
Bis(N-substituted oxamate)palladate(II) complexes as effective catalysts for sustainable Heck carbon–carbon coupling reactions in n-Bu4NBr as the solvent†
Francisco Ramón
Fortea-Pérez
a,
Berit L.
Rothenpieler
a,
Nadia
Marino
b,
Donatella
Armentano
*b,
Giovanni
De Munno
b,
Miguel
Julve
*a and
Salah-Eddine
Stiriba
*ac
aInstituto de Ciencia Molecular (ICMol), Universitat de València, C/ Catedrático José Beltrán 2, 46980 Paterna (València), Spain. E-mail: miguel.julve@uv.es; stiriba@uv.es
bDipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, 87036 Rende, Cosenza, Italy. E-mail: donatella.armentano@unical.it
cEquipe de Chimie Moléculaire, Matériaux et Modélisation – C3M, Faculté Polydisciplinaire de Safi, Université Cadi Ayyad, Safi, Morocco
First published on 14th September 2015
Abstract
Five bis(oxamato)palladate(II) complexes of the formulae (n-Bu4N)2[Pd(4-Fpma)2] (1), (n-Bu4N)2[Pd(4-Clpma)2] (2), (n-Bu4N)2[Pd(4-Brpma)2] (3), (n-Bu4N)2[Pd(4-Brpma)2]·H2O (3a), (n-Bu4N)2[Pd(4-MeOpma)2] (4) and (n-Bu4N)2[Pd(4-Isopma)2] (5) (n-Bu4N+ = tetra-n-butylammonium, 4-Fpma = N-4-fluorophenyloxamate, 4-Clpma = N-4-chlorophenyloxamate, 4-Brpma = N-4-bromophenyloxamate, 4-MeOpma = N-4-methoxyphenyloxamate and 4-isopma = N-4-isopropylphenyloxamate) have been easily prepared and characterized by spectroscopic methods and the crystal structures of two of them (3a and 4) have been determined by single crystal X-ray diffraction. Each palladium(II) ion in 3a and 4 is four-coordinate with two oxygen and two nitrogen atoms from two fully deprotonated oxamate ligands building a centrosymmetric square planar PdN2O2 surrounding. The values of the Pd–N and Pd–O bond lengths vary in the ranges 2.034(3)–2.043(4) and 1.999(4)–2.013(3) Å, respectively. The reduced bite of the chelating oxamate ligands [81.3(2)–81.7(1) (3a) and 81.61(7)° (4)] is at the origin of the mean distortion of the ideal square environment. The catalytic role of compounds 1–5 as structurally well-defined precatalysts for the Heck-vinylation of a series of aryl iodide/bromide derivatives in n-Bu4NBr as a benign nonaqueous ionic liquid (i.d. molten salt) has been examined and compared with some commercially available palladium(II/0) complexes. From this study, it appears that the oxamate-containing precatalysts 1–5 are not just ecologically benign, but also highly efficient, easily recoverable and reusable at least eight times without any relevant loss of catalytic activity or leaching from the ionic liquid medium.
Introduction
The synthetic method based on metal-catalyzed cross-coupling reactions where the substitution of an aryl, vinyl and alkyl halide/pseudohalide by a nucleophile takes place has been a subject of thorough investigation since the early 1970s.1,2 These impressive worldwide research efforts were rewarded with the Nobel Prize in chemistry to Richard F. Heck, Ei-ichi Negishi and Akira Suzuki, in 2010.3 One of the main reasons to pursue this type of chemistry is its great impact on the chemical industry and relevance for the design of new ligands, with consequences on other frontier research fields including supramolecular and polymer chemistry, molecular magnetism, bioinorganic chemistry, materials science, etc.4,5A good number of palladium-based systems with excellent catalytic performances in carbon–carbon cross-coupling reactions have been developed during the last five decades, their efficiency being largely examined. Without being exhaustive, representative examples are those with phosphine ligands,6 palladacycle catalysts,7 N-heterocyclic carbene-containing catalysts (NHCs),8 nanoparticles9 and immobilized palladium catalysts10 in both homogeneous and heterogeneous regimes. It deserves to be noted that, in most cases, the activity of homogeneous catalysts was found to be higher than that of their heterogeneous analogues.11
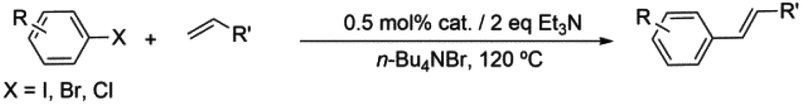
The search for new, ecologically benign procedures of the carbon–carbon cross-coupling type is still a challenge. These are some of the main conditions to be fulfilled: (i) low catalyst amount (ii) high catalyst activity and selectivity, (iii) mild reaction conditions for energy costs saving and (iv) efficient catalyst recycling and reuse with regular yields over a large number of runs. Keeping this in mind, part of our current research endeavours have focused onto the design and use of simple, cheap, and recyclable palladium(II)-based active catalysts together with environmentally friendly green solvents. In this context, we bet on ionic liquids as alternative green solvents,12 in particular on tetra-n-butylammonium bromide (n-Bu4NBr), a widely used molten salt.13 This appears as an appropriate medium for our anionic bis(N-substituted oxamate)palladate(II) systems (vide infra), especially due to the presence of the same cation in our palladium(II) systems and for electrostatic/stability reasons.14 By performing cross-coupling Heck reactions following an alternative green catalytic route with no ecologically harmful chemicals (phosphine ligands for instance) and avoiding difficult-to-handle carbene ligands, our idea to employ low-cost, versatile N,O-ligands15 to build new catalytic systems seems advantageous as both a sustainable synthetic organic methodology and a suitable strategy for catalyst recovery.12i,16 Noteworthily, chelating ligands containing oxygen or nitrogen as donor atoms have been shown to stabilize palladium complexes.17
Apart from their use in magnetic studies as diamagnetic spacers to build heterometallic compounds,18 oxamate-containing palladium(II) complexes exhibit cytotoxic activity against leukemia19 and they have also shown catalytic activity for the palladium-catalyzed Suzuki and Heck reactions.14,20 Focusing on their potential in catalysis, the present work deals with the design and spectroscopic characterization of five new bis(oxamato)palladate(II) complexes of the formulae (n-Bu4N)2[Pd(4-Fpma)2] (1), (n-Bu4N)2[Pd(4-Clpma)2] (2), (n-Bu4N)2[Pd(4-Brpma)2] (3), (n-Bu4N)2[Pd(4-Brpma)2]·H2O (3a), (n-Bu4N)2[Pd(4-MeOpma)2] (4) and (n-Bu4N)2[Pd(4-Isopma)2] (5) (n-Bu4N+ = tetra-n-butylammonium, 4-Fpma = N-4-fluorophenyloxamate, 4-Clpma = N-4-chlorophenyloxamate, 4-Brpma = N-4-bromophenyloxamate, 4-MeOpma = N-4-methoxyphenyloxamate and 4-isopma = N-4-isopropylphenyloxamate), the X-ray structure of 3a and 4 and the catalytic activity/recycling of 1–5 in the Heck cross-coupling reaction of aryl iodine/bromide derivatives with olefins using n-Bu4NBr ionic liquid as the solvent.
Experimental section
Material and general characterization
Aniline derivatives, K2[PdCl4], [Pd3(OAc)6], [PdCl2], n-Bu4NOH, ethyl chlorooxoacetate and all the solvents for the syntheses were purchased from commercial sources and used without further purification. [Pd(bda)2] (bda = dibenzylideneacetone) was prepared as previously reported.21 Infrared samples were prepared as KBr pellets, and spectra were recorded in the 4000–450 cm−1 range using a Nicolet 5700 FT-IR instrument. 1H- and 13C-NMR spectra were recorded on an Avance DRX 300 Bruker instrument at room temperature and referenced to the residual proton in the solvent for 1H NMR and the same solvent 13C signal for 13C NMR. The elemental analysis (C, H and N) was carried out on a EuroEA3000 analyzer by the Servei Central d'Instrumentació Científica at the University of Jaume I. The value of the Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cl (2) and Pd:Br (3) molar ratio (1:2) was determined through electron probe X-ray microanalysis by using a Philips XL-30 scanning electron microscope (SEM) from the Central Service for Support to Experimental Research (SCSIE) at the University of Valencia. All GC-MS spectra were recorded on an Agilent 6890N gas chromatograph equipped with capillary columns from the SCSIE at the University of Valencia, using perfluorotributylamine (PFTBA) as the internal standard.
:Cl (2) and Pd:Br (3) molar ratio (1:2) was determined through electron probe X-ray microanalysis by using a Philips XL-30 scanning electron microscope (SEM) from the Central Service for Support to Experimental Research (SCSIE) at the University of Valencia. All GC-MS spectra were recorded on an Agilent 6890N gas chromatograph equipped with capillary columns from the SCSIE at the University of Valencia, using perfluorotributylamine (PFTBA) as the internal standard.
Synthesis of the proligands
The preparation of the proligands as ethyl esters was carried out by following the procedure shown in Scheme 1. The corresponding aniline derivative (83 mmol) was dissolved in THF (250 mL) under a nitrogen atmosphere in a two neck round bottom flask equipped with a dropping funnel and a stirring bar. The solution was treated with ethyl chloroacetate (9.3 mL, 83 mmol) in the presence of triethylamine (12 mL, 83 mmol) at room temperature under continuous stirring for 30 minutes. The white Et3NCl precipitate was separated by filtration and the remaining pale yellow solution was removed under vacuum to afford an oily crude that rapidly became solid. The obtained white solid was suspended in water and filtered off, washed with a small amount of diethyl ether and dried under reduced pressure. Details of the yield and spectroscopic characterization are given in the ESI.†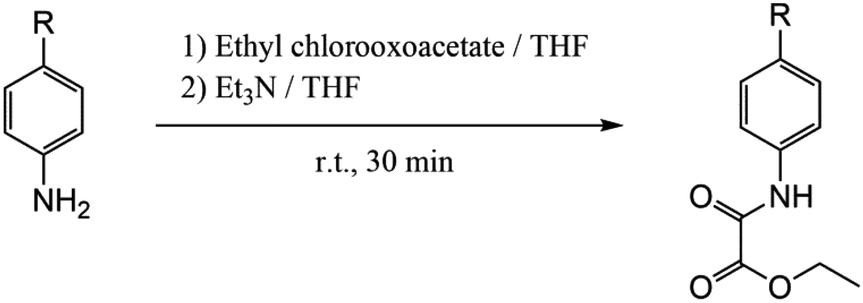 | ||
| Scheme 1 Preparative route of the proligands: R = F, Cl, Br, MeO and isopropyl for EtH-4-Fpma, EtH-4-Clpma, EtH-4-Brpma, EtH-4-MeOpma and EtH-4-Isopma, respectively. | ||
Synthesis of the complexes 1–5
The general synthetic method for the bis(N-substituted oxamato)palladate(II) complexes 1–5 is depicted in Scheme 2.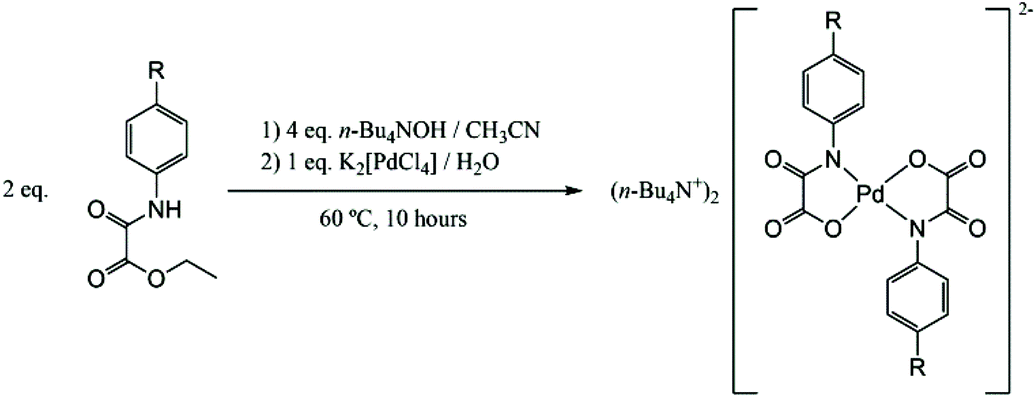 | ||
| Scheme 2 Preparative route of the bis(N-substituted oxamato)palladate(II) complexes. | ||
A methanolic solution of n-Bu4NOH (1.0 M, 1.5 mL, 1.2 mmol) was added directly to a two-neck round bottom flask equipped with a condenser containing a suspension of the corresponding N-substituted oxamate proligand (0.6 mmol) in 10 mL of acetonitrile at 60 °C. Then, an aqueous solution of K2[PdCl4] (100 mg, 0.3 mmol) was added dropwise to the resulting solution and the reaction mixture was heated at 60 °C for 10 hours. The obtained mixture was filtered and the acetonitrile was removed under reduced pressure. Dichloromethane (three times, 15 mL) was added to extract the complex from the aqueous solution. The recovered organic fractions were combined and concentrated under reduced pressure to afford a pale yellow solid. This solid was washed with n-hexane, collected by filtration and dried under vacuum. X-ray quality crystals as pale yellow prisms were grown for 3 (as 3a) and 4 by two different methods: slow vapor diffusion of ether into an acetonitrile solution (3a) or slow evaporation at room temperature of a water/acetonitrile mixture (2:1 v/v) (4) where a fraction of the corresponding solid was dissolved. The crystals of 3a appeared overnight whereas those of 4 were grown after two weeks.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (CDCl3) δ (ppm): 0.90–0.94 (t, J = 7.2 Hz, 24H, n-Bu4N+), 1.24–1.46 (m, J = 7.0 Hz, J = 6.9 Hz, 32H, n-Bu4N+), 2.93–2.99 (t, J = 13.1 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 6.63–6.66 (d, J = 8.9 Hz, J = 2.9 Hz, 4H, Haryl), 7.27–7.30 (d, J = 8.9 Hz, J = 2.9 Hz, 4H, Haryl). Anal. Calcd for C48H80F2N4O6Pd (954 g mol−1): C 60.46, H 8.46, N 5.88. Found: C 60.23, H 8.27, N 5.79%.
O); 1H NMR (CDCl3) δ (ppm): 0.91–0.96 (t, J = 7.2 Hz, 24H, n-Bu4N+), 1.28–1.40 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 1.46–1.56 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 3.08–3.14 (t, J = 13.1 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 7.02–7.05 (d, 8.9 Hz, J = 2.9 Hz, 4H, Haryl), 7.33–7.36 (d, J = 8.9 Hz, J = 2.9 Hz, 4H, Haryl). Anal. Calcd for C48H80Cl2N4O6Pd (987 g mol−1): C 58.44, H 8.17, N 5.68. Found: C 58.32, H 8.05, N 5.61%.
O); 1H NMR (CDCl3) δ (ppm): 0.90–0.95 (t, J = 7.2 Hz, 24H), 1.26–1.38 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 1.41–1.52 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 3.01–3.07 (t, J = 13.1 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 6.75–6.81 (t, J = 8.9 Hz, J = 2.9 Hz, 4H, Haryl), 7.31–7.36 (m, J = 8.9 Hz, J = 2.9 Hz, 4H, Haryl). Anal. Calcd for C48H80Br2N4O6Pd (1075 g mol−1): C 53.61, H 7.50, N 5.21. Found: C 53.48, H 7.41, N 5.15%. X-ray quality crystals of (n-Bu4N)2[Pd(4-Brpma)2]·H2O (3a) as pale yellow prisms were grown overnight by slow vapor diffusion of ether into an acetonitrile solution of a solid sample of 3 and its stoichiometry was determined by X-ray diffraction on single crystals.
O); 1H NMR (CDCl3) δ (ppm): 0.97–1.02 (t, J = 7.2 Hz, 24H, n-Bu4N+), 1.35–1.47 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 1.50–1.60 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 3.11–3.17 (t, J = 13.1 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 3.73 (s, 6H, OCH3), 6.71–6.74 (d, J = 8.8 Hz, J = 2.8 Hz, 4H, Haryl), 7.37–7.40 (d, J = 8.8 Hz, J = 2.8 Hz, 4H, Haryl). Anal. Calcd for (978 g mol−1): C 61.43, H 8.87, N 5.73. Found: C 61.28, H 8.71, N 5.65%. Single crystals of 4 as pale yellow prisms were grown by slow evaporation at room temperature of a water/acetonitrile (2:1 v/v) mixture where the obtained solid was dissolved. The crystals of 4 appeared within 2–4 weeks.
O); 1H NMR (CDCl3) δ (ppm): 0.97–1.02 (t, J = 7.2 Hz, 24H, n-Bu4N+), 1.19–1.21 (d, J = 7.0 Hz, 12H, CH3), 1.36–1.48 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 1.52–1.62 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 2.76–2.85 (m, 2H, CH), 3.15–3.21 (t, J = 13.1 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 7.02–7.05 (d, J = 8.8 Hz, J = 2.8 Hz, 4H, Haryl), 7.35–7.38 (d, 8.8 Hz, J = 2.8 Hz, 4H, Haryl). Anal. Calcd for C54H94N4O8Pd (1002 g mol−1): C 64.74, H 9.46, N 5.59. Found: C 64.55, H 9.31, N 5.42%.
O); 1H NMR (CDCl3) δ (ppm): 0.90–0.94 (t, J = 7.2 Hz, 24H, n-Bu4N+), 1.24–1.46 (m, J = 7.0 Hz, J = 6.9 Hz, 32H, n-Bu4N+), 2.93–2.99 (t, J = 13.1 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 6.63–6.66 (d, J = 8.9 Hz, J = 2.9 Hz, 4H, Haryl), 7.27–7.30 (d, J = 8.9 Hz, J = 2.9 Hz, 4H, Haryl). Anal. Calcd for C48H80F2N4O6Pd (954 g mol−1): C 60.46, H 8.46, N 5.88. Found: C 60.23, H 8.27, N 5.79%.
O); 1H NMR (CDCl3) δ (ppm): 0.91–0.96 (t, J = 7.2 Hz, 24H, n-Bu4N+), 1.28–1.40 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 1.46–1.56 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 3.08–3.14 (t, J = 13.1 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 7.02–7.05 (d, 8.9 Hz, J = 2.9 Hz, 4H, Haryl), 7.33–7.36 (d, J = 8.9 Hz, J = 2.9 Hz, 4H, Haryl). Anal. Calcd for C48H80Cl2N4O6Pd (987 g mol−1): C 58.44, H 8.17, N 5.68. Found: C 58.32, H 8.05, N 5.61%.
O); 1H NMR (CDCl3) δ (ppm): 0.90–0.95 (t, J = 7.2 Hz, 24H), 1.26–1.38 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 1.41–1.52 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 3.01–3.07 (t, J = 13.1 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 6.75–6.81 (t, J = 8.9 Hz, J = 2.9 Hz, 4H, Haryl), 7.31–7.36 (m, J = 8.9 Hz, J = 2.9 Hz, 4H, Haryl). Anal. Calcd for C48H80Br2N4O6Pd (1075 g mol−1): C 53.61, H 7.50, N 5.21. Found: C 53.48, H 7.41, N 5.15%. X-ray quality crystals of (n-Bu4N)2[Pd(4-Brpma)2]·H2O (3a) as pale yellow prisms were grown overnight by slow vapor diffusion of ether into an acetonitrile solution of a solid sample of 3 and its stoichiometry was determined by X-ray diffraction on single crystals.
O); 1H NMR (CDCl3) δ (ppm): 0.97–1.02 (t, J = 7.2 Hz, 24H, n-Bu4N+), 1.35–1.47 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 1.50–1.60 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 3.11–3.17 (t, J = 13.1 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 3.73 (s, 6H, OCH3), 6.71–6.74 (d, J = 8.8 Hz, J = 2.8 Hz, 4H, Haryl), 7.37–7.40 (d, J = 8.8 Hz, J = 2.8 Hz, 4H, Haryl). Anal. Calcd for (978 g mol−1): C 61.43, H 8.87, N 5.73. Found: C 61.28, H 8.71, N 5.65%. Single crystals of 4 as pale yellow prisms were grown by slow evaporation at room temperature of a water/acetonitrile (2:1 v/v) mixture where the obtained solid was dissolved. The crystals of 4 appeared within 2–4 weeks.
O); 1H NMR (CDCl3) δ (ppm): 0.97–1.02 (t, J = 7.2 Hz, 24H, n-Bu4N+), 1.19–1.21 (d, J = 7.0 Hz, 12H, CH3), 1.36–1.48 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 1.52–1.62 (m, J = 7.0 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 2.76–2.85 (m, 2H, CH), 3.15–3.21 (t, J = 13.1 Hz, J = 6.9 Hz, 16H, n-Bu4N+), 7.02–7.05 (d, J = 8.8 Hz, J = 2.8 Hz, 4H, Haryl), 7.35–7.38 (d, 8.8 Hz, J = 2.8 Hz, 4H, Haryl). Anal. Calcd for C54H94N4O8Pd (1002 g mol−1): C 64.74, H 9.46, N 5.59. Found: C 64.55, H 9.31, N 5.42%.
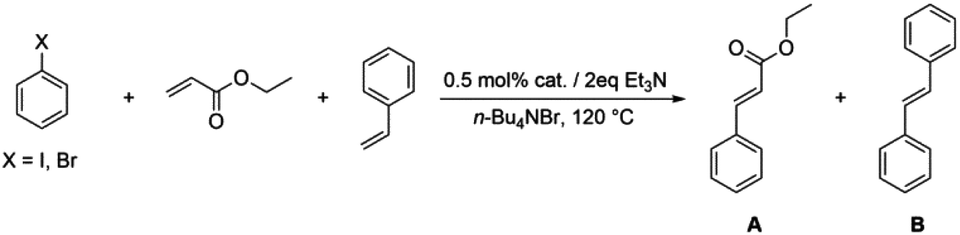
General procedure for the Heck reaction and recycling of catalysts
A test-tube with a screw cap and valve was charged with a magnetic stir bar and loaded with the precatalyst 0.25–0.5 mol%, the aryl halide (0.50 mmol), Et3N (1.00 mmol), the olefin (0.75 mmol), and 3–4 g of n-Bu4NBr. The mixture was heated under continuous stirring for a given time at 120 °C. The reaction was monitored by thin layer chromatography on silica gel. The reaction mixture was cooled and extracted with 5 mL of n-pentane. After the first extraction, the test-tube containing the palladium(II) catalyst in the ionic solvent was charged again with a fresh portion of aryl halide (0.50 mmol), Et3N (1.00 mmol) and the olefin (0.75 mmol) to start another run (a minimum of 3 runs and a maximum of 8 runs were performed). Each extract was characterized by gas chromatography-mass spectrometry (GC-MS). The catalytic products were purified by column chromatography and characterized by 1H, 13C and Dept-135 NMR spectroscopy. Yields were determined by GC-MS analysis using perfluorotributylamine as the internal standard. The isolated products of the first and final catalytic runs of each protocol were analyzed by 1H NMR and FT-IR spectroscopy to check the presence of typical signals and bands of the palladium(II) precatalysts. The lack of palladium traces in the isolated dried products was verified by SEM-EDX microscopy.X-ray crystallography
X-ray diffraction data on single crystals of 3a and 4 were collected with a Bruker-Nonius X8-APEXII CCD area detector system by using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The data were processed through the SAINT22 reduction and SADABS23 absorption software. A summary of the crystallographic data and structure refinement for the two compounds is given in Table 1. The structures of 3a and 4 were solved by direct methods and subsequently completed by Fourier recycling using the SHELXTL-2013 software package,24 then refined by the full-matrix least-squares refinements based on F2 with all the observed reflections. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms of the tetra-n-butylammonium cation and those of the N-substituted oxamate ligands were included at geometrically calculated positions and refined using a riding model. In 3a, one of the n-Bu4N+ cations was found disordered; two sets of atoms were defined for it, whose relative occupancy reached values of about 0.7 and 0.3 at convergence. The disorder was refined with the aid of similarity restraints on 1,2- and 1,3 distances as well as rigid-bond restraints, by using established methods.25 Disordered water molecules of crystallization were also found in 3a; five partially occupied sites were defined, for a total of one water molecule per Pd atom. These sites [O(1w), O(2w), O(2wA), O(3w) and O(3wA)] were only refined isotropically. The final geometrical calculations were performed using the XP utility within SHELXTL24 whereas the graphical manipulations were performed with the CrystalMaker software.26 Selected bond lengths and angles are listed in Tables 2 (3a) and 3 (4).| Compound | 3a | 4 |
|---|---|---|
| a R 1 = ∑||Fo| – |Fc||/∑|Fo|. b wR2 = {∑w(Fo2 − Fc2)2/∑[w(Fo2)2]}1/2 and w = 1/[σ2(Fo)2 + (mP)2 + nP] with P = (Fo2 + 2Fc2)/3, m = 0.1225 (3a) and 0.0475 (4), and n = 0.0000 (3a) and 1.5167 (4). | ||
| Formula | C144H246Br6N12O21Pd3 | C50H86N4O8Pd |
| f w | 3280.17 | 977.62 |
| Crystal system | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| a/Å | 13.524(2) | 9.7928(5) |
| b/Å | 16.287(3) | 16.3297(8) |
| c/Å | 20.967(3) | 16.0978(8) |
| α/° | 100.901(8) | 90 |
| β/° | 103.304(8) | 95.392(2) |
| γ/° | 99.809(8) | 90 |
| V/Å3 | 4301.1(13) | 2562.9(2) |
| Z | 1 | 2 |
| D c /g cm−3 | 1.266 | 1.267 |
| T/K | 296(2) K | 100(2) |
| μ/mm−1 | 1.763 | 0.416 |
| F(000) | 1710 | 1048 |
| Refl. Collected | 88674 |
60847 |
| Refl. indep. [Rint] | 18125 (0.0508) |
5228 (0.0661) |
| Refl. obs. [I > 2σ(I)] | 9794 | 5228 (0.0661) |
| Data/restraints/param. | 18125/808/1006 |
5228/0/286 |
| Goodness-of-fit on F2 | 1.005 | 1.200 |
| R 1 [I > 2σ(I)] (all) | 0.0587 (0.1231) | 0.0329 (0.0453) |
| wR2b [I > 2σ(I)] (all) |
0.865/−0.487 | 0.0830 (0.0957) |
| Δρmax, min/e Å−3 | 0.841/−0.499 | 0.461/−0.895 |
| a Symmetry codes used to generate equivalent atoms: (a) = −x + 1, −y + 1, −z + 1; (b) = −x, −y, −z, (c) = −x + 2, −y + 2, −z + 1. | |||
|---|---|---|---|
| Pd(1)–N(1A) | 2.034(3) | C(1A)–O(1A) | 1.288(5) |
| Pd(1)–O(1A) | 2.011(3) | C(1A)–O(2A) | 1.228(5) |
| C(2A)–O(3A) | 1.255(5) | ||
| Pd(2)–N(1B) | 2.038(3) | C(1B)–O(1B) | 1.288(5) |
| Pd(2)–O(1B) | 2.020(3) | C(1B)–O(2B) | 1.228(5) |
| C(2B)–O(3B) | 1.238(4) | ||
| Pd(3)–N(1C) | 2.046(4) | C(1C)–O(1C) | 1.292(8) |
| Pd(3)–O(1C) | 2.006(5) | C(1C)–O(2C) | 1.221(8) |
| C(2C)–O(3C) | 1.255(7) | ||
| O(1A)–Pd(1)–N(1A) | 81.5(1) | Pd(1)⋯Pd(2) | 11.759(2) |
| O(1Aa)–Pd(1)–N(1A) | 98.5(1) | Pd(1)⋯Pd(3) | 9.658(1) |
| O(1B)–Pd(2)–N(1B) | 81.7(1) | Pd(2)⋯Pd(3) | 18.793(3) |
| O(1Bb)–Pd(2)–N(1B) | 98.3(1) | Pd(3)⋯Pd(2d) | 10.483(2) |
| O(1C)–Pd(3)–N(1C) | 81.3(2) | ||
| O(1Cc)–Pd(3)–N(1C) | 98.7(2) | ||
a
| a Symmetry code used to generate equivalent atoms: (a) = −x + 2, −y, −z + 2. | |||
|---|---|---|---|
| Pd(1)–N(1) | 2.044(2) | C(1)–O(1) | 1.302(3) |
| Pd(1)–O(1) | 2.003(2) | C(1)–O(2) | 1.225(3) |
| C(2)–O(3) | 1.240(3) | ||
| O(1)–Pd(1)–N(1) | 81.61(7) | ||
| O(1a)–Pd(1)–N(1) | 98.39(7) | ||
CCDC reference numbers are 1402358 (3a) and 1402359 (4).
Results and discussion
Synthesis and IR spectroscopy
The N-substituted oxamate proligands were easily prepared as the respective ethyl ester derivatives by the straightforward condensation of the ethyl chlorooxoacetate with the corresponding 4-halo- or 4-methoxyaniline in THF, in the presence of Et3N as a base at room temperature. They were isolated in very satisfactory yields (ca. 80–96%). Their IR spectra exhibit a typical absorption peak at ca. 3000–3300 cm−1 for the N–H stretching vibration and two intense bands at ca. 1727 and 1677 cm−1 which are attributed to the stretching frequencies from the ester and amide groups, respectively. 1H and 13C NMR spectra support the presence of the N–H amide function as well as those corresponding to the ethyl ester group (–CH2CH3). The lack of the signals of the N–H amide and ethyl groups in the infrared spectra of the isolated complexes 1–5 together with the occurrence of two strong peaks at ca. 1638 and 1600 cm−1 support the deprotonation and hydrolysis of the proligands, resulting in the formation of N-substituted oxamate ligands which are subsequently coordinated to the Pd(II) ions. The X-ray structures of 3a and 4 (see below) fully support this spectroscopic evidence.Description of the structures 3a and 4
Compounds 3a and 4 crystallize in the triclinic space group P and the monoclinic space group P21/c respectively, and their crystal structures consist of centrosymmetric [Pd(4-Brpma)2]2− (3a) and [Pd(4-MeOpma)2]2− (4) complex anions [Fig. 1 (3a) and 2 (4)], organic n-Bu4N+ cations (3a and 4) and free water molecules (3a) held together by electrostatic forces. Three crystallographic independent complex anions are present in the asymmetric unit of 3a whereas only one occurs in 4.
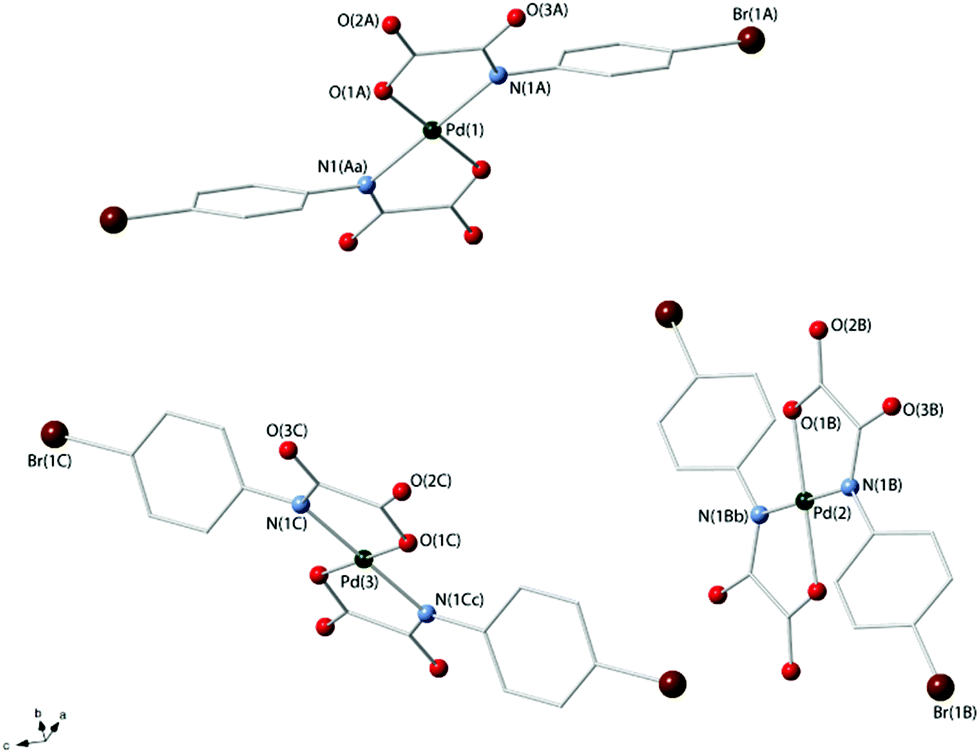 | ||
| Fig. 1 A perspective view of the three crystallographically independent [Pd(4-Brpma)2]2− units in 3a showing the atom numbering [symmetry codes: (a) −x + 1, −y + 1, −z + 1; (b) −x, −y, −z, (c) −x + 2, −y + 2, −z + 1]. | ||
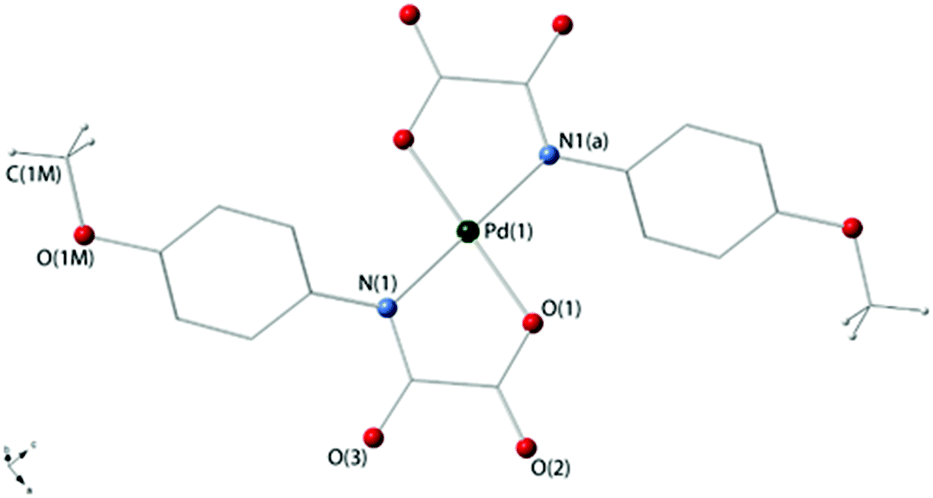 | ||
| Fig. 2 Perspective view of the anionic [Pd(4-MeOpma)2]2− entity in 4 showing the atom numbering [symmetry code: (a) = −x + 2, −y, −z + 2]. | ||
The palladium(II) ions in 3a [Pd(1), Pd(2) and Pd(3)] and 4 [Pd(1)] are four-coordinate with two amidate-nitrogen and two carboxylate-oxygen atoms from two bidentate oxamate ligands at each metal centre building a centrosymmetric square-planar surrounding. The reduced bite of the bidentate oxamate [81.5(1), 81.7(1) and 81.3(2)° at Pd(1), Pd(2) and Pd(3) in 3a and 81.61(7)° at Pd(1) in 4] mainly accounts for the deviations from the ideal geometry. Each Pd(II) ion is located exactly in the plane of the four donor atoms for symmetry reasons. The Pd–N/Pd–O bond lengths [see Tables 2 (3a) and 3 (4)] compare well with those found in the oxamato-containing palladium(II) complexes {[Na(H2O)]2trans-[Pd(2,6-Me2pma)2]}n (2,6-Me2pma = N-2,6-dimethylphenyloxamate), (n-Bu4N)2[Pd(2-Mepma)2]·2H2O (2-Mepma = N-2-methylphenyloxamate), (n-Bu4N)2[Pd(4-Mepma)2]·2H2O·MeCN (4-Mepma = N-4-methylphenyloxamate), [Pd(H2O)4][Pd(2,6-Me2pma)2]·2H2O, (n-Bu4N)2[Pd(2,6-Me2pma)2]·2CHCl3, Na[Pd(Hpba)]·2H2O [H4pba = 1,3-propylenebis(oxamic acid)], K2[Pd(opba)]·2H2O [H4opba = N,N′-1,2-phenylenebis(oxamic acid)], [Pd(NH3)4][Pd(opba)] and {[K4(H2O)(dmso)][(Pd2mpba)2]} [H4mpba = 1,3-phenylenebis(oxamic acid)] [average Pd–N/Pd–O distances of 2.020(1)/2.009(1), 2.0209(11)/2.0384(13), 2.0207(13)/2.0214(14), 2.023(3)/2.020(3), 2.03(2)/1.97(2), 1.970/2.040, 1.928/2.055, 1.942(10)/2.045(8) and 2.000(3)/2.026(2) Å, respectively].14,18–20,27
The values of the dihedral angle between the square plane at the palladium(II) ion and the mean plane of the oxamate groups (ϕ) are 6.53(8) [Pd(1)], 7.00(1) [Pd(2)] and 1.79(4)° [Pd(3)] in 3a and 7.9(1)° in 4, whereas those between each oxamate mean plane and the corresponding phenyl ring (Φ) are 53.8(2), 49.8(2) and 43.8(2)° [ligands A, B and C in Fig. 1] for 3a and 31.67(6)° for 4. The bond lengths of the peripheral C(1)–O(2) and C(2)–O(3) bond distances [values in the range 1.220(5)–1.255(4) (3a) and 1.225(3)–1.240(3) Å (4)] are somewhat shorter than the inner C(1)–O(1) bond [1.288(5)–1.294(5) (3a) and 1.302(3) Å (4)] in agreement with the greater double bond character of the free carbonyl groups.
The organic tetra-n-butylammonium cations in 3a and 4 exhibit the usual tetrahedral geometry and their bond lengths and angles are as expected. The complex anions in 3a are well-separated from each other by bulky n-Bu4N+ cations and they weakly interact through hydrogen bonds involving terminal oxygen atoms of the oxamate ligands and disordered water molecules of crystallization (O⋯O separations varying in the range 2.70–3.17 Å) giving rise to a supramolecular 1D motif developing along the crystallographic a-axis (Fig. 3a).
 | ||
| Fig. 3 (a) View along the b-axis of the 1D supramolecular motif in 3a. Projections of the crystal packing of 3a along the crystallographic a-axis excluding (b) and including (c) the tetra-n-butylammonium cations. | ||
The shortest values of the intermolecular palladium⋯palladium separation in 3 are 9.658(1) [Pd(1)⋯Pd(3)], 10.483(2) [Pd(3)⋯Pd(2d); symmetry code: (d) = x + 1, y + 1, z], 11.795(2) [Pd(1)⋯Pd(2)] and 18.793 Å [Pd(2)⋯Pd(3)]. In 4, instead, the [Pd(4-MeOpma)2]2− entities are pillared along the crystallographic a axis by means of weak C–H⋯O interactions and the cations are placed between the resulting anionic supramolecular columns.
The values of the shortest metal–metal separations in 4 are 9.7928(5) [Pd(1)⋯Pd(1b); (b) = x + 1, y, z] and 11.4651(4) Å [Pd(1)⋯Pd(1c); (c) = −x + 2, y − 0.5, −z + 1.5]. Views of fragments of the crystal packing of 3a and 4 are given in Fig. 3 and 4, respectively.
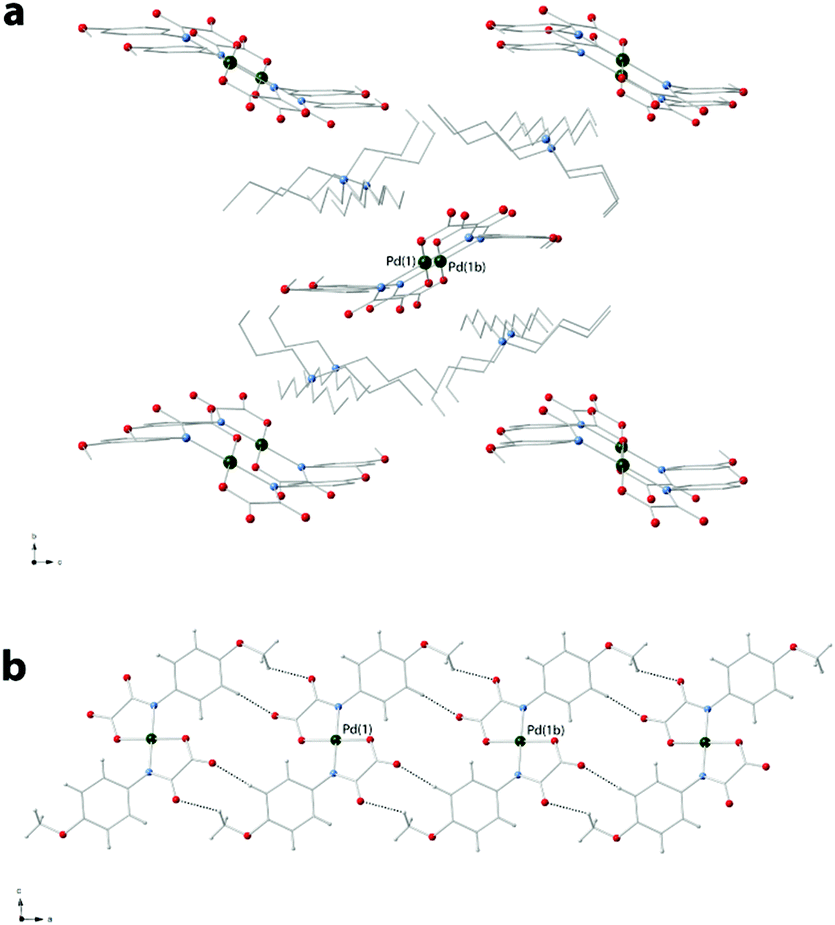 | ||
| Fig. 4 (a) A projection of the crystal packing of 4 along the crystallographic a axis. (b) A view of a supramolecular column of the [Pd(4-MeOpma)2]2− complex anions developing along the same direction. | ||
Catalytic study
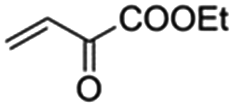
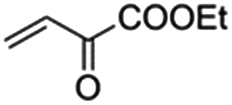
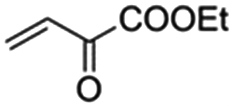
The bis(N-substituted oxamate)palladate(II) precatalysts 1–5 which exhibit different electron and steric properties have been checked for the Heck-vinylation of a series of aryl halides with various olefins using the ionic liquid n-Bu4NBr as the reaction medium. In this respect, it deserves to be noted that this ionic liquid has been previously used as an additive for this type of reaction, its role being qualified as essential.28 In our case, the catalyst and solvent recovery and recycling are achieved by working with anionic bis(N-substituted oxamate)palladate(II) complexes in tetra-n-butylammonium bromide as a green solvent instead, not just an additive. We carried out the Heck reactions with 0.5 and 0.25 mol% of catalyst with Et3N as the base at the minimum temperature required for the n-Bu4NBr to melt (120 °C). Very short reaction times were required to achieve high yields (see Tables 4 and S1†). The catalytic activity of 1–5 was investigated by using a variety of aryl halide derivatives together with a selection of olefins and the obtained results improve the previous findings of bis(N-substituted oxamate)palladate(II) in such carbon–carbon cross coupling reactions.14|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | Olefin | Cat. | Time (min) | Runs – Yieldb (%) | |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||||
| a Reaction conditions: 0.5 mmol iodobenzene, 0.75 mmol olefin, 0.5 mol% cat., 1 mmol of Et3N, 5–30 min, 120 °C in n-Bu4NBr. b Determined by GC-MS analysis using perfluorotributylamine (PFTBA) as the internal standard. | |||||||||||
| 1 |

|
1 | 5 | 99 | 99 | 99 | 99 | 99 | 99 | 99 | 99 |
| 2 |
|
2 | 5 | 99 | 99 | 99 | 99 | 99 | 99 | 99 | 99 |
| 3 |

|
3 | 5 | 99 | 99 | 99 | 99 | 99 | 99 | 99 | — |
| 4 |
|
4 | 5 | 98 | 99 | 99 | 99 | 99 | 99 | 98 | 92 |
| 5 |
|
5 | 5 | 92 | 99 | 99 | 99 | 99 | 98 | 98 | 96 |
| 6 |
|
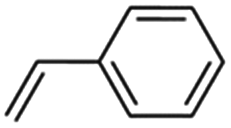
1 | 30 | 99 | 97 | 99 | 99 | 99 | 99 | 99 | 99 |
| 7 |
|
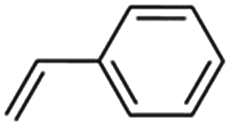
2 | 30 | 70 | 95 | 96 | 99 | 99 | 98 | 98 | 98 |
| 8 |
|
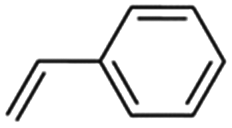
3 | 30 | 99 | 99 | 99 | 98 | 90 | 90 | 97 | 95 |
| 9 |
|
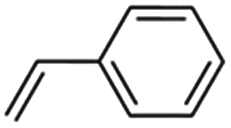
4 | 30 | 86 | 99 | 99 | 99 | 98 | 98 | 99 | 99 |
| 10 |
|
5 | 30 | 88 | 77 | 99 | 99 | 99 | 99 | 99 | 96 |
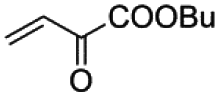
As shown in Table 4, high efficiency in recovery and recycling (at last 8 catalytic runs) is observed by using complexes 1–5 (0.5 mol%) for a Heck coupling of the active aryl iodides (0.5 mmol) and ethyl acrylate (0.75 mmol, entries 1–5) or styrene (0.75 mmol, entries 6–10) and Et3N (1 mmol). A consistent yield value of 99% is achieved in each run for 1–5 with very short reaction times and satisfactory TOF values (5 minutes, entries 1–5 in Table 4; TOF ca. 2376 h−1, see Table S1†). Yields about 99% are also obtained by using a less activated olefin as styrene although only if the reaction time is increased (30 minutes, entries 6–10 in Table 4; TOF ca. 396 h−1, see Table S1†). These short reaction times and the green solvent used allow one not only to perform reaction under mild reaction conditions but also to save energy costs.
When the amount of the catalyst is decreased from 0.5 to 0.25 mol%, the consistency of the measured yields in the vinylation of iodobenzene with ethyl acrylate is lost. However, in this case yields appear to be improved with the successive runs attaining values in the range 83–99% at the eighth run (see Table S2†). This demonstrates that the recovery and recycling of 1–5 happens without leaching phenomena. It deserves to be noted that the reaction time of 5 min is maintained with high TOF values (ca. 4800 h−1).
The behaviour of complexes 1–5 when working with a less reactive aryl halide such as bromobenzene is shown in Tables 5 and S3.† One can see therein the increase in the reaction time from 5 min (entries 1–5 in Table 4) to 3 hours (entries 1–5 in Table 5) using ethyl acrylate or from 30 minutes (entries 6–10 in Table 4) to 4 hours (entries 9–13 in Table 5) using the less active styrene. Moreover, under our reaction conditions, complexes 1–5 are more efficient than the commercial catalysts [PdCl2] and [Pd3(OAC)6] and even [Pd(dba)2]29 (entries 6–8 in Table 5). The fact that catalytic activity using bromobenzene (Table 5) is somewhat lower and more variable than that observed when using iodobenzene (Table 4) is due to the different dissociation energies of the C–Br and C–I bonds (81 and 65 kcal mol−1, respectively).30 An increase in the yield when using bromobenzene could be achieved by working at higher temperatures; however, we are limited by the decomposition temperature of the n-Bu4NBr molten salt,31i.e. ca. 130 °C. Having reached this point, one may consider when thinking about energy costs and pursuing green catalytic routes that it is worth the effort to carry out the reaction with cheaper chemicals for 4 hours (bromobenzene and styrene; entries 9–13 in Table 5) vs. using more expensive materials for 30 minutes (iodobenzene and styrene; entries 6–10 in Table 4).
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | Olefin | Catalyst | Time (h) | Runs – Yieldb (%) | |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||||
| a Reaction conditions: 0.5 mmol bromobenzene, 0.75 mmol olefin, 0.5 mol% cat., 1 mmol of Et3N, 3–4 h, 120 °C in n-Bu4NBr. b Determined by GC-MS analysis using perfluorotributylamine (PFTBA) as the internal standard. c Not tested. | |||||||||||
| 1 |
|
1 | 3 | 99 | 99 | 92 | 86 | 84 | 79 | 78 | 61 |
| 2 |
|
2 | 3 | 72 | 87 | 90 | 84 | 87 | 80 | 78 | 70 |
| 3 |
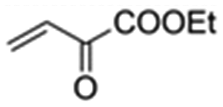
|
3 | 3 | 84 | 91 | 91 | 87 | 88 | 83 | 80 | 72 |
| 4 |
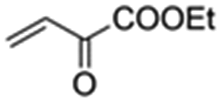
|
4 | 3 | 18 | 64 | 80 | 77 | 82 | 77 | 76 | 71 |
| 5 |
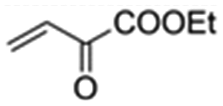
|
5 | 3 | 75 | 89 | 85 | 84 | 82 | 82 | 76 | 69 |
| 6 |

|
[PdCl2] | 3 | 0 | 0 | 0 | 0 | 0 | 0 | —c | —c |
| 7 |
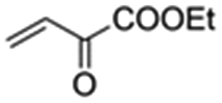
|
[Pd3(OAc)6] | 3 | 0 | 0 | 0 | 0 | 0 | 0 | —c | —c |
| 8 |
|
[Pd(dba)2] | 3 | 62 | 68 | 74 | 73 | 70 | 60 | —c | —c |
| 9 |

|
1 | 4 | 92 | 86 | 66 | 56 | 62 | 83 | 73 | 67 |
| 10 |
|
2 | 4 | 66 | 76 | 75 | 72 | 70 | 74 | 67 | 68 |
| 11 |

|
3 | 4 | 0 | 58 | 65 | 75 | 68 | 73 | 69 | 77 |
| 12 |
|
4 | 4 | 36 | 92 | 72 | 61 | 70 | 70 | 70 | 68 |
| 13 |
|
5 | 4 | 80 | 90 | 82 | 77 | 74 | 72 | 72 | 65 |
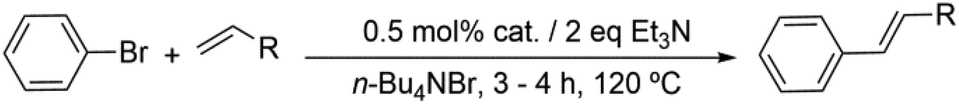
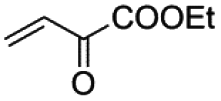
Similar results have been obtained with the use of different 4-monosubstituted aryl halides and olefins as shown in Tables 6 and S4.† Once again, very short reaction times (5 minutes) and high yields (ca. 90%) are observed when working with aryl iodide derivatives (entries 1–5 in Table 6). An increase of the reaction time to achieve the same good yields is required for the bromo derivatives (entries 7 and 8 in Table 6), as mentioned above. Finally, it deserves to be noted that this type of palladium(II) complex is unable to activate less active aryl halides such as chlorobenzenes (entry 9 in Table 6) which need higher temperatures to react (T > 120 °C),32 a feature that precludes the use of n-Bu4NBr as the solvent.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya | Aryl halide | Olefin | Time | Catalyst/Runs – Yieldb (%) | ||||
| 1 | 2 | 3 | 4 | 5 | ||||
| a Reaction conditions: 0.5 mmol aryl halide, 0.75 mmol olefin, 0.5 mol% cat.,1 mmol Et3N, 120 °C in (n-Bu4N)Br. b Determined by GC-MS analysis using perfluorotributylamine (PFTBA) as the internal standard. c Not tested. | ||||||||
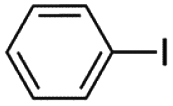
| 1 |
|
|
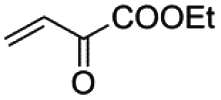
5 min | 75 | 88 | 97 | 90 | 92 |
| 2 |
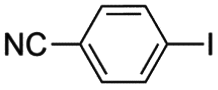
|
|
5 min | 89 | 99 | 99 | 89 | 88 |
| 3 |
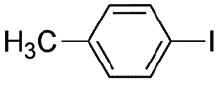
|
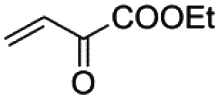
|
5 min | 90 | 85 | 96 | 99 | 99 |
| 4 |
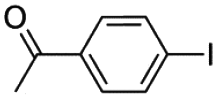
|
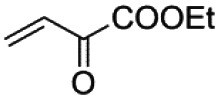
|
5 min | 93 | 93 | 98 | 99 | 99 |
| 5 |
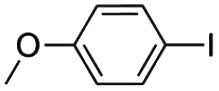
|
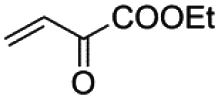
|
5 min | 93 | 92 | 99 | 99 | 99 |
| 6 |
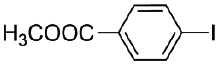
|
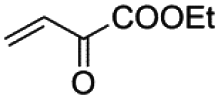
|
15 min | 99 | 99 | 95 | 99 | 99 |
| 7 |
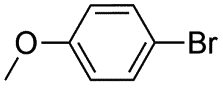
|
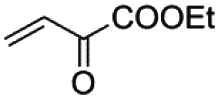
|
1.5 h | 99 | 99 | 99 | 99 | 99 |
| 8 |
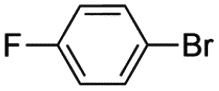
|
|
4 h | 92 | 94 | 93 | 91 | 94 |
| 9 |
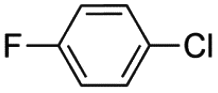
|
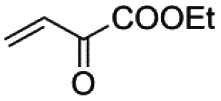
|
72 h | —c | 13 | —c | —c | —c |
The characteristic event that stops a Pd(II)-catalyzed carbon–carbon coupling reaction is the formation of inactive metallic palladium.33 Under our catalytic conditions, this occurs when working with commercial palladium catalysts such as [PdCl2] and [Pd3(OAc)6] (first two pictures in Fig. 5). However, the formation of the inactive palladium black does not occur if the reaction is performed with the oxamate-containing palladium(II) complexes of this work (last two pictures in Fig. 5).
 | ||
| Fig. 5 From left to right: [PdCl2], [Pd3(OAc)6], 3 and 4, after the Heck reaction of bromobenzene with ethyl acrylate in n-Bu4NBr at 120 °C for 3 h. | ||
Finally, the competing reaction between aryl halides and two different olefins (ethyl acrylate and styrene) was investigated. These reactions were carried out at two different times because the reaction with ethyl acrylate runs faster than that with styrene (Table 7). The obtained results clearly show a higher activity for ethyl acrylate compared to styrene in the Heck reaction by using oxamate-containing palladium(II) complexes in n-Bu4NBr, just the opposite trend with respect to that reported by Böhm and Herrmann in 2001 by using a phosphapalladacycle as the catalyst.34
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Aryl halide | Catalyst | Time | Ab (%) | Bb (%) | K o = B/A |
| a Reaction conditions: 1 mmol iodobenzene, 5 mmol ethyl acrylate, 5 mmol styrene, 0.5 mol% cat., 2 mmol Et3N, 120 °C in n-Bu4NBr. b Determined by GC-MS analysis using perfluorotributylamine (PFTBA) as the internal standard. | ||||||
| 1 |
|
2 | 6 min | 95 | 5 | 0.05 |
| 2 |
|
4 | 6 min | 99 | 0 | 0 |
| 3 |

|
2 | 30 min | 91 | 9 | 0.10 |
| 4 |
|
4 | 30 min | 88 | 12 | 0.14 |
| 5 |
|
2 | 3 h | 99 | 0 | 0 |
| 6 |
|
4 | 3 h | 80 | 0 | 0 |
| 7 |

|
2 | 4 h | 60 | 0 | 0 |
| 8 |
|
4 | 4 h | 60 | 0 | 0 |
Conclusions
In summary, five novel functional palladium(II)-oxamate complexes (1–5) with well-defined structures were synthesized, exhibiting several advantages over commercially available palladium(0) or palladium(II) catalysts. They are environmentally friendly chemicals (in contrast to harmful phosphines) and their easy and cheap preparation (compared mainly to the harder and time-consuming carbene systems) makes them very suitable green systems for catalytic cross-coupling purposes. They appear robust, even under air (controlled atmosphere – N2 or Ar – is commonly used with other palladium catalysts in carbon–carbon cross coupling reactions),16a without significant appearance of the non-catalytic palladium black species. They are also soluble and highly stable in water and remain unchanged under ambient conditions (Fig. S1†).The present study on the catalytic properties of 1–5 shows that they are suitable catalysts to work with iodo- and bromo-aryl derivatives with yields around ca. 80–99%.
Moreover, in comparison with previous studies,14 the change of the R substituent group at the para-position of the aryl portion of the oxamate ligand allows a decrease in the amount of the catalyst needed to carry out the reaction with high yields (0.5–0.25 mol% cat.) and in very short reaction times (a minimum of 5 min of catalytic reaction and a maximum of TON/TOF ca. 400/ca. 4800 h−1).
Thinking at addressing upcoming environmental concerns, the n-Bu4NBr solvent as a superior ionic liquid serves not only as an alternative, greener medium to perform Heck reactions, but also provides an easy work-up procedure in which various consecutive runs preclude the loss of the catalyst in the unloading and recharge of products and reactive elements. Thus, the combination of oxamate-containing palladium(II) complexes as structurally-well defined precatalysts with ionic liquids opens a new way to perform Heck reactions under greener conditions without loss of the catalytic activity.
Acknowledgements
Financial support from the Spanish Ministerio de Ciencia e Innovación (MCIIN) through Project CTQ2013-448449, the Generalitat Valenciana (PROMETEOII/2014/070 and ISIC/2012/002), the Italian Ministero dell'Istruzione dell'Università e della Ricerca Scientifica (MiUR) and the European Community's Seventh Framework Program (FP7 2007–2013) through the MATERIA Project (PONa3_00370) is acknowledged. F. R. F.-P. thanks the MCIIN for a predoctoral FPU grant and N. M. also thanks the European Commission, FSE (Fondo Sociale Europeo) and Calabria Region for a post-doctoral fellowship.Notes and references
- (a) K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374 CrossRef CAS; (b) R. J. P. Corriu and J. P. Masse, J. Chem. Soc., Chem. Commun., 1972, 144a RSC; (c) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS; (d) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed.
- Some selected examples: (a) R. F. Heck, Acc. Chem. Res., 1979, 12, 146 CrossRef CAS; (b) N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS PubMed; (c) M. Shibasaki, C. D. J. Boden and A. Kojima, Tetrahedron, 1997, 53, 7371 CrossRef CAS; (d) A. Suzuki, J. Organomet. Chem., 1999, 576, 147 CrossRef CAS; (e) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed; (f) W. A. Herrmann, V. P. W. Böhm and C.-P. Reisinger, J. Chem. Educ., 2000, 77, 92 CrossRef CAS; (g) F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 2419 CAS; (h) J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651 CrossRef CAS PubMed; (i) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed; (j) J. F. Hartwig, Nature, 2008, 41, 314 CrossRef PubMed.
- (a) E. Negishi, Angew. Chem., Int. Ed., 2011, 123, 6870 CrossRef PubMed; (b) E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738 CrossRef CAS PubMed; (c) A. Suzuki, Angew. Chem., Int. Ed., 2011, 123, 6854 CrossRef PubMed; (d) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed; (e) D. Astruc, Anal. Bioanal. Chem., 2011, 399, 1811 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (b) J.-P. Corbet and G. R. Migani, Chem. Rev., 2006, 106, 2651 CrossRef CAS PubMed; (c) V. L. Budarin, P. S. Shuttleworth, J. H. Clark and R. Luque, Industrial Applications of C-C Coupling Reactions, Bentham Science Publishers, 2010 Search PubMed.
- (a) P. J. Steel, Acc. Chem. Res., 2005, 38, 243 CrossRef CAS PubMed; (b) J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574 CrossRef CAS PubMed; (c) T. Gadzikwa, B.-S. Zeng, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2008, 3672 RSC; (d) Q. Meng, J. Gao, R. Li, L. Jiang, C. Wang, H. Zhao, C. Liu, H. Li and W. Hu, J. Mater. Chem., 2009, 19, 1477 RSC; (e) J. A. Lipton-Duffin, O. Ivasenko, D. F. Perepichka and F. Rosei, Small, 2009, 5, 592 CrossRef CAS PubMed; (f) M. Alyapyshev, V. Babain, N. Borisova, I. Eliseev, D. Kirsanov, A. Kostin, A. Legin, M. Reshetova and Z. Smirnova, Polyhedron, 2010, 29, 1998 CrossRef CAS PubMed; (g) P. Rattanatraicharoen, K. Yamabuki, T. Oishi and K. Onimura, Polym. J., 2011, 44, 224 CrossRef PubMed; (h) M. Jurícek, J. C. Barnes, E. J. Dale, W.-G. Liu, N. L. Strutt, C. J. Bruns, N. A. Vermeulen, K. C. Ghooray, A. A. Sarjeant, C. L. Stern, Y. Y. Botros, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 12736 CrossRef PubMed; (i) M. Castellano, R. Ruiz-García, J. Cano, J. Ferrando-Soria, E. Pardo, F. R. Fortea-Pérez, S.-E. Stiriba, M. Julve and F. Lloret, Acc. Chem. Res., 2015, 48, 510 CrossRef CAS PubMed.
- (a) S. Sankaranarayanapillai, L. Wang and W. R. Thiel, Adv. Synth. Catal., 2010, 352, 425 CrossRef PubMed; (b) C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694 RSC; (c) R. J. Lundgren and M. Stradiotto, Chem. – Eur. J., 2012, 18, 9758 CrossRef CAS PubMed; (d) N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916 RSC.
- (a) G.-C. Ge, D.-L. Mo, C.-H. Ding, L. X. Dai and X.-L. Hou, Org. Lett., 2012, 14, 5156 CrossRef PubMed; (b) G. R. Rosa and D. S. Rosa, RSC Adv., 2012, 2, 5080 RSC; (c) K. Karami, N. Rahimi and M. B. Shehni, Tetrahedron Lett., 2012, 53, 2428 CrossRef CAS PubMed; (d) S. J. Sabounchei and M. Ahmadi, Inorg. Chim. Acta, 2013, 405, 15 CrossRef CAS PubMed.
- (a) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC; (b) F. Godoy, C. Segarra, M. Poyatos and E. Peris, Organometallics, 2011, 30, 684 CrossRef CAS; (c) J. J. Dunsford and K. J. Cavel, Dalton Trans., 2011, 40, 9131 RSC; (d) C. Valente, S. Çalimsiz, K. H. Hou, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314 CrossRef CAS PubMed; (e) Y.-M. Liu, Y.-C. Lin, W.-C. Chen, J.-H. Cheng, Y.-L. Chen, G. P. A. Yap, S.-S. Sun and T.-G. Ong, Dalton Trans., 2012, 41, 7382 RSC.
- (a) N. J. S. Costa, P. K. Kiyohara, A. L. Monteiro, Y. Coppel, K. Philippot and L. M. Rossi, J. Catal., 2010, 276, 382 CrossRef CAS PubMed; (b) S. S. Zalesskiy and V. P. Ananikov, Organometallics, 2012, 31, 2302 CrossRef CAS; (c) H. Firouzabadi, N. Iranpoor, F. Kazemi and M. Cholinejad, J. Mol. Catal. A: Chem., 2012, 357, 154 CrossRef CAS PubMed; (d) M. Pérez-Lorenzo, J. Phys. Chem. Lett., 2012, 3, 167 CrossRef.
- (a) B. Karimi, D. Elhamifar, J. H. Clark and A. J. Hunt, Chem. – Eur. J., 2010, 16, 8047 CrossRef CAS PubMed; (b) M. H. G. Prechtl, J. D. Scholten and J. Dupont, Molecules, 2010, 15, 3441 CrossRef CAS PubMed; (c) B. Tamami, F. Farjadian, S. Ghasemi and H. Allahyari, New J. Chem., 2013, 37, 2011 RSC; (d) H. Li, M. Yang and Q. Pu, Microporous Mesoporous Mater., 2012, 148, 166 CAS; (e) A. S. Singh, U. B. Patil and J. M. Nagarkar, Catal. Commun., 2013, 35, 11 CrossRef CAS PubMed.
- (a) H.-U. Blaser, A. Indolese, A. Schnyder, H. Steiner and M. Studer, J. Mol. Catal. A: Chem., 2001, 173, 3 CrossRef CAS; (b) T. Rosner, A. Pfaltz and D. G. Blackmond, J. Am. Chem. Soc., 2001, 123, 4621 CrossRef CAS; (c) C. Rocaboy and J. A. Gladysz, Org. Lett., 2002, 4, 1993 CrossRef CAS PubMed; (d) C. Rocaboy and J. A. Gladysz, New J. Chem., 2003, 27, 39 RSC; (e) S. Paul and J. H. Clark, J. Mol. Catal. A: Chem., 2004, 215, 107 CrossRef CAS PubMed; (f) K. Yu, W. Sommer, J. M. Richardson, M. Weck and C. W. Jones, Adv. Synth. Catal., 2005, 347, 161 CrossRef CAS PubMed; (g) N. T. S. Phan, M. Van der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef CAS PubMed.
- (a) R. Sheldon, Chem. Commun., 2001, 2399 RSC; (b) C. M. Gordon, Appl. Catal., A, 2001, 222, 101 CrossRef CAS; (c) D. Zhao, M. Wu, Y. Kou and E. Min, Catal. Today, 2002, 74, 157 CrossRef CAS; (d) R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792 CrossRef PubMed; (e) D. Holbrey John, B. Turner Megan and D. Rogers Robin, Ionic Liquids as Green Solvents, American Chemical Society, Washington, DC, 2003, vol. 856 Search PubMed; (f) T. Welton, Coord. Chem. Rev., 2004, 248, 2459 CrossRef CAS PubMed; (g) V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615 CrossRef PubMed; (h) G. Imperato, B. König and C. Chiappe, Eur. J. Org. Chem., 2007, 1049 CrossRef CAS PubMed; (i) H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1 CrossRef CAS PubMed; (j) P. Mastrorilli, A. Monopoli, M. M. Dell'Anna, M. Latronico, P. Cotugno and A. Nacci, Top. Organomet. Chem., 2015, 51, 237 CrossRef.
- Some selected examples: (a) V. P. W. Bohm and W. A. Herrmann, Chem. – Eur. J., 2000, 6, 1017 CrossRef CAS; (b) F. Bellina and C. Chiappe, Molecules, 2010, 15, 2211 CrossRef CAS PubMed; (c) A. R. Hajipour and F. Rafiee, Appl. Organomet. Chem., 2011, 25, 542 CrossRef CAS PubMed; (d) V. Hornillos, A. W. van Zijl and B. L. Feringa, Chem. Commun., 2012, 48, 3712 RSC; (e) A. Ataei, S. Nadri, E. Rafiee, S. Jamali and M. Joshaghani, J. Mol. Catal. A: Chem., 2013, 366, 30 CrossRef CAS PubMed.
- F. R. Fortea-Pérez, I. Schlegel, M. Julve, D. Armentano, G. De Munno and S.-E. Stiriba, J. Organomet. Chem., 2013, 743, 102 CrossRef PubMed.
- (a) C. Zhou, J. Wang, L. Li, R. Wang and M. Hong, Green Chem., 2011, 13, 2100 RSC; (b) M. Bagherzadeh, M. Amini, A. Ellern and L. K. Woo, Inorg. Chim. Acta, 2012, 383, 46 CrossRef CAS PubMed.
- (a) A. Molnár, Chem. Rev., 2011, 111, 2251 CrossRef PubMed; (b) H.-P. Steinrück and P. Wasserscheid, Catal. Lett., 2015, 145, 380 CrossRef.
- (a) M. Guerrero, J. García-Antón, M. Tristany, J. Pons, J. Ros, K. Philippot, P. Lecante and B. Chaudret, Langmuir, 2010, 26(19), 155532 CrossRef PubMed; (b) G. Sánchez, J. García, M. Martínez, A. R. Kapdi, J. Pérez, L. García and J. L. Serrano, Dalton Trans., 2011, 40, 12676 RSC; (c) D. Penno, F. Estevan, E. Fernández, P. Hirva, P. Lahuerta, M. Sanaú and M. A. Úbeda, Organometallics, 2011, 30, 2083 CrossRef CAS.
- (a) W. X. C. Oliveira, M. A. Ribeiro, C. B. Pinheiro, W. C. Nunes, M. Julve, Y. Journaux, H. O. Stumpf and C. L. M. Pereira, Eur. J. Inorg. Chem., 2012, 34, 5685 CrossRef PubMed; (b) W. X. C. Oliveira, M. A. Ribeiro, C. B. Pinheiro, M. M. da Costa, A. P. S. Fontes, W. C. Nunes, D. Cangussu, M. Julve, H. O. Stumpf and C. L. M. Pereira, Cryst. Growth Des., 2015, 15, 1325 CrossRef CAS.
- W. X. C. Oliveira, M. M. da Costa, A. P. S. Fontes, C. B. Pinheiro, F. C. S. de Paula, E. H. L. Jaimes, E. F. Pedroso, P. P. de Souza, E. C. Pereira-Maia and C. L. M. Pereira, Polyhedron, 2014, 76, 16–21 CrossRef CAS PubMed.
- (a) F. R. Fortea-Pérez, N. Marino, D. Armentano, G. De Munno, M. Julve and S.-E. Stiriba, CrystEngComm, 2014, 16, 6971 RSC; (b) F. R. Fortea-Pérez, D. Armentano, M. Julve, G. de Munno and S.-E. Stiriba, J. Coord. Chem., 2014, 67, 4003 CrossRef PubMed.
- M. F. Rettig and P. M. Maitlis, Inorg. Synth., 2007, 17, 134 CrossRef PubMed.
- SAINT, Version 6.45, Bruker Analytical X-ray Systems Inc., Madison, WI, USA, 2003 Search PubMed.
- SADABS, Version 2.03, Bruker AXS Inc., Madison, WI, USA, 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- (a) P. Müller, Cryst. Rev., 2009, 15, 57 CrossRef PubMed; (b) P. Müller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, Crystal Structure Refinement: A Crystallographer's Guide to SHELXL, in IUCr Texts on Crystallography, ed. P. Müller, Oxford University Press, Oxford, U.K., 2006 Search PubMed.
- CrystalMaker, CrystalMaker Software, Bicester, England, 2015 Search PubMed.
- R. Kivekas, A. Pajunen, A. Navarrete and E. Colacio, Inorg. Chim. Acta, 1999, 284, 292 CrossRef CAS.
- (a) R. B. Bedford, M. E. Blake, C. P. Butts and D. Holder, Chem. Commun., 2003, 466 RSC; (b) C. Nájera, J. Gil-Moltó, S. Karlström and L. R. Falvello, Org. Lett., 2003, 5, 1451 CrossRef PubMed; (c) Q. Yao, E. P. Kinney and C. Zheng, Org. Lett., 2004, 6, 2997 CrossRef CAS PubMed; (d) C. Wolf and R. Lerebours, Org. Biomol. Chem., 2004, 2, 2161 RSC; (e) F. Alonso, I. P. Beletskaya and M. Yus, Tetrahedron, 2005, 61, 11771 CrossRef CAS PubMed; (f) F. Alonso, I. P. Beletskaya and M. Yus, Tetrahedron, 2008, 64, 3047 CrossRef CAS PubMed; (g) S. M. Islam, P. Mondal, A. S. Roy, S. Mondal and D. Hossain, Tetrahedron Lett., 2010, 51, 2067 CrossRef CAS PubMed; (h) W. Susanto, C.-Y. Chu, W. J. Ang, T.-C. Chou, L.-C. Lo and Y. Lam, Green Chem., 2012, 14, 77 RSC; (i) A. Ataei, S. Nadri, E. Rafiee, S. Jamali and M. Joshaghani, J. Mol. Catal. A: Chem., 2013, 366, 30 CrossRef CAS PubMed.
- S. S. Zalesskiy and V. P. Ananikov, Organometallics, 2012, 31, 2302 CrossRef CAS.
- V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047 CrossRef CAS.
- F. Bellina and C. Chiappe, Molecules, 2010, 15, 2211 CrossRef CAS PubMed.
- (a) S. S. Pröckl, W. Kleist and K. Köhler, Tetrahedron, 2005, 61, 9855 CrossRef PubMed; (b) P. Srinivas, P. R. Likhar, H. Maheswaran, B. Sridhar, K. Ravikumar and M. L. Kantam, Chem. – Eur. J., 2009, 15, 1578 CrossRef CAS PubMed; (c) W. Kleist and S. S. Pröckl, Catal. Lett., 2008, 125, 197 CrossRef CAS; (d) L. C. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581 CrossRef CAS PubMed.
- (a) C. S. Consorti, M. L. Zanini, S. Leal, G. Ebeling and J. Dupont, Org. Lett., 2003, 5, 983 CrossRef CAS PubMed; (b) A. H. M. de Vries, J. M. C. a Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285 CrossRef CAS PubMed; (c) G. Brasche, J. García-Fortanet and S. L. Buchwald, Org. Lett., 2008, 10, 2207 CrossRef CAS PubMed.
- V. P. Böhm and W. A. Herrmann, Chem. – Eur. J., 2001, 7, 4191 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format, detailed synthesis of the proligands, stability of the precatalyst in water (Fig. S1), data of palladium-catalyzed Heck C–C coupling reactions (Tables S1–S4) and 1H-NMR, 13C-NMR and Dept-135-NMR of the catalytic products (Fig. S2–S8). CCDC 1402358 (3a) and 1402359 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00093a |
| This journal is © the Partner Organisations 2015 |