the various interactions present within the crystal are hierarchically and pictorially depicted in Fig. 4, making use of the final atomic coordinates obtained for the fully refined structure of 1. As Fig. 4a reveals, two terminal dtbp ligands and two water molecules, in an all-trans configuration, occupy four of the total six coordination sites around the metal. The remaining two coordination sites are occupied by the nitrogen terminals of two different azopy ligands in a trans fashion, eventually leading to the formation of a linear 1D polymer as shown in Fig. 4b (the azopy ligands coordinately linking the metals are shown by green rods) (cf. also Fig. S1–S3†). It is obvious from Fig. 4b that several such polymers run parallel to each other, with every adjacent polymeric chain displaced from the first by half the length of the azopy ligand. The next step in the structure evolution of 1–4 is the interaction of water molecules on the metal with the azo (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) fragment of the coordinated azopy ligand through O–H⋯N hydrogen bonding (pink linker in Fig. 4c; O5⋯N2 of 2.981 Å), leading to the formation of a tightly woven 2D sheet (Fig. 4c). Thus the coordinated azobpy ligand uses all its four nitrogen centers to form the 2D grid structure (Fig. 4 and 5).
N) fragment of the coordinated azopy ligand through O–H⋯N hydrogen bonding (pink linker in Fig. 4c; O5⋯N2 of 2.981 Å), leading to the formation of a tightly woven 2D sheet (Fig. 4c). Thus the coordinated azobpy ligand uses all its four nitrogen centers to form the 2D grid structure (Fig. 4 and 5).
 | ||
| Fig. 4 Evolution of a 3D framework structure in compounds 1–4 through coordination and hydrogen bonding interactions. | ||
 | ||
| Fig. 5 The role of the coordinated azopy ligand in the crystal lattice: two-fold coordination and two-fold H-bond formation. | ||
In the final step, the second molecule of the azopy molecule present in the asymmetric part of the unit cell plays a significant role by engaging itself in hydrogen bonding interactions between the 2D grid sheets (purple rod) as shown in Fig. 4d. This interaction between the pyridinic nitrogen of the uncoordinated azopy and the coordinated water is extremely strong (N(3)⋯H(1WB)–O(5): 2.979 Å, 168.5°), thereby forming a three-dimensional robust framework structure. In addition to the above-described H-bonding interactions that lead to framework building, there are additional intramolecular H-bonding interactions involving phosphate oxygen atoms of dtbp with water hydrogen atoms (not depicted in Fig. 3 or 4) (see also ESI†).
Synthesis and characterization of coordination polymers 5–8 using 1,2-bis(4-pyridyl)ethylene (bpe) spacer
In terms of end-to-end (N to N) distance, both azopy and bpe ligands are essentially similar. However, it can be anticipated that the replacement of the –NN– central unit by a –CHCH– unit would bring about significant changes in the hydrogen bonding pattern and hence a different kind of supramolecular aggregation. The reaction of transition metal acetates with dtbp-H was carried out in the presence of ditopic bpe in a 1:2:2 ratio of the reactants in methanol at room temperature. The products obtained analyzed as {[M(dtbp)2(bpe)(H2O)2]·(bpe)}x (M = Mn (5); Co (6); Cu (7); Ni (8)) for the four metals used in this reaction are similar to 1–4 in composition. Compounds 5–8, which are air and moisture stable and are soluble in polar organic solvents, have been characterized by elemental analysis, FT-IR, TG-DTA analysis, UV-Vis and EPR spectroscopic methods. The IR spectra of 5–8 show broad absorption in the range 3214–3376 cm−1 assignable to νO-H of the coordinated water molecules. Strong absorption bands observed around 1184 and 1071 cm−1 are characteristic for ν(P–O–O) and νM–O–P vibrations (Fig. S13–S16†).
 | ||
| Fig. 6 Differences in the structures and hydrogen-bonding patterns of 1 and 5. | ||
 | ||
| Fig. 7 Formation of 2D grid structures by orthogonally bound 4,4′-bipyridine ligands in [M(dtbp)2(4,4′-bpy)2]n. | ||
The structure evolution in metal-dtbp-4,4′-bpy complexes [M(dtbp)2(4,4′-bpy)2]n(2H2O)n depicted in Fig. 7 clearly shows that both the bpy ligands present in the asymmetric part of the unit cell are fully bound to the metal, which in turn does not leave room for water to coordinate to the metal. Since the polymer growth along two directions at the metal center is orthogonal, a two-dimensional grid decorated with dtbp ligands above and below the grid plane is the final result. It is important to note here that all the three series of compounds (bpy, azopy, and bpe) were prepared under almost identical conditions starting from one equivalent of the metal source, two equivalents of dtbpH ligands, and two equivalents of the ditopic spacer ligand.
It is unclear why both the equivalents of 4,4′-bipyridine used in the reaction manage to enter the coordination sphere of the metal in [M(dtbp)2(4,4′-bpy)2]n(2H2O)n while only one equivalent of the azopy or bpe ligand enters the metal coordination sphere in 1–4 or 5–8, leaving the second equivalent of azopy or bpe in the lattice. This mode of coordination in 1–8 however provides the required room for two water molecules to enter the metal coordination sphere and complete a 3D framework structure. The differing roles of 4,4-bipyridine vis-à-vis azopy or bpe can be visualized by comparing the distances between the adjacent metal centers (manganese taken as a common example) in the 2D grid lattice as shown in Fig. 7. Two observations are obvious from this comparison: (1) the insertion of additional two atoms (–NN– or –CHCH–) in between the two pyridyl rings results in larger Mn⋯Mn separation in the structures (in the case of 1 and 5); (2) the presence of a water molecule between the ditopic spacer and metal along one of the two grid axes results in unequal Mn⋯Mn separations along the two directions (13.647 and 17.510 Å). Due to these reasons, a M4 fragment of [M(dtbp)2(4,4′-bpy)2]n appears as nearly a square in Fig. 8, while those of 1 and 5 appear as a distorted rectangle.
 | ||
| Fig. 8 Comparison of the molecular structure of [Mn(dtbp)2(bpy)2](H2O)2 (a) with [Mn(dtbp)2(azopy)(H2O)2](azobpy) (1) and [Mn(dtbp)2(bpe)(H2O)2](bpe) (5). | ||
One of the original objectives of choosing a spacer with azo and ethylene units has been to investigate the resulting products for any photoisomerization (azo group) or photodimerization (ethylene unit), in view of well established photoactive units containing molecules undergoing structural changes (from one form to the other) by photoirradiation.12 The incorporation of photoactive azo groups in 1–4 and ethylene group in 5–8 indeed can offer such possibilities in the present case. However, due to the formation of fairly dense 3D structures, such processes do not take place in compounds 1–8. Further, from the discussions above, it is clear that the adjacent ethylene groups are not properly aligned for any light induced dimerization reactions to take place in 5–8.
Synthesis and characterization of coordination polymers 9–11 using a 1,2-bis(pyridin-3-ylmethylene)-hydrazine (bph) spacer
To study the role of the length of the N,N′-ditopic donor ligand on the final structures of the coordination polymers isolated, a relatively new spacer ligand that contains a four atom chain in between the two pyridyl ligands, viz. 1,2-bis(pyridin-3-ylmethylene)hydrazine (bph), has been used in place of the bpe/azoby ligands described above. One-dimensional coordination polymers [{M(bph)(H2O)4}{(dtbp)2}] (M = Ni (9) and M = Co (10)) and [Cd(bph)3(dtbp)2]n (11) (Scheme 3) have been isolated as the products from the reaction of the respective metal perchlorate salts with dtbp-H and spacer bph in methanol–DMF medium. The crystals obtained for these compounds are air and moisture stable and are soluble in polar organic solvents such as DMSO. | ||
| Scheme 3 Synthesis of various types of 1D coordination polymers 9–11. | ||
Elemental analysis values obtained for 9–11 are consistent with the calculated formula, which has later been verified by single crystal X-ray diffraction studies (vide infra). The FT-IR spectra of 9–11 show characteristic absorptions around 2970, 1190 and 1060 cm−1 corresponding to the νasy(C–H), ν(P–O–O) and νM–O–P vibrations. The presence of a broad νO–H absorption around 3400 cm−1 is indicative of the presence of water either in the coordination sphere of the metal or in the lattice in 9 and 10 (Fig. S17–S19†). While compounds 9 and 10 are paramagnetic, the diamagnetic 11 yields a single resonance in the 31P NMR spectrum at −6.93 ppm indicating the presence of only one type of phosphorus in the complex (Fig. S8†). The 1H NMR spectrum of 11 shows well separated peaks for the protons of bph and dtbp ligands in a 3:2 ratio. A doublet and three multiplets appearing at 9.034, 8.718, 8.290 and 7.569 ppm correspond to the C–H pyridine protons of the bph ligand and a singlet appearing at 8.794 ppm corresponds to the CH–N protons. The tert-butyl protons of dtbp appear as a singlet at 1.33 ppm.
space group. The immediate coordination environment around the central metal ion for compounds 9–10 is shown in Fig. 9. Due to the close resemblance of structures of 9 and 10, a detailed description of the structure of compound 9 alone is presented here. The asymmetric part of the unit cell in 9 contains one nickel ion, four coordinated water molecules and one bph ligand, apart from the two deprotonated dtbp− anions in the lattice (Fig. 9). The metal ion adopts a distorted octahedral geometry with –N2O4 coordination environments and the metal ion is coordinated by four O-atoms of four water molecules and two N-atoms of two bph. Another terminal N-atom of bph is linked to other metal ions and leads to the formation of a 1-dimensional coordination polymer (Fig. 10).
 | ||
| Fig. 9 Repeating units in the 1D polymeric structure of 9 (top) and 10 (bottom) (hydrogen atoms on carbon are omitted for clarity). | ||
 | ||
| Fig. 10 1-Dimensional polymeric view of 9 (hydrogen atoms on carbon are omitted for clarity). | ||
Four equatorial coordination sites of metal ions are coordinated by water molecules through O-atoms (Ni1–O6: 2.070(2) and Ni–O7: 2.061(2) Å) and the remaining two axial sites are coordinated by bph through N-atoms (Ni1–N1: 2.095(2) Å). Two deprotonated dtbp units are sitting in the lattice through strong hydrogen bonding with the coordinated water molecules (O6–H6B⋯O1: 2.16(4) and O7–H7D⋯O5: 1.88(3) Å). The distance between P and O atoms (P1–O5: 1.503(2) and P1–O2: 1.487(2) Å) reveals that the deprotonated oxygen exhibits strong hydrogen bonding interactions with water molecules. Selected bond lengths and angles of 9 and 10 are given in Tables S9 and S10.†
O units of two adjacent molecules (O2⋯H17A–O17: 2.003(2) Å). Other bond lengths and angles of 11 are listed in Table S11.†
 | ||
| Fig. 11 1-Dimensional polymeric view of 11 (hydrogen atoms are omitted for clarity). | ||
Thermal studies
The presence of highly thermally labile dtbp groups on the framework structures 1–8 prompted a detailed investigation of their thermal decomposition behavior (see ESI; Fig. S20–S30†). The thermo gravimetric analysis of 1 reveals that the first weight loss observed in the range of 85–130 °C (9.5%) corresponds to the loss of two H2O molecules. The second major weight loss beyond 130 °C corresponds to the loss of four isobutene and two azopy moieties in succession to result in the formation of Mn(PO3)2 (Fig. S20†). Similarly, the TGA of 3 reveals the first weight loss of 4% in the temperature range of 100–135 °C for the loss of coordinated H2O molecules. The second major weight loss occurring until 205 °C corresponds to the loss of four isobutene molecules while the third weight loss which sets in beyond this temperature is responsible for the loss of two 4,4′-azobipyridine moieties to yield Cu(PO3)2 after a further water loss. The TGA data for all complexes are listed in Table S2.† Similarly, TGA of 9 and 10 reveals that the first weight loss occurs around 100–135 °C due to the loss of coordinated H2O molecules. The weight loss in the range of 150–300 °C corresponds to the loss of organic groups in 9–11 (Fig. 12).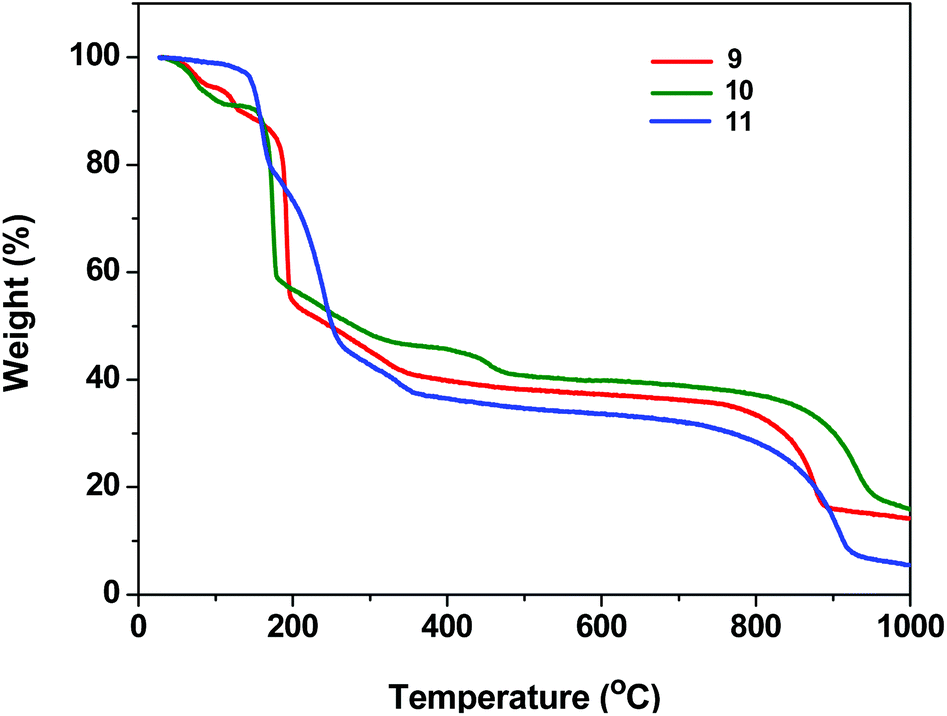 | ||
| Fig. 12 TGA curve for compounds 9–11 (N2, 10 °C min−1). | ||
Experimental section
Materials and methods
Commercial grade solvents have been purified employing conventional procedures and distilled prior to their use. Starting materials such as Mn(OAc)2·4H2O (E. Merck), Cu(OAc)2·H2O (E. Merck), Cd(ClO4)2·6H2O (Aldrich), 4-aminopyridine (Aldrich), Ni(OAc)2·6H2O (E. Merck), 1,2-bis(4-pyridyl)ethylene (bpe) (Aldrich) and Co(OAc)2·4H2O(Aldrich) used have been purchased from commercial sources and used without any further purification. 4,4′-Azobipyridine (azopy) and 1,2-bis(pyridin-3-ylmethylene)hydrazine have been synthesized following previously reported methods.13Elemental analyses have been performed on a Thermo Finnigan (FLASH EA 1112) microanalyzer at IIT-Bombay. Infrared spectra have been recorded on a Perkin Elmer FT-IR spectrometer as KBr diluted discs. The 1H and 31P NMR spectra were obtained on Bruker AS 300 and Varian 400S spectrometers using Me4Si (external) as the reference for 1H NMR and 85% H3PO4 (external) as the reference for 31P NMR spectral measurements. UV-vis spectra were obtained on Shimadzu UV-260 and UV-160A spectrophotometers. The EPR measurements have been performed on a Varian 109C E-line X-band EPR spectrometer fitted with a quartz Dewar for measurements at 77 K. The EPR spectra have been calibrated using tetracyanoethylene (tcne). Thermal analyses have been carried out on a Perkin Elmer Pyris Diamond thermal analysis system.
Synthesis of 5. M.p. 200 °C. Yield 0.0733 g (84% based on dtbp-H). IR data (KBr, cm−1): 3390(br), 2976(m), 1605(s), 1424(m), 1184(s), 1073(vs), 980(m), 828(w). Anal. Calcd for C40H60MnN4O10P2 (%): C 54.9, H 6.9, N 6.4. Found. C 55.1, H 7.1, N 6.0. TGA [temp. range, °C (% weight loss)]; 80–150 (5, –H2O); 150–232 (58, –isobutene, –bpe); 232–472 (6, –azopy); 472–950 (10, azopy); 850–1027 (–P2O5).
Synthesis of 6. M.p. 230 °C. Yield 0.069 g (80% based on dtbp-H). IR data (KBr, cm−1): 3376(br), 2972(m), 1604(s), 1424(m), 1186(s), 1071(vs), 979(m), 828(w). Anal. Calcd for C40H60CoN4O10P2 (%): C 54.7, H 6.9, N 6.7. Found. C 54.7, H 7.1, N 7.1. TGA [temp. range, °C (% weight loss)]; 75–150 (8, –H2O); 150–225 (44, –isobutene); 225–450 (6, –azoby), 450–940 (–bpe and –P2O5).
Synthesis of 7. M.p. 211 °C. Yield 0.074 g (85% based on dtbp-H). IR data (KBr): 3399(br), 2977(m), 1612(s), 1433(m), 1252(m), 1186(s), 1074(vs), 974(s), 834(s). Anal. Calcd for C40H60CuN4O10P2 (%): C 54.4, H 6.8, N 6.3. Found. C 54.44, H 6.75, N 6.34. TGA [temp. range, °C (% weight loss)]; 55–170 (8, –H2O); 170–220 (25, –isobutene); 220–455 (20, –isobutene and bpe); 450–900 (–P2O5).
Synthesis of 8. M.p. 190 °C. Yield 0.0.067 g (75% based on dtbp-H). IR data (KBr): 2973(m), 1609(s), 1425(m), 1184(s), 1071(s), 977(s), 829(s). Anal. Calcd for C40H60NiN4O10P2 (%): C 54.4, H 6.8, N 6.3. Found. C 53.59, H 6.74, N 5.66. TGA [temp. range, °C (% weight loss)]; 100–149 (10, –H2O and isobutene); 150–350 (50, –isobutene and bpe); 400–800 (–P2O5).
Synthesis of [{Ni(bph)(H2O)4}{(dtbp)2}]n (9). Green crystals have been obtained from Ni(ClO4)2·4H2O (96 mg, 0.025 mmol), dtbp-H (100 mg, 0.5 mmol) and bph (105 mg, 0.5 mmol). Yield 0.164 g (86% based on nickel perchlorate). IR data (KBr, cm−1): 3424 (br), 2974(m), 2926(m), 1636(s), 1425(m), 1194(s), 1063(s), 962(s), 827(m). Anal. Calcd for C28H54NiN4O12P2 (%): C 44.29, H 7.17, N 7.38. Found. C 44.30, H 8.06, N 7.26. TGA [temp. range, °C (% weight loss)]; 80–100 (7, –2H2O); 100–165 (8, –2H2O); 165–400 (46, L1 and isobutene); 400–900 (–P2O5).
Synthesis of [{Co(bph)(H2O)4}{(dtbp)2}]n (10). Purple crystals of 10 have been obtained from Co(ClO4)2·4H2O (96 mg, 0.025 mmol), dtbp-H (100 mg, 0.5 mmol) and bph (105 mg, 0.5 mmol). Yield 0.147 g (56% based on cobalt perchlorate). IR data (KBr, cm−1): 3424 (br), 2974(m), 2930(m), 1636(s), 1424(m), 1191(s), 1063(w), 965(m), 828(m). Anal. Calcd for C28H54CoN4O12P2 (%): C 44.27, H 7.17, N 7.38. Found. C 44.30, H 8.06, N 7.26. TGA [temp. range, °C (% weight loss)]; 80–145 (10, H2O); 145–181 (30, –isobutene); 181–600 (20, L1); 600–950 (–P2O5).
Synthesis of [Cd(bph)3(dtbp)2]n (11). Yellow crystals have been obtained from Cd(ClO4)2·6H2O (102 mg, 0.025 mmol), dtbp-H (100 mg, 0.5 mmol) and bph (105 mg, 0.5 mmol). Yield 0.169 g (58% based on metal perchlorate). IR data (KBr, cm−1): 3400 (br), 2976(m), 2931(m), 1629(s), 1419(m), 1188(s), 1076(s), 973(s), 827(m). Anal. Calcd for C52H66CdN12O8P2 (%): C 53.77, H 5.73, N 14.47. Found. C 53.34, H 5.87, N 13.91. 1H NMR (DMSO-d6, 500 MHz, ppm): δ 9.03 (d, J = 1 Hz, 2H, H1, pyridine), 8.79 (s, 2H, H5, CH
N), 8.71 (m, 2H, H4, pyridine), 8.29 (m, 2H, H3, pyridine), 7.56 (m, 2H, H2, pyridine) and 1.33 (s, 9H, C(CH3)3). 31P NMR (DMSO-d6, 202 MHz, ppm): δ −6.93 ppm. TGA [temp range, °C (% weight loss)]; 140–260 (50, –isobutene); 240–600 (21, L1); 600–920 (–P2O5).
| Compound | 1 | 2 | 3 | 4 |
5a |
9 |
10a |
11 |
|---|---|---|---|---|---|---|---|---|
| a Poor quality crystals – repeated attempts failed to improve the diffraction quality (R1 [I > 2σ(I)] = 0.1636 (5) and 0.1067 (10)). | ||||||||
| Formula | C36H56MnN8O10P2 | C36H56CoN8O10P2 | C36H56CuN8O10P2 | C36H56CdN8O10P2 | C40H60MnN4O10P2 | C28H54N4NiO12P2 | C28H44CoN4O12P2 | C52H68CdN12O9P2 |
| Formula weight | 877.77 | 881.76 | 886.37 | 935.23 | 873.80 | 759.40 | 749.54 | 1179.52 |
| Temperature (K) | 293(2) | 150(2) | 295(2) | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 1.54178 | 0.71073 | 0.71075 | 0.71075 | 0.71075 | 0.71075 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Space group | C2/c | C2/c | C2/c |
P |
P21/c |
P |
P |
P21/c |
| a (Å) | 29.7719(17) | 28.612(5) | 28.527 | 8.7071(3) | 14.296(7) | 7.026(3) | 7.23(2) | 9.877(4) |
| b (Å) | 13.6473(6) | 13.113(5) | 13.224 | 8.8842(3) | 13.946(6) | 10.737(5) | 11.02(4) | 23.870(9) |
| c (Å) | 11.1274(7) | 11.789(5) | 11.761 | 14.3210(3) | 22.881(11) | 13.782(6) | 14.09(4) | 24.139(9) |
| α (°) | 90 | 90.000(5) | 90 | 91.155(2) | 90 | 104.485(5) | 104.67(4) | — |
| β (°) | 109.043(7) | 91.344(5) | 91.26 | 92.669(2) | 93.456(8) | 96.215(8) | 96.20(2) | 97.363(9) |
| γ (°) | 90 | 90.000(5) | 90 | 102.119(2) | 90 | 103.909(3) | 104.44(7) | — |
| Volume (Å3) | 4273.7(4) | 4422(3) | 4435.7 | 1081.44(6) | 4554(4) | 961.3(7) | 1033(6) | 5644(4) |
| Z | 4 | 4 | 4 | 1 | 4 | 1 | 1 | 4 |
| D (calcd) (Mg m−3) | 1.364 | 1.324 | 1.327 | 1.436 | 1.275 | 1.312 | 1.205 | 1.388 |
| Cryst. size (mm3) | 0.2 × 0.2 × 0.2 | 0.24 × 0.22 × 0.17 | 0.38 × 0.22 × 0.12 | 0.31 × 0.28 × 0.23 | 0.20 × 0.09 × 0.04 | 0.04 × 0.12 × 0.24 | 0.12 × 0.21 × 0.32 | 0.08 × 0.21 × 0.32 |
| Abs coeff. (mm−1) | 0.447 | 0.521 | 1.904 | 0.641 | 0.416 | 0.646 | 0.547 | 0.508 |
| Data/restraints/parameters | 3753/3/259 | 3882/15/274 | 4460/69/298 | 3801/0/265 | 7966/113/514 | 3351/2/227 | 3519/0/214 | 9350/0/685 |
| Goodness-of-fit on F2 | 0.946 | 0.997 | 1.010 | 1.080 | 1.064 | 1.259 | 1.366 | 1.047 |
| R 1 [I > 2σ(I)] | 0.0474 | 0.0754 | 0.0800 | 0.0234 | 0.1636 | 0.0372 | 0.1067 | 0.0750 |
| R 2 [I > 2σ(I)] | 0.1265 | 0.2169 | 0.2314 | 0.0617 | 0.3587 | 0.1286 | 0.3254 | 0.1957 |
Conclusions
It has been shown in this contribution that both azopy and bpe behave very differently compared to 4,4′-bipyridine in the metallophosphate coordination polymer chemistry involving the thermally labile diester dtbp-H. The major structural difference in 1–8 is induced by the introduction of two coordinated water molecules around each metal, which in turn forces the second ditopic spacer out of the coordination sphere of the metal. This in turn facilitates very interesting hydrogen bonding interactions, which are often geometrically orthogonal, leading to the formation of a highly periodic 3D coordination polymeric framework. The use of a longer spacer ligand bph yields one-dimensional coordination polymers which are either cationic or surface N-functionalized. Among these the surface functionalized polymer 11 which contains two free N-donor sites at each repeating unit may serve as a useful starting point for building higher dimensional coordination polymers or even loading catalytically active metal ions such as palladium. Further, it is highly likely that other carefully chosen ditopic (and even tritopic) spacers in metal-dtbp chemistry will offer newer forms of coordination polymers which were hitherto unknown. We are currently investigating these possibilities.Acknowledgements
This work was supported by CSIR and DST, New Delhi and BRNS, Mumbai. R. M. thanks BRNS for the award of a DAE-SRC Outstanding Investigator Award enabling the purchase of a single crystal X-ray diffractometer.Notes and references
- (a) J. M. Lehn, Supramolecular Chemistry, Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed; (b) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734 CrossRef CAS PubMed; (c) M. Kiguchi, J. Inatomi, Y. Takahashi, R. Tanaka, T. Osuga, T. Murase, M. Fujita, T. Tada and S. Watanabe, Angew. Chem., Int. Ed., 2013, 52, 6202 CrossRef CAS PubMed; (d) Z. Zhu, A. C. Fahrenbach, H. Li, J. C. Barnes, Z. Liu, S. M. Dyar, H. Zhang, J. Lei, R. Carmieli, A. A. Sarjeant, C. L. Stern, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 11709 CrossRef CAS PubMed; (e) H. -J. Son, S. Jin, S. Patwardhan, S. J. Wezenberg, N. C. Jeong, M. So, C. E. Wilmer, A. A. Sarjeant, G. C. Schatz, R. Q. Snurr, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 862 CrossRef CAS PubMed.
- (a) G. B. Deacon and R. Phillips, J. Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS; (b) T. Osuga, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2012, 51, 12199 CrossRef CAS PubMed; (c) M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897 RSC; (d) S. Li, J. Huang, T. R. Cook, J. B. Pollock, H. Kim, K.-W. Chi and P. J. Stang, J. Am. Chem. Soc., 2013, 135, 2084 CrossRef CAS PubMed; (e) Z. Li, W. Liu, J. Wu, S. H. Liu and J. Yin, J. Org. Chem., 2012, 77, 7129 CrossRef CAS PubMed; (f) M. Beyler, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 2010, 132, 4409 CrossRef CAS PubMed; (g) M. Dinca and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766 CrossRef CAS PubMed; (h) M. Weston, A. A. DeLaquil, A. A. Sarjeant, O. K. Farha, J. T. Hupp and S. T. Nguyen, Cryst. Growth Des., 2013, 13, 2938 CrossRef CAS.
- (a) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS PubMed; (b) W. Morris, B. Volosskiy, S. Demir, F. Gandara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443 CrossRef CAS PubMed; (c) O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef PubMed; (d) M. O'Kee and O. M. Yaghi, Chem. Rev., 2012, 112, 675 CrossRef PubMed; (e) W. Bury, D. Fairen-Jimenez, M. B. Lalonde, R. Q. Snurr, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 739 CrossRef CAS; (f) C. Dey and R. Banerjee, Chem. Commun., 2013, 49, 6617 RSC; (g) T. Panda, T. Kundu and R. Banerjee, Chem. Commun., 2013, 49, 6197 RSC.
- (a) V. Chandrasekhar, R. K. Metre and R. S. Narayanan, Dalton Trans., 2013, 42, 8709 RSC; (b) R. Murugavel, S. Shanmugan and N. Gogoi, Inorg. Chem. Commun., 2010, 13, 1530 CrossRef CAS PubMed; (c) R. Murugavel and N. Gogoi, Bull. Mater. Sci., 2009, 32, 321 CrossRef CAS PubMed; (d) S. S. Iremonger, J. Liang, R. Vaidhyanathan and G. K. H. Shimizu, Chem. Commun., 2011, 47, 4430 RSC; (e) Q. Yue, J. Yang, G.-H. Li, G.-D. Li and J.-S. Chen, Inorg. Chem., 2006, 45, 4431 CrossRef CAS PubMed; (f) D. Kong, L. Yang, O. Xiang, A. V. Prosvirin, H. Zhao, J. H. Ross, Jr., K. M. Dunbar and A. Clearfield, Chem. Mater., 2004, 16, 3020 CrossRef CAS; (g) O. R. Evans, H. L. Ngo and W.-B. Lin, J. Am. Chem. Soc., 2001, 123, 10395 CrossRef CAS.
- (a) E. M. Pineda, F. Tuna, R. G. Pritchard, A. C. Regan, R. E. P. Winpenny and E. J. L. McInnes, Chem. Commun., 2013, 49, 3522 RSC; (b) Y. Zheng, M. Evangelisti, F. Tuna and R. E. P. Winpenny, J. Am. Chem. Soc., 2012, 134, 1057 CrossRef CAS PubMed; (c) A. N. Alsobrook, B. G. Hauser, J. T. Hupp, E. V. Alekseev, W. Depmeier and T. E. Albrecht-Schmitt, Cryst. Growth Des., 2011, 11, 1385 CrossRef CAS; (d) S. S. Iremonger, J. Liang, R. Vaidhyanathan, I. Martens, G. K. H. Shimizu, T. D. Daff, M. Z. Aghaji, S. Yeganegi and T. K. Woo, J. Am. Chem. Soc., 2011, 133, 20048 CrossRef CAS PubMed.
- (a) V. Chandrasekhar, P. Sasikumar and P. Thilagar, Organometallics, 2007, 26, 4386 CrossRef CAS; (b) C. G. Lugmair and T. D. Tilley, Inorg. Chem., 1998, 37, 1821 CrossRef CAS; (c) C. G. Lugmair, T. D. Tilley and A. L. Rheingold, Chem. Mater., 1997, 9, 339 CrossRef CAS; (d) V. Chandrasekhar, T. Senapati, E. C. Sanudo and R. Clerac, Inorg. Chem., 2009, 48, 6192 CrossRef CAS PubMed; (e) V. Chandrasekhar and L. Nagarajan, Dalton Trans., 2009, 6712 RSC.
- (a) R. Murugavel, S. Kuppuswamy, N. Gogoi and A. Steiner, Inorg. Chem., 2010, 49, 2153 CrossRef CAS PubMed; (b) R. Murugavel, S. Kuppuswamy, A. N. Maity and M. P. Singh, Inorg. Chem., 2009, 48, 193 CrossRef PubMed; (c) R. Pothiraja, S. Shanmugan, M. G. Walawalkar, M. Nethaji, R. J. Butcher and R. Murugavel, Eur. J. Inorg. Chem., 2008, 1834 CrossRef CAS; (d) R. Murugavel, S. Kuppuswamy, R. Boomishankar and A. Steiner, Angew. Chem., Int. Ed., 2006, 45, 5536 CrossRef CAS PubMed.
- (a) R. Murugavel, A. Choudhury, M. G. Walawalkar, R. Pothiraja and C. N. R. Rao, Chem. Rev., 2008, 108, 3549 CrossRef CAS PubMed; (b) P. Ramaswamy, S. Mandal and S. Natarajan, Inorg. Chim. Acta, 2011, 372, 136 CrossRef CAS PubMed; (c) R. Dey, B. Bhattacharya, E. Colacio and D. Ghoshal, Dalton Trans., 2013, 42, 2094 RSC; (d) J. A. Armstrong, E. R. Williams and M. T. Weller, Dalton Trans., 2013, 42, 2302 RSC; (e) M. Sathiyendiran and R. Murugavel, Chem. Lett., 2001, 84 Search PubMed; (f) C. G. Lugmair, T. D. Tilley and A. L. Rheingold, Chem. Mater., 1999, 11, 1615 CrossRef CAS; (g) C. G. Lugmair, T. D. Tilley and A. L. Rheingold, Chem. Mater., 1997, 9, 339 CrossRef CAS.
- (a) R. Murugavel, M. G. Walawalkar, M. Dan, H. W. Roesky and C. N. R. Rao, Acc. Chem. Res., 2004, 37, 763 CrossRef CAS PubMed; (b) R. Pothiraja, M. Sathiyendiran, R. J. Butcher and R. Murugavel, Inorg. Chem., 2005, 44, 6314 CrossRef CAS PubMed; (c) R. Murugavel, M. Sathiyendiran, R. Pothiraja, M. G. Walawalkar, T. Mallah and E. Riviere, Inorg. Chem., 2004, 43, 945 CrossRef CAS PubMed; (d) M. Sathiyendiran and R. Murugavel, Inorg. Chem., 2002, 41, 6404 CrossRef CAS PubMed.
- (a) R. Pothiraja, M. Sathiyendiran, A. Steiner and R. Murugavel, Inorg. Chim. Acta, 2011, 372, 347 CrossRef CAS PubMed; (b) R. Murugavel and N. Gogoi, J. Organomet. Chem., 2010, 695, 916 CrossRef CAS PubMed; (c) R. Pothiraja, M. Sathiyendiran, R. J. Butcher and R. Murugavel, Inorg. Chem., 2004, 43, 7585 CrossRef CAS PubMed; (d) R. Murugavel, M. Sathiyendiran and M. G. Walawalkar, Inorg. Chem., 2001, 40, 427 CrossRef CAS; (e) R. Howlader, M. G. Walawalkar and R. Murugavel, Inorg. Chim. Acta, 2013, 405, 147 CrossRef CAS PubMed.
- (a) R. Murugavel, S. Kuppuswamy, N. Gogoi, R. Boomishankar and A. Steiner, Chem. – Eur. J., 2010, 16, 994 CrossRef CAS PubMed; (b) R. Murugavel, S. Kuppuswamy, N. Gogoi, A. Steiner, J. Bacsa, R. Boomishankar and K. G. Suresh, Chem. – Asian J., 2009, 4, 143 CrossRef CAS PubMed; (c) R. Murugavel and S. Kuppuswamy, Inorg. Chem., 2008, 47, 7686 CrossRef CAS PubMed; (d) R. Murugavel, S. Kuppuswamy and S. Randoll, Inorg. Chem., 2008, 47, 6028 CrossRef CAS PubMed; (e) R. Murugavel and S. Kuppuswamy, Chem. – Eur. J., 2008, 14, 3869 CrossRef CAS PubMed; (f) R. Murugavel and S. Kuppuswamy, J. Chem. Sci., 2008, 120, 131 CrossRef CAS PubMed; (g) R. Murugavel and S. Kuppuswamy, Angew. Chem., Int. Ed., 2006, 45, 7022 CrossRef CAS PubMed; (h) A. Kalita, C. Roch-Marchal and R. Murugavel, Dalton Trans., 2013, 42, 9755 RSC.
- (a) G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755 RSC; (b) G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647 CrossRef CAS; (c) G. K. Kole, A. M. P. Peedikakkal, B. M. F. Toh and J. J. Vittal, Chem. – Eur. J., 2013, 19, 3962 CrossRef CAS PubMed; (d) D. K. Bucar, G. S. Papaefstathiou, T. D. Hamilton, Q. Chu, I. G. Georgiev and L. R. MacGillivray, Eur. J. Inorg. Chem., 2007, 4559 CrossRef CAS; (e) P. Rajakannu, F. Hussain, B. Shankar and M. Sathiyendiran, Inorg. Chem. Commun., 2012, 26, 46 CrossRef CAS PubMed.
- S. Verma, V. Vajpayee, S. M. Lee, H. J. Jung, H. Kim and K. W. Chi, Inorg. Chim. Acta, 2012, 387, 435 CrossRef CAS PubMed.
- CrysAlisPRO, Oxford Diffraction/Agilent Technologies UK Ltd, Yarnton, England Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Selected bond lengths and angles in tables and figures for 1–5 and 9–11. CCDC 942679–942683 and 1015310–1015312. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00149d |
| ‡ Authors have equally contributed. |
| This journal is © the Partner Organisations 2015 |