
Water adsorption properties of a Sc(III) porous coordination polymer for CO2 capture applications†‡
J. Raziel
Álvarez
a,
Ricardo A.
Peralta
a,
Jorge
Balmaseda
a,
Eduardo
González-Zamora
*b and
Ilich A.
Ibarra
*a
aInstituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n, CU, Del. Coyoacán, 04510, México D. F., Mexico. E-mail: argel@unam.mx
bDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, San Rafael Atlixco 186, Col. Vicentina, Iztapalapa, C. P. 09340, México D. F., Mexico
First published on 2nd November 2015
Abstract
Water adsorption at room temperature in NOTT-400 was investigated along with its ability to perform CO2 capture under relative humidity (RH) conditions. Thus, the CO2 capture was increased from 4.2 wt% (anhydrous conditions) to 10.2 wt% at 20% RH and 30 °C.
Metal–organic frameworks (MOFs) or porous coordination polymers (PCPs) are very promising candidates for gas sequestration, since these materials show high sorption selectivity towards adsorbates. This selectivity can be adjusted as a function of the size, shape and chemical composition of the pores.1 Even with their potential as porous capture materials, many PCPs are unstable when exposed to moisture.2,3 Water could either shift the bound ligand, leading to the collapse of the PCP structure, or block the binding adsorption sites and therefore, avoid the adsorption of other desired molecules.3 Thus, water stability has limited the use of PCPs in industrial settings because moisture is ubiquitous in the environment and it must be accounted for in adsorption, storage and separation systems. For example, natural gas streams are frequently saturated with water vapour that has to be removed (to ppm levels) before storage or use.4 Industrial flue gas, resulting from the combustions of fossil fuels, is saturated with water (5–7%).5
However, a significant number of PCPs have exhibited relatively good stability to water, for example: UiO-66,6 NOTT-401,7 MIL-100,8 MIL-101,9 MIL-5310 and InOF-1.11 Thus, understanding and predicting the adsorption properties of water in hydrostable PCPs is fundamental for the development of industrial applications. Doonan and co-workers12 showed a Cu(II) metal–organic framework (Cu(bcppm)H2O, H2bcppm = bis(4-(4-carboxyphenyl)-1H-pyrazolyl)methane) which is hydrostable and also performed a remarkable selective separation of CO2 from N2. Walton and co-workers have shown water adsorption isotherms for a series of PCPs (Mg-MOF-174, DMOF-1, UiO-66 and UMCM-1) and compared them with reference micro-mesoporous materials.13 Farrusseng et al.14 reported the structure property relationships of water adsorption in a comprehensive series of PCPs (MIL-53, MIL-68, MIL-101, MIL-125, etc.). Moreover, some of these water stable materials have been investigated for application in storage technologies for arid environments,15 heat-pumps and chillers,16 low-cost water capture15 and proton conductivity.17
The effect of water on the CO2 capture has only recently been investigated on PCPs.18 Water adsorption is often unfavourable for CO2 capture since water acts as a strong adsorptive competitor. However, LeVan,19 Matzger20 and Walton21 demonstrated that controlled water adsorption can enhance CO2 capture in PCPs. Llewellyn and co-workers22 investigated the CO2 adsorption in some PCPs under different relative humidities of water vapour. Then, UiO-66 did not show any enhanced CO2 uptake and for MIL-100(Fe), a remarkable 5-fold increase in CO2 uptake was achieved. Interestingly, Walton and co-workers23 showed that functional groups act as a directing agent for water in the pores, which allows for more efficient packing. Furthermore, Yaghi et al.15 proposed that the presence of these functional groups, within the porous coordination polymer, enhance the affinity of the material for water. Thus, in the present work we have chosen a material named NOTT-400.24a This material crystallises in the tetragonal space group I4122, showing a binuclear [Sc2(μ2-OH)] building block (Fig. S1, ESI‡). The coordination around each Sc(III) centre is octahedral from six O-donors: 4 from different carboxylate groups and 2 from two μ2-OH hydroxo groups which bridge two metal (Sc(III)) centres. NOTT-400 shows a three-dimensional framework structure with channel openings of approximately 8.1 Å (considering the van der Waals radii of the surface atoms).24a We report herein the water adsorption properties and the CO2 capture in the presence of water24b in NOTT-400.
First, the non-coordinated solvent molecules within the pores of the as-synthesised NOTT-400 were exchanged for acetone promoting accessibility to the desolvated framework after activation by heating. Thermogravimetric analysis (see Fig. S2, ESI‡) and bulk powder X-ray diffraction (PXRD) patterns (see Fig. S3, ESI‡) of the as-synthesised and desolvated NOTT-400 confirmed that the material constantly maintains its structural integrity after desolvation. N2 adsorption isotherms for activated NOTT-400 at 77 K were obtained in order to calculate the BET surface area (0.01 < P/P0 < 0.04) of 1356 m2 g−1 (see Fig. S8, ESI‡). Water isotherms were obtained on the activated samples of NOTT-400 (see the Experimental section). Fig. 1 shows the water adsorption isotherm of NOTT-400 at 30 °C. The adsorbed amount of water slowly increased with increasing pressure up to %P/P0 = 20. Thus, a rapid water uptake was observed in the pressure range from %P/P0 = 20 to 40. Finally, from %P/P0 = 40 to 90 there was a gradual weight increase, and the maximum water uptake was ∼44.9 wt%. The overall water isotherm exhibited a sigmoidal shape and a relatively strong hysteresis loop (at %P/P0 = 20–80) was observed with marked stepped profiles in the desorption phase (Fig. 1, open circles).
 | ||
| Fig. 1 Water adsorption isotherm at 30 °C of NOTT-400. Solid circles represent adsorption, and open circles show desorption. | ||
The pore openings in NOTT-400 (approximately 8.1 Å)24a are considerably much larger than the kinetic diameter of water (∼2.7 Å). Thus, in order to interpret this hysteresis loop, the arguments of ‘kinetic trap’, suggested by other research groups,25 seem not to be suitable. Instead, the observed hysteresis is most likely due to moderately strong host–guest interactions, which result in an enhanced affinity of NOTT-400 for water. We believe that such interactions arise from the fact that water molecules can form hydrogen bonds with the bridging hydroxo functional groups (μ2-OH), within the pores, as previously described.26
In addition, according to the crystal structure of NOTT-40024a there are 8 hydroxo groups (μ2-OH) per unit cell. Thus, at low water loadings these groups can interact with water molecules representing the first domain (from 0 to approximately 32% P/P0) of the water adsorption isotherm. The second domain, after the full coverage of the hydroxo functional groups with H2O molecules, occurs at higher loadings. Thus, the slope of the isotherm grew abruptly due to stronger intermolecular interactions between water molecules affording the condensation of water into the pores (Fig. S5, see ESI‡).
In order to measure the isosteric heat of adsorption for H2O, we recorded a water adsorption isotherm at 20 °C (see Fig. S9, ESI‡) and the enthalpy for H2O adsorption was then calculated by fitting both isotherms (30 and 20 °C) to a Clausius–Clapeyron equation (see ESI‡). The estimated enthalpy value was 46.8 kJ mol−1 which is a typical value for PCPs.21d
To test the water adsorption–desorption recyclability of NOTT-400 water sorption isotherms were measured, on the same sample, for 5 cycles at 30 °C (Fig. 2). These results showed no apparent decrease in capacity over five cycles and revealed the complete regeneration of the material solely by evacuating for only 30 min without any thermal activation. In order to investigate any sample degradation, we have carried out PXRD experiments on the NOTT-400 sample after these cycling experiments. Fig. S4 (see ESI‡) confirms that the crystallinity of the sample after each water adsorption–desorption experiment (5 cycles) was retained.
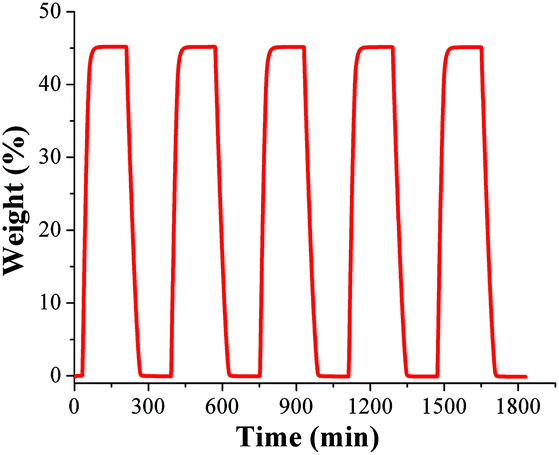 | ||
| Fig. 2 Water readsorption–desorption uptake on NOTT-400 at 30 °C. | ||
Dynamic and isothermal CO2 experiments were performed on NOTT-400 (see the Experimental section). In Fig. 3, a kinetic uptake experiment at 30 °C is shown. At this ambient temperature, the maximum amount of CO2 captured under anhydrous conditions is 4.4 wt%, rapidly reached after only 7 min and it was constant until the end of the experiment at 60 min. In addition, we run CO2 sorption experiments (adsorption/desorption) under static and isothermal conditions on NOTT-400 (see Fig. S10, ESI‡). The CO2 capture was 18.2 wt% which is significantly higher than under dynamic conditions (4.4 wt%). However, the central target of this paper is to exhibit, in a more realistic scenario, how NOTT-400 performs when it is exposed to a constant CO2 flow gas (60 mL min−1) and under water conditions.
 | ||
| Fig. 3 Kinetic uptake experiment performed at 30 °C with a CO2 flow of 60 mL min−1. | ||
Kinetic isotherm experiments at 30 °C and different relative humidities (10, 20, 35 and 60% RH) were carried out. We decided on these RH values based on the water adsorption isotherm (see Fig. 1): two values in the low loading region (10 and 20% RH); one value in the middle of the sigmoidal-shaped isotherm (35% RH) and finally one value in the high loading region (60% RH). First, an activated NOTT-400 sample (180 °C for 1 h and under a flow of N2 gas) was stabilised at 30 °C and 10% RH. After the equilibrium was reached a constant CO2 flow (60 mL min−1) was carried out (see Fig. 4, left).
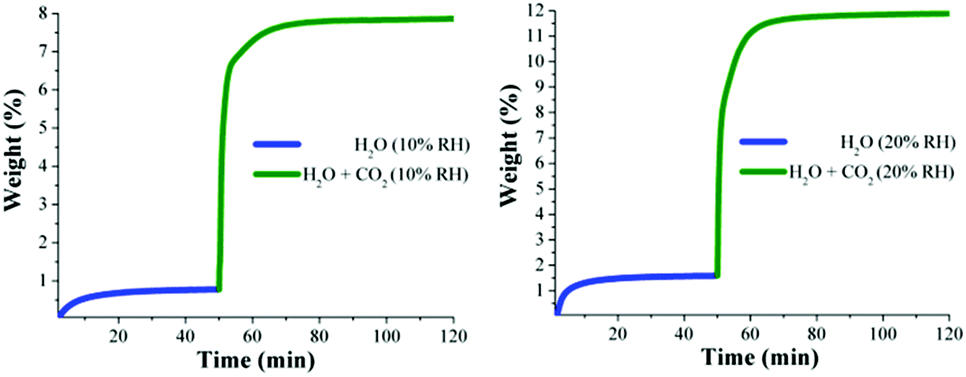 | ||
| Fig. 4 Kinetic uptake experiments carried out at 10 and 20% RH (both experiments at 30 °C); H2O (blue line) and H2O + CO2 (green line). | ||
In Fig. 4 (left) the gradual weight increase (only H2O) starts at 0 min and is stabilised at around 15 min. In contrast, under anhydrous conditions the CO2 uptake quickly reached stability (7 min, see Fig. 3). Since the diffusion coefficient of water is smaller than CO2, the vapour adsorption (water) process takes considerably more time to stabilise than the gas adsorption process in microporous materials.27 Then, from 15 min to 50 min the H2O uptake (0.7 wt%, which is in good agreement with the water adsorption isotherm; 0.67 wt%) was constant (plateau) and next, at 50 min the CO2 flow (60 mL min−1) was opened and a sharp weight gain, which reached stability at approximately 70 min, was observed (see Fig. 4, left). We hypothesise that the adsorbed amount of H2O is unchanged after the dosing of H2O/CO2 mixed gas, as we observed earlier.29 Then, from 70 min to 120 min (end of the experiment), the maximum amount of CO2 captured (considering the water uptake of 0.7 wt%) corresponded to 7.8 wt%. Therefore, the CO2 capture was approximately 2-fold increased with a 10% RH (from 4.2 wt% to 7.8 wt%) in comparison with anhydrous conditions.
Later, kinetic CO2 uptake experiments were performed on an activated sample of NOTT-400 at 30 °C and 20% RH. Fig. 4 (right) shows the gradual weight increase (H2O) starting at 0 min and stabilised at around 15 min. From 15 to 50 min the water uptake was constant and equal to 1.5 wt% (in good agreement with the water adsorption isotherm value of 1.47 wt%). Then, the CO2 flow was opened and abrupt uptake was observed (Fig. 4 right). After stabilisation (after 70 min) the total CO2 uptake was equal to 10.2 wt%. Thus, the CO2 capture was approximately 2.5-fold increased (from anhydrous conditions to 20% RH). This enhance in the CO2 uptake in the presence of water can be explained by CO2 confinement effects induced by water molecules.28
Similarly, more kinetic CO2 uptake isotherm experiments were carried out on the activated samples of NOTT-400 at 30 °C and 35% RH (see Fig. S6, ESI‡) and 30 °C and 60% RH (see Fig. S7, ESI‡). The stabilisation times were 40 and 55 min (see Fig. S6 and S7, ESI‡), respectively, and in both cases after the CO2 flow was started there was no weight increase, meaning that CO2 capture was not achieved at those relative humidities. At 35 and 60% RH, the water uptakes were 18.6 and 39.9 wt%, respectively. These values were in good agreement with the water adsorption isotherms (see Fig. 1), 18.72 and 39.88 wt%, respectively.
The porosity of NOTT-400 corresponds to the microporosity regime with a pore diameter of 8.1 Å.24a The remarkable result shown by Llewellyn and co-workers,22 increase in the CO2 uptake, was reported in a mesoporous material at 40% RH and 30 °C (MIL-100(Fe)).22 This mesoporous material includes two types of cages of free openings of ∼25 and 29 Å.22 We elucidated that at 35 and 60% RH the saturation of the micropores in NOTT-400, with H2O molecules, was complete and thus, the inclusion of CO2 molecules, into the micropores, was not possible. This explanation is supported by the experimental evidence that we previously reported29 in another microporous material.
Additionally, Paesani et al.30 investigated by computational infrared spectroscopy the behavior of water confined in a porous coordination polymer named MIL-53(Cr).31 Interestingly, MIL-53(Cr) is constructed with one-dimensional chains of corner-sharing CrO4(μ2-OH) octahedra, linked by 1,4-benzenedicarboxylate (BDC) ligands.31 Thus, they demonstrated30 that water molecules (at low water loadings) interact strongly with the framework, via hydrogen bonding between the μ2-OH functional group and H2O, whereas intermolecular interactions between H2O molecules become considerably stronger at higher loading. In other words, at low water loadings the MIL-53(Cr) channels provide a template for more efficiently packing the water molecules and therefore, these H2O molecules can then donate a hydrogen bond to the CO2 molecules enhancing the total CO2 uptake. These results correlate and support our experimental evidence.
Conclusions
In summary, the hydrostable Sc(III) coordination polymer, NOTT-400, has enabled an evaluation of how porous coordination polymers can be used in the sorption of water at ambient temperature. NOTT-400 carries out CO2 sequestration under relative humidity conditions. Finding the best partial saturation of H2O molecules (percentage of relative humidity) into the micropores of NOTT-400 is crucial to increase the CO2 uptake. Thus, after testing different relative humidity conditions (60, 35, 20 and 10% RH), we found that the maximum CO2 capture was obtained at 20% RH and 30 °C with a total amount of ∼10.2 wt%. Considerably, this CO2 capture, under humid conditions, represents a 2.5-fold increase in comparison with anhydrous conditions. The CO2 confinement effects induced by H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 can occur within the micropores of NOTT-400 enhancing the total CO2 uptake.
28 can occur within the micropores of NOTT-400 enhancing the total CO2 uptake.
Experimental section
Water adsorption experiments
Water vapour isotherms were recorded by a dynamic method, using air as a carrier gas, in a DVS Advantage 1 instrument from a surface measurement system (mass sensitivity: 0.1 μg, Relative Humidity (RH), accuracy: 0.5% RH, vapour pressure accuracy: 0.7% P/P0). The hydration–dehydration cycles were measured in the same instrument. The water uptake in weight percent (wt%) units was calculated as [(adsorbed amount of water)/(amount of adsorbent) × 100], consistent with the established procedures.CO2 capture experiments
Kinetic uptake experiments were performed by using a thermobalance (Q500 HR, from TA) at room temperature (30 °C) with a constant CO2 flow (60 mL min−1). Then, samples of NOTT-400 were placed inside the thermobalance and activated by heating from room temperature to 180 °C for 1 h under a flow of N2 gas. After the activated sample was cooled down, the desired temperature was set (30 °C) and a constant CO2 flow (60 mL min−1) was carried out. With a humidity-controlled thermobalance (Q5000 SA, from TA) kinetic uptake experiments at 30 °C with a constant CO2 flow (60 mL min−1) were performed on the activated samples (180 °C for 1 h under a flow of N2 gas) of NOTT-400.Acknowledgements
The authors thank Dr A. Tejeda-Cruz (X-ray; IIM-UNAM), CONACyT Mexico (212318), and PAPIIT UNAM Mexico (IN100415) for financial support. J. B. thanks SEP-CONACyT (154626) and UNAM-DGAPA-PAPIIT (IG-100315). E.G-Z. thanks CONACyT (156801 and 236879), Mexico for financial support. Thanks to U. Winnberg (ITAM and ITESM) for scientific discussions.Notes and references
- (a) S. Yang, G. S. B. Martin, G. J. J. Titman, A. J. Blake, D. R. Allan, N. R. Champness and M. Schröder, Inorg. Chem., 2011, 50, 9374–9384 CrossRef CAS PubMed; (b) A. J. Nuñez, L. N. Shear, N. Dahal, I. A. Ibarra, J. W. Yoon, Y. K. Hwang, J.-S. Chang and S. M. Humphrey, Chem. Commun., 2011, 47, 11855–11857 RSC; (c) I. A. Ibarra, K. E. Tan, K. V. M. Lynch and S. M. Humphrey, Dalton Trans., 2012, 41, 3920–3923 RSC.
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834–15842 CrossRef CAS PubMed.
- J. Cavinet, A. Feteeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC.
- A. M. Ribeiro, T. P. Sauer, C. A. Grande, R. F. P. M. Moreira, J. M. Loureiro and A. E. Rodriguez, Ind. Eng. Chem., 2008, 47, 7019–7026 CrossRef CAS.
- (a) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724–781 CrossRef PubMed; (b) R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- H. A. Lara-García, M. R. Gonzalez, J. H. González-Estefan, P. Sánchez-Camacho, E. Lima and I. A. Ibarra, Inorg. Chem. Front., 2015, 2, 442–447 RSC.
- K. A. Cychosz and A. J. Matzger, Langmuir, 2010, 26, 17198–17202 CrossRef CAS PubMed.
- D.-Y. Hong, Y. K. Hwang, C. Serre, G. Férey and J.-S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS.
- J. Liu, F. Zhang, X. Zou, G. Yu, N. Zhao, S. Fan and G. Zhu, Chem. Commun., 2013, 49, 7430–7432 RSC.
- J. Qian, F. Jiang, D. Yuan, M. Wu, S. Zhang, L. Zhang and M. Hong, Chem. Commun., 2012, 48, 9696–9698 RSC.
- W. M. Bloch, R. Babaro, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441–10448 CrossRef CAS PubMed.
- P. M. Schoenecker, C. G. Carson, H. Jasuja, C. J. J. Flemming and K. S. Walton, Ind. Eng. Chem. Res., 2012, 51, 6513–6519 CrossRef CAS.
- J. Cavinet, J. Bonnefoy, C. Daniel, A. Legrand, B. Coasne and D. Farrusseng, New J. Chem., 2014, 38, 31012–33111 Search PubMed.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- (a) F. Meunier, Appl. Therm. Eng., 2013, 61, 830–836 CrossRef; (b) C. Janiak and S. K. Henninger, Chimia, 2013, 67, 419–424 CrossRef CAS PubMed.
- M. Sadakiyo, H. Ōkawa, A. Shigematsu, M. Ohba, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 5472–5475 CrossRef CAS PubMed.
- (a) S.-i. Noro, R. Matsuda, Y. Hijikata, Y. Inubushi, S. Takeda, S. Kitagawa, Y. Takahashi, M. Yoshitake, K. Kubo and T. Nakamura, ChemPlusChem, 2015, 80, 1517–1524 CrossRef CAS; (b) D. Kim, Y.-H. Ahn and H. Lee, J. Chem. Eng. Data, 2015, 60, 2178–2186 CrossRef CAS; (c) J. A. Mason, T. M. McDonald, T.-H. Bae, J. E. Bachman, K. Sumida, J. J. Dutton, S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2015, 137, 4787–4803 CrossRef CAS PubMed.
- (a) J. Liu, A. I. Benin, A. M. B. Furtado, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2011, 27, 11451–11456 CrossRef CAS PubMed; (b) J. Liu, Y. Wang, A. I. Benin, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2010, 26, 14301–14307 CrossRef CAS PubMed.
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS PubMed.
- (a) H. Jasuja, Y.-G. Huang and K. S. Walton, Langmuir, 2012, 28, 16874–16880 CrossRef CAS; (b) H. Jasuja, J. Zang, D. S. Sholl and K. S. Walton, J. Phys. Chem. C, 2012, 116, 23526–23532 CrossRef CAS; (c) J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y.-G. Huang and K. S. Walton, J. Mater. Chem. A, 2013, 1, 5642–5650 RSC; (d) N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- E. Soubeyrand-Lenoir, C. Vagner, J. W. Yoon, P. Bazin, F. Ragon, Y. K. Hwang, C. Serre, J.-S. Chang and P. L. Llewellyn, J. Am. Chem. Soc., 2012, 134, 10174–10181 CrossRef CAS PubMed.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15606–15613 CrossRef CAS PubMed.
- (a) I. A. Ibarra, S. Yang, X. Lin, A. J. Blake, P. J. Rizkallan, H. Nowell, D. R. Allan, N. R. Champness, P. Hubberstey and M. Schröder, Chem. Commun., 2011, 47, 8304–8306 RSC; (b) M. R. Gonzalez, J. H. González-Estefan, H. A. Lara-García, P. Sánchez-Camacho, E. I. Basaldella, H. Pfeiffer and I. A. Ibarra, New J. Chem., 2015, 39, 2400–2403 RSC.
- (a) X. B. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012–1015 CrossRef CAS PubMed; (b) H. J. Choi, M. Dincă and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848–7850 CrossRef CAS PubMed.
- V. Haigis, F.-X. Coudert, R. Vuilleumier and A. Boutin, Phys. Chem. Chem. Phys., 2013, 15, 19049–19056 RSC.
- I. P. O'koye, M. Benham and K. M. Thomas, Langmuir, 1997, 13, 4054–4059 CrossRef.
- (a) N. L. Ho, F. Porcheron and R. J.-M. Pellenq, Langmuir, 2010, 26, 13287–13296 CrossRef CAS PubMed; (b) L. N. Ho, J. Perez-Pellitero, F. Porcheron and R. J.-M. Pellenq, Langmuir, 2011, 27, 8187–8197 CrossRef CAS PubMed; (c) L. N. Ho, S. Clauzier, Y. Schuurman, D. Farrusseng and B. Coasne, J. Phys. Chem. Lett., 2013, 4, 2274–2278 CrossRef CAS.
- R. A. Peralta, B. Alcántar-Vázquez, M. Sánchez-Serratos, E. González-Zamora and I. A. Ibarra, Inorg. Chem. Front., 2015, 2, 898–903 RSC.
- G. R. Medders and F. Paesani, J. Phys. Chem. Lett., 2014, 5, 2897–2902 CrossRef CAS PubMed.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louer and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Pedro Bosch who contributed with 38 years of brilliant research. |
| ‡ Electronic supplementary information (ESI) available: TGA data, PXRD data and kinetic CO2 experiments. See DOI: 10.1039/c5qi00176e |
| This journal is © the Partner Organisations 2015 |