
Removal of CO2 from CH4 and CO2 capture in the presence of H2O vapour in NOTT-401†
Hugo A.
Lara-García
,
Maximiliano R.
Gonzalez
,
Juan H.
González-Estefan
,
Pedro
Sánchez-Camacho
,
Enrique
Lima
and
Ilich A.
Ibarra
*
Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n, CU, Del. Coyoacán, 04510, México D. F., Mexico. E-mail: argel@unam.mx
First published on 15th April 2015
Abstract
From a binary equimolar gas-mixture of CO2 and CH4, NOTT-401 exhibits CO2 separation from CH4. By kinetic uptake experiments, NOTT-401 shows a maximum of 1.47 wt% CO2 capture at 30 °C and a significant 7-fold increase (∼9.90 wt%) in CO2 capture under 40% relative humidity.
Introduction
The use of natural gas as fuel is indeed very advantageous for the environment and is also economically attractive. If natural gas is used as a vehicular fuel, the reductions in CO, SO2 and CO2 emissions will be 97, 90 and 24%, respectively, and there will be no release of lead in exhaust gases. In addition, it is cheaper than gasoline or diesel. To date, natural gas supplies one-fourth of the energy needed in homes, factories, business, vehicles, industries and power plants around the world. It is estimated that the consumption of natural gas will grow by 50% over the next 10 years.1,2 The main constituent of natural gas is methane (CH4), which has the highest hydrogen to carbon (H/C) ratio among all hydrocarbon fuels.3 However, the quality of natural gas, coming from land fields and biogas plants, is considerably low with impurities like CO2 (20 to 35%), N2, H2O and H2S.4 Then, pre-combustion CO2 sequestration from natural gas is essential to maximize its energy content (CH4). In addition, post-combustion CO2 capture from plant flue gas is also crucial in order to control greenhouse emissions.5CO2 directly affects our environment by raising global temperature and causing acidification of the oceans.6 The main reason for the increasing CO2 levels is the accelerating global energy demands.7 These energy requirements are expanding fast due to rapid growth in world population, increase in standards of living and the development of technologies leading to a doubling in the energy demand over the last three decades.8 Thus, CO2 separation and capture have motivated many governments to invest in the development of new methods for efficiently and effectively capturing CO2.9 Typical absorption in aqueous alkanolamine solutions has been widely used and studied, but it has many major limitations as an adsorbent for industrial CO2 capture due to its heat instability and corrosion towards vessels and pipelines.10 Therefore, the use of porous solids for the adsorption of CO2 is an appropriate research area and the search for materials with a high adsorption capacity, structural stability, fast sorption kinetics and mild regeneration properties, remains a major challenge.
Porous coordination polymers (PCPs) or metal–organic frameworks (MOFs) are among the most promising candidates for gas separation, because their sorption selectivity towards small molecule adsorbates is directly tunable as a function of the topology and chemical composition of the micropores.11 Porous metal–organic materials showing high surface area and high pore volume normally show high CO2 storage capacities at room temperature and relatively high pressures.12 Although PCPs show high CO2 capacity and selectivity, many gas separation processes involve the exposure to water vapor.
However, a small number of PCPs have shown good stability to water, and water is most often unfavorable to gas separations.13 Along with those few examples, Hong et al.14 reported a water-stable PCP based on a binuclear [In2(μ2-OH)] building block (see Scheme S1, ESI†), InOF-1, constructed from a flexible BPTC4− ligand (H4BPTC = biphenyl-3,3′,5,5′-tetracarboxylic acid) which also showed a high CO2/N2 and CO2/CH4 selectivities (by using the experimental single-component gas adsorption isotherms). The effect of water on the CO2 capture has only recently been investigated on PCPs.15 Matzger and co-workers15b studied the effect of humidity on the performance of M/DOBDC (M = ZnII, NiII, CoII or MgII) by collecting N2/CO2/H2O breakthrough curves at different relative humidities. LeVan et al.16 found that a small amount of water did not decrease but may actually increase the CO2 capacity of PCPs.
Interestingly, Llewellyn and co-workers17 investigated the CO2 adsorption in some PCPs under different relative humidities. Indeed, HKUST-1 was shown to degrade in the presence of humidity, and UiO-66 did not show any enhanced CO2 uptake.17 In the case of MIL-100(Fe), a remarkable 5-fold increase in CO2 uptake was observed with increasing relative humidity (RH), 105 mg g−1 at 40% RH. In addition, Yaghi et al.18 showed that the presence of hydroxyl functional groups increase the affinity of the framework for water. Thus, in the present work we have chosen a material NOTT-40119 (Fig. 1) based on a binuclear [Sc2(μ2-OH)] building block (see Scheme S1, ESI†) which is the same building block as for the water-stable InOF-114 and possesses hydroxo functional groups (μ2-OH) to study the separation of a binary gas mixture (not a single-component gas) of CO2 and CH4, and we have successfully performed CO2 capture in the presence of water vapour.
 | ||
| Fig. 1 Space-filling view of the structure of NOTT-401 along the c-axis (scandium: green; sulphur: yellow; oxygen: red; carbon: grey; hydrogen: small grey).19 | ||
Experimental
Scandium triflate (0.057 g, 0.116 mmol) and thiophene-2,6-dicarboxylic acid, H2TDA, (0.01 g, 0.058 mmol) were dispersed in THF (4.0 ml), DMF (3.0 ml), H2O (1.0 ml) and HCl (36.5%, 2 drops) and sealed in a pressure tube. The clear solution was heated at 90 °C in an oil bath for 72 h. The tube was cooled to room temperature over a period of 12 h and the colorless crystalline product was separated by filtration, washed with DMF (5.00 ml) and dried in air. Yield: 71.1% (based on the ligand).The uncoordinated solvent molecules in the pores of the as-synthesized NOTT-401 were exchanged with acetone and this promotes accessibility to the desolvated framework after activation by heating. Thus, thermogravimetric (see Fig. S1, ESI†) analysis and bulk powder X-ray diffraction patterns (see Fig. S2, ESI†) of the as-synthesised and desolvated NOTT-401 confirmed that the material consistently retains its structural integrity upon solvent removal. N2 adsorption isotherms for activated NOTT-40119 at 77 K were used to calculate the BET surface area (0.01 < p/po < 0.04) of 1514 m2 g−1.
A catalytic reactor system (BEL-REA, BEL Japan; see Fig. S3, ESI†) was employed to evaluate the separation of CO2 from CH4. This catalytic reactor system allowed each sample of acetone-exchanged NOTT-401 to be activated (150 °C for 2 h) under a flow of N2 gas and then directly exposed to adsorbates (CO2 and CH4) in situ, and studied by FTIR spectroscopy over many cycles without physical manipulation or exposure to air.
Kinetic uptake experiments were performed by using a thermobalance (Q500 HR, from TA) at different temperatures with a constant CO2 flow (60 mL min−1). Then, acetone-exchanged samples of NOTT-401 were placed in the thermobalance and activated by heating from room temperature to 150 °C for 2 h and under a flow of N2 gas. After the activated sample was cooled down, the desired temperature was set and a constant CO2 flow (60 mL min−1) was started. With a humidity-controlled thermobalance (Q5000 SA, from TA) we carried out kinetic uptake experiments at 30 °C with a constant CO2 flow (60 mL min−1) on activated samples (150 °C for 2 h and under a flow of N2 gas) of NOTT-401.
Results
An acetone-exchanged sample of NOTT-401 (40 mg) was packed into the holder sample in the BEL-REA system (see Fig. S3, ESI†) and activated as described before (vide supra). Then, the system was allowed to cool down to room temperature (30 °C) and the activated NOTT-401 sample was exposed to a flow of the binary equimolar (0.13 mmol min−1) gas mixture of CO2 and CH4. This mixture corresponds to a more realistic composition in the field of gas-separation processes. Then, after stabilization of the gas flow within the sample, the resulting exhaust gases were analysed by FTIR spectroscopy (see Fig. S3, ESI†). Each FTIR spectrum was recorded every 40 seconds (∼0.66 min), until the detector was saturated, to obtain a total of 10 FTIR spectra (see Fig. S4, ESI†).The most characteristic FTIR bands for the CO2 and CH4 molecules were at 2349 cm−1 and 3016 cm−1, respectively, Fig. 2. In both cases, it is possible to monitor a continuous increase in the characteristic band intensities (Fig. 2), for CO2 and CH4 in time. Therefore, from spectrum 1 to spectrum 10 the intensity of the characteristic FTIR band is increasing while the transmittance is decreasing.
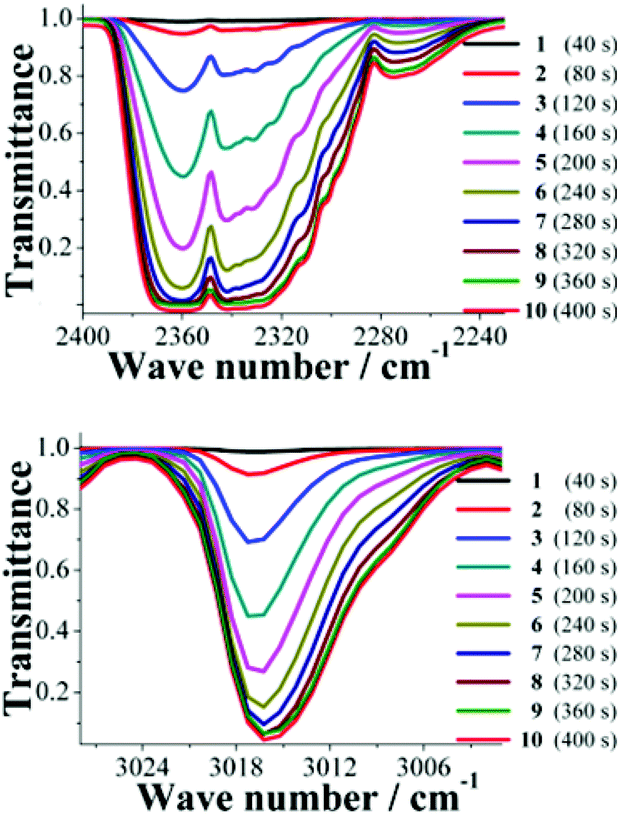 | ||
| Fig. 2 FTIR spectra of the resulting exit exhaust of the binary equimolar (0.13 mmol min−1) gas-mixture of CO2 and CH4 of: (top); the characteristic CO2 band and (bottom); the characteristic CH4 band. | ||
The intensities of these characteristic CO2 and CH4 FTIR bands were different. This suggests that the CO2 and CH4 molecules arrive at the FTIR detector at different times. By normalizing the intensities, considering their respective transmittances, it is possible to plot the increase in intensity of each scan for CO2 and CH4 simultaneously (Fig. 3, top). The normalized intensity, for each scan, of CH4 is higher than CO2, suggesting that the molecules of CH4 effectively arrive at the FTIR detector before the CO2 molecules. It is possible to rationalize this result as follows; when the binary gas mixture (CO2 and CH4) goes through the activated sample NOTT-401 this material retains CO2 stronger than CH4, and therefore, the CH4 gas molecules flow ‘faster’ inside the material and are detected earlier. To verify this hypothesis, we carried out three more experiments: first, an acetone-exchanged sample of NOTT-401 (40 mg) was packed into the BEL-REA system, activated and stabilized as described (see below) and a flow of only CO2 (0.13 mmol min−1) was set. Then, the exhaust gas was analyzed by FTIR spectroscopy and 10 scans were collected, until the detector was saturated (see Fig. S5, ESI†).
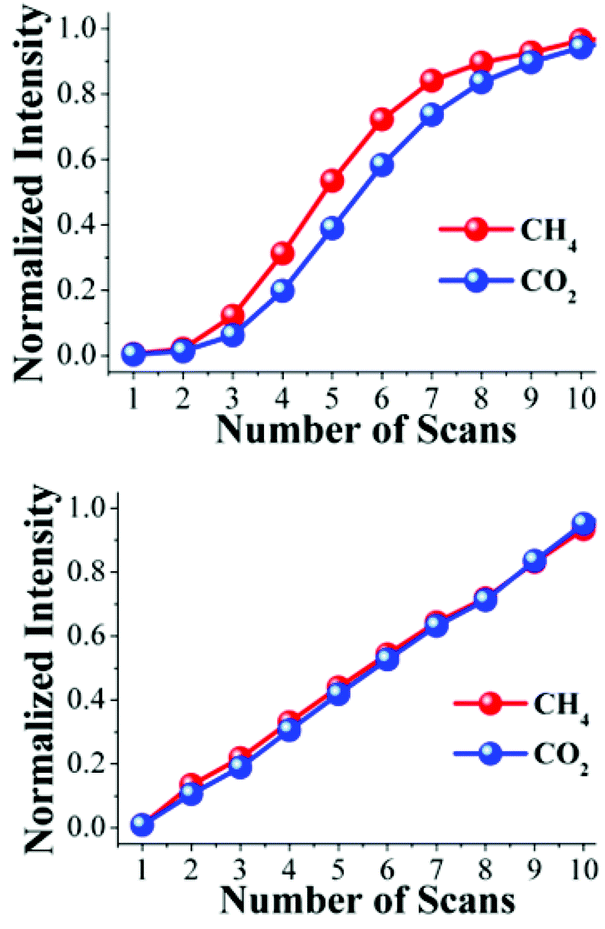 | ||
| Fig. 3 Normalized characteristic FTIR intensities of CO2 and CH4 as a function of the number of scans. (Top) FTIR intensities from a resulting exit exhaust of the binary equimolar (0.13 mmol min−1) gas-mixture of CO2 and CH4; (bottom) FTIR intensities from individual flows of CO2 and CH4. | ||
Second, another acetone-exchanged NOTT-401 (40 mg) sample was packed in the BEL-REA system, activated, stabilized and analyzed as described before. Then, the sample was exposed to a flow of only CH4 gas (0.13 mmol min−1). As in the previous experiment, 10 FTIR spectra were recorded from the exhaust gas until the detector was saturated (see Fig. S6, ESI†). Again, by normalizing the characteristic FTIR intensities it was possible to simultaneously plot the normalized intensity of each scan for CO2 and CH4 (Fig. 3, bottom).
The normalized intensities for CO2 and CH4 at each scan (from 1 to 10) were basically the same (Fig. 3, bottom) suggesting that when the resulting exhaust of each pure-gas component (not a gas mixture) is analyzed separately by FTIR spectroscopy, the molecules of CO2 and CH4 arrive at the same time at the FTIR detector. These results corroborate that NOTT-401 is more selective to CO2 than CH4 when a binary equimolar (0.13 mmol min−1) gas-mixture of CO2 and CH4 travels within an activated sample. We interpreted this selectivity as the time delay of the CO2 molecules in reaching the FTIR detector. By polynomial regressions of the normalized intensities on Fig. 3 (top), we estimated this delay to be ∼ 23 s (see Fig. S7 and S8, ESI†).
Finally a third experiment was carried out: in order to confirm that this delay was due to the adsorption selectivity shown by NOTT-401 (a microporous PCP) rather than any other phenomena, a non-porous material (PCM-1420) was packed in the BEL-REA system. PCM-14 is a dense coordination polymer that has shown to be a non-porous material when it is activated between 25 and 150 °C. Then, a sample of PCM-14 (40 mg) was activated at 150 °C for 2 h under a flow of N2 gas and then directly exposed to binary equimolar (0.13 mmol min−1) CO2 and CH4 gas mixture. The resulting exhaust gas was analyzed by FTIR spectroscopy and just 6 scans were recorded, until the detector was saturated. By normalizing the characteristic FTIR intensities, we plotted the normalized intensities of each CO2 and CH4 scans (see Fig. S9, ESI†). Interestingly, the normalized intensities for CO2 and CH4 at each scan (from 1 to 6) were essentially the same (see Fig. S9, ESI†) corroborating that the time delay is due to the microporosity of NOTT-401.
Dynamic and isothermal CO2 experiments were carried out on NOTT-401. Fig. 4, top, shows the kinetic uptake experiments from 30 °C to 100 °C. At 30 °C the material exhibited the maximum weight% gain, which represents the maximum amount of CO2 captured. This amount corresponds to 1.47 wt% which was rapidly reached (constant uptake) after just ∼300 s (5 min) and it was constant until the end of the experiment (3600 s or 60 min). At 40 °C the uptake was estimated to be 1.16 wt% which was also reached after around 300 s (Fig. 4, top). Clearly, while the temperature is increased (from 30 to 100 °C), the CO2 weight (%) gradually decreases (Fig. 4, top) to 0.09 wt% (at 100 °C) and interestingly, the kinetic uptake experiments did not show any difference in the equilibrium times. These equilibrium times have been previously observed in another microporous material NOTT-400.21
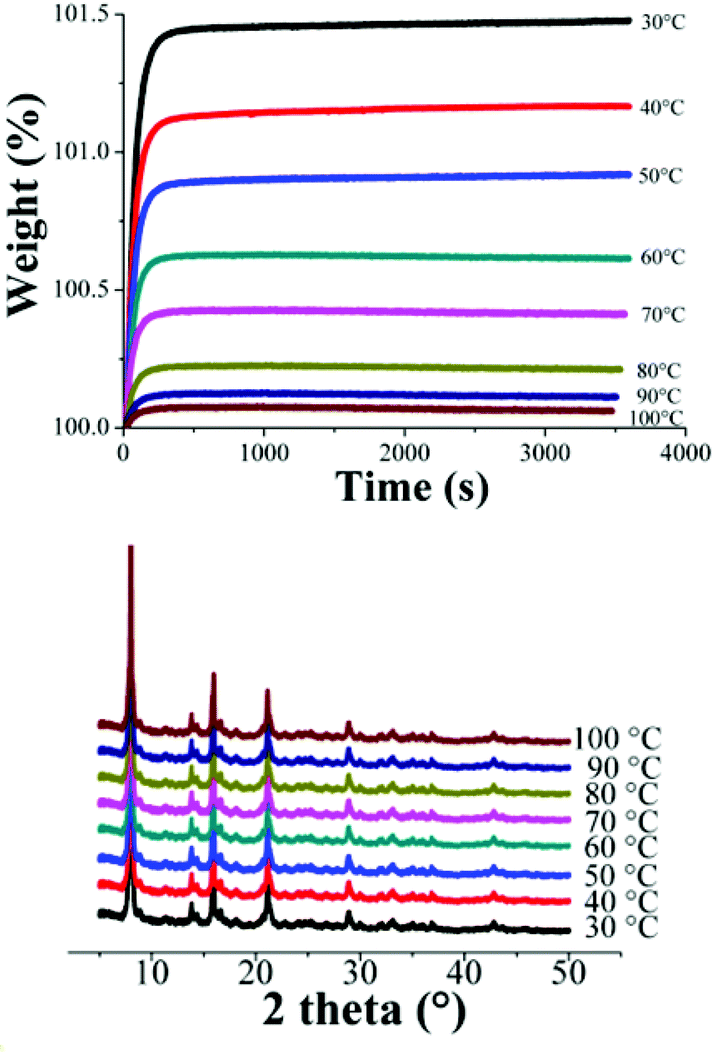 | ||
| Fig. 4 (Top) Kinetic uptake experiments performed at different temperatures (30, 40, 50, 60, 70, 80, 90 and 100 °C) with a CO2 flow of 60 mL min−1; (bottom) PXRD patterns of each NOTT-401 samples after the kinetic CO2 isotherms were carried out at different temperatures. | ||
In order to corroborate that this decrease is not due to sample degradation, we have carried out PXRD experiments on each sample after these CO2 capture experiments. Fig. 4 (bottom) confirms that the crystallinity of the samples after each CO2 capture experiments is retained.
Encouraged by the promising results that Hong et al. reported,14 by showing a water stable framework (InOF-1) with the same binuclear [M2(μ2-OH)] building block to NOTT-401, we explored the water stability of NOTT-401. Then, acetone-exchanged samples of NOTT-401 were exposed to air and soaked in distilled water. PXRD patterns of these samples (see Fig. S10, ESI†) confirmed structural stability of NOTT-401 in water. In addition, we calculated the BET surface areas (by N2 adsorption isotherms, 0.01 < p/po < 0.04) of samples NOTT-401 exposed to air and NOTT-401 soaked in distilled water of 1510 and 1516 m2 g−1, respectively. This demonstrates that NOTT-401 retains its surface area after water exposure.
This water-stability can be attributed to the presence of the hydroxo functional groups (inside the pores of NOTT-401) which has been previously shown19 and these functional groups, increase the affinity of the material for water. Indeed, Walton et al.22 proposed that the functional groups act as a directing agent for water in the pores, which allows for more efficient packing. Thus, after finding the best CO2 capture temperature (30 °C, Fig. 4, top), under anhydrous conditions, a kinetic isotherm experiment at 30 °C, with a constant CO2 flow, and a relative humidity (RH) of 40% was carried out. It was decided to run this experiment with a 40% RH based on the remarkable results that Llewellyn et al.17 had previously reported (5-fold increase in CO2 uptake for MIL-100(Fe)). An activated NOTT-401 sample (150 °C for 2 h and under a flow of N2 gas) was placed in a humidity-controlled thermobalance. After activation of the material, the equipment was stabilized at 40% RH (30 °C) and a constant CO2 flow (60 mL min−1) was started. Afterwards, we repeated this experimental procedure on a new activated NOTT-401 sample and set a constant N2 flow (60 mL min−1). Fig. 5 exhibits the kinetic uptake experiments at 30 °C and 40% RH for CO2 and N2. For both isotherms, it is clearly observed that the material shows a constant increase in weight (while the experiment is still continuing, see Fig. 5). This increase in weight is due to the contribution of H2O and CO2 or H2O and N2, respectively.
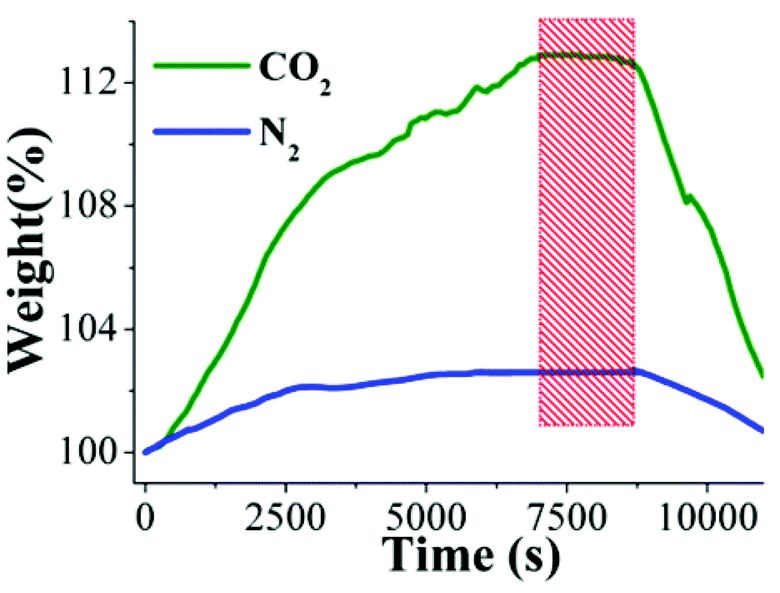 | ||
| Fig. 5 Kinetic uptake experiments carried out at 30 °C and 40% RH with CO2 (green line) and N2 (blue line) flows of 60 mL min−1, respectively. | ||
In order to find the maximum CO2 capture under 40% RH conditions, we need to differentiate the contribution of H2O to the weight increase. By just taking the difference of the two isotherms (CO2 and N2) we could obtain the CO2 capture at 40% RH. This is valid if the material does not capture any N2 at 30 °C. Consequently, by performing a kinetic uptake experiment on a new activated NOTT-401 sample at 30 °C without the presence of H2O vapor (0% RH) with a constant N2 flow (60 mL min−1) we obtained a N2 capture of approximately 0.01 wt%. This result is consistent with previous reports where the capture capacity of N2 in PCPs at room temperatures is basically negligible.23 In Fig. 5 it is shown that the gradual weight increase (for CO2/H2O and N2/H2O) starts at 0 s and stabilises at ∼7000 s (117 min). In contrast, under anhydrous conditions the CO2 uptake rapidly reaches stability (5 min, see Fig. 4, top). This equilibrium discrepancy is due to the nature of the vapour adsorption process that in general takes considerably more time to reach stability than the gas adsorption process in microporous materials.24 Then, from 7000 s until approximately 8700 s (145 min) both isotherms seem to reach a plateau where both uptakes are practically constant (Fig. 5, red rectangle). At 8700 s, the maximum amounts of CO2/H2O and N2/H2O captured are 12.60 wt% and 2.70 wt%, respectively and by simply taking the difference of these two values (since there is no N2 uptake at 30 °C) the CO2 capture in the materials is ∼9.90 wt%. Finally, from 8700 s to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s (Fig. 5) the flow of each gas and the relative humidity were stopped and the decrease in weight represents the gas and water vapour desorption.
000 s (Fig. 5) the flow of each gas and the relative humidity were stopped and the decrease in weight represents the gas and water vapour desorption.
Therefore, the CO2 capture was increased approximately 7-fold with a 40% RH. This increase in CO2 uptake in the presence of water can be explained by CO2 confinement effects induced by bulky molecules (H2O).25 In addition, we decided to carry a CO2 experiment (60 mL min−1) at 40% RH and 30 °C on an activated PCM-1420 sample (150 °C for 2 h, under a flow of N2 gas). Since PCM-14 is a non-porous coordination polymer, when activated between 25 and 150 °C, it offered a direct CO2 capture comparison to NOTT-401 (microporous material). Thus, from 0 s to 11000 s the maximum CO2 uptake (under 40% RH) was 0.8 wt% (see Fig. S11, ESI†). This result corroborated that there is no CO2 sequestration in a non-porous material when the relative humidity is 40% at 30 °C.
Conclusions
In conclusion, NOTT-401, a Sc(III) porous coordination polymer, exhibited CO2 separation from CH4, in a more realistic scenario, when this microporous material was exposed to a binary, CO2/CH4, equimolar gas-mixture. By running these equimolar (CO2/CH4) gas experiments on PCM-14, a non-porous coordination polymer, it was established that the intrinsic microporosity of NOTT-401 is responsible for the gas separation.By kinetic isotherm experiments, NOTT-401 exhibits a total CO2 amount of 1.47 wt% at 30 °C, which was rapidly reached after just approximately 300 s. While increasing the temperature of the kinetic isotherm experiments, the CO2 capture capacity of NOTT-401 decreased considerably to less than 0.1 wt% at 100 °C. Remarkably, NOTT-401 exhibits high stability towards humidity, which was corroborated by PXRD. We attributed this water stability, as previously reported,18,22 to the presence of hydroxo functional groups within the pores of NOTT-401. Due to this particularly high water stability, NOTT-401 performs CO2 uptake under relative humidity conditions (40% RH) and 30 °C, displaying a maximum CO2 capture of approximately 9.9 wt%.
Significantly, this CO2 capture, under humid conditions, represents a 7-fold increase in comparison to anhydrous conditions. PCM-14 showed non CO2 capture under RH conditions, suggesting that the microporosity provided by NOTT-401 is fundamental for this capture process. Since PCM-14 is a non-porous coordination polymer, when activated between 25 and 150 °C, the CO2 confinement effects induced by H2O25 in porous materials cannot take place unlike NOTT-401 where these effects occur within the micropores.
Acknowledgements
The authors thank Dr A. Tejeda-Cruz (X-ray; IIM-UNAM), CONACyT Mexico (212318), PAPIIT UNAM Mexico (IN100415) for financial support. M. R. G. thanks CONICET and BECAR Argentina for scholarship funding. Thanks to U. Winnberg (ITAM) for scientific discussions.Notes and references
- F. W. Lipfert, Air Pollution and Community Health: A Critical Review and Data Source Book, Van Nostrand Reinhold, New York, USA, 1994, p. 556 Search PubMed.
- P. D. Noyes, M. K. McElwee, H. D. Miller, B. W. Clark, L. A. Van Tiem, K. C. Walcott, K. N. Erwin and E. D. Levin, Environ. Int., 2010, 35, 971 CrossRef PubMed.
- T. Burchell and M. Rogers, SAE Tech. Pap. Ser., 2000, 2000 Search PubMed.
- M. Herout, J. Malat’ák, L. Kučera and T. Dlabaja, Res. Agric. Eng., 2011, 57, 137 Search PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef PubMed.
- J. T. Litynski, S. M. Klara, H. G. McIlvried and R. D. Srivastava, Environ. Int., 2006, 32, 128 CrossRef CAS PubMed.
- M. Z. Jacobson, Energy Environ. Sci., 2009, 2, 148 CAS.
- International Energy Agency (IEA), Key World Energy Statistics, OECD/IEA, France, 2013 Search PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, Z. T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed.
- (a) G. T. Rochelle, Science, 2009, 325, 1652 CrossRef CAS PubMed; (b) F. Karadas, M. Atilhan and S. Aparicio, Energy Fuels, 2010, 24, 5817 CrossRef CAS.
- (a) S. Yang, G. S. B. Martin, G. J. J. Titman, A. J. Blake, D. R. Allan, N. R. Champness and M. Schröder, Inorg. Chem., 2011, 50, 9374 CrossRef CAS PubMed; (b) A. J. Nuñez, L. N. Shear, N. Dahal, I. A. Ibarra, J. W. Yoon, Y. K. Hwang, J.-S. Chang and S. M. Humphrey, Chem. Commun., 2011, 47, 11855 RSC.
- (a) H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed; (b) A. M. Bohnsack, I. A. Ibarra, P. W. Hatfield, J. W. Yoon, Y. K. Hwang, J.-S. Chang and S. M. Humphrey, Chem. Commun., 2011, 47, 4899 RSC; (c) P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80 CrossRef CAS PubMed.
- (a) S. S. Nagarkar, A. K. Chaudhari and S. K. Ghosh, Inorg. Chem., 2012, 51, 572 CrossRef CAS PubMed; (b) H. J. Choi, M. Dincă, A. Daily and J. R. Long, Energy Environ. Sci., 2010, 3, 117 RSC.
- J. Qian, F. Jiang, D. Yuan, M. Wu, S. Zhang, L. Zhang and M. Hong, Chem. Commun., 2012, 48, 9696 RSC.
- (a) J. Liu, A. I. Benin, A. M. B. Furtado, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2011, 27, 11451 CrossRef CAS PubMed; (b) A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368 CrossRef CAS PubMed; (c) H. Jasuja, Y.-g. Huang and K. S. Walton, Langmuir, 2012, 28, 16874 CrossRef CAS PubMed; (d) H. Jasuja, J. Zang, D. S. Sholl and K. S. Walton, J. Phys. Chem. C, 2012, 116, 23526 CrossRef CAS; (e) J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y.-g. Huang and K. S. Walton, J. Mater. Chem. A, 2013, 1, 5642 RSC.
- J. Liu, Y. Wang, A. I. Benin, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2010, 26, 14301 CrossRef CAS PubMed.
- E. Soubeyrand-Lenoir, C. Vagner, J. W. Yoon, P. Bazin, F. Ragon, Y. K. Hwang, C. Serre, J.-S. Chang and P. L. Llewellyn, J. Am. Chem. Soc., 2012, 134, 10174 CrossRef CAS PubMed.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369 CrossRef CAS PubMed.
- I. A. Ibarra, S. Yang, X. Lin, A. J. Blake, P. J. Rizkallan, H. Nowell, D. R. Allan, N. R. Champness, P. Hubberstey and M. Schröder, Chem. Commun., 2011, 47, 8304 RSC.
- I. A. Ibarra, K. E. Tan, V. M. Lynch and S. M. Humphrey, Dalton Trans., 2012, 41, 3920 RSC.
- M. R. Gonzalez, J. H. González-Estefan, H. A. Lara-García, P. Sánchez-Camacho, E. I. Basaldella, H. Pfeiffer and I. A. Ibarra, New J. Chem., 2015, 39, 2400 RSC.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15613 CrossRef PubMed.
- (a) E. Haldoupis, S. Nair and D. S. Sholl, J. Am. Chem. Soc., 2012, 134, 4313 CrossRef CAS PubMed; (b) Y.-S. Bae, O. K. Farha, J. T. Hupp and R. Q. Snurr, J. Mater. Chem., 2009, 19, 2131 RSC.
- I. P. O'koye, M. Benham and K. M. Thomas, Langmuir, 1997, 13, 4054 CrossRef.
- N. L. Ho, F. Porcheron and R. J.-M. Pellenq, Langmuir, 2010, 26, 13287 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TGA data, PXRDP data, FTIR data, polynomial regressions and kinetic uptake experiments. See DOI: 10.1039/c5qi00049a |
| This journal is © the Partner Organisations 2015 |