
From solid-state metal alkoxides to nanostructured oxides: a precursor-directed synthetic route to functional inorganic nanomaterials
Jun
Zhao†
ab,
Yipu
Liu†
a,
Meihong
Fan†
a,
Long
Yuan
a and
Xiaoxin
Zou
*a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, China. E-mail: xxzou@jlu.edu.cn; chemistryzouxx@gmail.com
bCollege of Science, Hebei University of Science and Technology, Shijiazhuang 050018, China
First published on 12th December 2014
Abstract
Functional nanostructured oxides are important inorganic materials for various energy- and environment-related applications, such as photocatalysis and lithium ion batteries. To optimize their properties/functions, synthetic methods that can lead to nanomaterials with unique composition, morphology and size are highly desirable. In this review, we summarize recent research efforts towards the construction of nanostructured solid-state metal alkoxides-a family of inorganic–organic hybrid compounds and their conversion into functional inorganic nanomaterials. The chemical transformation from metal alkoxides to nanostructured oxides represents a novel precursor-directed synthetic route to functional inorganic nanomaterials. The uniqueness of this method mainly lies in: (i) the crystal/molecular structure of metal alkoxides which plays a crucial role in their nanosized structures; (ii) the use of metal alkoxides as precursor materials which determines the composition and (micro)structure of the finally-obtained oxide nanomaterials; and (iii) that this method can be employed to synthesize nanomaterials that cannot be readily achieved using other approaches.
 Jun Zhao | Jun Zhao was awarded her Ph.D in Inorganic Chemistry at Jilin University in June 2014. Now, she is a lecturer at Hebei University of Science and Technology. Her research interests focus on the design and synthesis of transition metal oxides with novel micro- and nano-structures for environmental and energy applications. |
 Yipu Liu | Yipu Liu received her bachelor's degree at Jilin University, Changchun, 2014. Now, she is working on her masters degree at State Key Laboratory of Inorganic Synthesis and Preparative Chemistry. Her main research interest is the development of highly active catalytic materials for electrochemical hydrogen evolution. |
 Meihong Fan | Meihong Fan received her bachelor’s degree at Anyang Normal University in 2013. She is now a postgraduate student at State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, Changchun. Her research interests focus on the synthesis of electrochemically active materials for water splitting. |
 Long Yuan | Long Yuan (1988) received his bachelor’s degree in chemical engineering from Northeast Dianli University in 2010. He is currently a graduate student at State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, Changchun. His research interests include synthesis and facet control of complex metal oxides and their surface dependent catalytic activity. |
 Xiaoxin Zou | Xiaoxin Zou was awarded a Ph.D. in Inorganic Chemistry from Jilin University (China) in 06/2011; and then moved to the University of California, Riverside and Rutgers, The State University of New Jersey as a Postdoctoral Scholar from 07/2011 to 10/2013. He has been an associate professor at Jilin University since 12/2013. His research interests focus on the design and synthesis of noble metal-free, nanostructured and/or nanoporous materials for water splitting and renewable energy conversion. |
1. Introduction
Nano/micro-sized architecture chemistry is an innovative, frontier branch in modern science and technology.1 Although bulk materials have already propped up the whole industrial revolution which drastically changed our lifestyle, times have changed and the properties of these materials hardly meet the demands of various modern technologies today. Little trickeries such as ball-milling technology turn up to overcome the deficiency of bulk materials. However, there is no abyss that can be filled up by a single pebble. Instead of pebbles, there should be a bridge to get through the challenge. In material science, it would be likely to reason that nanoscience can provide an opportunity to solve a dilemma: (i) based on nanofabrication techniques,2 scientists can firstly prepare various materials on a smaller scale.3 The reduction of the physical dimensions to the nanometre scale increases the surface to volume ratio and the number of active surface sites. These structural advantages are always related to the catalytic performance of the materials owing to the enhanced interaction between the catalysts and their surrounding media. When it comes to lithium-ion batteries, nanomaterials have already been confirmed to efficiently improve the deficiency of the charge-discharge volume swings of anode and cathode materials, and furthermore, to improve the cyclability as an additional benefit.4 (ii) Nanochemistry offers a possibility to enhance the electrical, optical, magnetic, thermal and mechanical properties of a material by controlling its size, shape, porosity and surface functional group.5 For example, in semiconductor nanocrystals, continuous energy levels of electronic bands can be transformed into discrete energy levels.6 In addition, increasing the percentage of exposed highly reactive facets, at the nanoscale, does improve the performance of materials in diverse applications such as in photocatalysis and in dye-sensitized solar cells.7 (iii) Besides presenting individual properties, nanoparticles can become a kind of structural unit, in other words, “artificial atoms” to produce a new type of materials which have imponderable collective properties.8 (iv) Through nanoengineering, we can devise nanomaterials which contain unique characteristics that bulk materials can never possess. This means that we are creating materials rather than finding them. The charming opportunities in nanoscience are appealing enough to ignite the imagination and efforts of scientists and chemical industries all over the world.9Owing to their attractive size- and morphology-enhanced properties/functions, inorganic nanomaterials have drawn tremendous interest in recent years. Thus, pursuing new synthetic methodologies that can lead to nanomaterials with well-controlled structures, shapes and compositions are essential to solve these problems.5,10 Among the numerous existing synthetic methods, the precursor-directed approach has been deemed as an effective way to achieve various inorganic nano-to-micro structures.11 Metal alkoxides, some of which have already been commercialised (e.g., tetraethyl orthosilicate, TEOS), have received much research interest as unique precursor materials.12 Unlike pure inorganic precursors, metal alkoxides benefit from the synergistic effects of their inorganic and organic components, which often play an important role in fabricating nanostructured functional materials. Until now, liquid metal alkoxides have been widely used as precursors for the preparation of metal oxide nanomaterials.13 By comparison, the use of solid-state metal alkoxides as a new type of precursor material, has only recently been a subject of research. However, their versatility as precursor materials has attracted wide attention. In this review, we focus on the recent development of the solid-state alkoxides, which were not discussed in any previous reviews. In particular, we summarize the preparation, characterization, and physicochemical properties of solid-state metal alkoxides. We also discuss the conversion of alkoxides, as well as the structure and properties of the resulting metal oxide nanomaterials. Finally, a short conclusion was given based on this method, together with an outlook for the further development of the preparation and applications of functional nanomaterials.
2. Metal elements involved in solid-state metal alkoxides
Metals that can form solid-state metal alkoxides are mainly located in the d-, ds-, f-, and p-blocks of the periodic table of elements (Fig. 1). The d-block elements include Ti, V, Mn, Fe, Co, Ni, Y, Mo, W, and the ds-block elements include Cu and Zn. The rare earth elements (i.e., the f-block elements such as Ce, Nd, Sm and Gd) and main group elements (i.e., the p-block elements such as In, Sn, Pb and Bi) were also reported as being able to form solid-state alkoxides. Most of the metals that are able to form alkoxides are transition metals with partially-filled d-/f-orbitals. Thus, they often possess extra electrons, and thus they can easily coordinate with the hydroxyl groups of alcohols to form stable compounds. However, alkali-metal or alkaline-earth metal-based alkoxides are rarely reported since they have a poor coordination ability and their simple oxides do not show impressive functions. In addition, metal alkoxides are, generally, difficult to form using noble-metals (such as Ru, Rh, Ag, Au, Ir and Pt) because of the tendency for these metal ions to be easily reduced back to metallic particles. In this sense, we could conclude that the formation of solid-state alkoxides for a wide range of transition metals and even several main group elements; and therefore, their controllable conversion might open up a new avenue for the construction of a variety of functional oxide nanostructures. | ||
| Fig. 1 Metal elements that are able to form solid-state metal alkoxides. The metals that can be coordinated by ethylene glycol, glycerol and benzyl alcohol are shown in red, blue and yellow, respectively. | ||
3. Alcohols involved in solid-state metal alkoxides
Searching the literature that we have already summarized, not every alcohol can react with metal ions to form solid-state metal alkoxides. Generally, the chemical nature of the alcohols, rather than their physical properties, is the key to obtaining desirable solid-state metal alkoxides. Table 1 presents information about the alcohols which may be used to form the required solid-state metal alkoxides. From this table we can roughly divide the above alcohols into two types: (i) polyalcohols (such as ethylene glycol,14,15 1,3-propanediol16,17 and glycerol18) that contain two or more hydroxyl groups; and (ii) aromatic alcohols (such as benzylalcohol19,20) which contain a phenyl group, instead of multiple hydroxyl groups. The polyalcohol hydroxyl group chelators have a strong propensity to coordinate with metal ions, and sometimes this can also increase the coordination number of the central metal ion in the resulting compound, thus resulting in stable structures. For example, commercially available liquid titanium(IV) isopropoxide has a coordination number of 4 for Ti, while titanium glycolate is a kind of solid-state metal alkoxide in which the coordination number of Ti is 6. Moreover, among the metal-oxygen bonds in metal alkoxides, the transformation from terminal oxygen atoms in titanium(IV) isopropoxide to the bridging oxygen atoms in titanium glycolate causes the formation of a solid-state polymeric structure. For the aromatic alcohol-based alkoxides, the π–π interaction of phenyl groups might be the main chemical driving force in the formation of solid-state alkoxides. In addition to using the special chemical features stated above, the low cost, non-toxicity and earth-abundance of these alcohols are also advantageous for their wide application.14| Name | Molecular Formula | Melting point (°C) | Boiling point (°C) | Density (m3 g−1) | Structure |
|---|---|---|---|---|---|
| a All of the information about the alcohols was obtained from the http://www.chemicalbook.com/ website. | |||||
| Ethylene glycol | C2H6O2 | −13 | 196–198 | 1.220 |
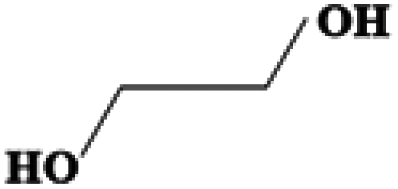
|
| 1,2-Propanediol | C3H8O2 | −60 | 187 | 1.036 |
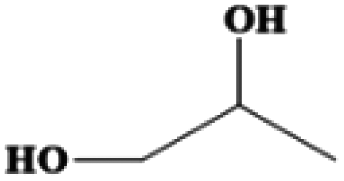
|
| 1,3-Propanediol | C3H8O2 | −32 | 214 | 1.053 |

|
| 1,2-Butanediol | C4H10O2 | −50 | 191–192 | 1.006 |
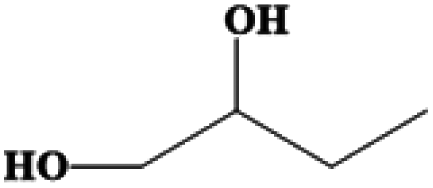
|
| 1,3-Butanediol | C4H10O2 | −54 | 203–204 | 1.005 |
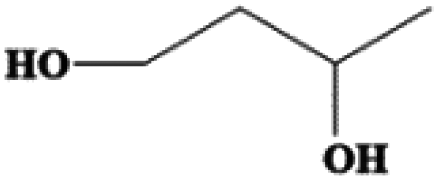
|
| 1,4-Butanediol | C4H10O2 | 20 | 230 | 1.017 |

|
| 2,3-Butanediol | C4H10O2 | 25 | 183–184 | 1.01 |
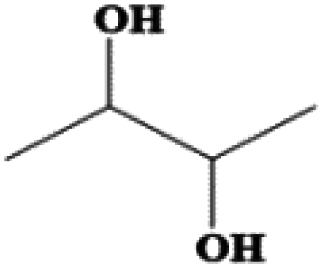
|
| Glycerol | C3H8O3 | 20 | 290 | 1.261 |
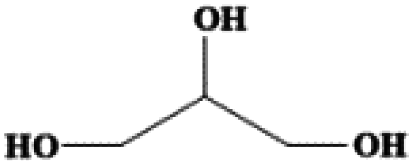
|
| Benzylalcohol | C7H8O | −15.3 | 205.7 | 1.042 |
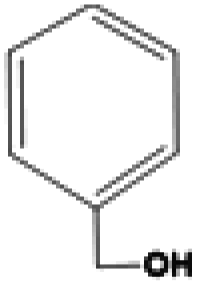
|
4. The characteristics of solid-state metal alkoxides
Metal alkoxides, especially commercial small-molecule liquid-state alkoxides, have been extensively used as important precursors for the preparation of oxide-ceramic materials.21 Compared to these small-molecule alkoxides, solid-state metal alkoxides exhibit some important characteristics that are beneficial for making oxide nanomaterials with unique compositions and/or structures.(i) Unlike many small-molecule alkoxides with extreme moisture sensitivity, solid-state alkoxides are generally more stable towards moisture. This enables the possibility of storing these molecules under ambient conditions, for further use without the need for special protective measures. In addition, this feature is also beneficial for the study of the structures and properties of solid-state alkoxides.
(ii) Generally, hydrolysis of small-molecule alkoxides is too fast to control.22 In contrast with liquid phase metal alkoxides, the reaction between solid-state alkoxides and water is mild, even above room temperature. Thus, it is more reasonable to prepare oxide nanomaterials with better structures and functions by controllable hydrolysis of solid-state alkoxides.
(iii) Some of the solid-state alkoxides are crystalline materials with well-defined crystal structures (e.g., titanium glycolate,14 vanadyl glycolate,23 cobalt monoglycerolate18b and zinc glycerolate19). Thus, we can analyse the relationship between the structure and the properties of these alkoxides (e.g., titanium glycolate).14Fig. 2 shows the crystal structure of titanium glycolate. Titanium glycolate contains infinite one-dimensional edge-sharing TiO6 octahedral chains with the unit cell parameters: a = 15.204, b = 7.568, c = 5.816 Å, β = 110.878°. The six oxygen atoms originate from four ethylene glycol ligands, with each ligand bridging two titanium atoms with one of its oxygen atoms, and the other oxygen is terminal. Because of the unique one-dimensional crystal structure, crystalline titanium glycolate always exhibits a rod- or wire-like morphology and it can be used as the precursor for the synthesis of 1D titanium oxide nanomaterials. (see section 5).
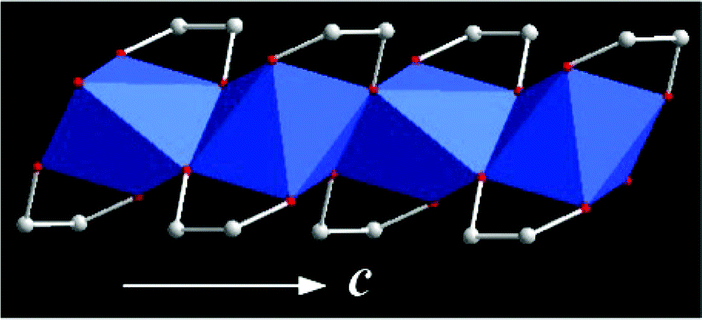 | ||
| Fig. 2 Crystal structure of titanium glycolate. View of the one-dimensional chain of titanium glycolate along the c axis. Carbon and oxygen atoms are denoted as white and red spheres, respectively. TiO6 is denoted as blue octahedra.14 | ||
(iv) Many solid-state alkoxides, intrinsically, have unique nanostructures, which are determined by crystal structure to a large extent. The composition can be tuned by doping, and the morphology can be controlled with suitable additives.
(v) Solid-state alkoxides can easily transfer their structure/composition features to their corresponding desired oxide nanomaterials. Thus, we can realize the target synthesis of oxide nanomaterials by design of alkoxide nanostructures to a certain extent.
5. Solid-state metal alkoxides and their derived oxide nanomaterials
In this section, we discuss the synthesis of nanostructured solid-state metal alkoxides and their conversion into functional inorganic nanomaterials, while emphasizing that (i) the crystal/molecular structure of metal alkoxides plays a crucial role in their nanosized structures; (ii) metal alkoxides, when used as precursor materials determine the composition and (micro)structure of the resulting oxide nanomaterials; and (iii) this precursor-directed method can be employed to prepare nanomaterials that cannot be readily achieved using other approaches.5.1 Titanium alkoxides and their derived TiO2 nanomaterials
TiO2 nanomaterials with micro- and nano-structures have potential application in many fields.24,25 Titanium alkoxides as one of the important inorganic–organic hybrids, mainly including titanium glycolate and titanium glycerolate, have been widely used as effective precursors to prepare functional TiO2 nanomaterials.The synthesis of crystalline titanium glycolate is based on the chemical reaction between a small-molecule titanium source (e.g., titanium(IV)isopropoxide) and ethylene glycol at elevated temperatures. Without any additives, high-quality titanium glycolates with 1D morphologies, such as nanorods, nanowires and nanorod self-assemblies can be obtained through tuning the reaction conditions (e.g. the concentration of the titanium source, reaction time, temperature and so on).26
Unlike crystalline titanium glycolates, amorphous titanium glycolates with a uniform sphere-like morphology were achieved at room temperature. In 2003, Xia et al. developed an acetone-assisted method to prepare amorphous titanium glycolate nanospheres with a tuneable particle size (Fig. 3).27 These mono-dispersed spheres could not be obtained by conventional sol–gel methods due to the fast hydrolysis rates of the Ti precursors, which makes the nucleation and growth steps indistinguishable. In this paper, the authors proposed that the ethylene glycolate precursor formed first without any precipitation, then the titanium glycolate spherical colloids formed when the ethylene glycolate precursor was poured into an acetone solution containing a small amount of water (∼0.3%). The morphology difference between crystalline and amorphous titanium glycolates further indicates the importance of crystal/molecular structure in determining nanosized structures.
 | ||
| Fig. 3 SEM images of titanium glycolate spherical colloids that were prepared using different molar concentrations of Ti(OBu)4 in acetone: (A) 1.21, (B) 1.03, (C) 0.86 and (D) 0.68 mM.27 Copyright © 2003 WILEY VCH Verlag GmbH & Co. KGaA Weinheim. | ||
Moreover, the composition of titanium glycolates can be altered within a certain range during the synthesis. For example, Zou et al. prepared transition-metal-doped titanium glycolates (M = Fe, Mn) through a solvothermal doping approach, which opened up a new synthetic strategy for producing non-stoichiometric heterometal alkoxides.15 This method has also been adopted to prepare vanadium doped titanium glycolate.28 Following a similar approach, Li et al. successfully doped Bi into amorphous titanium glycolates at room temperature based on Xia's method.29
To realize the conversion of as-obtained titanium glycolate precursors to TiO2 nanomaterials, heat-treatment as a “traditional” method has been widely used in the past several years. The heat-treatment process usually involves the direct calcination of the titanium glycolate precursors above 400 °C in air or inert atmosphere (e.g., N2). The annealing temperature is close to that of the combustion of the organic component of the alkoxides. When heat-treated in air, the organic component of the titanium glycolates will decompose into H2O and CO2. This leads to TiO2 nanomaterials with well-developed porosities and large surface areas.30 By contrast, the organic component of the titanium glycolates will partially convert into carbon in an inert atmosphere. This resulted in the formation of TiO2/C nanocomposites.31 The difference in the material formation when synthesized under different atmospheres, demonstrates that the existence of an organic component plays an important role in the synthesis of porous structures. In addition, the morphology of precursors can usually be maintained in the final product. For instance, Fe-doped TiO2 nanorods with porous structures can be obtained by calcining an Fe-doped titanium glycolate precursor, and the resulting material shows high photocatalytic activity for the degradation of phenol under ultraviolet (UV) irradiation.15
To decrease the amorphous titanium glycolate to TiO2 conversion temperature, Song et al. explored a simple approach which was based on the in situ hydrolysis of titanium glycolate precursor microspheres (Fig. 4).32 By simply treating the titanium glycolate precursor in water at reflux temperature, nanoporous TiO2 (anatase) with a large surface area (244.6 m2 g−1) could be easily obtained without the addition of any templating surfactant. For comparison, calcining the glycolate precursor at 500 °C only resulted in a low surface area TiO2 sample (29.1 m2 g−1). The nanoporous TiO2 showed considerable activity towards the extraction of As(V) ions and the photodegradation of Eosin B, owing to its higher specific surface area and highly porous structure. Moreover, Zou et al. reported the synthesis of a porous TiO2–Ag core–shell nanocomposite material with a large surface area by in situ hydrolysis of Sn2+-grafted titanium glycolate microspheres in the presence of Ag+ ions. The as-prepared nanocomposite material was shown to serve as an efficient self-cleaning surface-enhanced Raman scattering (SERS) substrate.33
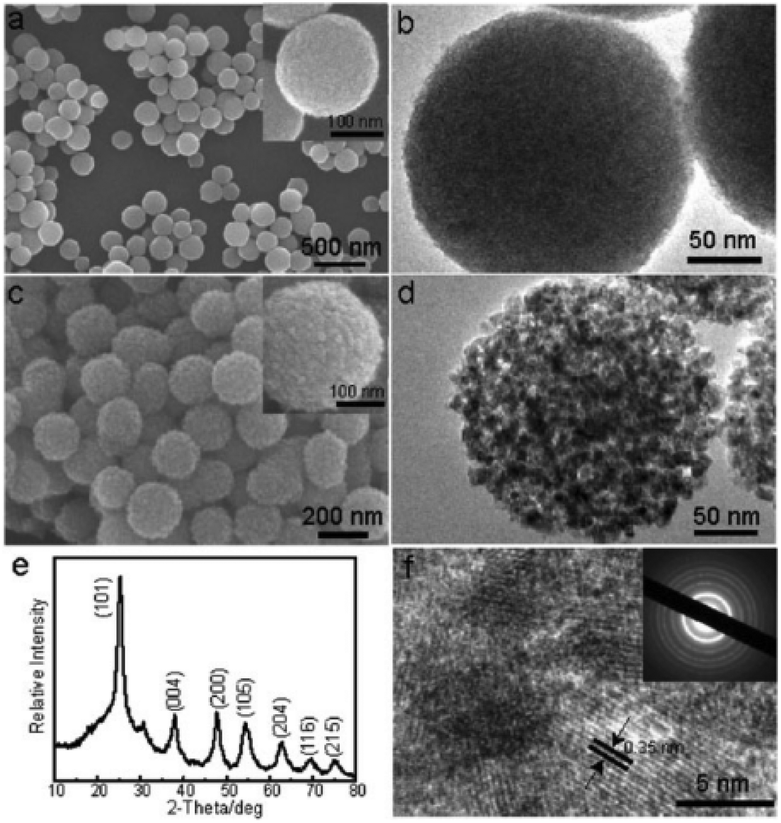 | ||
| Fig. 4 (a, c) SEM images and (b, d) TEM images of amorphous titanium glycolates and the resulting TiO2 material; (e) XRD pattern and (f) HRTEM image of the resulting TiO2 material.32 Reproduced with permission from The Royal Society of Chemistry. | ||
Titanium glycolates as kinds of organic–inorganic hybrid materials were proven to be photoactive towards UV light by Zou et al.34 Furthermore, it was also found that UV irradiation destroyed the structure of the titanium glycolates and removed their structural organic components, leading to the formation of TiO2 nanomaterials at room temperature.34 Compared with other thermally-driven conversion methods (see above), this light-induced synthetic strategy is green, facile, and controllable. It is worth noting that the as-obtained TiO2 nanomaterials have three main characteristics: the TiO2 material (i) is amorphous, differing from the crystalline TiO2 materials obtained by calcination; (ii) has large surface area owing to the nanoporous structure; and (iii) possesses a much richer surface density of hydroxyl groups than those in the thermally prepared TiO2 samples. Undoubtedly, this special TiO2 nanomaterial and subsequent materials obtained after its further processing will inevitably demonstrate the advantage in exploring this material for new functions. In this respect, Chen's group carried out a series of studies.34–39
5.1.1.1 Room-temperature spontaneous crystallization. The porous amorphous TiO2 materials obtained using a light-driven synthetic strategy still had some other special interesting aspects, one of which is the crystallization of anatase TiO2 from amorphous TiO2 at room temperature and atmospheric pressure (Fig. 5).35 This phase transition needs 80–90 days. It is believed that although the period for this transformation is longer than that required by conventional synthetic processes (e.g., thermally-driven procedures), this process is rather appealing because it is the first example of a room-temperature crystallization of a high-surface area anatase TiO2 material prepared from solvent-, additive-, and catalyst-free conditions. Obviously, this process is economically viable due to its energy-efficiency and cost-effectiveness. In addition, this spontaneous transformation was proven to be promoted by water molecules. Furthermore, the as-obtained porous TiO2 material possesses a large BET surface area of 400 m2 g−1, and a high photocatalytic activity for H2 evolution which is more than twice that of the benchmark P25 TiO2.
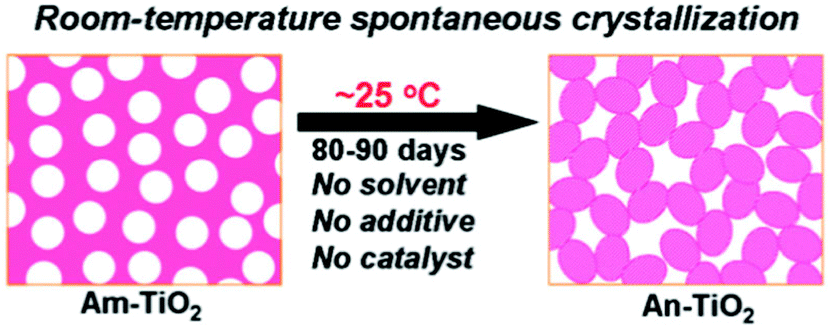 | ||
| Fig. 5 Schematic representation of the room-temperature spontaneous transformation from amorphous TiO2 (Am-TiO2) to anatase TiO2 (An-TiO2) without any solvent, additive and (or) catalyst. Temperature, 25 °C; time, 80–90 days.35 Reproduced with permission from The Royal Society of Chemistry. | ||
5.1.1.2 Photogenerated Ti3+ self-modified TiO2 materials. Ti3+ self-modified TiO2 materials are a significant research branch in the “defect engineering” of inorganic nanomaterials. Zou and coworkers have pointed out that the as-obtained porous amorphous TiO2 nanomaterial is one of the three main types of TiO2 materials that can be efficiently modified by photo-generated Ti3+.36 After UV irradiation, they discovered a phenomenon that the colour of the porous TiO2 could turn from white to intense blue under the protection of inert gas (N2), which demonstrated that a large number of Ti3+ species existed in the porous TiO2. The as-obtained Ti3+ modified TiO2 materials can effectively and selectively transform nitrobenzene into aniline. Importantly, the realization of this function is largely dependent on the characteristics of the materials. Ti3+ modified TiO2 materials with porous structures and large surface areas can attributed to the unique structure of titanium glycolate and its preparation using a light-driven method. In addition, photo-generated Ti3+ only exists on the TiO2 surface in water and cannot be exposed as it undergoes oxidation to Ti4+ when exposed to air. The Ti3+ charges were balanced by surface protons, rather than oxygen vacancies; and generation and consumption of Ti3+ is a proton-coupled process.
Following Zou's work, porous vanadium-doped TiO2 (V-TiO2) nanomaterial was obtained using the same method.28 The surface area and the amount of active hydrogen obviously increased in the obtained porous V-TiO2 nanomaterial. It was pointed out that the chemoselective hydrogenation of nitroarenes to aminoarenes can be easily achieved under ambient conditions using the as-obtained TiO2 materials (Fig. 6), which are proton-coupled electron transfer agents. The reduction reaction is instant (< 10 s) and highly selective. The rich H* species (i.e., coupled e−) and H+) produced from V-TiO2 (H*) were renewable during the reduction process through UV light irradiation of the reactant in methanol.
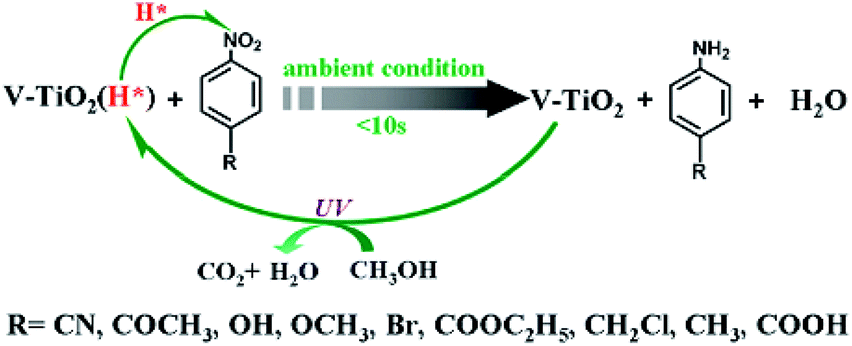 | ||
| Fig. 6 Schematic diagram for the selective hydrogenation of nitroarenes to aminoarenes by Ti3+ self-modified TiO2 materials and their regeneration process.28 Reproduced with permission from The Royal Society of Chemistry. | ||
5.1.1.3 Direct conversion of urea into graphitic carbon nitride. Mesoporous TiO2 nanospheres with a large surface area (460 m2 g−1) were successfully obtained using this light-assisted synthetic strategy, taking the amorphous titanium glycolate spheres as a precursor.37 Significantly, the as-obtained TiO2 nanomaterials can transform urea to graphitic carbon nitride under mild conditions. The authors claimed that this transformation was mainly attributed to a much richer surface density of hydroxyl groups on the large surface of TiO2. This work offers an implication about the importance of microstructure tuning on material properties.
5.1.1.4 Self-modified TiO2 materials for CO sensing and photocatalysis. In addition, thermal reduction can be used to prepare Ti3+ self-modified TiO2 material, by taking porous amorphous TiO2 (which was prepared through the UV-irradiation of titanium glycolate) as a reactant.38 Porous crystalline titania with heavily self-doped Ti3+ species were easily synthesized by heating a mixture of this porous amorphous TiO2 and urea at 700 °C in a nitrogen atmosphere, as shown in Fig. 7. Firstly, urea is converted to carbon nitride at c.a. 300 °C with the assistance of the porous TiO2 precursor, which in turn is coated by the as-formed C3N4 (see Fig. 7). When the temperature is elevated to 700 °C, the as-formed C3N4 decomposes and reacts with the porous TiO2, leading to the formation of Ti3+-TiO2 with a porous crystalline structure. Moreover, unlike photogenerated Ti3+, Ti3+ generated by thermal reduction of the as-obtained Ti3+-TiO2 was stable in air and showed unique functions. The resulting Ti3+ self-doped TiO2 nanomaterials can serve as efficient room-temperature gas-sensing materials for specific CO detection with fast response/recovery. The self-dopant (Ti3+) in the titania material has been proven to decrease the resistance of TiO2 significantly and to increase the chemisorbed oxygen species substantially, thus achieving excellent room-temperature sensing properties.
 | ||
| Fig. 7 Schematic representation of the preparation of Ti3+–TiO2 using porous amorphous TiO2 and urea as starting materials. With the increase of in the reaction temperature from room temperature (RT) to 700 °C, urea first converts into C3N4 on the TiO2 surface at around 300 °C, then the as-formed C3N4 totally decomposes at 700 °C and reacts with the porous TiO2, leading to the formation of Ti3+–TiO2 with a crystalline structure.38 Reprinted with permission from ref. 38. Copyright 2013 American Chemical Society. | ||
In addition, Zou et al. reported the synthesis of a TiO2 photocatalyst that was self-modified with a large number of oxygen vacancies using porous amorphous TiO2 as a reactant and imidazole as a reductant at 450 °C in air. This material was proven to exhibit satisfactory thermal and photo-stability, as well as improved photocatalytic activity for H2 evolution from water under visible light irradiation using methanol as a sacrificial reagent (Fig. 8).39
 | ||
| Fig. 8 (a) A typical time course plot of H2 evolution for self-modified TiO2 material under UV and visible light. (b) A typical time course plot of H2 evolution for this material under visible light irradiation. The reaction was continued for 70 h with the evacuation of H2 every 14 h (dashed line). Reaction conditions: catalyst 0.1 g; co-catalyst Pt (1 wt%); reaction solution aqueous methanol solution (25 vol%, 120 mL).39 Copyright © 2003 WILEY VCH Verlag GmbH & Co. KGaA Weinheim. | ||
 | ||
| Fig. 9 (A) SEM images and (B) HRTEM image of TiO2 microspheres with the corresponding TEM image (top) and fast Fourier transform image (bottom) shown in the inset.45 Reproduced with permission from The Royal Society of Chemistry. | ||
Besides the solvothermal method, a microemulsion-based approach was adopted to controllably prepare multi-hollow titanium glycerolates in a glycerol and isopropanol solvent mixture using diethylenetriamine (DETA) as an additive.46 Owing to this multi-hollow structure, the as-obtained TiO2 nanospheres as scattering centers showed a superior light-scattering effect as well as efficient electrolyte diffusion. As a result, a high DSSC efficiency (η) of 8.25% was achieved, corresponding to a 43% increase of η compared to that of P25 film.
5.2 Vanadium alkoxides and their derived V2O5 nanomaterials
Vanadium alkoxides, including vanadyl glycolates and vanadium glycolates, have been widely studied for the preparation of novel V2O5 nanomaterials, which are functional materials with a manifold of excellent performances and the potential for broad application.23,47–51 Vanadyl glycolate with the formula of VO(OCH2CH2O) is composed of one dimensional VO6 and VO5 chains (Fig. 10). This structure was successfully determined by Whittingham's group in 2003.23 Without any additives, the crystalline vanadyl glycolates were simply synthesized using solvothermal or reflux methods using the vanadium source (NH4VO3) and ethylene glycol as starting materials. Hollow vanadyl glycolate microspheres consisting of nanoparticles were prepared by Cao et al. without any surfactants.47 They proposed that NH4VO3 can react with ethylene glycol to release N2, and that the N2 microbubbles worked as a template to assist in the formation of hollow nanostructures. The thermally-treated material was also tested as a lithium-ion battery cathode and showed excellent cycle stability and a high rate performance owing to its structural features. In addition, nanostrip-like vanadyl glycolate48 and hierarchical vanadyl glycolate microspheres49 were also successfully prepared by other groups.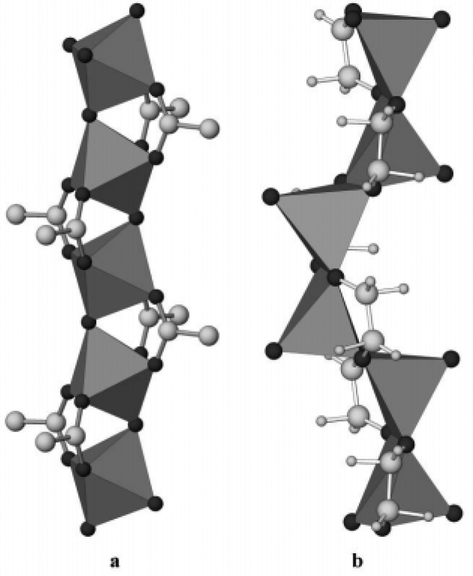 | ||
| Fig. 10 (a) The 1D chain of VO6 octahedra and (b) the VO5 square pyramids in the vanadyl glycolate structure.23 Reproduced with permission from The Royal Society of Chemistry. | ||
Vanadium glycolate is the other vanadium alkoxide precursor. Different from vanadyl glycolate, vanadium glycolate has no precise crystal structure, which is usually characteristic of a metal alkoxide. To pursue diversified nanostructures of V2O5, the synthesis of vanadium glycolate often requires the assistance of surfactants. For instance, in 2005, with the assistance of poly(vinylpyrrolidone) (PVP), Wan et al. synthesized hollow vanadium glycolate microspheres which were composed of nanorods.50 It was pointed out that the morphologies of the vanadium glycolate precursor were varied by changing the concentration of vanadium(III) acetylacetonate (V(acac)3) and PVP. The as-prepared vanadium glycolate hollow microspheres can be calcined into crystalline V2O5 without changing their morphology. The as-obtained vanadium hollow microspheres served as good lithium-ion battery cathode materials owing to their novel hollow morphology and porous structure.
Recently, in order to simplify the synthetic procedure, and to reduce economic cost, surfactant-free methods were very well implemented to obtain vanadium glycolate nanomaterials. Lou’s group prepared hollow vanadium glycolate microspheres consisting of hierarchical nanosheets using a simple template-free solvothermal method with VOC2O4 as a vanadium source.51 After thermal treatment in air, V2O5 hollow hierarchical microspheres (Fig. 11) were obtained. The as-synthesized material showed remarkable electrochemical performance with excellent rate capability and cycling stability as cathode materials for Li-ion batteries.
 | ||
| Fig. 11 (a) XRD pattern, (b) FESEM and (c, d) TEM images of the hierarchical V2O5 hollow microspheres obtained by annealing the as prepared V-glycolate hollow microspheres in air at 350 °C for 2 h.51 Reprinted with permission from ref. 51. Copyright © 2013 WILEY VCH Verlag GmbH & Co. KGaA Weinheim. | ||
5.3 Manganese alkoxides and their derived Mn-based oxide nanomaterials
In recent years, nanostructured manganese oxides have attracted a growing interest in the field of lithium ion batteries (LIBs) for their use as anode materials, essentially owing to their earth-abundance, non-toxicity, low cost and environmental friendliness.52 Among the manganese oxides, Mn2O3 materials with a high theoretical capacity of 1018 mA h g−1 and lower operating voltage are promising as an electrode material, but their capacity retention is generally poor.53 Recently, porous Mn2O3 nanoplates were prepared by Zhang et al. via a facile polyol solution method combined with a simple post-annealing process.54 The formation of porous Mn2O3 nanoplates benefited from the morphology-conserved transformation of Mn-glycolate precursor nanoplates. This material exhibited a higher reversible specific capacity of 813.7 mA h g−1 at a current density of 100 mA g−1 after 50 cycles. To address the low conductivity of Mn-based oxides, Lou et al. have successfully synthesized carbon nanofiber@MnO nanocomposites (CNF@MnO) with an enhanced lithium storage performance using a similar facile two-step alkoxide-assisted method (Fig. 12).55 The resulting nanocomposite exhibited better electrochemical performance with both high capacity and good cycling stability. The capacity could be stabilized at around 510 mA h g−1 with an ultra-long cycle life of 300 cycles.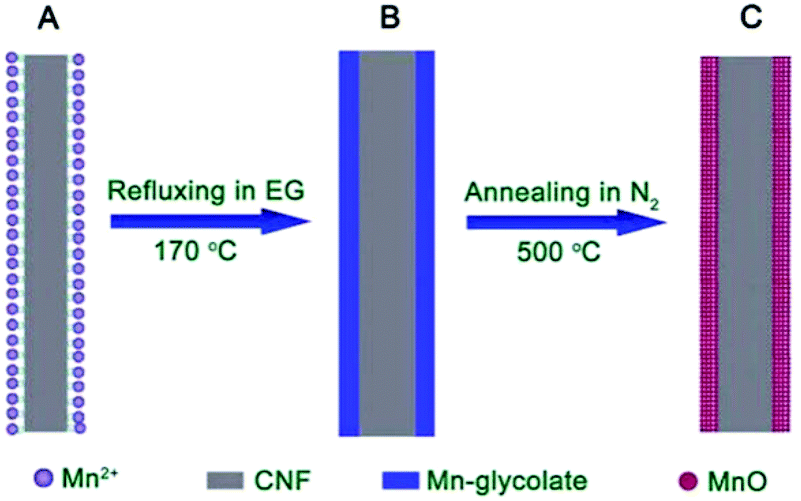 | ||
| Fig. 12 Schematic illustration of the formation of CNF@MnO coaxial nanocables: (A) CNF with functional groups complexed with Mn2+ on the surface; (B) CNF@Mn-glycolate precursor nanocables; (C) CNF@MnO coaxial nanocables after annealing at 500 °C for 4 h under N2.55 Reproduced with permission from The Royal Society of Chemistry. | ||
5.4 Iron alkoxides and their derived oxide nanomaterials
As an important class of solid-state metal alkoxides, iron alkoxides have also attracted great attention from researchers over the past few years, because their derived iron-based oxide nanomaterials (Fe2O3, Fe3O4) can be widely applied to many fields.56–61 Simply taking an iron source and ethylene glycol as raw materials, the as obtained iron glycolate does not have an exact crystal structure. However, with the assistance of additives, including urea,63–66 tetrabutylammonium bromide (TBAB),67 dimethylformamide (DMF),68 ethylenediamine (EDA),69 hexamethylenetetramine (HMTA),70–72N,N′-methylenebisacrylamide (MBA) and CH3COONa (NaAc),73 nanostructured iron alkoxides with special morphologies have been prepared in solvothermal systems.62–73 For example, Zhong et al. synthesized novel three-dimensional (3D) flower-like iron alkoxide through an ethylene glycol (EG)-mediated self-assembly process in the presence of urea and TBAB.67 After thermal treatment, iron oxides preserving the morphologies of their parent iron glycolate precursors can be obtained. It is pointed out that when the thermal treatment was performed in air, the as-obtained iron oxide was α-Fe2O3, while Fe3O4 was obtained in an inert atmosphere (e.g., N2, or Ar). The as-prepared iron oxide nanomaterials showed an excellent ability to remove heavy metal ions and other pollutants in water. In another important example, Lou’s group developed a simple ethylenediamine-mediated solvothermal method for the synthesis of iron alkoxide hollow microspheres composed of nanoplates.69 These iron alkoxide precursors can be readily converted into Fe3O4 hollow microspheres by thermal treatment under N2 atmosphere (Fig. 13). The resulting material was evaluated as an anode material for lithium-ion batteries and showed significantly improved lithium-storage capabilities with a very high reversible capacity of 580 mA h g−1 even after 100 cycles. | ||
| Fig. 13 (a) XRD pattern, (b, c) FESEM images, and (d–f) TEM images of the as-obtained Fe3O4 hollow microspheres. Image (f) is a magnified view of the area marked by a rectangle in (e).69 Copyright © 2013 WILEY VCH Verlag GmbH & Co. KGaA Weinheim. | ||
The microwave- or ultrasound-assisted hydrothermal method combined with calcination was also used to synthesize hierarchically structured iron oxides.74,75 For instance, Cao et al. prepared hierarchically nanostructured hollow spheres formed from the organization of nanosheets of a layer-structured iron alkoxide in the presence of sodium hydroxide (NaOH) and sodium dodecyl benzene sulfonate (SDBS).74 After thermal treatment, the as-obtained Fe3O4 or γ-Fe2O3 hollow spheres had a high drug loading capacity and showed favorable release properties for ibuprofen. In addition, Li et al. produced a flower-like hierarchical iron alkoxide precursor using an ultrasound-assisted hydrothermal method with the assistance of NH4Ac.76 The derived flower-like Fe3O4 microspheres with high BET surface areas (70.28 m2 g−1) exhibited fast adsorption rates and a high removal capacity for organic pollutants (such as Congo red). This magnetic material could be easily separated using a magnet, and provided a convenient way to remove organic pollutants in waste water treatment.
5.5 Cobalt alkoxides and their derived Co3O4 nanomaterials
For the purpose of investigating the stability and chemical nature of metal alkoxides, cobalt alkoxides were prepared in earlier years.77,78 The structure of cobalt glycerolate was first confirmed by Slade et al. in 1970.79 Using information from X-ray powder diffraction, they proposed the view that the basic unit of the cobalt glycerolate is a system of five-membered rings with a common C–O group, and that the whole structure contained double chains of such five-membered rings. The structure of cobalt glycolate was again proposed by Viau et al. in 2005, and they further investigated the magnetic properties of cobalt glycolates.80 Structural characterization showed that cobalt glycolate could be described as a stacking of layers made up of CoO6 octahedra sharing an edge and with the CH2CH2 moieties occupying the interlayer spacing. Besides the two types of cobalt alkoxides, the structures of other cobalt alkoxides have not been confirmed.As the focus turned from the basic properties of cobalt alkoxides to their morphology, which could be tuned by controlling the reaction conditions (type of alcohol, reagent concentration, and the addition of the surfactant), many works have been conducted to build unique morphologies.81–88 Larcher et al. first synthesized a series of cobalt alkoxides with a controllable morphology by changing the type of alcohol.82 Their results clearly showed that the morphology of the cobalt alkoxides had a strong relationship with the type of the alcohol: 1,3-propanediol had a tendency to form alkoxide octahedra, while 1,2-propanediol led to the formation of alkoxide nanospheres.
In addition, Viau et al. prepared nanoplate cobalt alkoxides by heating a mixture of cobalt acetate and ethanediol to 175 °C for 30 min, the orientation of which could be controlled by simply changing the ways it was deposited on a microscope support.80 Wan et al. modified the above method by adding PVP into the reaction system, demonstrating that the morphology of the cobalt alkoxides could transform from nanospheres to nanoplates and hierarchical structures.83 The reason that such hierarchical structures are formed is due to the stack of distorted plates which were first produced in the reaction system. It should be noted that the existence of PVP does not seem to have much impact upon the morphology. Chen et al. prepared porous hollow microspheres by nanoslicing without the addition of a surfactant.84 This result further confirmed that the crystal/molecular structure of metal alkoxides, intrinsically, played a crucial role in their nanosized structures. When the cobalt alkoxides were converted into Co3O4 using a simple calcination process, the morphologies of the alkoxides remained in some cases, while others showed charming changes. For example, Ma et al. obtained Co3O4 multishelled spheres by modulating the calcination condition of the cobalt glycolates, and the final materials exhibited a desired cycle stability and rate capacity when applied in lithium-ion batteries.85 Yu et al. prepared Co3O4 nanoplates which contained exposed (111) facets with a high reactivity towards the adsorption of heavy metal ions.86 Zhou et al. used a simple two-step method to synthesize a core–shell-type Co3O4 octahedron with a large surface area (190 m2 g−1) from a Co-1,3-propanediol precursor.16 This material was proven to have an enhanced photochemical water oxidation performance. Generally, all of the nanostructures stated above cannot be easily prepared by other methods, which further confirms the view that this precursor-directed method can be employed to prepare nanomaterials that cannot be readily achieved using other approaches.
Instead of directly calcining the cobalt alkoxides, Zhao et al. presented a hydrothermal treatment method for cobalt glycerolates. It is interesting that cobalt glycerolates can be used as templates, leading to the formation of a three-dimensional hierarchical spherical hollow structure without any additives.87 Further low-temperature calcination (200 °C) gave hollow Co3O4 microspheres composed of porous, ultrathin (<5 nm), single-crystal-like nanosheets (Fig. 14). The final Co3O4 material obtained showed an excellent performance in both electro- and photocatalytic water oxidation reactions.
 | ||
| Fig. 14 (A) SEM and (B and C) TEM images of hollow Co3O4 material. (D) HRTEM image of Co3O4 with the corresponding fast Fourier transform image in the inset.87 Reprinted with permission from ref. 87. Copyright 2014 Royal Society of Chemistry. | ||
5.6 Nickel alkoxides and their derived NiO nanomaterials
NiO nanomaterials have been commonly prepared from their hydroxide precursors.88 The use of strong bases (e.g., sodium hydroxide) in the reaction system might cause environmental concerns. Zhu et al. presented an alternative method for the preparation of NiO nanostructures using Ni glycolate as the precursor.89 Uniform, monodispersed Ni glycolate was synthesized in the presence of additives, sodium chloride and sodium acetate. The additives were proven to play important roles in the control of the dispersibility and uniformity of the glycolate. By calcining the alkoxide precursor at 300 °C, nanoflake assembled NiO nanospheres can be successfully obtained (Fig. 15). The resulting NiO nanospheres with a high surface area (222 m2 g−1) were shown to exhibit excellent adsorption for Congo red (a toxic dye) with a maximum capacity of 440 mg g−1. The improved adsorption performance could be correlated to the unique hierarchical structure and high surface area of the porous NiO nanospheres.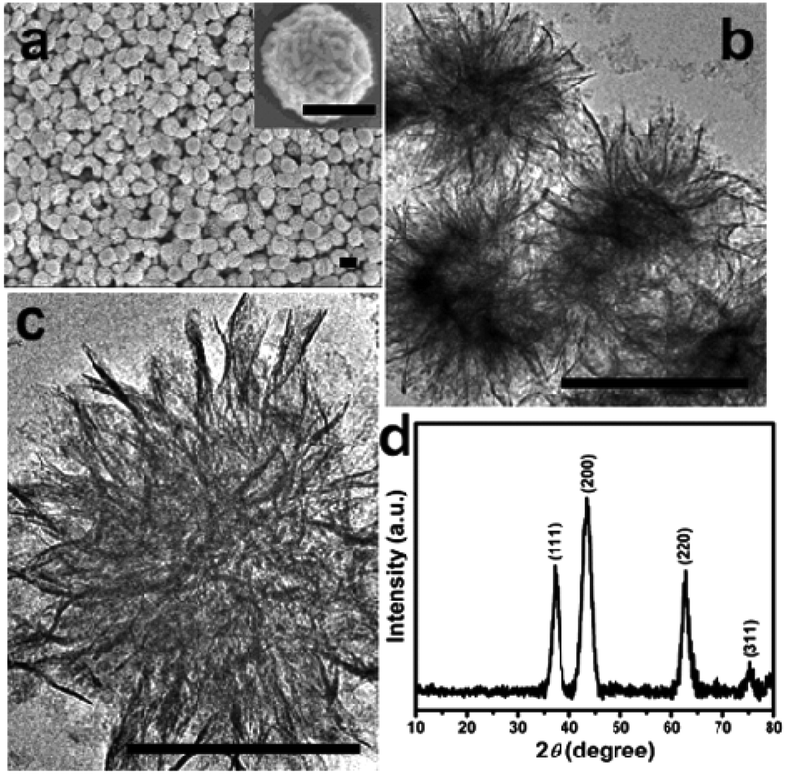 | ||
| Fig. 15 FESEM image (a), TEM images (b and c), and XRD pattern (d) of the NiO nanospheres (scale bar: 500 nm).89 Reprinted with permission from ref. 89. Copyright 2012 American Chemical Society. | ||
5.7 Copper alkoxides and their derived CuO nanomaterials
Copper alkoxide nanostructures were first obtained by Gao et al. in 2007.90 In this paper, the authors reported the synthesis of copper glycolate using a solvothermal system containing copper acetate and ethylene glycol. The morphology of copper glycolate (microplates, doughnut-like structures and multilayer microspheres) could be tuned by adjusting the concentration of the starting materials. The unique nanostructures of copper glycolate were preserved upon annealing at 400 °C to form CuO nanomaterials. For example, the BET surface area of doughnut-like CuO is up to 87 m2 g−1 and its removal ability for As(III) in water is about three-fold higher than that of its commercial counterpart. In another report, a novel 3D hierarchical cotton-candy-like copper glycolate was prepared by adding urea into the reaction system. Similarly, the CuO was synthesized by heating Cu glycolate in air. The as-obtained cotton-candy-like CuO with a high surface area (119 m2 g−1) was composed of highly porous nanofibers (Fig. 16). The adsorption capacity of this material for As(III) is about 12.9 mg g−1, which is obviously higher than doughnut-like CuO (5.7 mg g−1).41 | ||
| Fig. 16 (a, b) SEM images of the copper glycolate precursor and (c, d) TEM images of the CuO.41 Reprinted with permission from ref. 41. Copyright 2013 American Chemical Society. | ||
5.8 Zinc alkoxides and their derived ZnO nanomaterials
In general, the alkoxide-directed method is relatively safe, less-consumptive, easy to operate, and applicable to many metals and several kind of alcohols.91 Zn alkoxides, like Zn glycolate and Zn glycerolate, can also be easily synthesized from Zn salts and corresponding alcohols. Zhang et al. reported the synthesis of one-dimensional zinc glycolate nanorods having sharp or round tips.92 When exposed to moisture or washed with alcohol, the morphology of the nanorods changed greatly. They became thinner when washed with absolute alcohol; or were eroded when exposed to moisture. The authors proposed that ethylene glycol molecules were washed away or replaced by water molecules. Moreover, the same group synthesized octahedral-shaped zinc glycolate microparticles using an ultrasonic-assisted method.93 When heated at 350 °C or higher, octahedral ZnO microcrystals could be obtained. The above results also provide useful ways to synthesize ZnO with a tunable structure by controlling the surface reactivity of metal alkoxides.Glycerol can also react with zinc ions to form alkoxide nanostructures in place of ethylene glycol. For example, Zhao et al. synthesized zinc monoglycerolate using a mild solvothermal reaction system, in which zinc acetate dehydrate and glycerol were used as reactants, and isopropanol functioned as a solvent.94 Experimental results showed that isopropanol as the supporting solvent played an important role in the formation of the hierarchical zinc mono-glycerolate nanostructure. Two functions of isopropanol were proposed by the authors. They included: (i) decreasing the speed of the reaction; and (ii) controlling the crystal facets of zinc mono-glycerolate particles. Thermal treatment of zinc alkoxide at 400 °C led to the formation of hierarchical ZnO with a highly porous structure (Fig. 17). This porous ZnO material showed an excellent ethanol sensing ability with fast response and high sensitivity.
 | ||
| Fig. 17 (A) XRD pattern and (B, C, D) SEM images of the ZnO nanomaterials derived from zinc glycerolates.94 Reproduced with permission from The Royal Society of Chemistry. | ||
5.9 Indium, tin, lead, cerium, bismuth and tungsten alkoxides and their derived metal oxides
In recent years, In,40 Sn,95 Pb,95 Ce96 and Bi97 alkoxides were also synthesized by different groups. Wang et al. reported the simple synthesis of a 3D flower-like In-glycerolate nanostructure, and Xia's group synthesized Sn and Pb glycolate nanowires. In addition, Wan's group prepared Ce glycolate nanoflowers, and Nan's group synthesized Bi glycolate nanospheres. Through a simple thermal treatment, the above-mentioned alkoxides can be readily transformed into their corresponding metal oxide nanomaterials with potential applications in sensing, photocatalysis and water treatment. For example, the as-obtained SnO2 nanowires (Fig. 18) can be used as a room-temperature sensing material for CO and ethanol detection. It should be pointed out that most of the SnO2 materials do not have a sensing response at room temperature. | ||
| Fig. 18 (A and B) SEM images of SnO2 nanowires before (A) and after (B) calcination. (C and D) High-magnification SEM and TEM images of the SnO2 nanowires. The inset shows a typical SAED pattern of the nanowires, which could be indexed to SnO2.95 Reprinted with permission from ref. 95. Copyright 2003 American Chemical Society. | ||
In another paper, Zou et al.98 reported a W-benzyl alcohol nanohybrid, which could be thermally transformed into single crystalline WO3 nanoplates with an uneven surface. Due to the unique surface structure, this material exhibited excellent acetone sensing properties.
5.10 Yttrium, neodymium, samarium, gadolinium alkoxides
Besides tungsten, some rare earth elements (Y,99 Nd, Sm and Gd100) can also react with benzyl alcohol to form hybrid alkoxides. This method is described as a “benzyl alcohol route”, leading to ordered hybrid materials composed of metal oxide nanobuilding blocks with at least one dimension on the sub-nanometer scale.19 The π–π interaction of phenyl groups in benzyl alcohol molecules might be the main chemical driving force for the formation of solid-state alkoxides. For example, Karmaoui et al. synthesized crystalline samarium, neodymium and gadolinium nanohybrids using this simple one-pot reaction process.100 The as-obtained neodymium and gadolinium samples consisted of a lamellar structure oriented in the same direction, Nd2O3 or Gd2O3 equally spaced by an organic layer formed by benzoate molecules, and the periodic lamellar structure is kept together by simple π–π interactions between the phenyl rings. The gadolinium based nanohybrids showed outstanding optical emission properties when doped with terbium(III) and europium(III). However, there are no further studies on the conversion of these rare earth hybrid alkoxides into metal oxides. This should be worthy of further study for the development of new functions for the derived rare-earth metal oxides.6. Conclusions and outlook
In this article, we reviewed the recent developments in the synthesis of solid-state small molecule metal alkoxides with controllable composition, morphology and size, the transformation of metal alkoxides into oxide nanostructures, and the properties/functions of the resulting oxide nanomaterials. It has been demonstrated that the novel precursor-directed synthetic route to functional nanomaterials is simple, universal, and effective. Despite the many achievements that have been made in the field of solid-state metal alkoxides, as outlined in this review, there are still some difficulties that should be addressed in the future (Fig. 19). (i) Most metal alkoxides have no precise crystal structure (e.g., titanium glycerolate and iron glycolate), because it is very difficult to prepare large single crystals for further analysis. Therefore, this calls for more research work on the crystal engineering of metal alkoxides. (ii) The number of metal elements involved in solid-state metal alkoxides is 19, which only accounts for 20% of the total metal elements. So, it is necessary to expand solid-state metal alkoxide chemistry to the as yet unexplored metal elements (e.g., Cr, Sr). (iii) Although many metal alkoxides with novel structures have been prepared successfully, the formation mechanism/rules for them on both molecular and nanoscale levels are still unclear. It is necessary to investigate the reaction mechanism, probably through the integration of in situ characterization and theoretical calculations. (iv) The morphological and structural features of the resulting oxides are closely related not only to their metal alkoxides, but also to the post-treatment methods used. So, it is essential to establish a relationship between alkoxides and their derived oxides and explore new effective conversion methods with the aim of obtaining nanostructured oxides with fascinating properties. (v) In order to achieve the goal-directed design of metal alkoxides and their derived functional nanomaterials, more studies need to be carried out and carefully summarized.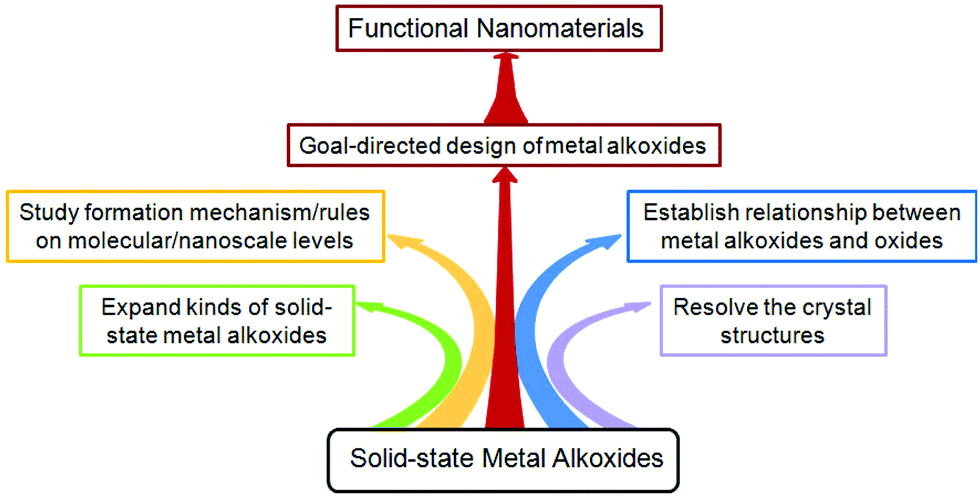 | ||
| Fig. 19 The research outlook for solid-state metal alkoxides. | ||
Acknowledgements
This work was supported by the NSFC (21371070, 21401066); the National Basic Research Program of China (2013CB632403); Jilin province science and technology development projects (20140101041JC, 20130204001GX); Graduate Innovation Fund of Jilin University (2014052).Notes and references
- T. Appenzeller, Science, 1991, 254, 1300 CAS.
- B. D. Gates, Q. Xu, M. Stewart, D. Ryan, C. G. Willson and G. M. Whitesides, Chem. Rev., 2005, 105, 1171 CrossRef CAS PubMed.
- M. Pagliaro, G. Palmisano, R. Ciriminna and V. Loddo, Energy Environ. Sci., 2009, 2, 838 CAS.
- P. Poizot, S. Laruelle, S. Grugeon and L. T. Dupont, Nature, 2000, 407, 496 CrossRef CAS PubMed.
- (a) V. F. Puntes, K. M. Krishnan and A. P. Alivisatos, Science, 2001, 291, 2115 CrossRef CAS PubMed; (b) E. T. Kang and Y. Zhang, Adv. Mater., 2000, 12, 1481 CrossRef CAS.
- S. C. Erwin, L. Zu, M. I. Haftel, A. L. Efros, T. A. Kennedy and D. J. Norris, Nature, 2005, 436, 91 CrossRef CAS PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638 CrossRef CAS PubMed.
- (a) G. Schedelbeck, Science, 1997, 278, 1792 CrossRef CAS; (b) G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- G. A. Ozin, Adv. Mater., 1992, 4, 612 CrossRef CAS.
- R. Garcia, R. V. Martinez and J. Martinez, Chem. Soc. Rev., 2006, 35, 29 RSC.
- (a) B. Geng, F. Zhan, C. Fang and N. Yu, J. Mater. Chem., 2008, 18, 4977 RSC; (b) H. B. Yao, X. Zhang, X. H. Wang, S. H. Yu and J. Li, Dalton Trans., 2011, 40, 3191 RSC; (c) L. Xu, Z. Li, Q. Cai, H. Wang, H. Gao, W. Lv and J. Liu, CrystEngComm, 2010, 12, 2166 RSC.
- R. C. Mehrotra, Proc. Indian Natn. Sci. Acad., 1986, 52, 954 CAS.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893 CrossRef CAS PubMed.
- D. Wang, R. Yu, N. Kumada and N. Kinomura, Chem. Mater., 1999, 11, 2008 CrossRef CAS.
- X. X. Zou, G. D. Li, M. Y. Guo, X. H. Li, D. P. Liu, J. Su and J. S. Chen, Chem. – Eur. J., 2008, 14, 11123 CrossRef PubMed.
- L. J. Zhou, Y. C. Zou, G. D. Li, X. X. Zou, J. Zhao, M. H. Fan, Y. P. Liu and D. J. Wang, RSC Adv., 2014, 4, 22951 RSC.
- D. Larcher, G. Sudant, R. Patriceand and J. M. Tarascon, Chem. Mater., 2003, 15, 3543 CrossRef CAS.
- (a) R. P. Eckberg, W. E. Hatfield and D. B. Losee, Inorg. Chem., 1974, 13, 740 CrossRef CAS; (b) T. W. Hambley and M. R. Snow, Aust. J. Chem., 1983, 36, 1249 CrossRef CAS.
- N. Pinna, J. Mater. Chem., 2007, 17, 2769 RSC.
- D. Chen, L. Gao, A. Yasumori, K. Kuroda and Y. Sugahara, Small, 2008, 4, 1813 CrossRef CAS PubMed.
- (a) D. C. Bradley, Chem. Rev., 1989, 89, 1317 CrossRef CAS; (b) C. D. Lakeman and D. A. Payne, Mater. Chem. Phys., 1994, 38, 305 CrossRef CAS.
- S. Acosta, R. Corriu, D. Leclercq, P. H. Mutinand and A. Vioux, J. Sol-Gel Sci. Technol., 1994, 2, 25 CrossRef CAS.
- C. Weeks, Y. Song, M. Suzuki, N. A. Chernova, P. Y. Zavalij and M. S. Whittingham, J. Mater. Chem., 2003, 13, 1420 RSC.
- (a) Y. Bai, I. Mora-Seró, F. D. Angelis, J. Bisquert and P. Wang, Chem. Rev., 2014, 114, 10095 CrossRef CAS PubMed; (b) X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed; (c) H. Chen, C. E. Nanayakkara and V. H. Grassian, Chem. Rev., 2012, 112, 5919 CrossRef CAS PubMed; (d) S. U. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243 CrossRef CAS PubMed.
- A. S. Campbell, C. Dong, A. Maloney, J. Hardinger, X. Hu, F. Meng, A. Guiseppe-Elie, N. Wu and C. Z. Dinu, Nano Life, 2014, 4, 145005 CrossRef.
- (a) X. Jiang, Y. Wang, T. Herricks and Y. Xia, J. Mater. Chem., 2004, 14, 695 RSC; (b) M. Wei, H. Zhou, Y. Konishi, M. Ichihara, H. Sugih and H. Arakawa, Inorg. Chem., 2006, 45, 5684 CrossRef CAS PubMed.
- X. C. Jiang, T. Herricks and Y. Xia, Adv. Mater., 2003, 15, 1205 CrossRef CAS.
- J. Su, X. X. Zou, G. D. Li, L. Li, J. Zhao and J. S. Chen, Chem. Commun., 2012, 48, 9032 RSC.
- H. Li, D. Wang, P. Wang, H. M. Fan and T. F. Xie, Chem. – Eur. J., 2009, 15, 12521 CrossRef CAS PubMed.
- S. H. Liu, H. P. Jia, L. Han, J. L. Wang, P. F. Gao, D. D. Xu, J. Yang and S. N. Che, Adv. Mater., 2012, 24, 3201 CrossRef CAS PubMed.
- X. X. Zou, G. D. Li, J. Zhao, J. Su, X. Wei, K. X. Wang, Y. N. Wangand and J. S. Chen, Int. J. Photoenergy., 2012, 2012, 720183 CrossRef PubMed.
- L. S. Zhong, J. S. Hu, L. J. Wan and W. G. Song, Chem. Commun., 2008, 1184 RSC.
- X. X. Zou, R. Silva, X. Huang, J. A-Sharab and T. Asefa, Chem. Commun., 2013, 49, 382 RSC.
- X. X. Zou, G. D. Li, K. X. Wang, L. Li, J. Su and J. S. Chen, Chem. Commun., 2010, 46, 2112 RSC.
- J. Su, X. X. Zou, G. D. Li, Y. M. Jiang, Y. Cao, J. Zhao and J. S. Chen, Chem. Commun., 2013, 49, 8217 RSC.
- J. Su, X. Zou and J. S. Chen, RSC Adv., 2014, 4, 13979 RSC.
- X. X. Zou, G. D. Li, Y. N. Wang, J. Zhao, C. Yan, M. Y. Guo, L. Li and J. S. Chen, Chem. Commun., 2011, 47, 1066 RSC.
- J. Su, X. X. Zou, Y. C. Zou, G. D. Li, P. P. Wang and J. S. Chen, Inorg. Chem., 2013, 52, 5924 CrossRef CAS PubMed.
- X. X. Zou, J. K. Liu, S. Su, F. Zuo, J. S. Chen and P. Y. Feng, Chem. – Eur. J., 2013, 19, 2866 CrossRef CAS PubMed.
- C. Wang, D. Chen and X. L. Jiao, J. Phys. Chem. C, 2009, 113, 7714 CAS.
- X. Y. Yu, R. X. Xu, C. Gao, T. Luo, Y. Jia, J. H. Liu and X. J. Huang, ACS Appl. Mater. Interfaces, 2012, 4, 1954 CAS.
- J. Das, F. S. Freitas, I. R. Evans, A. F. Nogueira and D. Khushalani, J. Mater. Chem., 2010, 20, 4425 RSC.
- (a) G. H. Tian, Y. J. Chen, W. Zhou, K. Pan, C. G. Tian, X. R. Huang and H. G. Fu, CrystEngComm, 2011, 13, 2994 RSC; (b) G. Tian, K. Pan, Y. Chen, J. Zhou, X. Miao, W. Zhou, R. Wang and H. Fu, J. Power Sources, 2013, 238, 350 CrossRef CAS PubMed.
- Z. F. Bian, J. Zhu, J. G. Wang, S. X. Xiao, C. Nuckolls and H. X. Li, J. Am. Chem. Soc., 2012, 134, 2325 CrossRef CAS PubMed.
- J. Zhao, X. X. Zou, J. Su, P. P. Wang, L. J. Zhou and G. D. Li, Dalton Trans., 2013, 42, 4365 RSC.
- J. Jiang, F. Gu, W. Shao and C. Li, Ind. Eng. Chem. Res., 2012, 51, 2838 CrossRef CAS.
- E. Uchaker, N. Zhou, Y. W. Li and G. Z. Cao, J. Phys. Chem. C, 2013, 117, 1621 CAS.
- P. Ragupathy, S. Shivakumara, H. N. Vasan and N. Munichandraiah, J. Phys. Chem., 2008, 112, 16700 CrossRef CAS.
- H. E. Wang, D. S. Chen, Y. Cai, R. L. Zhang, J. M. Xu, Z. Deng, X. F. Zheng, Y. Li, I. Bello and B. L. Su, J. Colloid Interface Sci., 2014, 418, 74 CrossRef CAS PubMed.
- A. M. Cao, J. S. Hu, H. P. Liang and L. J. Wan, Angew. Chem., Int. Ed., 2005, 44, 4391 CrossRef CAS PubMed.
- A. Pan, T. Zhu, H. B. Wu and X. W. Lou, Chem. – Eur. J., 2013, 19, 494 CrossRef CAS PubMed.
- Y. Deng, L. Wan, Y. Xie, X. Qin and G. Chen, RSC Adv., 2014, 4, 23914 RSC.
- M. W. Xu, Y. B. Niu, S. J. Bao and C. M. Li, J. Mater. Chem. A, 2014, 2, 3749 CAS.
- Y. J. Zhang, Y. Yan, X. Y. Wang, G. Li, D. R. Deng, L. Jiang, C. C. Shu and C. R. Wang, Chem. – Eur. J., 2014, 20, 6126 CrossRef CAS PubMed.
- G. Q. Zhang, H. B. Wu, H. E. Hostera and X. W. Lou, Energy Environ. Sci., 2014, 7, 302 CAS.
- E. Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758 RSC.
- M. B. Gawande, P. S. Branco and R. S. Varm, Chem. Soc. Rev., 2013, 42, 3371 RSC.
- W. Yan, H. L. Lien, B. E. Koelc and W. X. Zhang, Environ. Sci. Processes Impacts, 2013, 15, 63 CAS.
- B. I. Kharisov, H. V. R. Dias, O. V. Kharissova, V. M. Jiménez-Pérez, B. O. Pérez and B. M. Flores, RSC Adv., 2012, 2, 9325 RSC.
- N. A. Frey, S. Peng, K. Cheng and S. Sun, Chem. Soc. Rev., 2009, 38, 2532 RSC.
- Y. Pan, X. Du, F. Zhao and B. Xu, Chem. Soc. Rev., 2012, 41, 2912 RSC.
- X. H. Ma, X. Y. Feng, C. Song, B. K. Zou, C. X. Ding, Y. Yu and C. H. Chen, Electrochim. Acta, 2013, 93, 131 CrossRef CAS PubMed.
- S. W. Cao and Y. J. Zhu, J. Phys. Chem. C, 2008, 112, 12149 CAS.
- Q. Gao, A. W. Zhao, Z. B. Gan, W. Y. Tao, D. Li, M. F. Zhang, H. Y. Guo, D. P. Wang, H. H. Sun, R. R. Maa and E. Liu, CrystEngComm, 2012, 14, 4834 RSC.
- G. J. Su, Y. X. Liu, L. Y. Huang, H. J. Lu, S. Liu, L. W. Li and M. H. Zheng, Chemosphere, 2014, 99, 216 CrossRef CAS PubMed.
- J. J. Zhang, Y. L. Chen, Y. F. Sun, T. Huang and A. S. Yu, RSC Adv., 2013, 3, 20639 RSC.
- L. S. Zhong, J. S. Hu, H. P. Liang, A. M. Cao, W. G. Song and L. J. Wan, Adv. Mater., 2006, 18, 2426 CrossRef CAS.
- J. S. Xu and Y. J. Zhu, CrystEngComm, 2011, 13, 5162 RSC.
- B. Wang, H. B. Wu, L. Zhang and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 4165 CrossRef CAS PubMed.
- Y. C. Chen, K. Zhang, Y. L. Min, Y. G. Zhang and R. Zhang, Mater. Chem. Phys., 2010, 123, 378 CrossRef CAS PubMed.
- S. Jin, H. Deng, D. Long, X. J. Liu, L. Zhan, X. Liang, W. Qiao and L. Ling, J. Power Sources, 2011, 196, 3887 CrossRef CAS PubMed.
- J. S. Xu and Y. J. Zhu, J. Colloid Interface Sci., 2012, 385, 58 CrossRef CAS PubMed.
- J. S. Xu, Y. J. Zhu and F. Chen, J. Solid State Chem., 2013, 199, 204 CrossRef CAS PubMed.
- S. W. Cao, Y. J. Zhu, M. Y. Ma, L. Li and L. Zhang, J. Phys. Chem. C, 2008, 112, 1851 CAS.
- Y. Jung, Y. H. Son and J. K. Lee, RSC Adv., 2012, 2, 5877 RSC.
- X. Y. Li, Z. J. Si, Y. Q. Lei, X. N. Li, J. K. Tang, S. Y. Song and H. J. Zhang, CrystEngComm, 2011, 13, 642 RSC.
- R. C. Mehrotra, J. Non-Cryst. Solids, 1988, 100, 1 CrossRef CAS.
- H. Pfalzgraf and L. G. Coord, Chem. Rev., 1998, 178–180, 967 Search PubMed.
- P. G. Slade, E. W. Radoslovich and M. Raupach, Acta Crystallogr., 1971, 27, 2432 CrossRef CAS.
- N. Chakroune, G. Viau, S. Ammar, N. Jouini, P. Gredin, M. J. Vaulay and F. Fievet, New J. Chem., 2005, 29, 355 RSC.
- F. L. Pratt, P. J. Baker, S. J. Blundell, T. Lancaster, M. A. Green and M. Kurmoo, Phys. Rev. Lett., 2007, 99, 17202 CrossRef CAS.
- D. Larcher, G. Sudant, R. Patrice and J. M. Tarascon, Chem. Mater., 2003, 15, 3543 CrossRef CAS.
- A. M. Cao, J. S. Hu, H. P. Liang, W. G. Song, L. J. Wan, X. L. He, X. G. Gao and S. H. Xi, J. Phys. Chem. B, 2006, 110, 15858 CrossRef CAS PubMed.
- Y. C. Chen, L. Hu, M. Wang, Y. L. Min and Y. G. Zhang, Colloids Surf., A, 2009, 336, 64 CrossRef CAS PubMed.
- X. Wang, X. L. Wu, Y. G. Guo, Y. Zhong, X. Q. Cao, Y. Ma and J. N. Yao, Adv. Funct. Mater., 2010, 20, 1680 CrossRef CAS.
- X. Y. Yu, Q. Q. Meng, T. Luo, Y. Jia, B. Sun, Q. X. Li, J. H. Liu and X. J. Huang, Scientific reports, 2013, 3, 2886 Search PubMed.
- J. Zhao, Y. C. Zou, X. X. Zou, T. Y. Bai, Y. P. Liu, R. Q. Gao, D. J. Wang and G. D. Li, Nanoscale, 2014, 6, 7255 RSC.
- (a) Y. Wang, Q. Zhu and H. Zhang, Chem. Commun., 2005, 5231 RSC; (b) X. Li, S. Xiong, J. Li, J. Bai and Y. Qian, J. Mater. Chem., 2012, 22, 14276 RSC.
- T. Zhu, J. S. Chen and X. W. Lou, J. Phys. Chem. C, 2012, 116, 6873 CAS.
- A. M. Cao, J. D. Monnell, C. Matranga, J. M. Wu, L. L. Cao and D. Gao, J. Phys. Chem. C, 2007, 111, 18624 CAS.
- N. Mira, M. SalavatiN and F. Davar, Chem. Eng. J., 2012, 181–182, 779 CrossRef PubMed.
- J. W. Zhang, P. L. Zhu, Z. W. Li, J. M. Chen, Z. S. Wu and Z. J. Zhang, Nanotechnology, 2008, 19, 165605 CrossRef PubMed.
- J. W. Zhang, P. L. Zhu, J. H. Li, J. M. Chen, Z. S. Wu and Z. J. Zhang, Cryst. Growth Des., 2009, 9, 2329 CAS.
- J. Zhao, X. X. Zou, L. J. Zhou, L. L. Feng, P. P. Jin, Y. P. Liu and G. D. Li, Dalton Trans., 2013, 42, 14357 RSC.
- Y. L. Wang, X. C. Jiang and Y. N. Xia, J. Am. Chem. Soc., 2003, 125, 16176 CrossRef CAS PubMed.
- L. S. Zhong, J. S. Hu, A. M. Cao, Q. Liu, W. G. Song and L. J. Wan, Chem. Mater., 2007, 19, 1648 CrossRef CAS.
- X. Xiao, R. P. Hu, C. Liu, C. L. Xing, C. Qian, X. X. Zuo, J. M. Nan and L. S. Wang, Appl. Catal., B, 2013, 140–141, 433 CrossRef CAS PubMed.
- X. X. Zou, G. D. Li, P. P. Wang, J. Zhao, L. J. Zhou, Y. N. Wang and J. S. Chen, Dalton Trans., 2012, 41, 9773 RSC.
- N. Pinna, G. Garnweitner, P. Beato, M. Niederberger and M. Antonietti, Small, 2005, 1, 1 CrossRef.
- M. Karmaoui, R. A. Sá Ferreira, A. T. Mane, L. D. Carlos and N. Pinna, Chem. Mater., 2006, 18, 4493 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © the Partner Organisations 2015 |